

Abstract
Activating BRAF mutants and fusions signal as RAS-independent constitutively active dimers with the exception of BRAF V600 mutant alleles which can function as active monomers1. Current RAF inhibitors are monomer selective, they potently inhibit BRAF V600 monomers but their inhibition of RAF dimers is limited by induction of negative cooperativity when bound to one site in the dimer1–3. Moreover, acquired resistance to these drugs is usually due to molecular lesions that cause V600 mutants to dimerize4–8. We show here that PLX8394, a new RAF inhibitor9, inhibits ERK signaling by specifically disrupting BRAF-containing dimers, including BRAF homodimers and BRAF-CRAF heterodimers, but not CRAF homodimers or ARAF-containing dimers. Differences in the amino acid residues in the N-terminal portion of the kinase domain of RAF isoforms are responsible for this differential vulnerability. As a BRAF-specific dimer breaker, PLX8394 selectively inhibits ERK signaling in tumors driven by dimeric BRAF mutants, including BRAF fusions and splice variants and as well BRAF V600 monomers, but spares RAF function in normal cells in which CRAF homodimers can drive signaling. Our work suggests that drugs with these properties will be safe and useful for treating tumors driven by activating BRAF mutants or fusions.
Main
PLX8394 is a new RAF inhibitor that inhibits ERK signaling in tumors driven by BRAF V600 mutants and also in some models driven by dimer-dependent BRAF mutants or fusions1,9–13. However, the mechanisms underlying these properties are unclear. We studied PLX8394 and 6 other RAF inhibitors in cells in which ERK signaling is driven by different mechanisms: receptor tyrosine kinase (RTK) activation of WT RAS/RAF (primary keratinocytes), NRAS Q61R activation of WT RAF dimers (SK-MEL-2), BRAF V600E monomers (SK-MEL-239), and p61 BRAF V600E homodimers (SK-MEL-239 C4). RAF inhibitors used included PLX8394, Group 1 drugs that selectively inhibit BRAF monomers (vemurafenib, dabrafenib and encorafenib (LGX818)), and Group 2 drugs (BGB659, TAK632 and LY3009120), recently described1 inhibitors of RAF dimers that are unaffected by negative cooperativity (Supplementary Fig. 1a). They inhibit mutant RAF dimers and monomers at similar doses in tumors1,14,15.
The cell lines were exposed to increasing concentrations of each of the drugs for 1hr (Fig. 1 and Supplementary Fig. 1b). BRAF V600E monomer-driven ERK phosphorylation (p-ERK) (SK-MEL-239) was potently inhibited by group 1 drugs; but 10–60 fold higher concentrations were required to inhibit BRAF V600E dimer-driven p-ERK (SK-MEL-239 C4) (IC75 comparison). Group 1 drugs caused significant ERK activation (>200%) in WT RAS/RAF cells (Keratinocytes) and mutant NRAS cells (SK-MEL-2) at concentrations that inhibit ERK in BRAF V600E cells (SK-MEL-239). Approximately 300–1,000-fold higher concentrations were required for 50% inhibition of ERK activation in these cells (SK-MEL-2 and Keratinocytes) (Supplementary Table 1). Consistent with these data, these compounds potently inhibit the growth of BRAF V600E SK-MEL-239 cells, but 10–100 fold higher concentrations were required to inhibit mutant (SK-MEL-239 C4) or wild type (SK-MEL-2 and Keratinocytes) dimer-dependent cells (Supplementary Fig.1b). In contrast, Group 2 drugs inhibited p-ERK in SK-MEL-239 and SK-MEL-239 C4 cell, whereas, their inhibition of ERK signaling and cell proliferation driven by WT RAF requires about 10 fold-higher concentrations (Fig. 1, Supplementary Table 1 and Supplementary Fig. 1b). They only weakly activate ERK signaling in keratinocytes and SK-MEL2 cells at low concentrations. Thus, Group 1 drugs are expected to effectively inhibit BRAF V600 monomer-driven signaling and tumor growth with a wide therapeutic index and have little utility in tumors driven by mutant BRAF dimers. These predictions have been born out in clinic. In contrast, Group 2 drugs (not yet tested clinically) should effectively inhibit both BRAF monomers and dimers, but may have a narrower therapeutic index.
Figure 1. Effects of RAF inhibitors on pERK in cell lines with wild type or mutant RAS and BRAF.
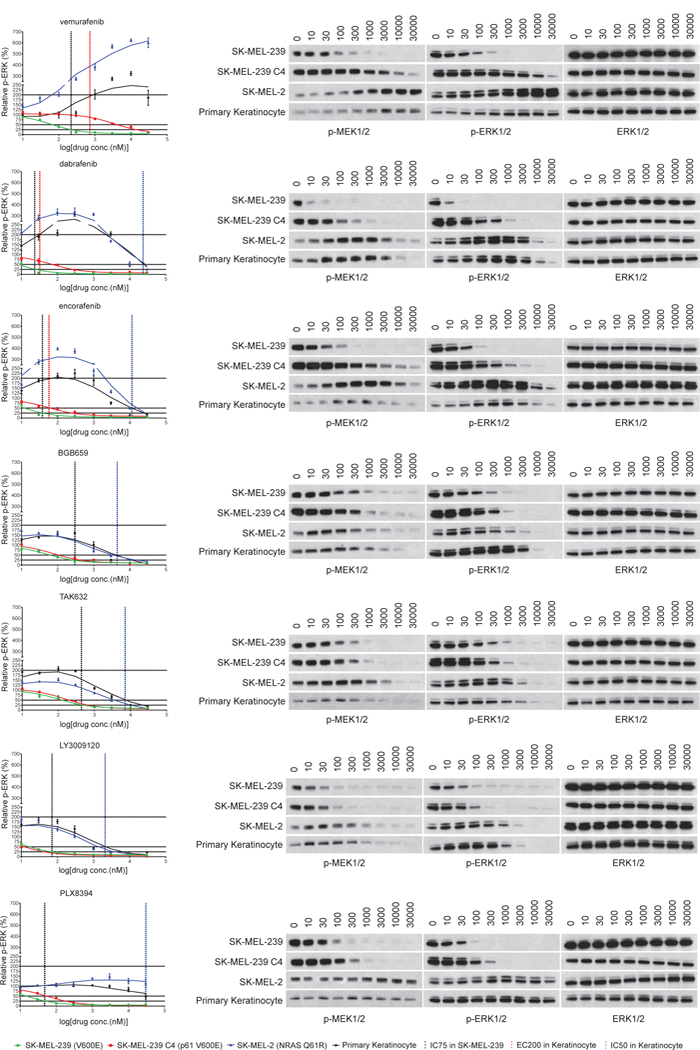
The indicated cell lines with wild type or mutant RAS/BRAF were treated with 0,10,30,100, 300, 1000, 3000, 10,000 and 30,000nM concentrations of different RAF inhibitors for 1 hr. Whole cell lysates were collected. The ERK pathway was assayed by Western blot with indicated antibodies. The p-ERK/total ERK level in each sample was quantified by densitometry and then normalized to the p-ERK/total ERK level in untreated cells. The p-ERK response curves were generated using Prism6. One time Western blot results are shown on the right panel. The experiments results were repeated 4 times, independently. The dots and error bars in the curves represent the mean+s.d. values (n=4). In the graphs, the black color vertical lines indicate the concentrations of each drug required to inhibit 75% p-ERK/total ERK in SK-MEL-239 cells; the red color vertical lines indicate the concentrations of each drug to cause 200% induction of p-ERK/total ERK in Keratinocytes; the blue color vertical lines indicate the concentrations of each drug required to inhibit 50% p-ERK/total ERK in Keratinocytes. The functional concentrations of each compound defined in the graphs are listed in Supplementary Table 1. For gel source data, see Supplementary Figure 8.
The pattern of responses elicited by PLX8394 was different than that of either group. PLX8394 inhibited p-ERK in SK-MEL-239 and SK-MEL-239 C4 cells with IC75s of 39 and 158 nM, respectively, and had almost no effect in keratinocytes or SK-MEL-2 cells (IC50s for p-ERK and cell growth are >20uM) (Fig.1, Supplementary Fig. 1b and Supplementary Table 1). Thus, it inhibits mutant BRAF dimers at 5 times the concentration required to inhibit monomers; but, uniquely, it neither activates nor effectively inhibits WT RAF signaling. This profile suggests that, unlike current inhibitors, PLX8394 could effectively treat tumors driven by mutant BRAF monomers or dimers at concentrations unlikely to cause ERK-dependent toxicity.
Inhibition of RAF dimers by Group 1 inhibitiors is limited by induction of negative cooperativiy of binding to the second site of the dimer after binding to the first site1. We used this property to identify compounds (Group 2) that bind to both sites at similar concentrations. To do this we utilized encorafenib, a drug that binds to the BRAF V600E monomer and to first site of RAF dimers with similar IC50s of 14nM and to the second site with an IC50 of 287 nM (a 20 fold difference). The off-rate of encorafenib from BRAF V600E monomers is quite long (>24hrs)1. When cells with activated WT or mutant RAF dimers are pretreated with high concentrations of encorafenib for 1 hour and the drug is then washed out, the compound dissociates from one binding site with a short half-life (<20 min) but remains bound to the other site for up to 24 hours. In cells with WT RAF, the half-bound WT RAF dimers are hyperactivated, whereas the activity of the half-bound mutant BRAF-dimer approximates that of unbound dimers1. We have used cells with encorafenib half-bound dimers (obtained after washout of drug at 1hr) to determine the concentrations at which other drugs inhibit the second site, and used this technique to identify dimer inhibitors1. Here, we used this strategy again, to determine the binding of PLX8394 to the second site of the mutant BRAF dimer when the other site is occupied. SK-MEL-239 and SK-MEL-239 C4 were pretreated with 3uM encorafenib for 1hr and then the drug was washed out. pMEK/pERK remained inhibited in BRAF V600E monomer expressing SK-MEL-239 cells but was restored to basal level in SK-MEL-239 C4 cells because of dissociation of drug from the second site of the p61 dimers1 (Supplementary Fig. 2a). Vemurafenib did not potently inhibit these dimes because it is subject to negative cooperativity. In contrast, BGB659 is capable of inhibiting dimers and the second site because it is not subject to negative cooperativity. PLX8394 fits neither pattern. It inhibits p61 dimers at 100~300nM, but is unable to inhibit the second site when the first is occupied, even at concentrations much higher than those that inhibit p61 dimer-driven ERK signaling (Supplementary Fig. 2b). This suggests that, PLX8394 inhibits mutant BRAF dimer-driven signaling despite being subject to negative cooperativity. Thus, its inhibition of dimers does not require its binding to both sites.
We asked whether PLX8394 affects levels of RAF dimers. Different RAS-dependent or -independent RAF dimers were expressed in 293H (NRAS Q61K) cells. PLX8394 markedly decreased the levels of RAS-dependent full length BRAF/BRAF or BRAF/CRAF dimers and RAS-independent BRAF dimers (p61 BRAF) with IC50s of 100–300nM (Fig. 2a, Supplementary Fig. 2c). By contrast, it had no effect on RAS-dependent full length CRAF dimers or RAS-independent truncated CRAF (Cat C) homodimers at concentrations as high as 10uM (Fig. 2a). It also didn’t disrupt RAS-dependent ARAF homo-or heterodimers (Supplementary Fig. 2d). Reduction in BRAF-containing dimers occurred within 15 minutes after drug treatment, and was not associated with decreased expression of BRAF or CRAF proteins (Supplementary Fig. 2e). None of the other inhibitors tested here disrupted dimers (Supplementary Fig. 2f). Thus, PLX8394 selectively disrupts BRAF/BRAF and BRAF/CRAF dimers and its inhibition of ERK signaling in SK-MEL-239 C4 is likely due to disruption of p61 BRAF V600E dimers.
Figure 2. PLX8394 selectively disrupts BRAF homodimers and BRAF/CRAF heterodimers by binding to only one protomer in the dimers.
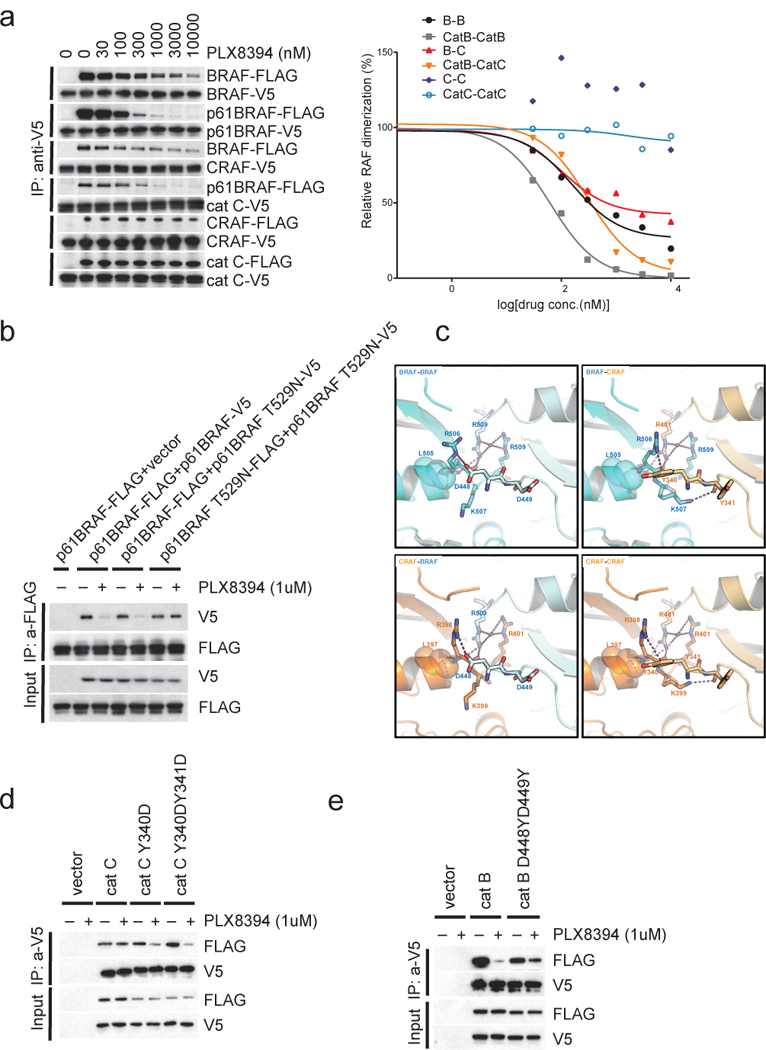
(a) Immunoprecipitation (IP) assays were performed with anti-V5 agarose on cell lysates from 293H (NRAS Q61K) cells that ectopically co-express BRAF-V5 and BRAF-FLAG, p61 BRAF-V5 and p61 BRAF-FLAG, CRAF-V5 and BRAF-FLAG, catC-V5 and p61 BRAF-FLAG, CRAF-V5 and CRAF-FLAG or catC-V5 and catC-FLAG. Prior to the IP experiment, cells were treated with increasing doses of PLX8394 for 1hr as indicated. The immunoprecipitated proteins were assayed by Western blot with anti-V5 and anti-FLAG antibody (left panel). The interaction between FLAG and V5 tagged proteins were represented by the levels of FLAG tagged and V5 tagged proteins pulled down, quantified by densitometry and normalized to levels in the untreated cells. The curves of protein interactions in response to the drug treatments were generated using Prism6 (right panel). The dots represent the relative FLAG tagged protein and V5 tagged protein interaction levels as indicated. The experiments were repeated 3 times, independently. (b) The indicated pairs of FLAG- and V5-tagged proteins were ectopically co-expressed in 293H cells for 24hrs. Then the cells were treated with 1μM PLX8394 for 1hr. IPs were then performed on lysates from these cells with anti-FLAG agarose. The Input and IP samples were assayed by Western blot with anti-FLAG/V5 antibodies as indicated. The experiments were repeated 4 times, independently. (c) Dimer interfaces highlighting the key structural differences between BRAF and CRAF. If PLX8394 binds to the promoter on the left (Protomer 1), its interaction with L505 (BRAF) or L397 (CRAF) is expected to push the C-terminal end of αC helix out, thereby perturbing the interactions formed by the two basic residues (R506 and K507 in BRAF and R398 and K399 in CRAF). Whether this structural alteration results in dimer disruption is determined by the partner protomer (Protomer 2). If Protomer 2 is CRAF, Y340 forms a strong cation-π interaction with R506 (BRAF) or R398 (CRAF). The interface is further stabilized by the interaction between the backbone of Y341 and K507 (BRAF) or K399 (CRAF). The energy barrier to overcome these concerted set of interactions is high. However, if Protomer 2 is BRAF, D448 forms a salt bridge with R506 (BRAF) or R398 (CRAF). The backbone of the D449 is in a conformation not conducive to interaction with K507 (BRAF) or K399 (CRAF), and the energy barrier to break the dimer is lower. (d) and (e) The indicated FLAG- and V5-tagged proteins were ectopically expressed in 293H cells for 24hrs. Then the cells were treated with 1μM PLX8394 for 1hr. The cells were lysed and the lystates were subjected to IP with anti-V5 agarose. The Input and IP samples were assayed by Western blot with anti-FLAG/V5 antibodies. These experiments were all repeated 3 times, independently. For gel source data, see Supplementary Figure 9.
To determine whether binding of PLX8394 to one site in the dimer is sufficient to cause its disruption, we introduced the T529N gatekeeper mutation into one of the tagged protomers of p61 BRAF dimers. We found that disruption of p61 BRAF homodimers was unaffected if only one protomer could bind drug, but prevented when both protomers contained T529N (Fig. 2b). This was the case for disruption of RAS-driven WT BRAF homodimers as well (Supplementary Fig. 3a, Lane 1–6). In contrast, whereas the T529N BRAF gatekeeper mutation did not prevent the disruption of RAS-driven BRAF/CRAF heterodimers by PLX8394, the T421M CRAF mutation did (Supplementary Fig. 3a, Lane 7–14). Thus, disruption of BRAF homodimers by PLX8394 requires drug binding to either of the protomers, but disruption of BRAF/CRAF heterodimers by PLX8394 specifically requires binding to CRAF.
PLX8394 selectively disrupts BRAF-containing dimers without affecting CRAF homodimers. The published BRAF and CRAF structures were analyzed to identify residues that make extensive contacts across the dimer interface (Supplementary Table 2). Major differences between the BRAF-BRAF and CRAF-CRAF dimer interfaces are centered on the two residues situated at the N-terminus of the kinase domain, D448/D449 in BRAF and Y340/Y341 in CRAF and, more specifically, the interactions they form with the two basic residues near the C-terminus of the αC-helix (R506/K507 in BRAF and R398/K399 in CRAF). As suggested previously9, PLX8394 interacts with L505 on the αC-helix of BRAF (and by inference, L397 in CRAF). This interaction is expected to push the C-terminal end of the αC-helix out, thereby perturbing the cross-dimer interactions associated with the two basic residues. However, the structural change in the αC-helix of the inhibitor-bound protomer (Protomer 1) is likely to have a different effect on the integrity of the dimer depending on the inhibitor-free partner (Protomer 2) (Fig. 2c). When Protomer 2 is BRAF, D448 from Protomer 2 forms a salt bridge with R506 (BRAF) or R398 (CRAF) from Protomer 1. This is replaced with a strong cation-π interaction when the Protomer is CRAF and the aspartate is replaced by tyrosine 340 (Fig. 2c). This interface is further stabilized by the interaction between the backbone of Y341 (CRAF) from Protomer 2 and K507 (BRAF) or K399 (CRAF) from Protomer 1. We predicted that the energy barrier to break the dimer is significantly higher when the inhibitor-free Promoter (Protomer 2) is CRAF because of the additional dimer-stabilizing interactions. This would explain the reduced sensitivity of CRAF-CRAF dimers to disruption by PLX8394 as well as the asymmetric effect on BRAF-CRAF heterodimers depending on whether PLX8394 binds to BRAF or CRAF. In support of this idea, introduction of Y340D and Y341D into CRAF to create a BRAF-like dimer interface sensitizes the mutant CRAF homodimers to disruption by the drug (Fig. 2d). In contrast, D448Y and D449Y substitutions in BRAF reduce the sensitivity of BRAF homodimers to disruption (Fig. 2e). Y340D/Y341D enhanced activation of truncated CRAF dimers (RAS binding domain deleted) and Y340A/Y341A decreased their activity (Supplementary Fig. 3b). However, D448Y/D449Y in BRAF has almost no effect on the activation of truncated BRAF dimers (RAS binding domain deleted) (Supplementary Fig. 3c). These observations are consistent with previous studies that suggested the phosphorylation of Y340 and Y341 in CRAF regulate CRAF activation16–19. However, we could not detect the phosphorylation of CRAF at Y340 and Y341 with commercially available antibodies or mass spectrometry in homodimers isolated from cells. We have confirmed that neither the RAS-dependent full length nor the RAS-independent truncated CRAF homodimer are disrupted by PLX8394 (Fig. 2a). It is also difficult to infer the features of the ARAF dimer interface given the absence of detailed structural information. In ARAF, the sites analogous to CRAF Y340/Y341 are also tyrosines (Y301/Y302), so it is possible that the insensitivities of ARAF and CRAF dimers to drug have similar explanations.
Thus, unlike group 2 inhibitors, PLX8394 inhibits BRAF dimers by directly disrupting dimerization rather than binding to both protomers. The doses of PLX8394 required to inhibit p61 BRAF V600E dimers (100–300nM) or p61 WT BRAF dimers (300–1000nM) are higher than that required to inhibit BRAF V600E monomers (30–100 nM); In contrast, PLX834 activated ERK signaling in cells with activated CRAF homodimers (Fig. 3a). Our model predicts that binding of PLX8394 to one protomer will not disrupt the CRAF dimer, but rather will induce negative cooperativity and transactivate the unbound protomer. We asked whether replacement of the cation-π interaction in CRAF dimers with the BRAF salt bridge interaction would sensitize the former to the drug. This turned out to be the case, the Y340D mutation or Y340D/Y341D double mutation sensitized CRAF homodimers (cat C) to inhibition by PLX8394, supporting the proposed mechanism (Fig. 3b). This observation is consistent with the observation that Y340D or Y340D/Y341D mutations allow the drug to break the CRAF homodimers (Fig. 2d).
Figure 3. PLX8394 inhibits ERK signaling driven by BRAF monomers or dimers, but activates ERK signaling driven by CRAF homodimers.
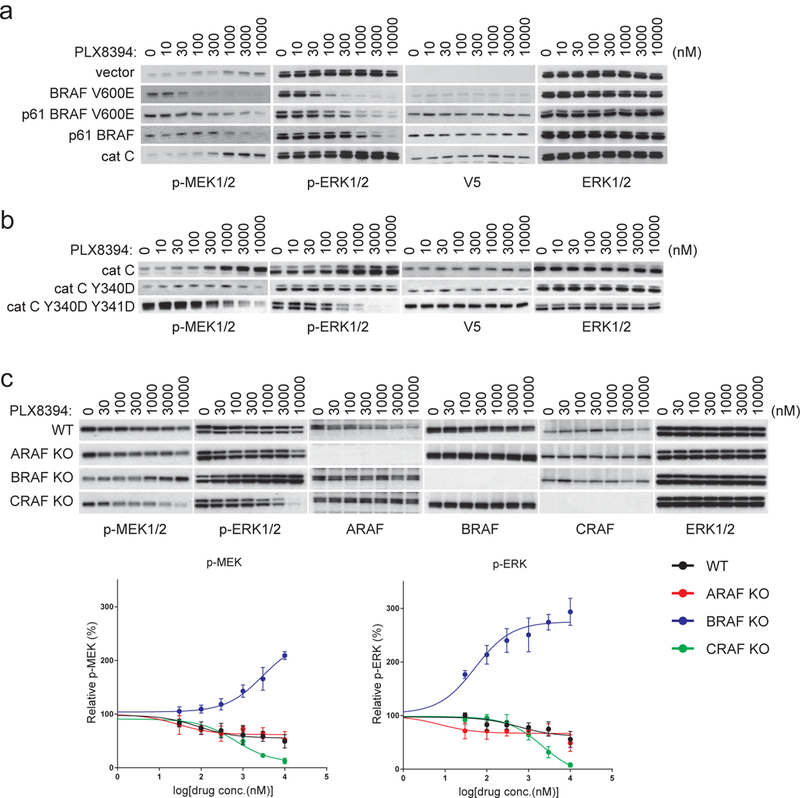
(a) SKBR3 cells were transfected with pcDNA3 vector plasmid or plasmids encoding the indicated RAF proteins. After 24 hrs, the vector transfected cells were treated with DMSO vehicle; other cells were treated with 1uM lapatinib (to suppress endogenous RAF activity1). Then all of the cells were treated with PLX8394 at increasing doses as indicated for 1hr. Expression and/or phosphorylation of the indicated proteins were assayed by Western blot. The experiments were repeated for 3 times, independently. (b) The SKBR3 cells ectopically expressing the indicated proteins were treated and assayed as described in (a). The experiments results were repeated for 4 times, independently. (c) Control wild type Mouse Embryonic Fibroblast (MEF) cells and ARAF, BRAF or CRAF knockout MEF cells were treated with increasing doses of PLX8394 for 1hr. Expression and/or phosphorylation of the indicated proteins were assessed by Western blot. The p-MEK/MEK and p-ERK/ ERK level in each sample were quantified by densitometry and then normalized to the levels in untreated cells. The response curves shown here were generated using Prism6. The dots and error bars in the curves represent the mean+s.d. (n=3). The immunoblotting data shown here are from one time experiment, but the graph represented the results from three replicates. For gel source data, see Supplementary Figure 10.
The selectivity of PLX8394 suggests that its effects in cells will depend on the relative abundance of the different RAF dimers. It will inhibit ERK signaling in cells in which activated BRAF-BRAF or BRAF-CRAF dimers are in excess, whereas it will activate the signaling, when CRAF homodimers and/or ARAF containing dimers predominate. In cells with WT RAS and RAF, levels of each type of dimers will depend on expression levels of the RAF isoforms. In HeLa cells (WT RAS and RAF), we engineered to express increasing amounts of BRAF or CRAF. BRAF overexpression sensitized ERK signaling to inhibition by PLX8394, whereas the drug activated the pathway in cells with CRAF overexpression (Supplementary Fig. 4a). Similarly, in MEFs in which BRAF was knocked out, the drug induced ERK signaling, whereas it inhibited signaling in CRAF-knockout cells; ARAF knockout had minimal effects (Fig. 3c). We generated MEFs in which ERK signaling was driven by specific RAF homodimers by expressing A-, B-, or CRAF in RAF-null MEFs. Vemurafenib activated ERK signaling in all three (Supplementary Fig. 4b). In contrast, ERK signaling was inhibited by PLX8394 in cells expressing only BRAF; and activated in those expressing CRAF or ARAF (Supplementary Fig. 4c).
We thus predict the effects of PLX8394 in tumor cells as a function of genotype and levels of expression of RAF isoforms. In cells with WT RAF, ERK signaling is driven by RAS-induced homo– and heterodimerization of the three RAF isoforms. When all are expressed, PLX8394 disrupts BRAF/BRAF and BRAF/CRAF dimers; but transactivates CRAF homodimers and ARAF dimers. In these cells, the opposing effects of PLX8394 on different dimers tend to prevent significant inhibition or paradoxical activation. Tumors in which ERK signaling is driven by RTKs or mutant RAS are likely to have significant levels of CRAF homodimers and are thus unlikely to respond to this drug. On the other hand, the drug is unlikely to cause toxicity in normal tissue for the same reason.
In contrast, activating BRAF mutants cause ERK-dependent feedback inhibition of RAS activity and therefore, of WT RAF dimers. In these tumors, ERK signaling is dominated by mutant BRAF monomers (Class1 mutants) or homodimers (Class 2 mutants and fusions)1,20. ERK activation and the growth of these tumor cells should be sensitive to a BRAF dimer disrupter such as PLX8394 that also inhibits active BRAF monomers. The situation is more complex in tumors with Class 3 BRAF mutants. In these tumors, RAS activation is required for ERK pathway activation. Although the majority of RAF activity in these cells comes from mutant BRAF/WT CRAF dimers20–22, activated CRAF homodimers are also present. Thus ERK activation in these tumors is likely to be less sensitive to PLX8394 and vary as a function of expression levels of the mutant.
To test these hypotheses, we examined the effects of PLX8394 on tumor cells with different BRAF mutant dimers. It disrupts both Class 2 BRAF mutant homodimers and Class 3 BRAF mutant heterodimers (Supplementary Fig. 5a). ERK signaling driven by Class 1 (BRAF V600 mutants) monomers (A375, SK-MEL-239 and MDST-8) was most sensitive (30–100nM); whereas inhibition in those expressing Class 2 BRAF mutants (SK-MEL-239 C4, SK-MEL-246, JVM-3, and 22RV1) or ectopically expressing a BRAF fusion (KIAA1549-BRAF), required higher concentrations (100–1000nM) (Supplementary Fig. 5b-c). Class 3 mutants driven signaling (H508 and H1666) was least sensitive, requiring 1–10uM. PLX8394 had almost no effect on ERK signaling or caused slight activation in cells with WT RAF driven by either WT or mutant RAS (A549, SK-MEL-285, BEAS-2B and melanocytes). The inhibition of cell growth of BRAF mutant cells occurred in dose ranges that inhibit ERK signaling (Fig. 4a). These finding are different from those observed with vemurafenib (group1 drug) and BGB659 (group2 drug) (Fig. 1, Fig. 4a and Supplementary Fig. 5b), and confirm our model.
Figure 4. PLX8394 preferentially inhibits ERK signaling and cell growth in tumors driven by RAS-independent BRAF mutants.
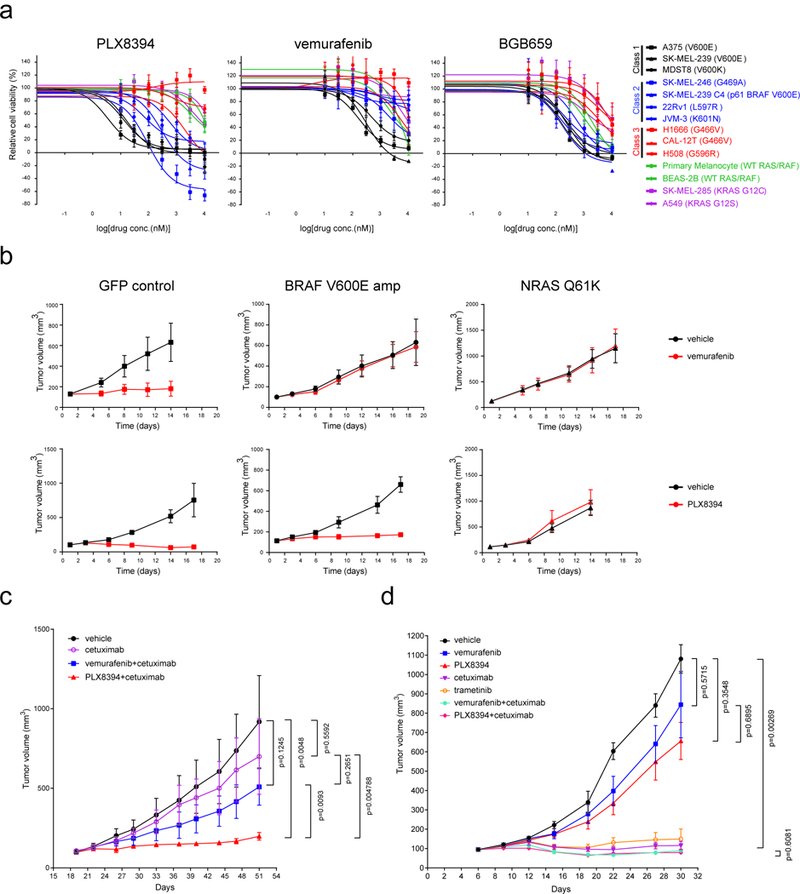
(a) A panel of cell lines (indicated in Supplementary Fig. 5b) was treated with the indicated RAF inhibitors for 3 days at 0, 10, 30, 100, 300, 1,000, 3,000 and 10,000 nM. Cell viability was determined by ATP-Glo assay. Dose dependent inhibition curves were generated using Prism6 (mean+s.d. are represented by the dots and error bars, n=8). (b) 5 million of A375 cells with doxycycline induced expression of GFP control, BRAF V600E or NRAS Q61K were injected s.c. into nude mice fed with doxycycline containing food to generate tumors. When tumors reached 100 mm3, the animals were fed with food containing doxycycline and treated with vehicle, vemurafenib (75mg/kg, BID) or PLX8394 (50mg/kg, BID) as indicated. Tumor sizes are represented by mean+s.d. in the graphs (n=5). (c) & (d) PDXs were established from tumor biopsy specimens from colorectal cancer patients. The tumor specimens and PDX tumor samples were sequenced with MSK-IMPACT. BRAF K601E mutant (c) and G466V (d) were detected as shown in Supplementary Table 3. No RAS or NF1 alterations were detected in these samples. Drug response was monitored by measurement of the tumor sizes. Vemurafenib was given at 75 mg/kg BID, PLX8394 50 mg/kg BID, and Cetuximab (50 mg/kg) IP twice weekly. The curves of tumor growth were generated using Garphpad Prism6. Tumor sizes are shown as mean+s.d. in the graphs (n=5, p-value was calculated by two-tailed unpaired t-test).
We examined the in vivo activity of PLX8394 in several BRAF mutant-driven tumor models. The drug was well tolerated in nude mice at doses up to 100mg/kg daily. In melanoma xenografts, the highest drug concentrations were achieved 7hrs after a single dose of 50mg/kg (Supplementary Fig. 6a). Maximum inhibition of signaling was observed 2–7hrs after dosing, returning to baseline by 24 hrs. We therefore used 50 mg/kg BID in subsequent studies. The drug was tested in models of dimer-dependent acquired resistance of BRAF V600E tumors to vemurafenib that we established previously1. In A375 cells, we induced overexpression of BRAF V600E (mimicking V600E amplification) or mutant NRAS with doxycycline. Both mechanisms cause vemurafenib resistance by inducing dimers. BRAF V600E overexpression drives BRAF V600E homodimers, whereas mutant NRAS drives all types of RAF dimers including CRAF homodimers. Consistent with our mechanism, both lesions caused resistance to vemurafenib, whereas ERK signaling and tumor growth were sensitive to PLX8394 in BRAF V600E amplified cells, but not in those with mutant NRAS (Fig. 4b and Supplementary Fig. 6b-c).
The effects of the drug were also tested in two colorectal cancer PDX models: one with a Class 2 BRAF mutants (K601E), another with a Class 3 BRAF mutant (G466V)20 (Supplementary Table 3). In the K601E PDX model, tumor growth was insensitive to vemurafenib and partially inhibited by PLX8394 (Supplementary Fig. 6d). Previous work suggested that incomplete inhibition of the growth of BRAF mutant colorectal tumors by RAF inhibitors is due to reactivation of RAS by feedback reactivation of EGFR signaling8. As shown in Fig. 4c and Supplementary Fig. 6d-e, ERK signaling and the growth of these tumors were potently inhibited by cetuximab combined with PLX8394, but not by cetuximab with vemurafenib or cetuximab alone. This suggests that PLX8394 more effectively suppresses mutant BRAF driven tumors when the reactivation of RAS-driven WT RAF dimers is also suppressed. In contrast, in the model expressing Class 3 BRAF G466V mutant, the ERK signaling and tumor growth are sensitive to cetuximab treatment which suppresses RAS activation20, but are insensitive to either vemurafenib or PLX8394 alone. In this case, combining vemurafenib or PLX8394 with cetuximab don’t improve efficacy compared to cetuximab alone (Fig. 4d and Supplementary Fig. 6f). These data are consistent those observed in cell lines (Figure. 4A and Supplementary Fig. 5b).
Our study shows that the unique properties of PLX8394 (Supplementary Table 4) are due to its ability to selectively disrupt BRAF homo- and BRAF/CRAF heterodimers. These findings suggest that this drug will be effective for the treatment of tumors driven by Class1 or 2 BRAF mutants and BRAF fusions. In addition, the drug will be useful for treating RAS-independent, BRAF dimer-dependent acquired resistance to current RAF inhibitors. The drug will not be effective for the treatment of resistance mechanisms based on RAS activation or activation of CRAF homodimers by any other means. It should have a wide therapeutic window, since it doesn’t inhibit WT CRAF dimers (Supplementary Fig. 7). This work together with previous studies1,2,20,23 show that an understanding of both the oncogenic mechanisms of different mutant alleles and the specific properties of drugs are necessary to précisely target BRAF mutations in human cancers.
Online Methods
Compounds
BGB659 was obtained from Beigene. Vemurafenib and PLX8394 were from Plexxikon. Lapatinib and dabrafenib were from GlaxoSmithKline. TAK632 was from Selleckchem. encorafenib was from Novartis. LY3009120 was purchased from Active Biochem, All these drugs were dissolved in DMSO to yield 10mM stock and stored at −20 °C Doxycycline (D3072) and 4-hydroxytamoxifen (4-OHT) (H7904) were purchased from Sigma Aldrich, Puromycin (A1113802) and Hygromycin (10687010) stock solution was from Thermo Fisher Scientific.
Cell lines and culture conditions
Cell lines A375: CRL-1619, 22Rv1: CRL-2505, H1666: CRL-5885, H508: CCL-253, H661: HTB-183, SKBR3: HTB-30, A549: CCL-185, BEAS-2B: CRL-9609, primary epidermal melanocytes: PCS-200–012 and primary epidermal keratinocytes: PCS-200–011 was obtained from American Type Culture Collection. SK-MEL-239, SK-MEL-239 C4, SK-MEL-2, SK-MEL-30, SK-MEL-246 and SK-MEL-285 were from MSKCC cell collection. CAL-12T: ACC433 and JVM-3: ACC18 were from DSMZ. 293H cells (#11631017) were purchased from ThermoFisher Scientific. MDST8 cells were from Millipore Sigma (#99011801). The conditional RAF knockout MEF cell line was from M. Barbacid lab.
The primary keratinocytes were cultured in Dermal cell Basal Medium (PCS-200–030) with keratinoctyes growth kit (PCS-200–040). The primary melanocytes were cultured in Dermal cell Basal Medium (PCS-200–030) with melanocyte growth kit (PCS-200–041). The BEAS-2B cells were culture with the BEBM medium obtained from Lonza/Clonetics Corporation (Catalog No. CC-3170). SK-MEL-2, SK-MEL-264, SK-MEL-30, SK-MEL-285, H1666, H661, A431, 22RV1, CAL-12T and H508, were cultured in RPMI, 10%FBS. SKBR3 in DMEM/F12+10%FBS, and other cell lines in DMEM with glutamine, antibiotics, 10%FBS. Inducible expression cells were maintained in medium with 50 ug/ml Hygromycin and 0.2 ug/ml Puromycin. All cell lines tested negative for mycoplasma. Genetic alterations in cell lines from the MSKCC cell collection were confirmed by IMPACT sequencing.
Antibodies
Western blot and immunoprecipitation (IP) were performed as described1,20. Antibodies used include: anti-p217/p221-MEK1/2 (p-MEK1/2) (#9154, lot#14 ), anti-p202/p204-ERK1/2 (p-ERK1/2) (#4370, lot# 17), anti-MEK1/2 (#4694, lot# 6), and anti-ERK1/2 (#4696, lot# 16) from Cell Signaling; anti-V5 (46–1157, lot# 1902786) from Invitrogen; anti-BRAF (cat# sc-5284, lot# C0116), anti-Cyclin D1 (cat# sc-718, lot# G1411 ) and anti-ARAF (cat# sc-166771, lot# A2617 ) from Santa Cruz Biotechnology; anti-FLAG (cat# F1804, lot# SLBT7654) from Sigma; anti-CRAF (cat# 610152, lot#7208706) from BD Transduction Laboratories; and anti-RAS (cat# 1862335, Lot # S1259698) from the active RAS pull-down and detection kit (Thermo Fisher Scientific, cat# 16117). For immunoprecipitations of tagged proteins, we used: anti-V5 agarose affinity gel (Sigma, cat# A7345, lot# SLBR7667V) and anti-FLAG M2 affinity gel (Sigma, cat# F1804, lot# SLBT7654), protein G agarose gel (Thermo Fisher Scientific cat# 15920010).The secondary antibodies used for Western blot are: goat anti-rabbit, HRP (Sigma, cat# A4914, lot# SLBM7730U) and donkey anti-mouse, HRP (Amersham, cat# NXA931, lot# 9708064).The primary antibodies used in Western Blot are diluted in 1:1,000, secondary antibodies are diluted in 1:5,000. The antibodies used in IP assay, are diluted in 1:200. All these antibodies have been validated in the previous studies1,5,6,20.
Plasmids
All the constructs for the transit expression of WT, mutant or truncated BRAF, ARAF and CRAF proteins were generated as previously described1,6,20. Plasmids for retroviral based inducible expression system were provided by Scott Lowe (MSKCC.) The BRAF and NRAS genes were sub-cloned into TTIGFP-MLUEX vector the tet-regulated promoter. Mutations were introduced using the site-directed Mutagenesis Kit (Stratagene).
Transfection
Cells were seeded in 60 mm or 100 mm plates and transfected the following day using Lipofectamine 2000 (ThermoFisher Scientific, #11668019) according to the instructions. A DNA (ug) to lipofectamine (ul) ratio of 1:3 was employed.
Generation of inducible expression cells
Retrovirus encoding rtTA or the ARAF, BRAF CRAF or NRAS genes (wild type or mutants) was packaged in Phoenix-AMPHO cells obtained from ATCC. Medium containing virus was filtered with 0.45 PVDF filters followed by incubation with the target cells for 6 hrs. Cells were then maintained in virus free media for 2 days. Cells were selected using Puromycin (2 μg/ml) or Hygromycin (250 μg/ml) for 3 days. Positive infected cell populations were further sorted using GFP as a marker after overnight exposure to 1μg/ml doxycycline. GFP positive cells were then cultured and expanded in medium without doxycycline but with antibiotics. According to this method, the inducible expression of NRAS Q61K was introduced into 293H cells, the inducible expression of BRAF V600E or NRAS Q61K was introduced into A375 cells and the inducible expression of wild type ARAF, BRAF or CRAF was introduced into RAF-less MEF cells.
The RAF-less cells were acquired from the conditional RAF knockout cells (Araflox/lox; Braflox/lox; Craflox/lox; RERT ert/ert MEF cells). The inducible genes were transduced into the cells in the absence of 4-OHT. After puromycin and hygromycin selections, the cells carrying the inducible RAF genes were infected with Adeno-Cre particles (multiplicity of infection = 100) and cultured in the medium containing 1uM 4-OHT and 50ng/ml doxycycline. The GFP positive cells were sorted after 2 weeks culture. The loss of endogenous expression of RAF genes and gain of expression of doxycycline induced RAF genes were confirmed by genomic PCR and western blotting assay.
Cell growth assay
Cells were seeded into 96 well plates at 1000 cells per well. Cell growth was quantified using the ATP-Glo assay (Promega, G7572) every 24 hrs. For each condition, 8 replicates at each concentration were measured. IC50 values were calculated using Graphpad Prism 6.
Patient Derived Xenograft and Animal study
Tumor samples were collected from MSK patients (Patient with BRAF G466V tumor: 55yo, Female, (Right) colon cancer, PDX collected after treatment with standard chemotherapy; Patient with BRAF K601E tumor: 41yo, Female, (Left) colon cancer, PDX collected after treatment with standard chemotherapy; no patient has received RAF inhibitor treatment when the samples are collected). Tumor tissue was transplanted orthotopically into NSG mice to make the PDX (IRB protocols 06–107, 14–091 (13)). The generated PDXs were subjected to high coverage next generation sequencing with the MSK-IMPACT24 assay, and the genomic alterations in the PDXs were verified against those in the metastasis resected from the patient (IRB protocol 12–245). Once tumor became visible in the mouse, it was transplanted and expanded in mice. The tumor tissue was implanted subcutaneously in the flank of 4–6 week old NSG female mice and treatment of the mice began when tumor reached 100–150mm3 in size. Mice were randomized (n=5 mice per group) in the colorectal cancer (CRC) PDX experiment to receive drug treatment or vehicle as control. Studies were performed in compliance with institutional guidelines under an IACUC approved protocol and all analysis of human tissues was conducted under institutional Review Board/Privacy Board approved protocol. Investigators were not blinded when assessing the outcome of the in vivo experiments.
Measurements of PLX8394 concentration in tumor samples
The tumor samples were collected at different time points after PLX8394 treatment. The fresh tumor samples (50–150mg) were frozen rapidly in ethanol-dry ice bath. Tumor tissues were homogenized, processed by protein precipitation and analyzed by LC-MS/MS method with a calibration range of 15–50000 ng/mL using internal standard. A standard curve was generated by adding known amounts of PLX8394 to an untreated homogenized sample. Quality control samples covering concentrations across the calibration range were included to ensure accuracy.
Statistics and Reproducibility
Results shown in the graphs are mean values ± s.d., n≥3 in all the graphs. All cellular experiments were repeated at least three times. For the in vivo experiments, mice were randomly assigned to each treatment groups. The sample sizes were pre-determined based on the previous studies. In each treatment groups, 5 animals were enrolled. A two-tailed unpaired Student’s t-test was applied for comparison between groups, when required; all analyzed samples were included for statistical analysis. Investigators were not blinded when assessing the outcome of the in vivo experiments.
Supplementary Material
Acknowledgments
We are grateful to Laura M Desrochers, Ingo Mellinghoff and Andrea Ventura for useful discussions. We thank Scott Lowe for the vectors of the retrovirus-based inducible expression system. This research was supported by grants to N.R. from the National Institutes of Health (NIH) (P01 CA129243; R35 CA210085); the Melanoma Research Alliance (237059 and 348724); The Commonwealth Foundation for Cancer Research and The Center for Experimental Therapeutics at Memorial Sloan Kettering Cancer Center; and the Stand Up To Cancer – American Cancer Society Lung Cancer Dream Team Translational Research Grant (SU2C-AACR-DT17–15). Support was also received from the NIH MSKCC Cancer Center Support Grant P30 CA008748 and T32 CA009207 (J. T.). Additional funding was provided by a Conquer Cancer Foundation of the American Society of Clinical Oncology Young Investigator Award (J.T.) and Career Development Award (R.Y.). We would like to acknowledge the support of the Arlene and Joseph Taub Foundation and of Paula and Thomas McInerney, without which this work would not have been possible. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was also supported by grants Spanish Ministry of Economy and Competitiveness (SAF2011–30173 and SAF2014–59864-R) to M.B. M.B. is the recipient of an Endowed Chair from the AXA Research Fund.
The data that support the findings of this study are available from the corresponding author upon request.
N.R. is on the SAB and receives research funding from Chugai, on the SAB and owns equity in Beigene, and Fortress. N.R. is also on the SAB of Daiichi-Sankyo, Astra-Zeneca-MedImmune and F-Prime, and is a past SAB member of Millenium-Takeda, Kadmon, Kura, and Araxes. N.R. is a consultant to Novartis, Boehringer-Ingelheim, Tarveda and Foresight and consulted in the last three years with Eli Lilly, Merrimack, Kura Oncology, Araxes, and Kadman. N.R. owns equity in ZaiLab, Kura Oncology, Araxes and Kadman. N.R. collaborates with Plexxikon.
RY has received research support from Array BioPharma, Genentech, GlaxoSmithKline and Norvatis; received travel expenses from Array BioPharma and served as advisory board for GlaxoSmithKline.
DMH has consulted for Atara Biotherapeutics, Chugai Pharma, CytomX Therapeutics, Boehringer Ingelheim, AstraZeneca, Pfizer, Bayer, Debiopharm Group, and Genetech and has received research funding from AstraZeneca, Puma Biotechnology, and Loxo Oncology.
Y.Z., C.Z., A. R. and G. B. are employees of Plexxikon Inc.
Footnotes
Availability of Data
Source data of all the Western Blotting in this work are provided in the Supplementary Figure 7~15 in the Supplementary information.
Ethical compliance
We declare compliance with all relevant ethical regulations.
Reporting Summary
Further information on research design is available in the Life Sciences Reporting Summary linked to this article.
Competing Financial Interests
References
- 1.Yao Z, et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 28, 370–383 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karoulia Z, et al. An Integrated Model of RAF Inhibitor Action Predicts Inhibitor Activity against Oncogenic BRAF Signaling. Cancer Cell 30, 485–498 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang H, et al. RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical melanoma models. Cancer Res 70, 5518–5527 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Nazarian R, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 468, 973–977 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poulikakos PI, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 480, 387–390 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poulikakos PI, Zhang C, Bollag G, Shokat KM & Rosen N RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464, 427–430 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi H, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun 3, 724 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yaeger R, et al. Mechanisms of acquired resistance to BRAF V600E inhibition in colon cancers converge on RAF dimerization and are sensitive to its inhibition. Cancer Res (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang C, et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 526, 583–586 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Basile KJ, Le K, Hartsough EJ & Aplin AE Inhibition of mutant BRAF splice variant signaling by next-generation, selective RAF inhibitors. Pigment Cell Melanoma Res 27, 479–484 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jain P, et al. CRAF gene fusions in pediatric low-grade gliomas define a distinct drug response based on dimerization profiles. Oncogene 36, 6348–6358 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okimoto RA, et al. Preclinical efficacy of a RAF inhibitor that evades paradoxical MAPK pathway activation in protein kinase BRAF-mutant lung cancer. Proc Natl Acad Sci U S A 113, 13456–13461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sievert AJ, et al. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci U S A 110, 5957–5962 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakamura A, et al. Antitumor activity of the selective pan-RAF inhibitor TAK-632 in BRAF inhibitor-resistant melanoma. Cancer Res 73, 7043–7055 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Peng SB, et al. Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti-tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell 28, 384–398 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Fabian JR, Daar IO & Morrison DK Critical tyrosine residues regulate the enzymatic and biological activity of Raf-1 kinase. Mol Cell Biol 13, 7170–7179 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stokoe D & McCormick F Activation of c-Raf-1 by Ras and Src through different mechanisms: activation in vivo and in vitro. EMBO J 16, 2384–2396 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takahashi M, Li Y, Dillon TJ, Kariya Y & Stork PJS Phosphorylation of the C-Raf N-region promotes Raf dimerization. Mol Cell Biol (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chong H, Lee J & Guan KL Positive and negative regulation of Raf kinase activity and function by phosphorylation. EMBO J 20, 3716–3727 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yao Z, et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 548, 234–238 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nieto P, et al. A Braf kinase-inactive mutant induces lung adenocarcinoma. Nature 548, 239–243 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heidorn SJ, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140, 209–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karoulia Z, Gavathiotis E & Poulikakos PI New perspectives for targeting RAF kinase in human cancer. Nat Rev Cancer 17, 676–691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Reference (Online Methods-only)
- 24.Cheng DT, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 17, 251–264 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.