

Abstract
Background
The sodium channel, Nav1.5, encoded by SCN5A, undergoes developmentally regulated splicing from inclusion of exon 6A in the fetal heart to exon 6B in adults. These mutually exclusive exons differ in 7 amino acids altering the electrophysiological properties of the Nav1.5 channel. In myotonic dystrophy type 1, SCN5A is mis‐spliced such that the fetal pattern of exon 6A inclusion is detected in adult hearts. Cardiac manifestations of myotonic dystrophy type 1 include conduction defects and arrhythmias and are the second‐leading cause of death.
Methods and Results
This work aimed to determine the impact of SCN5A mis‐splicing on cardiac function. We used clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR‐associated protein 9 (Cas9) to delete Scn5a exon 6B in mice, thereby redirecting splicing toward exon 6A. These mice exhibit prolonged PR and QRS intervals, slowed conduction velocity, extended action potential duration, and are highly susceptible to arrhythmias.
Conclusions
Our findings highlight a nonmutational pathological mechanism of arrhythmias and conduction defects as a result of mis‐splicing of the predominant cardiac sodium channel. Animals homozygous for the deleted exon express only the fetal isoform and have more‐severe phenotypes than heterozygotes that also express the adult isoform. This observation is directly relevant to myotonic dystrophy type 1, and possibly pathological arrhythmias, in which individuals differ with regard to the ratios of the isoforms expressed.
Keywords: alternative splicing, arrhythmia, conduction, myotonic dystrophy cardiomyopathy, SCN5A
Subject Categories: Genetically Altered and Transgenic Models, Animal Models of Human Disease, Arrhythmias, Basic Science Research, Pathophysiology
Clinical Perspective
What Is New?
Adults with mis‐splicing of the cardiac sodium channel gene, SCN5A, toward the fetal isoform, as found in myotonic dystrophy type 1, may be at an increased risk of arrhythmias and conduction defects.
Programmed electrical stimulation in mice expressing the fetal isoform of Scn5a by clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR‐associated protein 9 (Cas9) deletion of exon 6B produces arrhythmias and conduction defects that are indicative of sick sinus syndrome.
What Are the Clinical Implications?
Increased expression of the fetal relative to the adult isoform of Scn5a contributes to pathological arrhythmias in myotonic dystrophy type 1 and possibly in other pathological states.
Introduction
Aberrant sodium influx in cardiomyocytes is associated with arrhythmias and cardiac dysfunction contributing to sudden cardiac death.1, 2, 3 Mutations in the SCN5A gene encoding the Nav1.5 sodium channel are associated with cardiac diseases, including Brugada syndrome, long QT syndrome, and cardiac conduction defects, which are significant contributors to sudden cardiac death.4, 5 Analysis of SCN5A mutations have increased our understanding of the role of the sodium channel in disease, whereas the contributions of SCN5A splice variants remain largely unexplored. SCN5A undergoes a conserved developmentally regulated splicing transition from the inclusion of exon 6A during the fetal stages of heart development (RefSeq NM_001099404) to inclusion of exon 6B in adults (RefSeq NM_000335; Figure 1A).6, 7 Exon 6 encodes domain I segments 3 and 4 (DI:S3/4) and includes the voltage‐sensing transmembrane domain in which mutation can cause cardiac dysfunction.8, 9, 10 A notable difference is a change at position 211 from a positively charged lysine in exon 6A to a negatively charged aspartate in 6B. Experiments in vitro demonstrated that this difference in exon 6A causes slower channel kinetics, greater transient charge (Na+) influx, and slower recovery from inactivation compared with exon 6B.11, 12 Thus, the exon 6A Scn5a variant can have consequential effects such as potentiating dysfunction of a mutation associated with severe fetal arrhythmia.13
Figure 1.
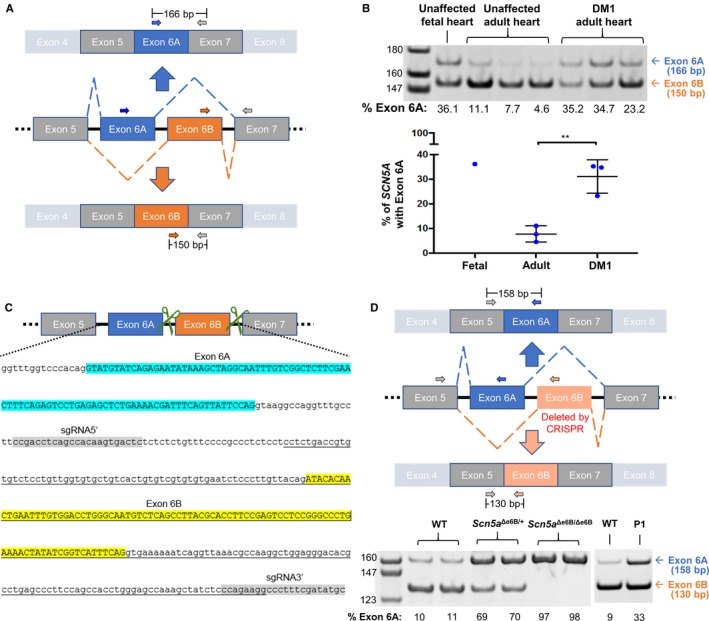
Mis‐splicing of SCN5A in heart tissue from DM1 patients and in exon 6B–deleted mice. A, Schematic representation of SCN5A exon 6 splice variants with 3 primers (2 forward and 1 reverse) used for RT‐PCR of human heart samples. B, RT‐PCR of RNA from unaffected fetal and adult hearts and from DM1 adult human heart tissues using primers shown in (A). Percent of exon 6A inclusion was determined by [exon 6A/(exon 6A+6B)]×100 and represented in a graph. Levels of exon 6A inclusion in fetal and adult heart are consistent with previous studies.6, 7 A significant difference in Scn5a exon 6A expression was found in DM1 hearts compared to unaffected individuals (P=0.006), also consistent with previous studies.6, 7 C, Schematic representation with genome sequence of mouse Scn5a containing exons 6A and 6B demonstrating the CRISPR/Cas9 approach to delete exon 6B. Green scissors represent guide RNAs targeting the intronic regions flanking exon 6B. In the genomic sequence, exon 6A is highlighted in blue, exon 6B is highlighted in yellow, and the 2 guide RNAs are highlighted in gray. The underlined sequence indicates the deleted genomic sequence. D, Schematic representation of 3 primers to distinguish exon 6A and 6B by RT‐PCR of heart tissue isolated from wild‐type (WT), heterozygotes (Scn5a Δe6B/+) and homozygotes (Scn5a Δe6B/Δe6B) mice with males and females showing no differences (n=3 for each group). Percent of exon 6A inclusion was calculated as in (B). RT‐PCR from an adult wild and postnatal day 1 (P1) type mice is shown to demonstrate that the Scn5a developmental splicing event is conserved in mouse and humans. **P<0.01. bp indicates base pair; CRISPR, clustered regularly interspaced short palindromic repeat; DM1, dystrophy type 1.
In healthy adults, ≈5% to 10% of total SCN5A mRNA includes exon 6A with >90% containing exon 6B. In adults with myotonic dystrophy type 1 (DM1), SCN5A mRNA is significantly mis‐spliced toward the fetal splicing pattern (Figure 1B).6, 7 DM1 is a progressive multisystemic disease and the most common form of muscular dystrophy in adults affecting 1 in 8000 individuals worldwide.14 DM1 is caused by an expansion of a CTG trinucleotide repeat in the 3′‐untranslated region of the DMPK (myotonic dystrophy protein kinase) gene.15 The expanded allele is transcribed and processed into mRNAs that contain hundreds to thousands of CUG repeats, which have a transdominant effect that disrupts developmentally regulated alternative splicing by disrupting the functions of RNA binding proteins, such as muscleblind‐like (MBNL) and CUG‐BP and ETR‐3‐like factors (CELF).16 Consequentially, retention of the fetal pre‐mRNA splicing patterns for a subset of genes is a molecular hallmark of DM1.17, 18
Cardiac‐related events in DM1 are the second‐leading cause of death in the disease.19, 20 Up to 80% of DM1 individuals have cardiac conduction defects, most notably prolonged PR and QRS intervals and second‐ and third‐degree atrioventricular block.21, 22 Rhythmic abnormalities are also present, including ventricular and supraventricular arrhythmias. In a DM1 mouse model that contains the human DMPK gene with the trinucleotide expansion, conduction defects were found that were correlated with abnormalities in the sodium current; however, the basis for the effect was not identified.23 In wild‐type (WT) mice, redirected splicing using AAV‐delivered U7 snRNA antisense sequence produced cardiac conduction defects, but the extent of the abnormalities and arrhythmias were not characterized.7 Splicing of Scn5a exon 6 is partially regulated by MBNL and optical mapping of hearts from Mbnl −/−, Mbnl2 +/− mice demonstrated decreased conduction velocities and longer action potential durations with inducible atrioventricular block and ventricular arrhythmias.6, 24 Total levels of Scn5a protein were unaffected in Mbnl1 −/−, Mbnl2 +/− mice, but Scn5a mRNA splicing was not examined.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
CRISPR‐Mediated Deletion Allele Design and Reagent Production
To delete the alternate exon 6B of Scn5a, 2 single‐guide RNAs (sgRNAs) were selected by the BCM Mouse Embryonic Stem Cell Core (BCM mES Core) using the Wellcome Trust Sanger Institute Genome Editing website (http://www.sanger.ac.uk/htgt/wge/) positioned to flank the genomic region containing the alternative exon 6B of Scn5a (5′ sgRNA: http://www.sanger.ac.uk/htgt/wge/crispr/551982233 and 3′ sgRNA: http://www.sanger.ac.uk/htgt/wge/crispr/551982280). Once complexed with Cas9, these sgRNAs could mediate double‐strand breaks for nonhomologous end joining (NHEJ) repair to remove genomic sequence in between the 2 sgRNA sites in Scn5a. sgRNAs were synthesized by the BCM mES Core using DNA templates for in vitro transcription. DNA templates were produced using overlapping oligonucleotides in a high‐fidelity PCR reaction.25 PCR products were first purified using the QiaQuick PCR purification kit and used as a template for in vitro transcription of the sgRNA with the MEGAshort script T7 kit (AM1354; Thermo Fisher Scientific, Waltham, MA). Following in vitro transcription, RNA was purified using the MEGAclear Transcription Clean‐Up Kit (AM1908; Thermo Fisher Scientific). All samples were analyzed by Nanodrop to determine concentration and visualized using the Qiaxcel Advanced System using the RNA QC V2.0 kit to check the quality of RNA product before storage at −80°C. Cas9 mRNA was purchased from Thermo Fisher Scientific (A25640). All sgRNAs were reanalyzed by Nanodrop before assembling the microinjection mixtures, which consisted of Cas9 mRNA (100 ng/μL) and sgRNA (10 ng/μL, each) in a final volume of 60 μL 1×PBS (RNAse‐free).
Microinjection of CRISPR/Cas9 Reagents
FVB/NJ female mice, 24 to 32 days old, were injected with 5 IU/mouse of pregnant mare serum, followed 46.5 hours later with 5 IU/mouse of human chorionic gonadotropin. Females were then mated to FVB/NJ males. All animals were from The Jackson Laboratory (Bar Harbor, ME). Fertilized oocytes were collected at 0.5 dpc for microinjection. The BCM Genetically Engineered Mouse Core microinjected the sgRNA/clustered regularly interspaced short palindromic repeat (CRISPR)‐associated protein 9 (Cas9)/ssOligo mixture into the cytoplasm of at least 100 pronuclear stage zygotes. Injected zygotes were transferred into pseudopregnant ICR females on the afternoon of the injection, ≈25 to 32 zygotes per recipient female. To determine whether double‐strand breaks induced by Cas9 had resulted in NHEJ repair to create a null allele, N0 mice were genotyped by standard PCR. Two primers ≈100 to 200 bases outside the 2 sgRNA sites were designed to amplify a smaller deletion amplicon compared with the WT amplicon.
RNA Isolation and Reverse‐Transcription PCR
Institutional review board approval was obtained for all animal studies and procedures were followed in accord with institutional guidelines. Ventricular and atrial heart tissues were dissected from euthanized mice, and RNA was isolated by guanidinium thiocyanate‐phenol‐chloroform extraction using TRIzol (15596‐018; Invitrogen, Carlsbad, CA). RNAs were then reverse transcribed to cDNA (4368813; Thermo Fisher Scientific) followed by PCR amplification using primers of interest. PCR products were loaded on a 13% acrylamide gel and results were analyzed on a molecular imager (Bio‐Rad ChemiDoc XRS+ System; Bio‐Rad Laboratories, Hercules, CA).
Human RT‐PCR primers were designed using a forward primer on exon 5 (5′ CTTCACCGCCATTTACACCT 3′) and reverse primers on exon 6A (5′ AAGAGCCGACAAATTGCCTA 3′) and exon 6B (5′ CCCAGGTCCACAAATTCAGT 3′). Mouse RT‐PCR primers were designed using forward primers on exon 6A (5′ GTCGGCTCTTCGAACTTTCA 3′) and exon 6B (5′ CGGGCCCTGAAAACTATATC 3′) along with a reverse primer on exon 7 (5′ CCAATGAGGGCAAAGACACT 3′). The expected size for human exon 6A amplicon is 166 base pairs and for exon 6B is 150 base pairs. The expected size for mouse exon 6A amplicon is 158 base pairs and for exon 6B is 130 base pairs.
Electrocardiography
Surface ECGs were captured from leads 1 to 3 using a Mouse Monitor S (Indus Instruments, Houston, TX). Two‐ to 3‐month‐old male and female mice were anesthetized in an induction chamber using 2.5% isoflurane and maintained during data collection with 1.5% isoflurane. Body temperature was monitored by a rectal probe. Data were collected for 1 minute per mouse and annotated using PowerLab software for analysis.
Echocardiography
In vivo cardiac function and morphology was assessed using a Vevo 2100 ultrasound machine equipped with a 40 MHz transducer‐MS550S (Visualsonics, Toronto, Ontario, Canada). Mice were anesthetized in an induction chamber using 2.5% isoflurane and then transferred to a heated ECG platform for heart rate monitoring during the imaging procedure. Body temperature was monitored by rectal probe and was maintained at 37°C. While imaging, anesthesia was maintained by a nosecone with 1% isoflurane. Standard B‐mode (2‐dimensional) and M‐mode images were taken in the short‐axis position at the level of the papillary muscles for each animal. Data analysis was performed by an experienced imager using the Visualsonics VevoLab analysis package. Two M‐mode tracings were analyzed per animal with an average being taken for each.
Intracardiac Programmed Electrical Stimulation
Experiments were followed as previously described.26 Briefly, mice were anesthetized using 2% isofluorane in 0.5 L/min 100% O2 and placed in a supine position with limbs taped onto ECG electrodes incorporated in a heating board (Indus Instruments). An incision was made to the right of the midline near the clavicle to access the right jugular vein for insertion of a 1.1F octapolar catheter (EPR‐800; Millar Instruments, Houston, TX) into the right atrium. Atrial pacing performed using 2‐ms current pulses delivered by an external stimulator (STG‐3008; Multi Channel Systems MCS GmbH, Reutlingen, Germany). Sinus node recovery time was defined as the interval between the last stimulus in the pacing train and the onset of first spontaneous sinus beat, as measured by applying a 15 second atrial pacing train at a basic cycle length of 100 ms. Atrioventricular effective refractory period was defined as the longest interval at which premature stimulation delivered to the atrium is followed by a His potential, but not QRS, complex after applying a series of atrial pacing trains at basic cycle length of 100 ms (S1) coupled with premature stimulus (S2), where the S1 to S2 interval is progressively reduced by 2 ms each pacing train from 70 to 20 ms. To determine inducibility of atrial arrhythmias, a burst pacing protocol was used where a 2‐second burst with a cycle length of 40 ms was applied followed by successive 2‐second bursts that are 2‐ms shorter in cycle length than the previous, down to 20 ms. With no arrhythmogenic substrate, sinus rhythm will resume following pacing. Burst pacing protocols were done in triplicate, and an arrhythmia was considered confirmed if evoked in at least 2 out of the 3.
Optical Mapping
Mice were anesthetized using 2% isofluorane with 1.0 L/min 100% O2 followed by intraperitoneal injection of heparin (100 units). A mid‐sternal incision was made, and the heart was quickly removed and washed in cold oxygenated (95% O2, 5% CO2) Tyrode's solution. Aorta was cannulated to a custom made 21‐gauge cannula using a 6‐0 black‐braided silk suture. After cannulation, the heart was retrogradely perfused with Tyrode's solution. Perfusion rate was maintained at 2 to 5 mL/min to keep the aortic pressure between 80 and 120 mm Hg. An electrode (Harvard Apparatus, Holliston, MA) was placed on the surface of right atria to conduct the pacing stimulations generated by PowerLab 26T (AD Instruments, Sydney, Australia). Hearts were loaded with the Blebbistatin (B0560‐5 mg, 50 μL of 2.5 mg/mL in DMSO; Sigma‐Aldrich, St. Louis, MO) to eliminate motion artifact and stained with voltage‐sensitive dye di‐4‐ANEPPS (D‐1199, 20 μL of 2.5 mg/mL in DMSO; Invitrogen). An LED light was used as the light source (excitation wavelength, 530 nm). The emitted fluorescence V m signal was long passed (>590 nm, 590FG05‐50, Andover Corporation Optical Filter; Andover Corporation, Salem, NH) and obtained through the MiCAM0 CMOS camera (SciMedia, Costa Mesa, CA). Surface ECG (ADInstruments) was recorded by LabChart. The atria and ventricle of the heart were burst paced at 10‐Hz frequency. Activation map and conduction velocity was calculated through Rhythm (developed by Dr Effimov's laboratory at University of Washington at Saint Louis).
Statistical Analysis
Statistical calculations were done using the Kruskal–Wallis test in Prism software (version 7.0b; GraphPad Software Inc, La Jolla, CA). Graphs are represented with the median and interquartile range. Sample sizes for ECG, echocardiography, and pacing studies are: 8 WT, 8 Scn5a Δe6B/+, and 6 Scn5a Δe6B/Δe6B. Sample sizes for optical mapping are: 4 WT, 4 Scn5a Δe6B/+, and 5 Scn5a Δe6B/Δe6B. *P<0.05; **P<0.01; ***P<0.001.
Results
To determine the extent to which mis‐splicing of SCN5A contributes to the diverse cardiac features of DM1, we used CRISPR/Cas9 to delete Scn5a exon 6B and redirect splicing to exon 6A (Figure 1C). Redirected splicing was validated by RT‐PCR of heart tissue from WT mice that express ≈10% Scn5a exon 6A mRNA, mice heterozygous for exon 6B deletion (Scn5a Δe6B/+) that have ≈70% Scn5a exon 6A, and mice homozygous for the deletion (Scn5a Δe6B/Δe6B) that exclusively express Scn5a with exon 6A (Figure 1D). Skipping of exon 6A is not detected by RT‐PCR in Scn5a Δe6B/+ or Scn5a Δe6B/Δe6B mice, indicating that all detectable mRNA from the deletion allele contains exon 6A (data not shown). In addition, RT‐PCR analysis of RNA from WT adult hearts showed the same 1:9 exon 6A/6B ratio in all 4 chambers (data not shown).
To determine the effects associated with expression of different levels of Scn5a exon 6A in adults, we assessed physiological differences in the heart of both Scn5a Δe6B/Δe6B and Scn5a Δe6B/+ mice in comparison with their WT littermates. We first measured cardiac conduction in anesthetized 2‐ to 3‐month‐old mice using surface ECG. Results showed a significant decrease in basal heart rate in Scn5a Δe6B/Δe6B mice with a trend in Scn5a Δe6B/+ (Figure 2A). PR interval was significantly prolonged in Scn5a Δe6B/Δe6B mice, but not in Scn5a Δe6B/+ mice (Figure 2B), whereas QRS interval was significantly prolonged in both (Figure 2C), consistent with cardiac features of DM1. The abnormal phenotypes observed in homozygous Scn5a Δe6B/Δe6B mice are more prominent and robust compared with heterozygous Scn5a Δe6B/+ mice, suggesting a direct correlation between the amount of exon 6A expression to cardiac abnormalities in adult mice. An interesting observation is the variability found in the corrected QT interval in both groups compared to WT littermates (Figure 2D). Statistical analysis using the F test to determine differences in variance was significant in both Scn5a Δe6B/Δe6B and Scn5a Δe6B/+ mice compared with WT (P=0.002 and 0.001, respectively). This variability may be indicative of QT dispersion, which suggests abnormalities with repolarization and correlates with the onset of arrhythmias.27 We next determined cardiac contractility and morphology using M‐mode echocardiography. Cardiac features of DM1 manifest primarily through abnormal conduction rather than issues with contractility.28 Consistent with DM1, both Scn5a Δe6B/Δe6B and Scn5a Δe6B/+ mice showed no significant differences in ejection fraction (Figure 2E) or fractional shortening (Figure 2F) and no significant morphological differences (Figure 2G and 2H).
Figure 2.
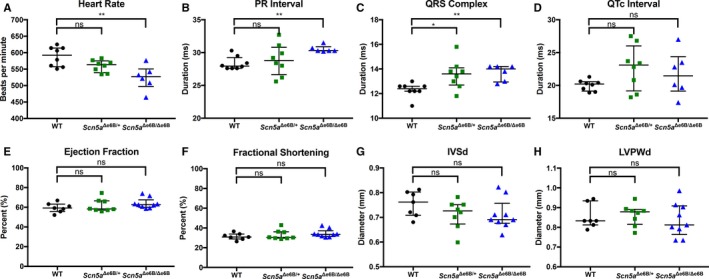
Electrocardiography and echocardiography of adult Scn5a Δe6B/Δe6B and Scn5a Δe6B/+ mice. Scn5a Δe6B/Δe6B and Scn5a Δe6B/+ mice compared with wild‐type (WT) littermates: (A) heart rate (P=0.002 and 0.143, respectively); (B) PR interval (P=0.008 and 0.384); (C) QRS interval (P=0.004 and 0.017); (D) corrected QT interval (P=0.389 and 0.114); (E) ejection fraction (P=0.112 and 0.759); (F) fractional shortening (P=0.139 and 0.845); (G) interventricular septal end diastole (P=0.141 and 0.254); and (H) left ventricular posterior wall end diastole (P=0.393 and 0.813). n=8 wild type, 8 Scn5a Δe6B/+, and 6 Scn5a Δe6B/Δe6B. *P<0.05, **P<0.01.
To help ensure observed phenotypes were not a result of CRISPR/Cas9 off‐target effects, 2 independent founder lines from the CRISPR/Cas9 deletion of Scn5a exon 6B were generated and mice from both founder lines show similar ECG abnormalities. Furthermore, the Scn5a Δe6B/+ mice were backcrossed for 7 generations to out‐cross off‐target loci and the backcrossed mice also show similar phenotypes. Last, the CRISPR/Cas9 deletion of Scn5a exon 6B was reproduced in C57BL/6NJ mice, which also show similar ECG phenotypes as observed in FVB/NJ, thereby demonstrating reproducibility, and provides further evidence that observed phenotypes are not likely to be a result of off‐target effects.
We performed in vivo intracardiac pacing on anesthetized Scn5a Δe6B/Δe6B and Scn5a Δe6B/+ mice to evaluate their susceptibility to arrhythmias compared with WT littermates (Figure 3A). We observed significant delays in sinoatrial node function of Scn5a Δe6B/Δe6B mice but not Scn5a Δe6B/+ mice as indicated by measuring the sinus node recovery time, corrected by subtracting the basic cycle length (Figure 3B). The atrioventricular effective refractory period was significantly prolonged in both groups (Figure 3C). In addition to sinus node recovery time and atrioventricular effective refractory period, intracardiac pacing also revealed various inducible arrhythmias in Scn5a Δe6B/Δe6B mice. One Scn5a Δe6B/Δe6B mouse died unexpectedly following overdrive pacing. Of the 5 surviving Scn5a Δe6B/Δe6B mice, we observed sinus bradycardia at baseline (5 of 5) with either premature atrial complexes (4 of 5; Figure 3D), sinus pause (4 of 5; Figure 3E), or spontaneous reversion from bradycardia (3 of 5; Figure 3F). Similar arrhythmias were inducible in Scn5a Δe6B/+ mice, but none were inducible in WT littermates (data not shown). Ventricular tachycardia was inducible in only 1 of the 5 Scn5a Δe6B/Δe6B mice and none in the WT.
Figure 3.
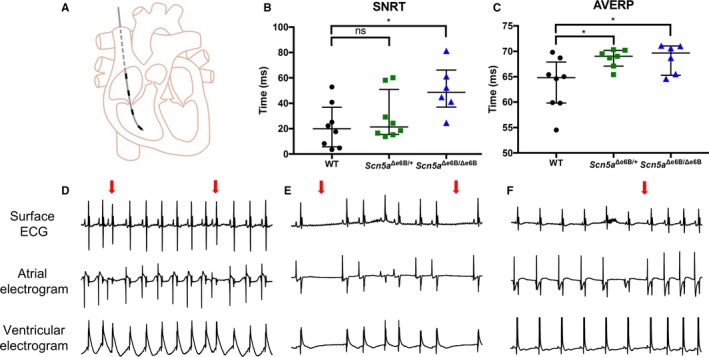
oElectrophysiological studies with programmed electrical stimulation in vivo. A, Schematic representation of electrical stimulation showing catheter with 4 electrodes inserted through the right jugular vein into the right atrium and right ventricle. Top electrode is used for atrial pace (“stimulating”) with the other 3 leads capture (“recording”) electrical activity in the heart. B, Duration of the sinoatrial node recovery time (SNRT) corrected for basic cycle length in Scn5a Δe6B/Δe6B and Scn5a Δe6B/+ mice compared with wild‐type (WT) littermates (P=0.013 and 0.465, respectively). C, Duration of the atrioventricular effective refractory period (AVERP) in Scn5a Δe6B/Δe6B and Scn5a Δe6B/+ mice compared with WT littermates (P=0.021 and 0.032, respectively). D, Representative intracardiac ECG tracing with premature atrial complex indicated by red arrow (4/5 Scn5a Δe6B/Δe6B, 0/8 WT). E, Representative intracardiac ECG tracing with sinus pauses indicated by red arrows (4/5 Scn5a Δe6B/Δe6B, 0/8 WT). F, Representative intracardiac ECG tracing with spontaneous reversion from bradycardia with the reversion indicated by the red arrow (3/5 Scn5a Δe6B/Δe6B, 0/8 WT). n=8 wild type, 8 Scn5a Δe6B/+, and 6 Scn5a Δe6B/Δe6B except for inducibility of arrhythmias where 1 died. *P<0.05.
To better understand the cardiac dysfunction observed in vivo, we performed optical mapping followed by pacing studies ex vivo on Langendorff‐perfused hearts isolated from adult Scn5a Δe6B/Δe6B and Scn5a Δe6B/+ mice to follow spatiotemporal conductive features. Optical mapping revealed significant delays in activation and propagation of conduction, with the activation map showing an average of 10 ms throughout the heart of WT mice (Figure 4A), but a delay to 14 ms in Scn5a Δe6B/+ mice (Figure 4B) and 27 ms in Scn5a Δe6B/Δe6B mice (Figure 4C). Action potential duration was significantly prolonged in Scn5a Δe6B/Δe6B and trending in Scn5a Δe6B/+ mice (Figure 4D) as well as significantly decreased ventricular conduction velocity in both groups (Figure 4E). These findings are consistent with optical mapping studies on Mbnl1−/−, Mbnl2+/− hearts by Chou et al, suggesting Scn5a mis‐splicing as a candidate for cardiac defects observed in Mbnl knockouts.24 No arrhythmias were present at baseline in any hearts isolated from WT littermates (0 of 4; Video S1) or in hearts from Scn5a Δe6B/+ (0 of 3). Arrhythmias found at baseline in Scn5a Δe6B/Δe6B hearts include atrioventricular block (3 of 5; Figure 4F) and afterdepolarizations (3 of 5; Figure 4G) that were either present simultaneously (Video S2) or separately (Video S3). Premature atrial complexes were also present at baseline in Scn5a Δe6B/Δe6B hearts (2 of 5; Video S4). A prolonged action potential duration was noticeable in all Scn5a Δe6B/+ hearts compared with WT (Video S5). Programmed electrical stimulation ex vivo was tolerated in WT mice (Video S6), but Scn5a Δe6B/Δe6B hearts demonstrate perturbed ventricular repolarization (Video S7) and also demonstrate instances where atrioventricular block prevented pacing of the ventricles (Video S8). Pacing in hearts from Scn5a Δe6B/+ showed atrioventricular block (Video S9).
Figure 4.
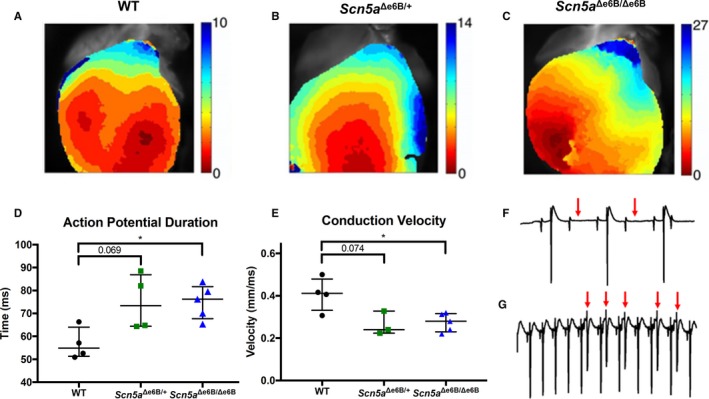
Optical mapping with programmed electrical stimulation ex vivo. Representative activation map from optical mapping in (A) wild‐type (WT), (B) Scn5a Δe6B/+ mice, and (C) Scn5a Δe6B/Δe6B mice, with color bar showing time from red to blue in milliseconds. D, Action potential duration in Scn5a Δe6B/Δe6B and Scn5a Δe6B/+ mice (P=0.028 and 0.069, respectively). E, Ventricular conduction velocity in Scn5a Δe6B/Δe6B and Scn5a Δe6B/+ mice (P=0.045 and 0.074, respectively). F, Representative tracing from ECG leads on hearts isolated ex vivo showing atrioventricular block (0/3 WT, 4/5 Scn5a Δe6B/Δe6B). Red arrows indicate the absence of a ventricular depolarization (large spikes) following atrial depolarization (small spikes). G, Representative tracing showing delayed afterdepolarizations (0/3 WT, 4/5 Scn5a Δe6B/Δe6B). Red arrows indicate ventricular afterdepolarizations. n=4 wild type, 4 Scn5a Δe6B/+, and 5 Scn5a Δe6B/Δe6B. *P<0.05.
Discussion
We have extensively characterized cardiac conduction defects associated with SCN5A splice variants. Atrioventricular conduction disorders and ventricular tachycardia have been proposed as mechanisms of sudden cardiac death in DM1.29, 30, 31, 32 A previous study by Lazarus et al involving invasive electrophysiological studies with intracardiac pacing in DM1 patients reported prolonged conduction with frequent inducible ventricular and supraventricular arrhythmias.33 They also report a low incidence (18%) of DM1 patients having inducible ventricular tachyarrhythmias. Both of these observations were reproducible in our mice. A noteworthy observation in our study is the loss of significance in Scn5a Δe6B/+ mice compared with Scn5a Δe6B/Δe6B mice in the PR interval and sinus node recovery time, 2 parameters that relate to the atria.
Previous patch clamp studies indicate slower kinetics of activation and inactivation as well as a decrease in peak sodium current of the Scn5a exon 6A splice variant in comparison with its exon 6B counterpart.7, 11 Overall, the slower kinetics of activation and inactivation of exon 6A allows more time for Na+ influx, resulting in greater transient charge influx as well as a decrease in peak sodium current. Many cardiac phenotypes observed with Scn5a mis‐splicing in our mice could be attributed to such electrophysiological differences, which serves as a trigger and substrate for arrhythmogenesis.11 A decrease in peak sodium current with Scn5a exon 6A splice variant also leads to a lower maximum upstroke velocity (dV/dtmax) and can cause a decrease in conduction velocity, consistent with our findings. Decreased conduction velocity and increased action potential duration are 2 determinants in producing unidirectional block by prolonging refractoriness, potentially resulting in re‐entrant arrhythmias.34 Afterdepolarizations and late ventricular potentials are features of abnormal repolarization associated with DM1 and significantly elevate the risk of sudden cardiac death.35
There are certain factors from our study that should be taken into consideration when evaluating its implications for DM1. Most notably, the percent of Scn5a expressing exon 6A in our mice is significantly greater than those found in most of DM1 patients (≈70% exon 6A inclusion in our Scn5a Δe6B/+ mice and 100% in our Scn5a Δe6B/Δe6B mice compared with ≈35% in DM1 patients, though up to ≈70% exon 6A inclusion has been reported in DM1).7 This exaggerated mis‐splicing of Scn5a in our mice is useful to elucidate the contribution of the exon 6A variant toward arrhythmias and conduction abnormalities, but unable to shed insight on whether a threshold for SCN5A mis‐splicing exists that serves as a risk factor for cardiac abnormalities in DM1. This is further complicated by other mis‐splicing events in the DM1 heart that may deleteriously contribute or perhaps help compensate cardiac abnormalities. Nonetheless, our findings corroborate and expand upon the connection between splice variants of SCN5A to cardiac conduction defects and arrhythmias, which have significant implications for DM1. Further investigations into this splicing event as well as other abnormal SCN5A splicing, such as a truncated variant found in heart failure, may provide the opportunity of discovering or repurposing therapeutic options.36
Sources of Funding
Pang is funded by the NIH (T32HL07676, F31HL140879). Cooper is funded by the NIH (R01HL045565, R01AR045653, and R01AR060733) and the Muscular Dystrophy Foundation. Wehrens is funded by the NIH (R01HL089598, R01HL091947, and R01HL117641), American Heart Association (13EIA14560061), and Saving Tiny Hearts Foundation.
Disclosures
None.
Supporting information
Video S1. Optical mapping of electrical conduction in wild‐type mice.
Video S2. Optical mapping of electrical conduction in Scn5a Δe6B/Δe6B mice with AV block and early afterdepolarizations.
Video S3. Optical mapping of electrical conduction in Scn5a Δe6B/Δe6B mice with early afterdepolarizations.
Video S4. Optical mapping of electrical conduction in Scn5a Δe6B/Δe6B mice with premature atrial complex.
Video S5. Optical mapping of electrical conduction in Scn5a Δe6B/+ mice at baseline with prolonged action potential duration.
Video S6. Optical mapping of electrical conduction in WT mice with programmed electrical stimulation.
Video S7. Optical mapping of electrical conduction in Scn5a Δe6B/Δe6B mice with programmed electrical stimulation.
Video S8. Optical mapping of electrical conduction in Scn5a Δe6B/Δe6B mice with programmed electrical stimulation showing AV block and EADs.
Video S9. Optical mapping of electrical conduction in Scn5a Δe6B/+ mice with programmed electrical stimulation showing AV block.
Acknowledgments
We would like to thank the Mouse Phenotyping Core at Baylor College of Medicine for assistance with electrocardiography and echocardiography with their funding from the NIH UM1HG006348 and NIH R01DK114356 and the Mouse Embryonic Stem Cell Core and Genetically Engineered Mouse Core at Baylor College of Medicine for generating the deletion of Scn5a exon 6B in mice. Resources accessed through the cores were supported by NIH‐NCI grant CA125123 to the Dan L. Duncan Cancer Center.
(J Am Heart Assoc. 2018;7:e010393 DOI: 10.1161/JAHA.118.010393.)
References
- 1. Klaver EC, Versluijs GM, Wilders R. Cardiac ion channel mutations in the sudden infant death syndrome. Int J Cardiol. 2011;152:162–170. [DOI] [PubMed] [Google Scholar]
- 2. Chugh SS, Reiner K, Teodorescu C, Evanado A, Kehr E, Al Samara M, Mariani R, Gunson K, Jui J. Epidemiology of sudden cardiac death: clinical and research implications. Prog Cardiovasc Dis. 2008;51:213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Adabag AS, Luepker RV, Roger VL, Gersh BJ. Sudden cardiac death: epidemiology and risk factors. Nat Rev Cardiol. 2010;7:216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ruan Y, Liu N, Priori SG. Sodium channel mutations and arrhythmias. Nat Rev Cardiol. 2009;6:337–348. [DOI] [PubMed] [Google Scholar]
- 5. Wang Q, Shen J, Li Z, Timothy K, Vincent GM, Priori SG, Schwartz PJ, Keating MT. Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum Mol Genet. 1995;4:1603–1607. [DOI] [PubMed] [Google Scholar]
- 6. Wahbi K, Algalarrondo V, Becane HM, Fressart V, Beldjord C, Azibi K, Lazarus A, Berber N, Radvanyi‐Hoffman H, Stojkovic T, Behin A, Laforet P, Eymard B, Hatem S, Duboc D. Brugada syndrome and abnormal splicing of SCN5A in myotonic dystrophy type 1. Arch Cardiovasc Dis. 2013;106:635–643. [DOI] [PubMed] [Google Scholar]
- 7. Freyermuth F, Rau F, Kokunai Y, Linke T, Sellier C, Nakamori M, Kino Y, Arandel L, Jollet A, Thibault C, Philipps M, Vicaire S, Jost B, Udd B, Day JW, Duboc D, Wahbi K, Matsumura T, Fujimura H, Mochizuki H, Deryckere F, Kimura T, Nukina N, Ishiura S, Lacroix V, Campan‐Fournier A, Navratil V, Chautard E, Auboeuf D, Horie M, Imoto K, Lee KY, Swanson MS, Munain AL, Inada S, Itoh H, Nakazawa K, Ashihara T, Wang E, Zimmer T, Furling D, Takahashi MP, Charlet‐Berguerand N. Splicing misregulation of SCN5A contributes to cardiac‐conduction delay and heart arrhythmia in myotonic dystrophy. Nat Commun. 2016;7:11067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shen C, Xu L, Han S, Dong Z, Zhao X, Wang S, Qian S, Li B, Ma X, Wang P, Zhu H, Zou Y, Fan Z, Ge J, Sun A. Novel idiopathic DCM‐related SCN5A variants localized in D1‐S4 predispose electrical disorders by reducing peak sodium current density. J Med Genet. 2017;54:762–770. [DOI] [PubMed] [Google Scholar]
- 9. Nair K, Pekheltsi R, Harris L, Care M, Morel C, Farid T, Backx PH, Szabo E, Nanthakumar K. Escape capture bigeminy: phenotypic marker of cardiac sodium channel voltage sensor mutation R222Q. Heart Rhythm. 2012;9:1681–1688. [DOI] [PubMed] [Google Scholar]
- 10. Beckermann TM, McLeod K, Murday V, Potet F, George AL Jr. Novel SCN5A mutation in amiodarone‐responsive multifocal ventricular ectopy‐associated cardiomyopathy. Heart Rhythm. 2014;11:1446–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Onkal R, Mattis JH, Fraser SP, Diss JK, Shao D, Okuse K, Djamgoz MB. Alternative splicing of Nav1.5: an electrophysiological comparison of ‘neonatal’ and ‘adult’ isoforms and critical involvement of a lysine residue. J Cell Physiol. 2008;216:716–726. [DOI] [PubMed] [Google Scholar]
- 12. Baptista‐Hon DT, Robertson FM, Robertson GB, Owen SJ, Rogers GW, Lydon EL, Lee NH, Hales TG. Potent inhibition by ropivacaine of metastatic colon cancer SW620 cell invasion and NaV1.5 channel function. Br J Anaesth. 2014;113(suppl 1):i39–i48. [DOI] [PubMed] [Google Scholar]
- 13. Murphy LL, Moon‐Grady AJ, Cuneo BF, Wakai RT, Yu S, Kunic JD, Benson DW, George AL Jr. Developmentally regulated SCN5A splice variant potentiates dysfunction of a novel mutation associated with severe fetal arrhythmia. Heart Rhythm. 2012;9:590–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ranum LP, Day JW. Myotonic dystrophy: RNA pathogenesis comes into focus. Am J Hum Genet. 2004;74:793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, Sohn R, Zemelman B, Snell RG, Rundle SA, Crow S, Davies J, Shelbourne P, Buxton J, Jones C, Juvonen V, Johnson K, Harper PS, Shaw DJ, Housman DE. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. [DOI] [PubMed] [Google Scholar]
- 16. Lee JE, Cooper TA. Pathogenic mechanisms of myotonic dystrophy. Biochem Soc Trans. 2009;37:1281–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chau A, Kalsotra A. Developmental insights into the pathology of and therapeutic strategies for DM1: back to the basics. Dev Dyn. 2015;244:377–390. [DOI] [PubMed] [Google Scholar]
- 18. Kuyumcu‐Martinez NM, Cooper TA. Misregulation of alternative splicing causes pathogenesis in myotonic dystrophy. Prog Mol Subcell Biol. 2006;44:133–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Groh WJ, Groh MR, Saha C, Kincaid JC, Simmons Z, Ciafaloni E, Pourmand R, Otten RF, Bhakta D, Nair GV, Marashdeh MM, Zipes DP, Pascuzzi RM. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N Engl J Med. 2008;358:2688–2697. [DOI] [PubMed] [Google Scholar]
- 20. Lazarus A, Varin J, Babuty D, Anselme F, Coste J, Duboc D. Long‐term follow‐up of arrhythmias in patients with myotonic dystrophy treated by pacing: a multicenter diagnostic pacemaker study. J Am Coll Cardiol. 2002;40:1645–1652. [DOI] [PubMed] [Google Scholar]
- 21. Fragola PV, Luzi M, Calo L, Antonini G, Borzi M, Frongillo D, Cannata D. Cardiac involvement in myotonic dystrophy. Am J Cardiol. 1994;74:1070–1072. [DOI] [PubMed] [Google Scholar]
- 22. Petkovich NJ, Dunn M, Reed W. Myotonia dystrophica with AV dissociation and Strokes‐Adams attacks. Am Heart J. 1964;68:391–396. [DOI] [PubMed] [Google Scholar]
- 23. Algalarrondo V, Wahbi K, Sebag F, Gourdon G, Beldjord C, Azibi K, Balse E, Coulombe A, Fischmeister R, Eymard B, Duboc D, Hatem SN. Abnormal sodium current properties contribute to cardiac electrical and contractile dysfunction in a mouse model of myotonic dystrophy type 1. Neuromuscul Disord. 2015;25:308–320. [DOI] [PubMed] [Google Scholar]
- 24. Chou CC, Chang PC, Wei YC, Lee KY. Optical mapping approaches on muscleblind‐like compound knockout mice for understanding mechanistic insights into ventricular arrhythmias in myotonic dystrophy. J Am Heart Assoc. 2017;6:e005191 DOI: 10.1161/JAHA.116.005191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bassett AR, Tibbit C, Ponting CP, Liu JL. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep. 2013;4:220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li N, Wehrens XH. Programmed electrical stimulation in mice. J Vis Exp. 2010;39:1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malik M, Batchvarov VN. Measurement, interpretation and clinical potential of QT dispersion. J Am Coll Cardiol. 2000;36:1749–1766. [DOI] [PubMed] [Google Scholar]
- 28. Lindgvist P, Morner S, Olofsson BO, Backman C, Lundblad D, Forsberg H, Henein MY. Ventricular dysfunction in type 1 myotonic dystrophy: electrical, mechanical, or both? Int J Cardiol. 2010;143:378–384. [DOI] [PubMed] [Google Scholar]
- 29. Hiromasa S, Ikeda T, Kubota K, Hattori N, Coto H, Maldonado C, Kupersmith J. Ventricular tachycardia and sudden death in myotonic dystrophy. Am Heart J. 1988;115:914–915. [DOI] [PubMed] [Google Scholar]
- 30. Cannom DS, Wyman MG, Goldreyer BN. Clinical and induced ventricular tachycardia in a patient with myotonic dystrophy. J Am Coll Cardiol. 1984;4:625–628. [DOI] [PubMed] [Google Scholar]
- 31. Grigg LE, Chan W, Hond HG, Vohra JK, Downey WF. Ventricular tachycardia and sudden death in myotonic dystrophy: clinical, electrophysiologic and pathologic features. J Am Coll Cardiol. 1985;6:254–256. [DOI] [PubMed] [Google Scholar]
- 32. Tamura K, Tsuji H, Matsui Y, Masui A, Hikosaka M, Karakawa M, Iwasaka T, Inada M. Sustained ventricular tachycardias associated with myotonic dystrophy. Clin Cardiol. 1996;19:674–677. [DOI] [PubMed] [Google Scholar]
- 33. Lazarus A, Varin J, Ounnoughene Z, Radvanyi H, Junien C, Coste J, Laforet P, Eymard B, Becane HM, Weber S, Duboc D. Relationship among electrophysiological findings and clinical status, heart function, and extent of DNA mutation in myotonic dystrophy. Circulation. 1999;99:1041–1046. [DOI] [PubMed] [Google Scholar]
- 34. Makielski JC. Late sodium current: a mechanism for angina, heart failure, and arrhythmia. Trends Cardiovasc Med. 2016;26:115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Milner MR, Hawley RJ, Jachim M, Lindsay J, Fletcher RD. Ventricular late potentials in myotonic dystrophy. Ann Intern Med. 1991;115:607–613. [DOI] [PubMed] [Google Scholar]
- 36. Noyes AM, Zhou A, Gao G, Gu L, Day S, Andrew Wasserstrom J, Dudley SC. Abnormal sodium channel mRNA splicing in hypertrophic cardiomyopathy. Int J Cardiol. 2017;249:282–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Optical mapping of electrical conduction in wild‐type mice.
Video S2. Optical mapping of electrical conduction in Scn5a Δe6B/Δe6B mice with AV block and early afterdepolarizations.
Video S3. Optical mapping of electrical conduction in Scn5a Δe6B/Δe6B mice with early afterdepolarizations.
Video S4. Optical mapping of electrical conduction in Scn5a Δe6B/Δe6B mice with premature atrial complex.
Video S5. Optical mapping of electrical conduction in Scn5a Δe6B/+ mice at baseline with prolonged action potential duration.
Video S6. Optical mapping of electrical conduction in WT mice with programmed electrical stimulation.
Video S7. Optical mapping of electrical conduction in Scn5a Δe6B/Δe6B mice with programmed electrical stimulation.
Video S8. Optical mapping of electrical conduction in Scn5a Δe6B/Δe6B mice with programmed electrical stimulation showing AV block and EADs.
Video S9. Optical mapping of electrical conduction in Scn5a Δe6B/+ mice with programmed electrical stimulation showing AV block.