

Abstract
Background
Lymphatic vessels interconnect with blood vessels to form an elaborate system that aids in the control of tissue pressure and edema formation. Although the lymphatic system has been known to exist in a heart, little is known about the role the cardiac lymphatic system plays in the development of heart failure.
Methods and Results
Mice (C57BL/6J, male, 8 to 12 weeks of age) were subjected to either myocardial ischemia or myocardial ischemia and reperfusion for up to 28 days. Analysis revealed that both models increased the protein expression of vascular endothelial growth factor C and VEGF receptor 3 starting at 1 day after the onset of injury, whereas a significant increase in lymphatic vessel density was observed starting at 3 days. Further studies aimed to determine the consequences of inhibiting the endogenous lymphangiogenesis response on the development of heart failure. Using 2 different pharmacological approaches, we found that inhibiting VEGF receptor 3 with MAZ‐51 and blocking endogenous vascular endothelial growth factor C with a neutralizing antibody blunted the increase in lymphatic vessel density, blunted lymphatic transport, increased inflammation, increased edema, and increased cardiac dysfunction. Subsequent studies revealed that augmentation of the endogenous lymphangiogenesis response with vascular endothelial growth factor C treatment reduced inflammation, reduced edema, and improved cardiac dysfunction.
Conclusions
These results suggest that the endogenous lymphangiogenesis response plays an adaptive role in the development of ischemic‐induced heart failure and supports the emerging concept that therapeutic lymphangiogenesis is a promising new approach for the treatment of cardiovascular disease.
Keywords: heart failure, lymphangiogenesis, lymphatics, myocardial ischemia
Subject Categories: Ischemia, Heart Failure, Inflammation
Clinical Perspective
What Is New?
This study is one of the first to demonstrate that chronic myocardial ischemia and myocardial ischemia‐reperfusion both stimulate an endogenous lymphangiogenesis response, despite different kinetics in the induction of vascular endothelial growth factor C.
This study is one of the first to demonstrate that the endogenous lymphangiogenesis response is initiated during the first week after the onset of myocardial ischemia‐reperfusion injury.
This is the first study to demonstrate that inhibition of the endogenous lymphangiogenesis response exacerbates ischemic‐induced heart failure.
What Are the Clinical Implications?
The findings of the current study demonstrate that the local delivery of vascular endothelial growth factor C using a hydrogel is a viable delivery strategy to attenuate the development of ischemic‐induced heart failure.
The findings of the current study support the emerging concept that therapeutic lymphangiogenesis is a promising new approach for the treatment of heart failure.
Introduction
Lymphatic vessels exist throughout the body in the same manner as blood vessels.1 They interconnect with blood vessels to form an elaborate system that functions in interstitial fluid drainage, lipid absorption, and immune cell responses.2 Lymphatic vessels originate from veins during embryonic development before undergoing extensive expansion and remodeling to form an ordered network consisting of lymphatic capillaries, precollectors, and collecting vessels that serve to return lymphatic fluid back to the blood circulation.2 Lymphangiogenesis, or the growth of lymphatic vessels from preexisting vessels, is the major, if not the exclusive, mode of lymphatic growth. Signaling via vascular endothelial growth factor C (VEGF‐C) and VEGF receptor 3 (VEGFR3) is perhaps the most central pathway for lymphangiogenesis. Disruption in lymphatic vessel formation during development (VEGF‐C– or VEGFR3‐deficient mice3, 4) leads to death. Additionally, the loss of lymphatic function in humans (hereditary disease, lymphatic damage, or surgical removal of lymph nodes) leads to lymphedema.5, 6 This and other, more recent, evidence has led to the understanding that lymphatic vessels are not simply passive conduits for interstitial fluid, but rather are essential for multiple physiological activities.1
The heart contains an elaborate network of lymphatic vessels, which serve to collect and return macromolecules, proteins, electrolytes, and fluid from the interstitial space to the circulation.7, 8, 9 As a result, the cardiac lymphatic system aids in the control of tissue pressure and edema formation.8 Interference with cardiac lymphatic flow (ie, through obstruction) predisposes the heart to edema, inflammation, fibrosis, and infection.10 Additionally, impairments in cardiac lymphatic flow have deleterious effects on cardiac dysfunction in the setting of myocardial ischemia.11 Despite these well‐documented actions, little is known about the role the cardiac lymphatic system plays in the development of heart failure. Recent studies have begun to focus on this issue with the demonstration that myocardial ischemia initiates an endogenous lymphangiogenesis response.12, 13 Moreover, treatment with VEGF‐C augments endogenous lymphangiogenesis and leads to improvements in cardiac function.12, 13 Thus, it appears that therapeutic lymphangiogenesis serves as a promising new treatment of heart failure.
Although these findings have laid the foundation for the concept of therapeutic lymphangiogenesis, there are several issues related to the kinetics and functional significance of the response that need to be addressed. Additionally, the consequences of inhibiting the endogenous lymphangiogenesis response have not been explored. Herein, we addressed these issues in well‐defined murine models of ischemic‐induced heart failure.
Materials and Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Animals
C57BL/6J mice (male; 8–12 weeks of age) were used in all experiments. Sex influences the development of cardiovascular disease.14 As such, we only used male mice in our studies. This allowed for the evaluation of ischemic‐induced lymphangiogenesis in a well‐controlled experimental system. All experimental protocols were approved by the Institute for Animal Care and Use Committee at T3 Laboratories and Emory University and conformed to the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health (publication 86‐23, revised 1996), and with federal and state regulations.
Materials
Recombinant human VEGF‐CCys156Ser (752‐VC‐025/CF) was purchased from R&D Systems (Minneapolis, MN). The VEGFR3 inhibitor, MAZ‐51 (No. 676 492) was purchased from Millipore Sigma (Burlington, MA). VEGF‐C neutralization antibody (52 393) was purchased from GeneTex, Inc (Irvine, CA). Gelatin‐based hydrogels were obtained from MedGEL (Kodaira, Tokyo, Japan).15, 16 5‐Bromo‐2′‐deoxyuridine (No. B5002) was purchased from Sigma‐Aldrich.
In Vitro Cell Culture
Lymphatic endothelial cells and human umbilical vein endothelial cells (HUVECs) were purchased from Lonza. Lymphatic endothelial cells were cultured in endothelial cell growth medium‐2MV (No. cc4176; Lonza). HUVECs were cultured in endothelial cell growth medium plus growth media (No. cc5035; Lonza). Cells (10 000/well) were plated in 96‐well plates and maintained in corresponding media until 80% confluent. Cells were then maintained in endothelial cell basal medium‐2 containing 0.5% fetal bovine serum for 24 hours. Cells were then exposed to 100 ng/mL of VEGF‐CCys156Ser or VEGF‐A. Cell proliferation was evaluated 72 hours later by use of the PreMix WST‐1 Cell Proliferation Assay System (TAKARA Bio Inc), according to the manufacturer's instructions.
Gelatin‐Based Hydrogel Preparation
Gelatin‐based hydrogels were prepared according to the manufacturer's instruction. In brief, to prepare gelatin hydrogels, 10 μL of an aqueous solution containing VEGF‐CCys156Ser (125 μg/mL),17 MAZ‐51 (25 mg/mL),18 VEGF‐C neutralizing antibody (NAb; 0.5 mg/mL), or vehicle was dropped onto the freeze‐dried hydrogels. The hydrogels were incubated at room temperature for at least 30 minutes. The hydrogels are designed to release their content as they degrade, with ≈90% being released by 14 days. This results in the following doses: VEGF‐CCys156Ser, 3.214 μg/kg per day; MAZ‐51, 0.643 mg/kg per day; and VEGF‐C NAb, 0.0218 mg/kg per day. After the onset of reperfusion, the gelatin hydrogels were placed on the surface of myocardium before the closing of the chest.
Heart Failure Protocols
Heart failure was induced by either permanent ligation of the left coronary artery or subjecting mice to 60 minutes of left coronary artery occlusion, followed by reperfusion for up to 4 weeks. Surgical ligation of the left coronary artery was performed under anesthesia (ketamine, 100 mg/kg; sodium pentobarbital, 20 mg/kg), as previously described.19, 20 All animals received prophylactic antibiotic therapy with cefazolin (20 mg/kg) and buprenorphine (0.05 mg/kg) for pain. A total of 185 mice were included in the present study after accounting for animal deaths. All mice were randomly assigned to the treatment groups. For the experiments examining the proliferation of lymph endothelial cells, 5‐bromo‐2′‐deoxyuridine (30 mg/mL)21 was injected intraperitoneally once daily after the onset of myocardial ischemia until the time of euthanasia.
Echocardiograph Analysis
Transthoracic echocardiography was performed at baseline and 4 weeks after reperfusion using the Vevo 2100 with a 38‐MHz linear array scanhead.19
Infarct Size Analysis
Myocardial infarct size was evaluated at 24 hours of reperfusion, as previously described.22 For these experiments, mice were subjected to 60 minutes of ischemia, followed by reperfusion. Gelatin hydrogels containing vehicle, MAZ‐51, VEGF‐C NAb, or VEGF‐CCys156Ser were placed on the surface of myocardium before the closing of the chest.
Western Blot Analysis
Whole cell fractions were obtained, as previously described.22 Protein concentrations were measured with the DC protein assay (Bio‐Rad Laboratories, Hercules, CA). Equal amounts of protein were loaded into lanes of Criterion TGX (Tris‐Glycine eXtended) Stain‐Free PAGE gels (BioRad). The gels were electrophoresed and activated using a ChemiDoc MP Visualization System (BioRad). The protein was then transferred to a polyvinylidene difluoride membrane. The membranes were then imaged using a ChemiDoc MP Visualization System to obtain an assessment of proper transfer and to obtain total protein loads. The membranes were then blocked and probed with primary antibodies overnight at 4°C. Immunoblots were next processed with secondary antibodies (Cell Signaling) for 1 hour at room temperature. Immunoblots were then probed with a Super Signal West Dura kit (Thermo Fisher Scientific) to visualize signal, followed by visualization using a ChemiDoc MP Visualization System (BioRad). Data were analyzed using Image Lab (BioRad). The total protein images were used as loading controls. For each protein of interest, the portion of the protein load image corresponding to the molecular weight of the protein of interest was used as the loading control.23
Histological Analysis and CD31 Staining
Hearts were harvested and fixed in 10% formalin and embedded in paraffin. Slices were cut at 7 μm and stained with Masson trichrome (Millipore Sigma, Burlington, MA).17 Fibrosis area was quantitatively analyzed with National Institutes of Health Image software. Immunohistochemistry was performed to visualize vascular density with a commercially available kit (Blood Vessel Staining Kit; Millipore).24 Primary antibody against CD31 (Abcam; ab28364; 1:50) was used. Digital images were obtained with a microscope, and CD31‐positive vessels were counted using ImageJ. CD31‐positive vessels per mm2 were calculated to evaluate the number of vessels per field.
Immunofluorescence
Frozen sections (7 μm in thickness) were prepared and stained with a lymphatic vessel endothelial hyaluronan receptor 1 (LYVE1) antibody to detect lymph vessels.25 These methods were followed by incubation with secondary antibodies. Coverslips were mounted using Vectashield H‐1500‐4′, 6‐diamidino‐2‐phenylindole–containing medium (Vector Laboratories).25 Images were acquired on a Leica DM6000. B lymphocytes were detected by staining sections with antibodies against CD45R (B220) and immunoglobulin M. Proliferating lymph endothelial cells were evaluated by staining sections with antibodies against 5‐bromo‐2′‐deoxyuridine and LYVE1.
Inflammatory Cytokines
The levels of tumor necrosis factor‐α, interleukin‐1β, and interleukin‐6 were evaluated in heart homogenates using ELISA kits (eBioscience; No. 88‐7324‐22, No. 88‐7013‐22, and No. 88‐7064‐22, respectively), according to the manufacturer's instructions.
Hyaluronic Acid Measurements
The levels of hyaluronic acid were evaluated in heart homogenates using an ELISA kit (Hyaluronan Quantikine ELISA Kit; No. DHYAL0; R&D Systems), according to the manufacturer's instructions.
Gravimetry
Cardiac water content was evaluated by the wet weight–dry weight method after desiccation of the heart tissue for 5 days at 65°C, as previously described.13
Lymph Vessel Function
To evaluate lymphatic flow, a fluorescently labeled macromolecule (2000‐kDa fluorescein isothiocyanate–dextran; 2 mg/mL; Pierce Thermo)26 was injected intramuscularly into the apex of the heart. Because of its size, the tracer is taken up by the lymphatics. After 30 minutes, a blood sample was collected to determine how much of the tracer had moved through the cardiac lymphatics into the circulation. The arbitrary units of fluorescence intensity for each sample were measured with a plate reader. The amount of tracer (μg) in each sample was calculated using a standard curve of fluorescein isothiocyanate–dextran.
Statistical Analysis
All data are expressed as mean±SEM. The data were first evaluated for normal distribution using the D'Agostina and Pearson omnibus normality test. Subsequent statistical significance was evaluated as follows: (1) an unpaired Student t test for comparison between 2 means; and (2) a 1‐way ANOVA with a Tukey test or Dunnett's multiple comparison test as the post hoc analysis for comparison among ≥3 groups. For the echocardiography data, a 2‐way repeated‐measures ANOVA with a Bonferroni test as the post hoc analysis was used. The following comparisons were made separately: (1) baseline versus postbaseline measurements for each group, (2) differences between each group's baseline measurements, and (3) differences between each group's postbaseline measurements. The P value for these evaluations was adjusted by applying the Bonferroni correction for multiple comparisons. P<0.05 denoted statistical significance, and P values were 2 sided. All statistical analysis was performed using Prism 5 (GraphPad Software Inc).
Results
Kinetics of Lymphangiogenesis Early After the Onset of Myocardial Ischemia
Previous studies report that myocardial ischemia induces an endogenous lymphangiogenesis response.12, 13 However, the kinetics of the response have not been evaluated during the early period after the onset of ischemia. We, therefore, addressed this issue herein. For these experiments, mice were subjected to permanent myocardia ischemia and followed up for up to 7 days. First, we assessed the protein expression of VEGF‐C and VEGFR3 in heart homogenates obtained from mice subjected to various periods of ischemia (Figure 1). The expression of VEGF‐C was significantly increased 1 day after the onset of ischemia. This increase persisted for up to 7 days of ischemia. The expression of VEGFR3 was increased from 3 to 7 days of ischemia. Next, we evaluated the remodeling of the cardiac lymphatics by focusing on the lymph density in the subendocardium (an area that experiences a robust increase in lymphatic density in response to myocardial ischemia13). Our analysis revealed a significant increase in the number of LYVE1‐positive cells starting at 3 days of ischemia that persisted for up to 7 days of ischemia (Figure 2A and 2B). To investigate if the observed increase in LYVE1‐positive cells was indicative of lymphatic cell proliferation, we treated a subset of mice with 5‐bromo‐2′‐deoxyuridine starting after the onset of myocardial ischemia. Analysis revealed a significant increase in the number of LYVE1‐positive cells labeled with 5‐bromo‐2′‐deoxyuridine starting at 3 days of ischemia, with a gradual increase noted at 7 days of ischemia (Figure 2C). Finally, we also evaluated the diameter of lymphatic precollectors in the epicardium as an evaluation of lymphatic drainage capacity.13 Our analysis revealed a significant increase in lumen area starting at 1 day of ischemia that persisted for up to 7 days of ischemia (Figure 2A and 2D).
Figure 1.
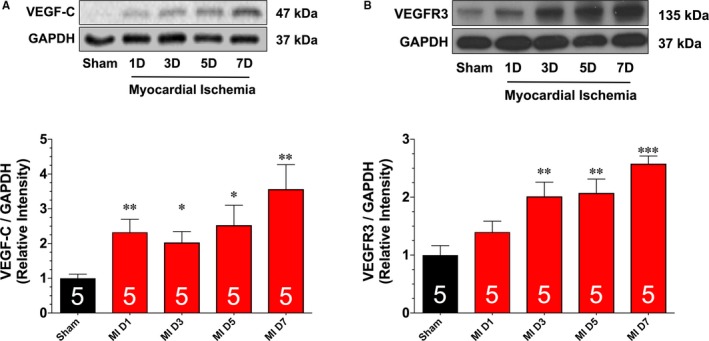
Representative immunoblots and analysis of vascular endothelial growth factor C (VEGF‐C; A) and VEGF receptor 3 (VEGFR3; B) protein expression levels in samples collected from hearts subjected to different periods of ischemia. Numbers in bars indicate sample sizes. Values are means±SEM. D indicates day; MI, myocardial ischemia. *P<0.05, **P<0.01, and ***P<0.001 vs sham.
Figure 2.
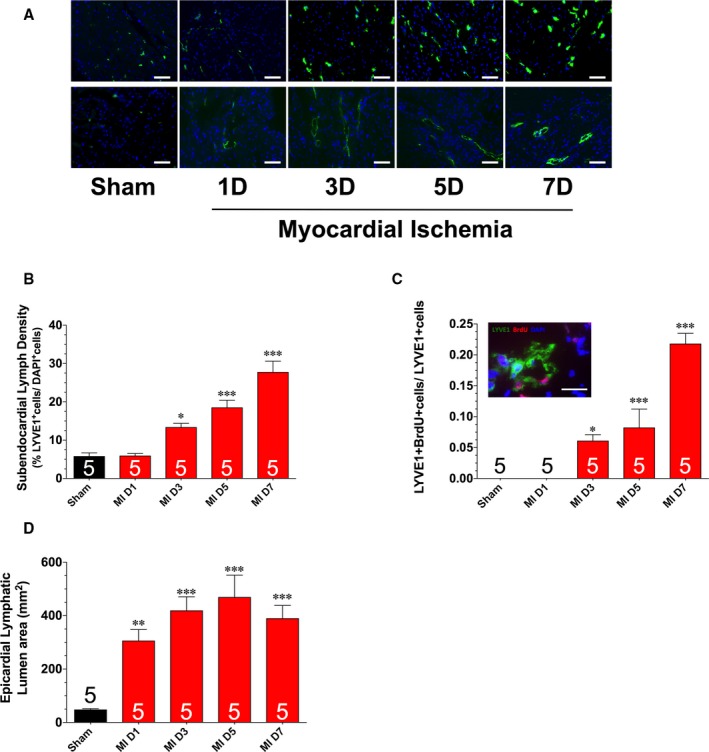
A, Representative images of left ventricular sections stained with an anti‐LYVE1 antibody to denote LYVE1‐positive cells (top panels) and lymphatic collecting vessel luminal area (bottom panels). B, Summary of LYVE1‐positive cells. C, Summary of LYVE1‐positive cells labeled with 5‐bromo‐2′‐deoxyuridine (BrdU). D, Lymphatic lumen area. Samples were collected from hearts subjected to different periods of ischemia. Bars: 50 μm (A); 20 μm (C). Values are means±SEM. D indicates day; DAPI, 6‐diamidino‐2‐phenylindole; MI, myocardial ischemia. *P<0.05, **P<0.01 and ***P<0.001 vs sham.
Kinetics of Lymphangiogenesis in the Setting of Myocardial Ischemia‐Reperfusion Injury
The next series of experiments evaluated the kinetics of the lymphangiogenesis response in a more clinically relevant model of myocardial ischemia‐reperfusion injury. For these experiments, mice were subjected to 60 minutes of myocardial ischemia, followed by up to 7 days of reperfusion. First, we assessed the protein expression of VEGF‐C and VEGFR3 in heart homogenates obtained from mice subjected to ischemia and to various periods of reperfusion (Figure 3). The expression of VEGF‐C was significantly increased 1 day after reperfusion. This increase persisted for up to 7 days of reperfusion, with a peak elevation observed at 3 days of reperfusion. The expression of VEGFR3 was likewise increased from 1 to 7 days of reperfusion. However, the increase was similar at all times evaluated. Additional analysis revealed a significant increase in the number of LYVE1‐positive cells starting at 3 days of reperfusion that persisted for up to 7 days of reperfusion (Figure 4A and 4B). Finally, we noted a significant increase in epicardial lymphatic lumen area starting at 3 days of reperfusion that persisted for up to 7 days of reperfusion (Figure 4A and 4C).
Figure 3.
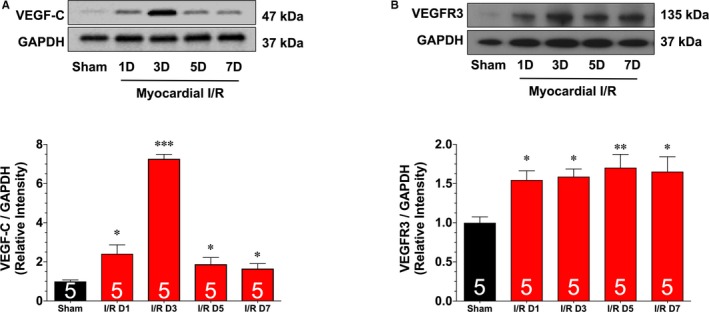
Representative immunoblots and analysis of vascular endothelial growth factor C (VEGF‐C; A) and VEGF receptor 3 (VEGFR3; B) protein expression levels in samples collected from hearts subjected to 60 minutes of myocardial ischemia and different periods of reperfusion. Values are means±SEM. D indicates day; I/R, ischemia and reperfusion. *P<0.05, **P<0.01, and ***P<0.001 vs sham.
Figure 4.
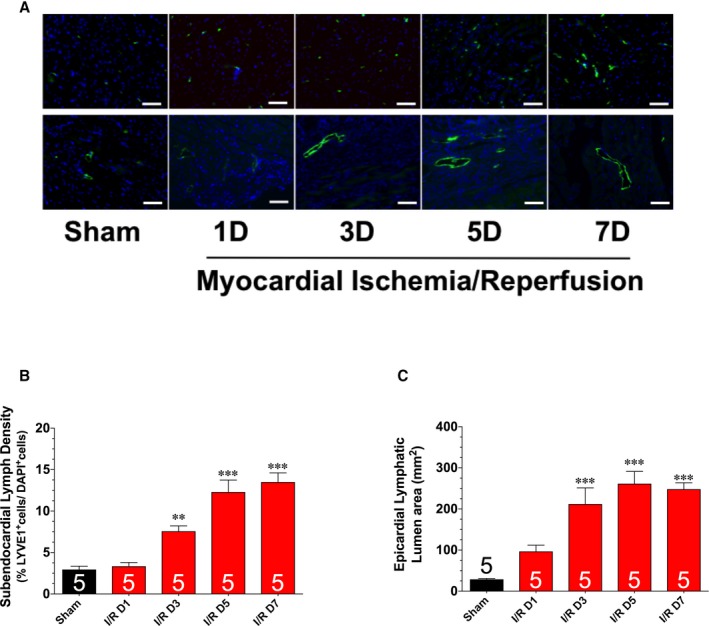
A, Representative images of left ventricular sections stained with an anti‐LYVE1 antibody to denote LYVE1‐positive cells (top panels) and lymphatic collecting vessel luminal area (bottom panels). Summary of LYVE1‐positive cells (B) and lymphatic lumen area (C). Samples were collected from hearts subjected to 60 minutes of myocardial ischemia and different periods of reperfusion. Bar=50 μm. Values are means±SEM. D indicates day; I/R, ischemia and reperfusion. **P<0.01 and ***P<0.001 vs sham.
MAZ‐51 and VEGF‐C NAb Impair the Endogenous Lymphangiogenesis Response Induced by Myocardial Ischemia and Reperfusion
As noted, previous studies have reported that treatment with VEGF‐C augments endogenous lymphangiogenesis and leads to improvements in cardiac function in rat models of permanent myocardial ischemia and myocardial ischemia‐reperfusion.12, 13 However, there is not any information available linking the consequences of inhibiting the endogenous lymphangiogenesis response to the development of heart failure. We, therefore, addressed this issue using 2 different pharmacological approaches. In the first set of experiments, mice were subjected to 60 minutes of ischemia, followed by reperfusion. At the time of reperfusion, a hydrogel containing the VEGFR3 inhibitor, MAZ‐51, was placed on the surface of the infarcted myocardium before the closing of the chest cavity. Mice treated with MAZ‐51 displayed a significant decrease in subendocardial lymph density at 7 days of reperfusion when compared with vehicle‐treated mice (Figure 5A and 5B). In view of this attenuation of the endogenous lymphangiogenic response, we investigated the effects of MAZ‐51 on cardiac lymphatic function. For this assessment, we injected a fluorescently labeled macromolecule (fluorescein isothiocyanate–dextran) intramuscularly into the apex of the heart at 7 days of reperfusion. Because of its size, the tracer is taken up by the lymphatics.26 After 30 minutes, a blood sample was collected to determine how much of the tracer had moved through the cardiac lymphatics into the circulation. Our analysis revealed that MAZ‐51 blunted the lymphatic transport when compared with vehicle‐treated mice (Figure 5D). The decrease in lymphatic transport was further confirmed by the enhanced accumulation of hyaluronic acid (surrogate of lymphatic drainage27) in the MAZ‐51–treated hearts (Figure 5E).
Figure 5.
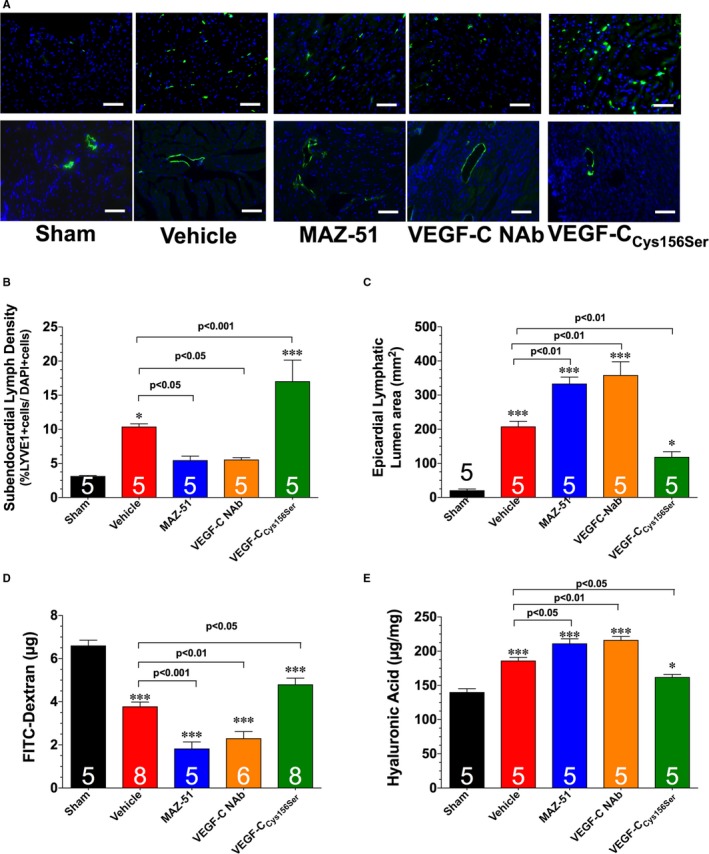
A, Representative images of left ventricular sections stained with an anti‐LYVE1 antibody to denote LYVE1‐positive cells (top panels) and lymphatic collecting vessel luminal area (bottom panels). Summary of LYVE1‐positive cells (B) and lymphatic lumen area (C). D, Levels of circulating fluorescein isothiocyanate (FITC)–dextran. E, Levels of cardiac hyaluronic acid. Samples were collected from hearts subjected to 60 minutes of myocardial ischemia and 1 week of reperfusion. Different groups of mice were treated with vehicle, MAZ‐51, vascular endothelial growth factor C (VEGF‐C) neutralizing antibody (NAb), or VEGF‐CCys156Ser. Bar=50 μm. Values are means±SEM. *P<0.05 and ***P<0.001 vs sham.
Given that the lymphatic system functions in the clearance of inflammatory cells and edema,28, 29, 30 we next evaluated the relationship in the development of lymphatic vessels and the inflammatory response after MAZ‐51 treatment. First, we observed a significant increase in the diameter of epicardial lymphatic precollectors in MAZ‐51–treated hearts when compared with vehicle‐treated mice (Figure 5A and 5C). This was associated with the accumulation of B lymphocytes (detected as B220 and immunoglobulin M double‐positive cells)31 and an increase in the levels of tumor necrosis factor‐α, interleukin‐1β, and interleukin‐6, as well as an increase in cardiac water content (Figure 6). In the second set of experiments, different groups of mice had a hydrogel containing a VEGF‐C NAb placed on the surface of the infarcted myocardium. In a similar manner to MAZ‐51, treatment with VEGF‐C NAb led to a decrease in subendocardial lymph density (Figure 5A and 5B), a decrease in lymphatic transport (Figure 5D and 5E), an increase in the diameter of epicardial lymphatic precollectors (Figure 5A and 5C), an increase in the inflammatory response (Figure 6A through 6D), and an increase in water content (Figure 6F) at 7 days of reperfusion.
Figure 6.
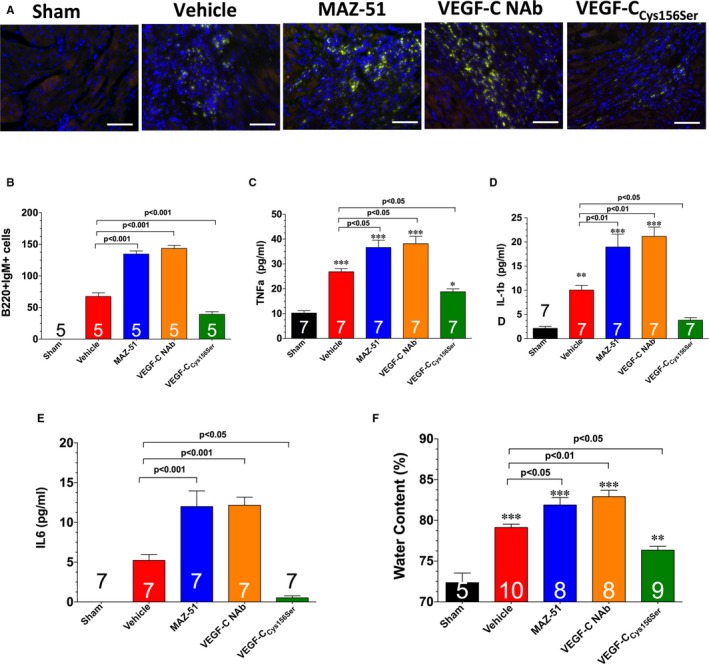
A, Representative images of left ventricular sections stained with B220 and immunoglobulin M to denote inflammatory cells. Bar=50 μm. B, Summary of B220‐ and immunoglobulin M–positive cells. Levels of tumor necrosis factor (TNF)‐α (C), interleukin‐1β (D), and interleukin‐6 (E). F, Cardiac water content. Samples were collected from hearts subjected to 60 minutes of myocardial ischemia and 1 week of reperfusion. Different groups of mice were treated with vehicle, MAZ‐51, vascular endothelial growth factor C (VEGF‐C) neutralizing antibody (NAb), or VEGF‐CC ys156Ser. Values are means±SEM. *P<0.05, **P<0.01, and ***P<0.001 vs sham.
Blocking Endogenous Lymphangiogenesis Response Exacerbates Ischemic‐Induced Heart Failure
Postischemic cardiac remodeling and dysfunction are closely linked to inflammation.32 We, therefore, investigated the effects of MAZ‐51 and VEGF‐C NAb treatment on cardiac remodeling and dysfunction. Analysis at 28 days of reperfusion revealed that both treatments increased infarct scar size, increased ventricular hypertrophy, increased left ventricular dilatation, and decreased left ventricular function (Figure 7).
Figure 7.
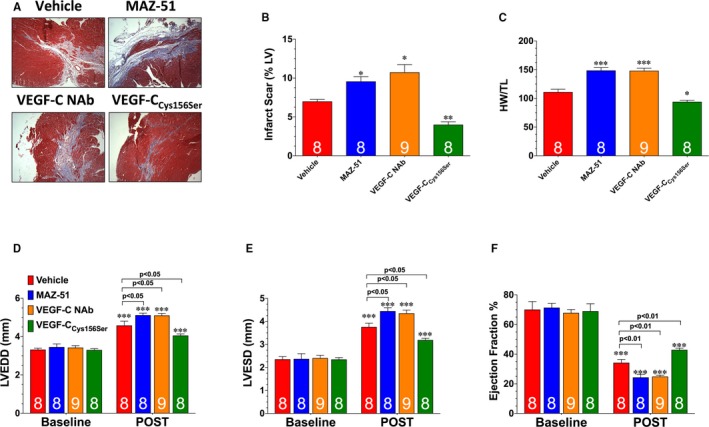
A, Representative images of heart sections stained with Masson's trichrome to denote infarct scar. B, Infarct scar area as a percentage of left ventricular area. C, Heart weight/tibia length (HW/TL) ratios. Left ventricular (LV) end‐diastolic diameter (LVEDD; D), LV end‐systolic diameter (LVESD; E), and LV ejection fraction (F) were measured in groups of mice using echocardiography images 4 weeks after myocardial ischemia and reperfusion (POST). Values are means±SEM. NAb indicates neutralizing antibody; VEGF‐C, vascular endothelial growth factor C. *P<0.05, **P<0.01, and ***P<0.001 vs vehicle (B and C); ***P<0.001 vs baseline (D‐F).
Enhancing Endogenous Lymphangiogenesis Response Attenuates Ischemic‐Induced Heart Failure
Next, we sought to determine the therapeutic potential of a VEGF‐C releasing hydrogel in the setting of myocardial ischemia‐reperfusion injury. For these experiments, mice were subjected to 60 minutes of ischemia, followed by reperfusion. At the time of reperfusion, a hydrogel containing VEGF‐CCys156Ser (a mutant form that specifically binds to VEGFR333) was placed on the surface of the infarcted myocardium before the closing of the chest cavity. Mice treated with VEGF‐CCys156Ser displayed a significant increase in subendocardial lymph density at 7 days of reperfusion when compared with vehicle‐treated mice (Figure 5A and 5B). This was associated with an increase in lymphatic transport (Figure 5D and 5E); a decrease in the diameter of epicardial lymphatic precollectors (Figure 5A and 5C); a decrease in the accumulation of B lymphocytes (Figure 6A and 6B); a decrease in the levels of tumor necrosis factor‐α, interleukin‐1β, and interleukin‐6 (Figure 6C through 6E); and a decrease in cardiac water content (Figure 6F) when compared with vehicle‐treated mice. Further analysis at 28 days of reperfusion revealed that VEGF‐CCys156Ser treatment decreased infarct scar size, decreased ventricular hypertrophy, decreased left ventricular dilatation, and increased left ventricular function (Figure 7).
Treatment With Hydrogels Containing MAZ‐51, VEGF‐C NAb, or VEGF‐CCys156Ser Does Not Alter Acute Myocardial Infarction
To determine if the observed changes in lymphangiogenesis and cardiac function were attributable to alterations in the initial development of myocardial infarction, subsequent groups of mice were euthanized at 24 hours of reperfusion. Analysis revealed that mice treated with hydrogels containing vehicle, MAZ‐51, VEGF‐C NAb, or VEGF‐CCys156Ser displayed a similar area at risk relative to left ventricle, as well as similar infarct areas relative to the area at risk and left ventricle (Figure 8A through 8C).
Figure 8.
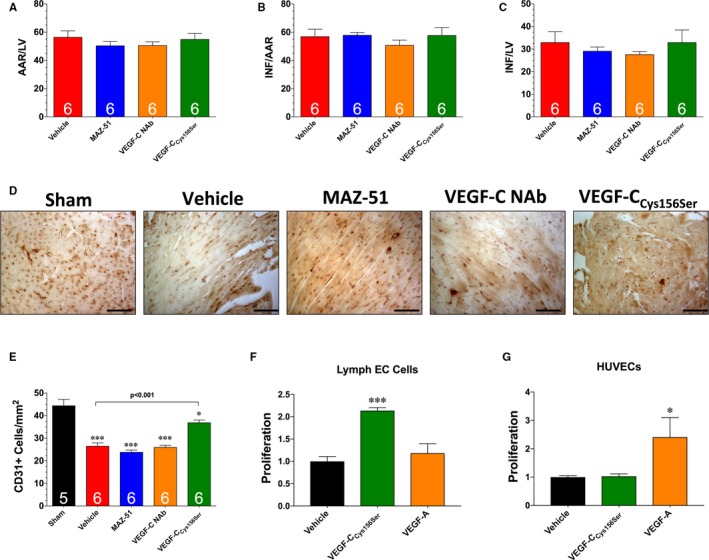
A, Myocardial area at risk (AAR) as a percentage of total left ventricle (LV). B, Infarct size (INF) as a percentage of AAR. C, INF as a percentage of LV. D, Representative images of heart sections stained with CD31. Bar=50 μm. E, Number of CD31‐positive cells per mm2. Proliferation of lymph endothelial cells (ECs; F) and human umbilical vein ECs (HUVECs; G) in response to treatment with 100 ng/mL of either vascular endothelial growth factor C (VEGF‐C)Cys156Ser or VEGF‐A. Values are means±SEM of 3 independent experiments of 3 biological replicates. NAb indicates neutralizing antibody. *P<0.05 and ***P<0.001 vs sham (E); *P<0.05 and ***P<0.001 vs vehicle (F and G).
VEGF‐CCys156Ser Attenuates Ischemia‐Reperfusion–Induced Blood Vascular Rarefication
Additional studies were performed to determine if treatment with hydrogels containing MAZ‐51, VEGF‐C NAb, or VEGF‐CCys156Ser affected blood vessel density after myocardial ischemia‐reperfusion. For these experiments, mice were subjected to 60 minutes of ischemia, followed by reperfusion for 7 days. Analysis revealed that myocardial ischemia‐reperfusion caused a significant decrease in the number of CD31+ vessels in the hearts of vehicle‐treated mice (Figure 8D and 8E). Hearts treated with MAZ‐51 or VEGF‐C NAb displayed a similar decrease as observed in the vehicle‐treated mice. In contrast, treatment with VEGF‐CCys156Ser attenuated the ischemia‐reperfusion–induced vascular rarefication. In view of these changes, further studies were conducted to determine if VEGF‐CCys156Ser could directly alter vascular endothelial cell proliferation. For this evaluation, cultured lymph endothelial cells and HUVECs were stimulated with 100 ng/mL of either VEGF‐CCys156Ser or VEGF‐A (Figure 8F and 8G). Analysis revealed that VEGF‐CCys156Ser induced proliferation in lymph endothelial cells, but not in HUVECs. In contrast, VEGF‐A induced proliferation in HUVECs, but not in lymph endothelial cells. This suggests that the effects of VEGF‐CCys156Ser on ischemia‐reperfusion–induced vascular rarefication are likely indirect.
Discussion
The molecular and cellular events initiated within hours to days after the onset of myocardial infarction dictate the ultimate consequences of the injury. Therefore, it is important to understand early signaling events that contribute to myocardial damage and repair.34 After the onset of ischemic injury, cardiomyocytes undergo irreversible injury, leading to cell death. The initiation of a highly regulated inflammatory response consisting of neutrophils and monocytes/macrophages occurs within hours after the insult to remove dead cells and matrix debris.35, 36, 37 This initial inflammatory phase is followed by the proliferation of vascular cells and matrix‐depositing fibroblasts, the formation of granulation tissue, and the replacement of this tissue with a collagen‐rich scar. These events serve to stabilize the infarct scar and prevent cardiac rupture. However, if these events are left unchecked, the infiltrating inflammatory cells can secrete enzymes, free radicals, and cytokines into the uninjured, remote myocardium, resulting in matrix destabilization, infarct expansion, and left ventricle dilatation.35, 36, 37 This ultimately contributes to the development of heart failure. As a result, the temporal sequence of events occurring after the onset of ischemic injury must be finely tuned to promote healing while at the same time minimizing adverse remodeling.34
In recent years, there has been a growing interest in lymphatic research because of several key breakthroughs into the biological characteristics of lymphatic endothelial cells. First, a critical breakthrough to a functional understanding was achieved with the discovery of the main lymphangiogenic growth factor, VEGF‐C, and its receptor, VEGFR3.38 This was followed by the identification of proteins that discriminate between endothelial cells of the blood and lymphatic vessel lineages (ie, LYVE‐1), which can serve as selective markers.39 These discoveries lead to the gradual understanding that lymphatic vessels serve an active role in disease processes ranging from inflammation to metastatic spreading of cancer.28, 29, 30 Moreover, stimulation of lymphangiogenesis in diseased settings with VEGF‐C or adipose‐derived regenerative cells has been shown to reduce edema and inflammation.17, 40, 41 This has led to the idea that therapeutic lymphangiogenesis is a viable clinical option for several diseased states.
The functional significance of the cardiac lymphatic system was first demonstrated by the observation that surgical ligation of a lymphatic vessel in the canine heart induced edema, cardiac fibrosis, and cardiac dysfunction.10 This was further substantiated by the observations that obstructing lymphatic flow after myocardial ischemia exacerbated ischemic‐induced edema, fibrosis, and cardiac dysfunction.11 However, until recently, few advances in our understanding of the physiological role of the cardiac lymphatic system have been made. In the past several years, 2 elegant studies have independently demonstrated that myocardial ischemia induces an endogenous lymphangiogenesis response.12, 13 In the current study, we confirmed these results in mouse models of myocardial ischemia and ischemia‐reperfusion injury. In our study, we focused on the kinetics of the endogenous lymphangiogenesis response during the early periods after the onset of either ischemia or reperfusion. Although we found a similar response in regard to an increase in the protein expression of VEGF‐C and VEGFR3 preceding an increase in subendocardial lymph density, there was a subtle difference between the 2 models. Specifically, we observed a difference in the kinetics of VEGF‐C expression. In the ischemia model, VEGF‐C was elevated to a similar level at all times evaluated. In contrast, the expression of VEGF‐C increased dramatically at 3 days of reperfusion before declining to a lower level by 5 days of reperfusion. Currently, the underlying cause for this difference is not known. However, the difference did not seem to influence the endogenous lymphangiogenesis response, because a similar pattern and onset of an increase in subendocardial lymph density was observed in both models. Our findings are in slight contrast with those previously reported.12, 13 Specifically, the previous studies reported that the endogenous lymphangiogenesis response occurs weeks after the onset of myocardial ischemia. Currently, we do not completely understand the conflicting observations about the timing of the response. However, the previous studies used a rat model of myocardial ischemia and focused on later time points. Herein, we used a mouse model and focused on the early periods after the onset of ischemia or reperfusion. Despite the differences in timing, all 3 studies clearly indicate that the response occurs.
In agreement with the 2 previous studies,12, 13 we also found that treatment with a mutated version of VEGF‐C augmented the endogenous lymphangiogenesis response and attenuated ischemic‐induced cardiac dysfunction. Specifically, we found that VEGF‐CCys156Ser treatment increased ischemic‐induced subendocardial lymph density and decreased inflammation and edema at 7 days of reperfusion. This was associated with a reduction in scar formation and improvement in cardiac function at 28 days of reperfusion. Importantly, these effects were found to be independent of the initial development of infarction. More important, we provide direct evidence that blocking the endogenous lymphangiogenesis response exacerbates cardiac injury and dysfunction after myocardial ischemia‐reperfusion. As noted, signaling via VEGF‐C and VEGFR3 is the most central pathway for lymphangiogenesis. Herein, we found that inhibiting VEGFR3 with MAZ‐51 blunted the ischemic‐induced increase in subendocardial lymph density, increased inflammation/edema, increased scar formation, and increased cardiac dysfunction. Likewise, targeting endogenous VEGF‐C with an NAb disrupted the endogenous lymphangiogenesis response and exacerbated cardiac injury. Together, these data suggest that the endogenous lymphangiogenesis response plays an adaptive role in response to myocardial ischemia‐reperfusion injury.
There is some evidence to suggest that remodeling of the lymphatic system in response to myocardial ischemia leads to transient lymphatic transport dysfunction.13 Specifically, these impairments were noted to contribute to the development of chronic myocardial edema and inflammation. However, augmentation of the endogenous lymphangiogenesis response has consistently proved effective in reducing cardiac dysfunction after myocardial ischemia. Together with our new evidence that blocking the response exacerbates myocardial injury, it can be suggested that the initial response plays a beneficial role. However, it seems that the response is not sufficient to alleviate the development of heart failure. As such, more work is needed to determine the most effective means to augment or complement the response during the early period after the onset of myocardial ischemia (ie, early reperfusion).
We anticipate that the clinical use of therapeutic lymphangiogenesis will not be achieved by systemic delivery, but rather local delivery. As such, the use of hydrogels could be a viable delivery strategy. Because of their highly tunable chemical, physical, and mechanical properties, hydrogels have been widely used as a tool in regenerative medicine.42, 43 In regards to the heart, hydrogel‐based materials have been used as a structural/mechanical support for the injured myocardium44 and have also been used as a means to deliver small molecules.45 The ability to fine‐tune the properties of the hydrogel provides the unique opportunity to control the delivery of pharmacological agents.43 Herein, we used a hydrogel to deliver VEGF‐CCys156Ser to the injured myocardium by placing the gel on the surface of the heart. Although this strategy might not be clinically feasible, it does show proof of concept that the delivery mechanism is viable. As such, future approaches, such as injecting the VEGF‐C–containing hydrogel directly into the myocardium, are warranted.
Although the current study demonstrates that the endogenous lymphangiogenesis response plays an adaptive role in the development of ischemic‐induced heart failure, there are a few alternative mechanisms that need to be noted. First, although VEGF‐C has been described to be a relatively specific growth factor for the lymphatic system, there is some evidence that VEGF‐C can induce proliferation of blood vessels (ie, angiogenesis).46, 47 Herein, we found that treatment with VEGF‐CCys156Ser attenuated ischemia‐reperfusion–induced blood vascular rarefication. Although this evidence does indicate that VEGF‐CCys156Ser alters the density of blood vessels in the heart, we believe that this evidence is not indicative of angiogenesis. This is supported by the finding that treatment with MAZ‐51 or VEGF‐C NAb did not cause further blood vascular rarefication when compared with vehicle‐treated hearts and the finding that VEGF‐CCys156Ser did not induce proliferation in HUVECs. It is more likely that treatment with VEGF‐CCys156Ser creates an environment whereby less blood vessel dropout occurs. Alternatively, the environment in the VEGF‐CCys156Ser–treated heart could be more conducive for angiogenesis to occur. Either way, the attenuation of blood vascular rarefication certainly contributes to the beneficial effects of VEGF‐CCys156Ser. With that being said, more work is warranted to determine the mechanism by which VEGF‐CCys156Ser attenuated ischemia‐reperfusion–induced blood vascular rarefication. Second, there is also some evidence from the literature that blood can flow through lymphatic vessels.48 Intriguingly, the hemodynamic forces created by the flow of blood through the lymphatic vessels can reprogram the lymphatic vessels to become blood vessels. As such, it is possible that ischemic‐induced lymphangiogenesis can create an alternative means to deliver blood to the damaged tissue. Whether this can occur in the heart or in the setting of ischemia‐reperfusion injury is unknown. Additionally, technical limitations related to the visualization of lymphatic vessels in the mouse heart and the findings that reprogrammed lymphatic vessels maintain their identity in vivo (ie, LYVE1‐positive staining)48 make the evaluation of this reprogramming difficult. As such, future studies need to be conducted in large animal models to determine if this is a mechanism by which VEGF‐CCys156Ser and/or lymphangiogenesis protect the heart from the development of ischemic‐induced heart failure. Third, there is evidence that VEGFR3 is expressed on cardiomyocytes and myofibroblasts.49 As such, it is possible that treatment with MAZ‐51, VEGF‐C NAb, or VEGF‐CCys156Ser could have directly affected signaling in the cardiomyocytes and/or fibroblasts during the remodeling phase. As such, future studies are needed to fully understand how VEGF‐C/VEGFR3 signaling in these different cell types affects the development of ischemic‐induced heart failure.
In summary, the findings of the current study provide evidence that the endogenous lymphangiogenesis response is induced within 3 days after myocardial ischemia. Moreover, we provide evidence that the response plays an adaptive role in the development of ischemic‐induced heart failure by enhancing lymphatic drainage. Thus, our findings support the emerging concept that therapeutic lymphangiogenesis is a promising new approach for the treatment of cardiovascular disease.13
Sources of Funding
This work was funded by grants from the American Heart Association (15POST25610016 to Shimizu, 17GRNT33670975 to Naqvi, and 16GRNT31190016 to Calvert) and the National Institutes of Health (R01DK115213 and R01HL136915 to Calvert). This work was also supported by funding from the Carlyle Fraser Heart Center of Emory University Hospital Midtown (Calvert).
Disclosures
None.
(J Am Heart Assoc. 2018;7:e009565 DOI: 10.1161/JAHA.118.009565.)
References
- 1. Kerjaschki D. The lymphatic vasculature revisited. J Clin Invest. 2014;124:874–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zheng W, Aspelund A, Alitalo K. Lymphangiogenic factors, mechanisms, and applications. J Clin Invest. 2014;124:878–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Karkkainen MJ, Haiko P, Sainio K, Partanen J, Taipale J, Petrova TV, Jeltsch M, Jackson DG, Talikka M, Rauvala H, Betsholtz C, Alitalo K. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol. 2004;5:74–80. [DOI] [PubMed] [Google Scholar]
- 4. Veikkola T, Jussila L, Makinen T, Karpanen T, Jeltsch M, Petrova TV, Kubo H, Thurston G, McDonald DM, Achen MG, Stacker SA, Alitalo K. Signalling via vascular endothelial growth factor receptor‐3 is sufficient for lymphangiogenesis in transgenic mice. EMBO J. 2001;20:1223–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. 2007;8:464–478. [DOI] [PubMed] [Google Scholar]
- 6. Alitalo K, Tammela T, Petrova TV. Lymphangiogenesis in development and human disease. Nature. 2005;438:946–953. [DOI] [PubMed] [Google Scholar]
- 7. Levick JR, Michel CC. Microvascular fluid exchange and the revised starling principle. Cardiovasc Res. 2010;87:198–210. [DOI] [PubMed] [Google Scholar]
- 8. Aspelund A, Robciuc MR, Karaman S, Makinen T, Alitalo K. Lymphatic system in cardiovascular medicine. Circ Res. 2016;118:515–530. [DOI] [PubMed] [Google Scholar]
- 9. Jones D, Min W. An overview of lymphatic vessels and their emerging role in cardiovascular disease. J Cardiovasc Dis Res. 2011;2:141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kline IK, Miller AJ, Katz LN. Cardiac LYMPH flow impairment and myocardial fibrosis: effects of chronic obstruction in dogs. Arch Pathol. 1963;76:424–433. [PubMed] [Google Scholar]
- 11. Kline IK, Miller AJ, Pick R, Katz LN. The effects of chronic impairment of cardiac lymph flow on myocardial reactions after coronary artery ligation in dogs. Am Heart J. 1964;68:515–523. [DOI] [PubMed] [Google Scholar]
- 12. Klotz L, Norman S, Vieira JM, Masters M, Rohling M, Dube KN, Bollini S, Matsuzaki F, Carr CA, Riley PR. Cardiac lymphatics are heterogeneous in origin and respond to injury. Nature. 2015;522:62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Henri O, Pouehe C, Houssari M, Galas L, Nicol L, Edwards‐Levy F, Henry JP, Dumesnil A, Boukhalfa I, Banquet S, Schapman D, Thuillez C, Richard V, Mulder P, Brakenhielm E. Selective stimulation of cardiac lymphangiogenesis reduces myocardial edema and fibrosis leading to improved cardiac function following myocardial infarction. Circulation. 2016;133:1484–1497; discussion 1497. [DOI] [PubMed] [Google Scholar]
- 14. Murphy E, Lagranha C, Deschamps A, Kohr M, Nguyen T, Wong R, Sun J, Steenbergen C. Mechanism of cardioprotection: what can we learn from females? Pediatr Cardiol. 2011;32:354–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tabata Y, Nagano A, Ikada Y. Biodegradation of hydrogel carrier incorporating fibroblast growth factor. Tissue Eng. 1999;5:127–138. [DOI] [PubMed] [Google Scholar]
- 16. Yamamoto M, Takahashi Y, Tabata Y. Controlled release by biodegradable hydrogels enhances the ectopic bone formation of bone morphogenetic protein. Biomaterials. 2003;24:4375–4383. [DOI] [PubMed] [Google Scholar]
- 17. Shimizu Y, Shibata R, Shintani S, Ishii M, Murohara T. Therapeutic lymphangiogenesis with implantation of adipose‐derived regenerative cells. J Am Heart Assoc. 2012;1:e000877 DOI: 10.1161/JAHA.112.000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Benedito R, Rocha SF, Woeste M, Zamykal M, Radtke F, Casanovas O, Duarte A, Pytowski B, Adams RH. Notch‐dependent VEGFR3 upregulation allows angiogenesis without VEGF‐VEGFR2 signalling. Nature. 2012;484:110–114. [DOI] [PubMed] [Google Scholar]
- 19. Shimizu Y, Nicholson CK, Lambert JP, Barr LA, Kuek N, Herszenhaut D, Tan L, Murohara T, Hansen JM, Husain A, Naqvi N, Calvert JW. Sodium sulfide attenuates ischemic‐induced heart failure by enhancing proteasomal function in an NRF2‐dependent manner. Circ Heart Fail. 2016;9:e002368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Calvert JW, Elston M, Nicholson CK, Gundewar S, Jha S, Elrod JW, Ramachandran A, Lefer DJ. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia‐induced heart failure in mice. Circulation. 2010;122:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Naqvi N, Li M, Calvert JW, Tejada T, Lambert JP, Wu J, Kesteven SH, Holman SR, Matsuda T, Lovelock JD, Howard WW, Iismaa SE, Chan AY, Crawford BH, Wagner MB, Martin DI, Lefer DJ, Graham RM, Husain A. A proliferative burst during preadolescence establishes the final cardiomyocyte number. Cell. 2014;157:795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Calvert JW, Jha S, Gundewar S, Elrod JW, Ramachandran A, Pattillo CB, Kevil CG, Lefer DJ. Hydrogen sulfide mediates cardioprotection through NRF2 signaling. Circ Res. 2009;105:365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barr LA, Shimizu Y, Lambert JP, Nicholson CK, Calvert JW. Hydrogen sulfide attenuates high fat diet‐induced cardiac dysfunction via the suppression of endoplasmic reticulum stress. Nitric Oxide. 2015;46:145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, Murohara T, Predmore BL, Gojon G Sr, Gojon G Jr, Wang R, Karusula N, Nicholson CK, Calvert JW, Lefer DJ. H(2)s protects against pressure overload‐induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation. 2013;127:1116–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shimizu Y, Shibata R, Ishii M, Ohashi K, Kambara T, Uemura Y, Yuasa D, Kataoka Y, Kihara S, Murohara T, Ouchi N. Adiponectin‐mediated modulation of lymphatic vessel formation and lymphedema. J Am Heart Assoc. 2013;2:e000438 DOI: 10.1161/JAHA.113.000438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kashiwagi S, Hosono K, Suzuki T, Takeda A, Uchinuma E, Majima M. Role of Cox‐2 in lymphangiogenesis and restoration of lymphatic flow in secondary lymphedema. Lab Invest. 2011;91:1314–1325. [DOI] [PubMed] [Google Scholar]
- 27. Cui Y, Liu K, Monzon‐Medina ME, Padera RF, Wang H, George G, Toprak D, Abdelnour E, D'Agostino E, Goldberg HJ, Perrella MA, Forteza RM, Rosas IO, Visner G, El‐Chemaly S. Therapeutic lymphangiogenesis ameliorates established acute lung allograft rejection. J Clin Invest. 2015;125:4255–4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alitalo K. The lymphatic vasculature in disease. Nat Med. 2011;17:1371–1380. [DOI] [PubMed] [Google Scholar]
- 29. Bruyere F, Noel A. Lymphangiogenesis: in vitro and in vivo models. FASEB J. 2010;24:8–21. [DOI] [PubMed] [Google Scholar]
- 30. Karaman S, Detmar M. Mechanisms of lymphatic metastasis. J Clin Invest. 2014;124:922–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zouggari Y, Ait‐Oufella H, Bonnin P, Simon T, Sage AP, Guerin C, Vilar J, Caligiuri G, Tsiantoulas D, Laurans L, Dumeau E, Kotti S, Bruneval P, Charo IF, Binder CJ, Danchin N, Tedgui A, Tedder TF, Silvestre JS, Mallat Z. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med. 2013;19:1273–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Joukov V, Kumar V, Sorsa T, Arighi E, Weich H, Saksela O, Alitalo K. A recombinant mutant vascular endothelial growth factor‐C that has lost vascular endothelial growth factor receptor‐2 binding, activation, and vascular permeability activities. J Biol Chem. 1998;273:6599–6602. [DOI] [PubMed] [Google Scholar]
- 34. Rainer PP, Hao S, Vanhoutte D, Lee DI, Koitabashi N, Molkentin JD, Kass DA. Cardiomyocyte‐specific transforming growth factor beta suppression blocks neutrophil infiltration, augments multiple cytoprotective cascades, and reduces early mortality after myocardial infarction. Circ Res. 2014;114:1246–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jung K, Kim P, Leuschner F, Gorbatov R, Kim JK, Ueno T, Nahrendorf M, Yun SH. Endoscopic time‐lapse imaging of immune cells in infarcted mouse hearts. Circ Res. 2013;112:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339:161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Frangogiannis NG. The mechanistic basis of infarct healing. Antioxid Redox Signal. 2006;8:1907–1939. [DOI] [PubMed] [Google Scholar]
- 38. Lohela M, Bry M, Tammela T, Alitalo K. VEGFS and receptors involved in angiogenesis versus lymphangiogenesis. Curr Opin Cell Biol. 2009;21:154–165. [DOI] [PubMed] [Google Scholar]
- 39. Tammela T, Alitalo K. Lymphangiogenesis: molecular mechanisms and future promise. Cell. 2010;140:460–476. [DOI] [PubMed] [Google Scholar]
- 40. Szuba A, Skobe M, Karkkainen MJ, Shin WS, Beynet DP, Rockson NB, Dakhil N, Spilman S, Goris ML, Strauss HW, Quertermous T, Alitalo K, Rockson SG. Therapeutic lymphangiogenesis with human recombinant VEGF‐C. FASEB J. 2002;16:1985–1987. [DOI] [PubMed] [Google Scholar]
- 41. Kataru RP, Jung K, Jang C, Yang H, Schwendener RA, Baik JE, Han SH, Alitalo K, Koh GY. Critical role of CD11b+ macrophages and VEGF in inflammatory lymphangiogenesis, antigen clearance, and inflammation resolution. Blood. 2009;113:5650–5659. [DOI] [PubMed] [Google Scholar]
- 42. Hastings CL, Roche ET, Ruiz‐Hernandez E, Schenke‐Layland K, Walsh CJ, Duffy GP. Drug and cell delivery for cardiac regeneration. Adv Drug Deliv Rev. 2015;84:85–106. [DOI] [PubMed] [Google Scholar]
- 43. Slaughter BV, Khurshid SS, Fisher OZ, Khademhosseini A, Peppas NA. Hydrogels in regenerative medicine. Adv Mater. 2009;21:3307–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fujimoto KL, Ma Z, Nelson DM, Hashizume R, Guan J, Tobita K, Wagner WR. Synthesis, characterization and therapeutic efficacy of a biodegradable, thermoresponsive hydrogel designed for application in chronic infarcted myocardium. Biomaterials. 2009;30:4357–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zustiak SP, Wei Y, Leach JB. Protein‐hydrogel interactions in tissue engineering: mechanisms and applications. Tissue Eng Part B Rev. 2013;19:160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cao Y, Linden P, Farnebo J, Cao R, Eriksson A, Kumar V, Qi JH, Claesson‐Welsh L, Alitalo K. Vascular endothelial growth factor c induces angiogenesis in vivo. Proc Natl Acad Sci USA. 1998;95:14389–14394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chien MH, Ku CC, Johansson G, Chen MW, Hsiao M, Su JL, Inoue H, Hua KT, Wei LH, Kuo ML. Vascular endothelial growth factor‐C (VEGF‐C) promotes angiogenesis by induction of COX‐2 in leukemic cells via the VEGF‐R3/JNK/AP‐1 pathway. Carcinogenesis. 2009;30:2005–2013. [DOI] [PubMed] [Google Scholar]
- 48. Chen CY, Bertozzi C, Zou Z, Yuan L, Lee JS, Lu M, Stachelek SJ, Srinivasan S, Guo L, Vicente A, Mericko P, Levy RJ, Makinen T, Oliver G, Kahn ML. Blood flow reprograms lymphatic vessels to blood vessels. J Clin Invest. 2012;122:2006–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhao T, Zhao W, Chen Y, Liu L, Ahokas RA, Sun Y. Differential expression of vascular endothelial growth factor isoforms and receptor subtypes in the infarcted heart. Int J Cardiol. 2013;167:2638–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]