

Abstract
Background
Dilated cardiomyopathy (DCM) is a common cause of heart failure in adult and pediatric patients, but the underlying mechanism may vary in adults and children, with few studies conducted to date. The objective of the present study was to determine whether differential remodeling of the extracellular matrix contributes to the differences between pediatric and adult DCM hearts.
Methods and Results
Explanted hearts were procured from adult (age, 46–61 years) and pediatric (age, 2–8) patients with DCM‐related heart failure and nonfailing control hearts. Fibrillar and nonfibrillar extracellular matrix (proteoglycans, glycosaminoglycans, glycoprotein), their regulatory enzymes (matrix metalloproteinases, disintegrin and metalloproteinases, and disintegrin and metalloproteinases with a thrombospondin domain), and their inhibitors (tissue inhibitor of metalloproteinases) were assessed. Pediatric DCM hearts exhibited less fibrosis compared with adult DCMs. Total glycosaminoglycans increased similarly in both DCM groups but exhibited a significantly lower affinity for transforming growth factor‐β in adult DCMs versus pediatric DCMs, resulting in increased bioavailability of transforming growth factor‐β1 and a significantly higher activity of the Smad2/3 pathway in adult DCMs. Glycosylated biglycan and versican, and cleaved thrombospondin‐1 increased in both DCMs. Protein expression of disintegrin and metalloproteinases with thrombospondin domains (‐1, ‐2, ‐4, ‐7) and disintegrin and metalloproteinases (‐12, ‐15, ‐17, ‐19) were altered differently in pediatric and adult control and failing hearts. Total matrix metalloproteinase activity increased in both DCMs. Tissue inhibitor of metalloproteinase levels were altered similarly with heart failure in both age groups, and only tissue inhibitor of metalloproteinase 3 decreased in both DCM groups.
Conclusions
Differential remodeling of glycosaminoglycans in pediatric DCMs versus adult DCMs could underlie the enhanced activation of the transforming growth factor‐β pathway, leading to more fibrosis in adult DCM hearts. The distinct remodeling of the fibrillar and nonfibrillar extracellular matrix between pediatric and adult DCM hearts highlights a distinct pathophysiological basis for these cohorts.
Keywords: cardiomyopathy, heart failure, remodeling
Subject Categories: Fibrosis, Myocardial Biology, Pathophysiology
Clinical Perspective
What Is New?
Composition of the fibrillar and nonfibrillar extracellular matrix is different in the failing adult heart compared with the failing pediatric heart.
Myocardial fibrosis is a prominent feature of the failing adult but not the failing pediatric heart.
Glycosaminoglycans are important components of proteoglycans and sequester growth factors in the extracellular matrix.
Total glycosaminoglycan content is increased similarly in adult and pediatric failing hearts.
Affinity of glycosaminoglycans to sequester transforming growth factor‐β is suppressed to a greater extent in the adult failing hearts, which could underlie the greater fibrosis in these hearts.
What Are the Clinical Implications?
Myocardial fibrosis is a central feature of failing dilated cardiomyopathy hearts in adults, and limiting myocardial fibrosis using anti–transforming growth factor‐β treatment is a potential therapeutic strategy.
By identifying that failing pediatric hearts do not develop fibrosis, linked to lower bioavailability of transforming growth factor‐β, this study provides novel insight into the phenotype of the failing pediatric heart.
The differential extracellular matrix remodeling could also partly explain why pediatric patients with heart failure are less responsive to therapies used to treat adults with heart failure.
Heart failure (HF) is an important cause of morbidity and mortality in adult and pediatric patients, and, in both cases, idiopathic dilated cardiomyopathy (DCM) is one of the most common underlying causes. In the pediatric age group, DCM is the most common underlying cause of HF resulting in cardiac transplantation.1, 2 Because of a paucity of clinical trials in children with HF, current guidelines for the management of pediatric HF due to DCM are often based on the information extrapolated from clinical trials in adults.3 This approach does not take into account the age‐related intrinsic differences and the biological and pathological factors that drive this disease in these 2 markedly different patient groups. Consistent with this notion, while treatments for HF in adult patients have reduced mortality, the same therapies (eg, angiotensin‐converting enzyme inhibitors and β‐blockers) have not shown definite benefits for pediatric patients.4, 5 Therefore, it is important to identify whether adult and pediatric DCM are biologically distinct disease entities6 with age and maturation specific features in the heart, which may modify the treatment response to HF therapy in pediatric patients.
DCM in adults is associated with extensive and progressive adverse structural remodeling of the left ventricle, eventually leading to clinical HF. While numerous studies have explored the alterations in cardiomyocyte function in the adult failing heart, the noncellular component of the myocardium, the extracellular matrix (ECM), has remained less investigated, particularly in the pediatric patient population. In addition to its fibrillar structure, ECM is composed of nonfibrillar components such as glycoproteins, proteoglycans, and glycosaminoglycans that allow the ECM to serve as an extracellular reservoir for growth factors, hormones, and cytokines.7 Proteoglycans (eg, syndecan, versecan, perlecan, decorin, and aggrecan) consist of a core protein to which ≥1 linear glycosaminoglycan chains are covalently attached. Glycosaminoglycans are unbranched polysaccharides including chondroitin sulfate, heparan sulfate, dermatan sulfate, and hyaluronic acid. Alterations in the nonfibrillar ECM in the failing heart and how this may differ in children with DCM is not well understood.
Physiological remodeling of the ECM is mediated by matrix metalloproteinases (MMPs) that degrade the existing matrix proteins, allowing their replacement with newly synthesized proteins. The activity of MMPs is negatively regulated by their inhibitor, tissue inhibitor of metalloproteinases (TIMPs).8 Recent studies have revealed diverse functions of MMPs,9 as well as non–MMP‐dependent functions for TIMPs10, 11; therefore, changes in these proteins as they happen in heart disease may impact multiple aspects of remodeling in the diseased hearts. In addition to MMPs, other metalloproteinases such as disintegrin and metalloproteinases (ADAMs) and ADAMs with a thrombospondin domain (ADAMTS) can contribute to proteolytic processing of proteoglycans and glycoproteins in the myocardium. In this study, we investigated whether the remodeling of fibrillar and nonfibrillar ECM differs in the pediatric failing heart compared with that in adult hearts.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Human Tissue Procurement From Nonfailing Controls and Patients With HF
Adult and pediatric failing heart specimens (DCM) were procured from patients with nonischemic DCM undergoing cardiac transplantation. Adult nonfailing control (NFC) heart samples (ejection fraction ≥60%) were obtained from donors with no history of heart disease who were unsuitable for transplant because of failure of ABO blood groups or human leukocyte antigen blood type matching through the regional organ procurement agency, the Human Organ Procurement and Exchange program, University of Alberta, Edmonton, Canada. Pediatric NFC specimens were procured from pediatric patients with congenital heart disorders (tetralogy of Fallot, double outlet right ventricle, pulmonary atresia), with normal systolic left ventricular and right ventricular function (median ejection fraction, 60.5%; interquartile range, 51–75.2). These were obtained via clinically indicated surgical resection at the time of corrective surgery (median age, 1 year; interquartile range, 1–7). Failing adult and pediatric DCM hearts were collected through the Human Explanted Heart Program.12 The study was approved by the institutional review committee/human research ethics board at the University of Alberta. Informed consent and assent was obtained from participants (or next of kin). The procured explanted failing (or NFC) hearts were excised following cold cardioplegia, placed in a bag of cold saline surrounded by ice. Hearts were then collected by 2 trainees within 5 to 10 minutes of its excision and were immediately processed. Hearts were dissected into right and left atrium, septum, and left and right ventricular free walls, which were further divided into apical, basal, and midwall regions. Tissues were immediately flash‐frozen and stored at −140°C for molecular analyses; formalin‐fixed (room temperature) or optimal cutting temperature–mount frozen (stored at −80°C) were used for histochemical analyses. For this study, only tissue from left ventricular anterior free wall at the midventricular position (≈2 cm below the aortic valve) was used following removal of the epicardial fat. Demographic details and clinical information for adult and pediatric patients are listed in Table S1. In this study, 15 NFC and 24 DCM adult hearts, and 12 NFC and 15 DCM pediatric hearts were analyzed.
Glycosaminoglycan Analyses
Glycosaminoglycan contents were measured using a modified previously described protocol.13 Briefly, an equal amount of frozen mid–left ventricular free wall tissue (50 mg) was homogenized in extraction buffer (50 mmol/L Tris‐HCl, pH 7.9, 10 mmol/L NaCl, 3 mmol/L MgCl2, and 1% TritonX‐100) at 4°C. The extracts were treated with proteinase K (final concentration of 50 μg/mL) at 56°C overnight, followed by proteinase K inactivation (incubation at 90°C, 30 minutes), and DNase treatment (30 U/mL at 37°C, overnight). Samples were then centrifuged (13 000g, 20 minutes) and supernatants were collected. For total sulfated glycosaminoglycan quantification, 20 μL of each sample treated with proteinase K/DNase (in triplicates) was incubated with 200 μL of 1,9‐dimethylmethylene blue (DMMB, Sigma‐Aldrich) solution (1.6% w/v DMMB, 0.3% w/v glycine, 0.16% w/v NaCl in 0.01 M acetic acid) and was read at 525 nm, along with a chondroitin sulfate standard curve. For quantification of heparan sulfate content, proteinase K/DNase–treated samples were incubated with chondroitinase ABC (20 mU/sample, 2 hours at 37°C). Samples (20 μL, in triplicate) were incubated with DMMB solution and read at 525 nm, along with a heparan sulfate standard curve. For quantification of chondroitin sulfate, proteinase K/DNase‐treated samples were incubated with a heparinase I, II, and III cocktail using 2.5 mU/sample for 24 hours at 37°C. Samples (20 μL, in triplicate) were incubated with 200 μL of DMMB solution and read at 525 nm, along with a chondroitin sulfate standard curve.
Glycosaminoglycans Affinity and Binding Assay to TGF‐β1
The glycosaminoglycan–transforming growth factor (TGF)‐β1 affinity assay was performed using a previously reported protocol.13 Total glycosaminoglycans were extracted from proteinase K/DNase–treated left ventricular tissue and quantified as previously mentioned. Precoated heparan sulfate 96‐well plates were blocked with 3% BSA in PBS for 1 hour. Recombinant TGF‐β1 (10 ng/mL) was incubated simultaneously with or without 100 ng of extracted glycosaminoglycans from pediatric or adult hearts for 2 hours followed by thorough washing. The amount of bound TGF‐β1 was detected with a primary antibody, followed by peroxidase‐labeled secondary antibody and detected with 3,3′,5,5′‐tetramethylbenzidine substrate.
Protein Extraction and Analyses
Flash‐frozen left ventricular tissues were freeze‐crushed in tissue lysis buffer containing EDTA‐free protease inhibitor cocktails and were processed for protein extraction, and gelatin zymography and immunoblotting were performed as previously described.12 Antibodies used for immunoblotting were as follows: anti‐ADAM12 (ab115203; Abcam), ADAM15 (ab124698; Abcam), ADAM17 (ab19027; Millipore), ADAM19 (ab104800; Abcam), ADAMTS1 (ab113847; Abcam), ADAMTS2 (ab125226; Abcam), ADAMTS4 (ab185722; Abcam), ADAMTS5 (ab41037; Abcam), ADAMTS7 (ab45044; Abcam), ADAMTS9 (ab32565; Abcam), TIMP1 (ab1827; Abcam), TIMP2 (ab1828; Abcam), TIMP3 (250885; Ab‐Biotech), TIMP4 (ab58425; Abcam), versican (ab171887; Abcam), osteopontin (ab8448; Abcam), TGF‐β1 (3711S, Cell Signaling Technology), phospho‐Smad2/3 (8828S, Cell Signaling Technology), total Smad2/3 (3102S, Cell Signaling Technology), biglycan (sc‐100857; Santa Cruz Biotechnology), decorin (sc‐73896; Santa Cruz Biotechnology), lumican (sc‐166871; Santa Cruz Biotechnology), tenascin X (sc‐271594; Santa Cruz Biotechnology), γ‐sacrcoglycan (sc‐515628; Santa Cruz Biotechnology), osteoglycin (sc‐365228; Santa Cruz Biotechnology), thrombospondin‐1 (ab85762, Abcam), and GAPDH (#2118; Cell Signaling Technology). Band intensities were quantified using densitometry analysis software (ImageQuant TL 7.0; GE) and values were normalized to GAPDH expressions for each sample.
Histological Analyses
Each heart sample was formalin‐fixed and paraffin‐embedded or flash‐frozen in mounting compound (optimal cutting temperature). Histochemical staining for collagen was performed with Picrosirius Red staining or trichrome staining as previously stated.12 For Alcian blue staining, all formalin‐fixed tissue sections were rehydrated and incubated with Alcian blue solution (0.1% w/v Alcian blue 8x, in 5% w/v aluminum sulfate solution, pH 2.5) for 30 minutes, followed by rinsing in distilled water for 5 minutes. The slides were then counterstained with a nuclear fast red solution for 5 minutes followed by rinsing in distilled water, dehydrated, and mounted.
Statistical Analyses
All statistical analyses were performed using SPSS software (IBM). Data distribution was determined by the Shapiro–Wilk statistic test. Nonparametric data were analyzed using the Kruskal–Wallis test followed by the Dunn test, and presented as median, maximum, and minimum using box‐and‐whisker graphs. Parametric data were analyzed using 1‐way ANOVA followed by the Tukey test and presented as mean±SEM in bar graphs. Statistical significance was recognized at P<0.05.
Results
Clinical Profile of Pediatric and Adult Patients With HF
Clinical information for pediatric (median age, 4 years; interquartile range, 3–6) and adult patients (median age, 41 years; interquartile range, 40–61) with nonischemic DCM HF is summarized in Table S1. Ejection fraction was comparable between the 2 failing groups (median, 16.9% in children and 22% in adults) and the 2 NFC hearts (median ≥60%). Among the adult patients with DCM, 70.8% received an angiotensin‐converting enzyme inhibitor or an angiotensin receptor blocker and 75% received β‐blockers, compared with 60% and 53.3%, respectively, for pediatric patients with DCM.
More Severe Interstitial Fibrosis in Adult DCM Than in Pediatric DCM Hearts
Based on a high degree of interaction predicted between the ECM proteins, ECM‐degrading proteases, and their inhibitors by STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) analysis (Figure 1A), and the reported importance of fibrosis in adult DCM,12, 14, 15 we investigated whether pediatric failing hearts would exhibit the same degree of fibrosis as adult failing hearts. Fibrillar collagen was visualized by Picrosirius Red (Figure 1B) and trichrome (Figure 1C) staining and quantified from Picrosirius Red images (Figure 1D). Both DCM groups showed increased collagen content compared with their corresponding NFCs. However, the degree of fibrosis was significantly higher in adult compared with pediatric failing hearts (Figure 1B through 1D).
Figure 1.
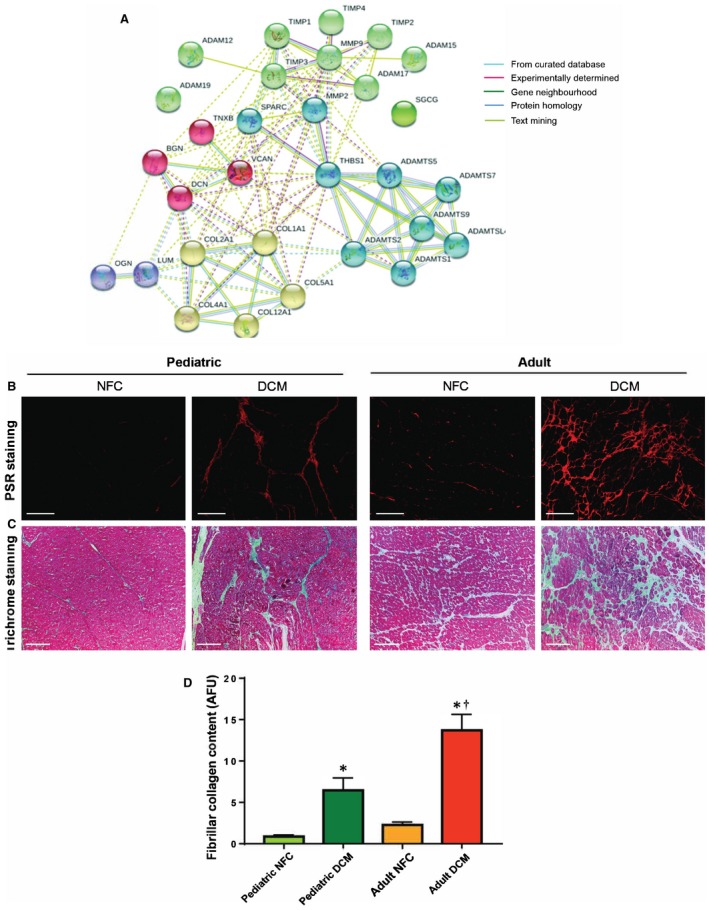
High‐confidence interaction among extracellular matrix (ECM)–related proteins and greater fibrosis in adult dilated cardiomyopathy (DCM) hearts compared with pediatric DCM hearts. A, STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) analysis shows high‐confidence interaction clusters (k‐means=5 clusters indicated by node color) formed by ECM‐related proteins. B through D, Representative images of Picrosirius Red (PSR) (B) and Masson trichrome–stained (C) left ventricular free wall from pediatric and adult control and DCM myocardium. Scale bar: 100 μm. D, Averaged quantification of collagen content (from PSR‐stained sections) in indicated groups (mean±SEM). n=6 hearts per group. *P<0.05 compared with the corresponding control, † P<0.05 compared with the corresponding pediatric group. ADAM indicates a disintegrin and metalloprotease; ADAMTS, a disintegrin and metalloproteinase with thrombospondin motifs; AFU, arbitrary fluorescence units; BGN, biglycan; COL, collagen; DCN, decorin; LUM, lumican; MMP, matrix metalloprotease; NFC, nonfailing control; OGN, osteoglycan; SGCG, sarcoglycan gamma; SPARC, secreted protein acidic and cysteine‐rich; THBS1, thrombosondin‐1; TIMP, tissue inhibitors of metalloprotease; TNXB, tenascin x; VCAN, versican.
Glycosaminoglycan Levels Are Increased in Both Pediatric and Adult DCM Hearts
Glycosaminoglycans are polysaccharides that constitute the side branches of proteoglycans and are one of the major nonfibrillar components of the myocardial ECM. Biochemical assessment of total sulfated glycosaminoglycans and its 2 major components, chondroitin sulfate and heparan sulfate, showed similar levels in the control groups, which increased significantly in the failing DCM hearts in both age groups (Figure 2A through 2C). Results of histochemical staining further confirmed accumulation of glycosaminoglycans in the myocardium of the failing hearts from pediatric and adult patients (Figure 2D). Therefore, the glycosaminoglycan content of the ECM was comparably elevated in the failing hearts of both pediatric and adult patients.
Figure 2.
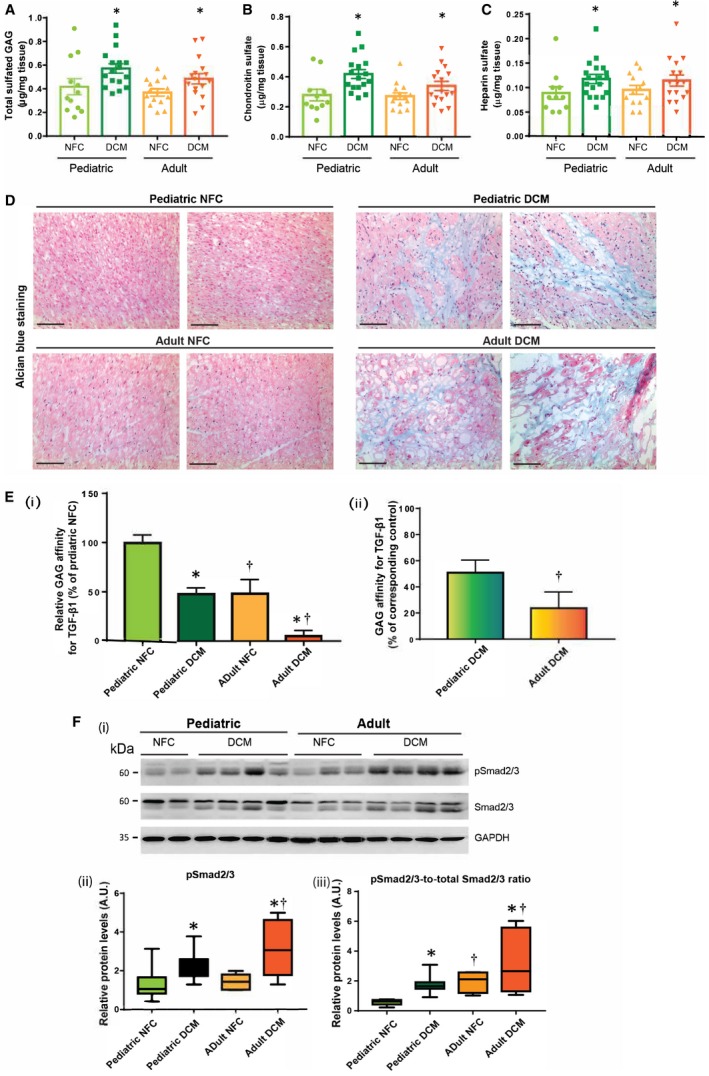
Remodeling of glycosaminoglycans in pediatric versus adult dilated cardiomyopathy (DCM) hearts. Quantification of total sulfated glycosaminoglycan (A), and its constituents, chondroitin sulfate (B) and heparan sulfate (C) in indicated groups. D, Representative images of Alcian blue–stained myocardium showing total proteoglycans/glycosaminoglycans in pediatric and adult nonfailing control (NFC) and DCM myocardium. Scale bar: 100 μm. n=6 per group. E, (i) Relative affinity of glycosaminoglycans extracted from pediatric and adult NFC and DCM hearts, for transforming growth factor (TGF)‐β1. (ii) Percent decrease in glycosaminoglycans–TGF‐β affinity in pediatric and adult DCM hearts compared with the corresponding NFC hearts. Data represent mean±SEM. F, Representative Western blots (i), and averaged quantification for phosphorylated Smad2/3 (p‐Smad2/3) (ii), and phospho‐to‐total ratio for Smad2/3 (iii). GAPDH served as the loading control. n=12 to 16 per group. Data represent median±interquartile range. *P<0.05 compared with the corresponding control, † P<0.05 compared with the corresponding pediatric group. AU indicates arbitrary units.
Binding Affinity of Glycosaminoglycans is Differently Altered in Pediatric Versus Adult DCM Hearts
Since glycosaminoglycans are responsible for sequestration of growth factors and cytokines in the interstitium,7, 16 we determined whether their binding affinity was altered differently in pediatric compared with adult failing hearts. This could imply an important growth factor sequestration effect, which may protect the developing heart. Using an established in vitro affinity assay,13 we found that age is an essential factor in glycosaminoglycan binding affinity for TGF‐β1, a major profibrotic cytokine. Glycosaminoglycans from adult nonfailing hearts showed a markedly lower affinity for TGF‐β1 compared with pediatric control hearts, and while this binding affinity was decreased with the disease in both age groups, this reduction was significantly greater in the adult DCM compared with the pediatric DCM hearts (Figure 2Ei and 2Eii). To confirm that this reduced binding affinity for TGF‐β1 translates into increased bioavailability of TGF‐β1 and enhanced activity of its downstream signaling pathway, we assessed the activity of the Smad2/3 pathway. Compared with controls, phospho‐Smad2/3 levels increased in the failing hearts of both age groups, but this rise was significantly greater in adult DCM compared with pediatric DCM hearts (Figure 2F). These data collectively suggest that the more drastic reduction in the binding affinity of glycosaminoglycans to TGF‐β in adult failing hearts results in elevated TGF‐β bioavailability, resulting in more pronounced fibrosis in these hearts compared with pediatric failing hearts.
Differential Expression of Proteoglycan and Glycoproteins in Nonfailing and Failing Pediatric and Adult Hearts
Next, we examined whether proteoglycans, which are associated with myocardial ECM homeostasis, are altered differently in pediatric versus adult DCM hearts. Versican is a chondroitin sulfate proteoglycan, highly expressed in the ventricles, and can be present in the glycosylated or nonglycosylated form. We found that the nonglycosylated core protein levels were significantly lower in failing adult compared with failing pediatric hearts (Figure 3A black arrow, Bi). Glycosylated versican, however, showed an opposite pattern with a rise in the adult failing hearts although it did not reach statistical significance (Figure 3A white arrows, 3Bi). Biglycan, decorin, lumican, and osteoglycin are members of the small leucine‐rich proteoglycans family. Biglycan protein levels were comparable in nonfailing hearts but were significantly decreased only in adult DCM hearts (Figure 3A, black arrow, 3Bii). Meanwhile, glycosylated biglycan (white arrow) increased significantly in both pediatric and adult failing hearts (Figure 3A, 3Bii). The increased glycosylation of versican and biglycan could be associated with the remodeling process of these failing hearts, while levels of core versican and biglycan remain relatively unaltered. Decorin remained unaltered among all groups (Figure 3A, 3Biii). Osteoglycin levels were higher in adult compared with pediatric control hearts, similar in the 2 failing heart groups (Figure 3A, 3Biv). Lumican levels were higher in pediatric compared with adult DCM hearts (Figure 3A, 3Bv).
Figure 3.
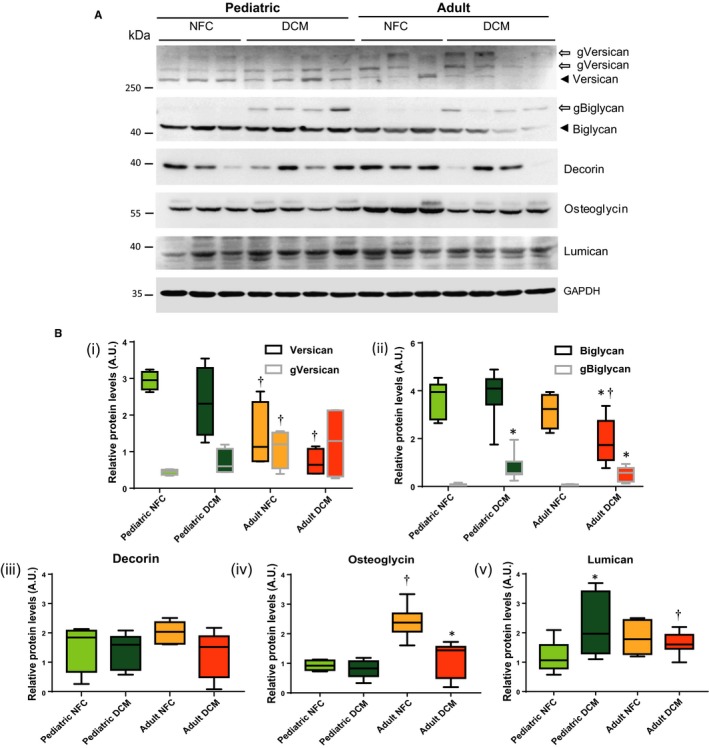
Proteoglycans are differentially regulated in pediatric and adult hearts. A, Representative Western blots for versican, biglycan, decorin, osteoglycin, and lumican. Black arrowheads indicate the core protein, while arrows indicate the glycosylated forms. B, Quantification of the protein levels for (i) versican core protein (black border box and whisker) and glycosylated form (gVersican, grey border box and whisker), (ii) biglycan (black border box and whisker) and glycosylated form (gBiglycan, grey border box and whisker), (iii) decorin, (iv) osteoglycin, and (v) lumican. GAPDH served as the loading control. n=12 to 16 per group. Data represent median±interquartile range. *P<0.05 compared with the corresponding control, † P<0.05 compared with the corresponding pediatric group. AU indicates arbitrary units; DCM, dilated cardiomyopathy; NFC, nonfailing control.
Glycoproteins and matricellular proteins comprise the nonstructural component of the ECM, but nevertheless are critical in determining the stability of the structural matrix proteins. Analysis of the key cardiac matricellular proteins revealed that thrombospondin‐1 was higher in control adult than pediatric hearts, whereas its cleaved form was increased significantly in both DCM groups (Figure 4A, 4Bi). Tenascin‐X was present at markedly higher levels in pediatric compared with adult DCM hearts, although it did not change with the disease in either age group (Figure 4A, 4Bii). Secreted protein acidic and cysteine‐rich levels decreased only in pediatric DCM hearts (Figure 4A, 4Biii). Osteopontin and γ‐sarcoglycan levels were not significantly different between pediatric and adult nor nonfailing and failing hearts (Figure 4A, 4Biv through 4Bv).
Figure 4.
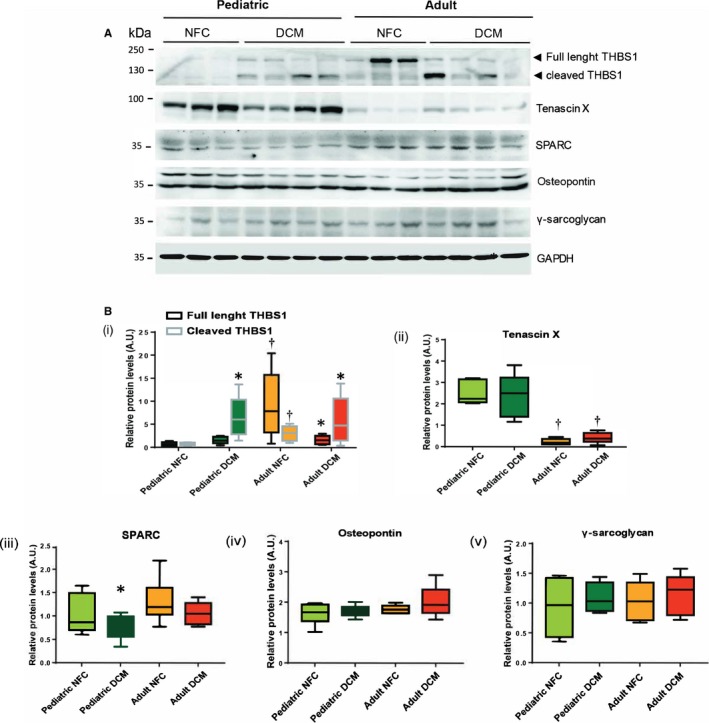
Protein levels of matricellular/glycoproteins in pediatric and adult nonfailing control (NFC) and dilated cardiomyopathy (DCM) hearts. A, Representative Western blots for thrombosondin‐1 (THBS1), tenascin‐x, secreted protein acidic and cysteine‐rich (SPARC), osteopontin, and γ‐sarcoglycan. B, Quantification of protein levels for indicated proteins (i through v). GAPDH serves as loading control. n=12 to 16 per group. Data represent median±interquartile range. *P<0.05 compared with the corresponding control, † P<0.05 compared with the corresponding pediatric group.
Divergent Modulation of ADAMs, ADAMTSs, and MMPs in Failing Pediatric Versus Adult Hearts
Proteoglycans can be degraded by proteases such as ADAMs, ADAMTSs, and MMPs.17, 18 We investigated whether the ADAMs and ADAMTSs being expressed in the heart18 were altered in the failing heart of pediatric versus adult patients. Among ADAMs, ADAM12 and ADAM17 increased significantly in adult compared with pediatric failing hearts, whereas ADAM15 decreased in both pediatric and adult failing hearts to comparable levels (Figure 5A, 5Bi through 5Biii). ADAM19 exhibited a different pattern in pediatric versus adult hearts. It increased significantly in the pediatric failing hearts but decreased in adult failing hearts compared with the corresponding NFCs (Figure 5A, 5Biv). ADAMTS1 was present at a higher level in nonfailing adults compared with nonfailing pediatric hearts, although it increased significantly in both failing groups to comparable levels (Figure 6A, 6Bi). Pediatric failing hearts exhibited higher levels of ADAMTS2 and ADAMTS7, whereas ADAMTS4 decreased significantly only in adult failing hearts and ADAMTS5 and ADAMTS9 remained comparable among all groups (Figure 6A, 6Bii through 6Bvi).
Figure 5.
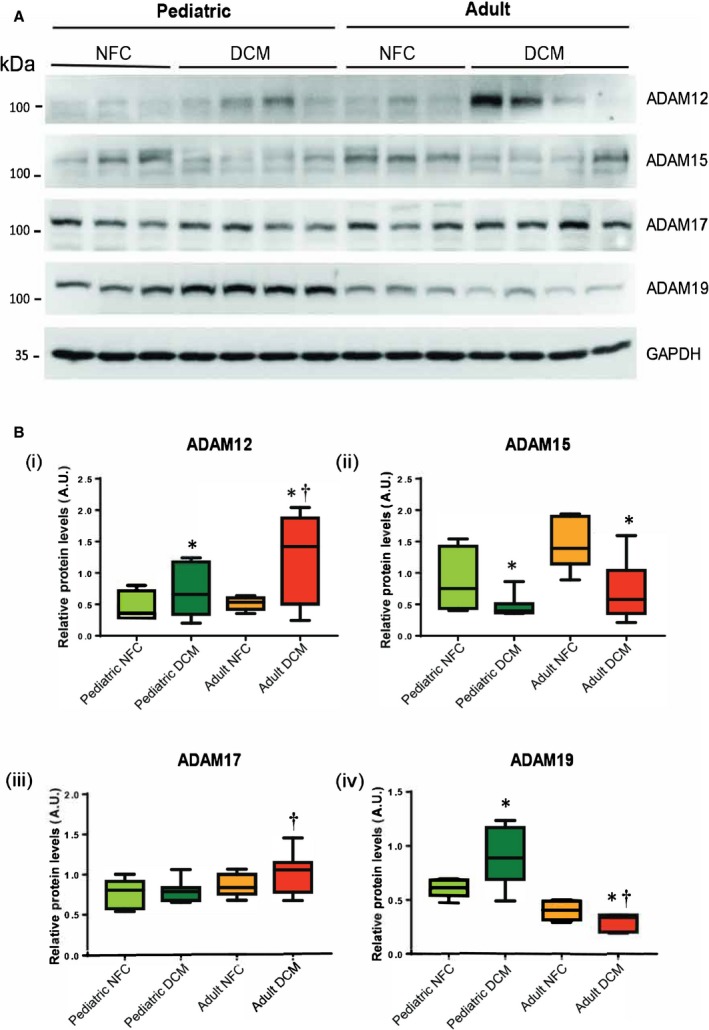
A disintegrin and metalloproteinases (ADAMs) in pediatric and adult control and failing hearts. A, Representative Western blots for myocardial ADAMs 12, 15, 17, and 19. B, Averaged quantification of protein levels for each ADAM (i through iv). GAPDH represents the loading control. n=12 to 16 per group. Data represent median±interquartile range. *P<0.05 compared with corresponding control, † P<0.05 compared with the corresponding pediatric group. AU indicates arbitrary units; DCM, dilated cardiomyopathy; NFC, nonfailing control.
Figure 6.
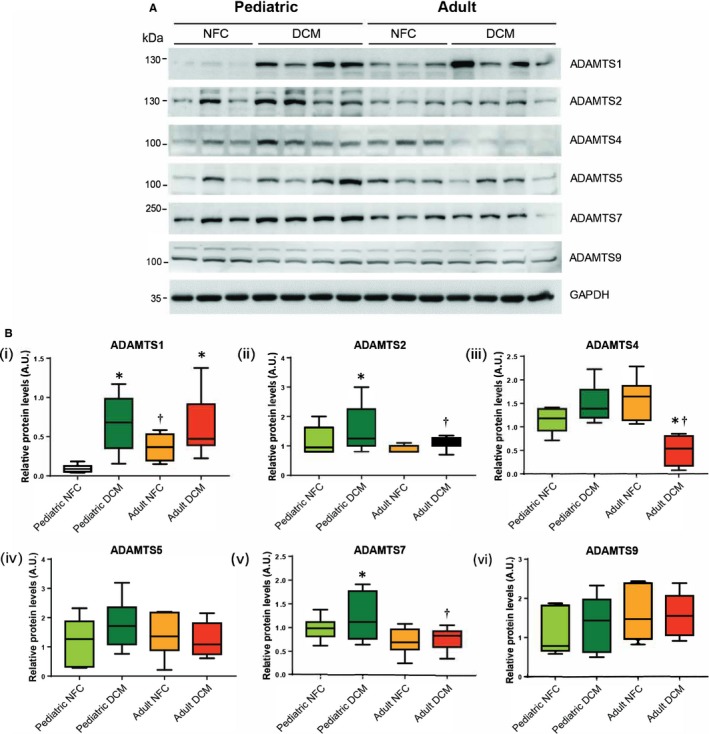
Alterations in a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTSs) protein expression in pediatric and adult control and failing hearts. Representative Western blots (A) and averaged protein quantification (B) for ADAMTSs 1, 2, 4, 5, 7, and 9. GAPDH served as the loading control. n=12 to 16 per group. Data represent median±interquartile range. *P<0.05 compared with corresponding control, † P<0.05 compared with corresponding pediatric group. AU indicates arbitrary units; DCM, dilated cardiomyopathy; NFC, nonfailing control.
Using a fluorescent‐based MMP activity assay, we found that total proteolytic activity was similarly enhanced in both pediatric and adult failing hearts compared with controls (Figure 7A). Further analyses using in vitro gelatin zymography showed that HF‐related rise in MMP9 was greater in adults compared with pediatric patients (Figure 7B, 7Ci). The increase in proMMP2 levels did not reach statistical significance in the failing hearts of either group (Figure 7B, 7Cii). Interestingly, pediatric hearts exhibited markedly higher levels of cleaved MMP2, which did not change with disease, whereas in adult hearts, cleaved MMP2 increased in the failing hearts although at a much lower level (Figure 7B, 7Ciii).
Figure 7.
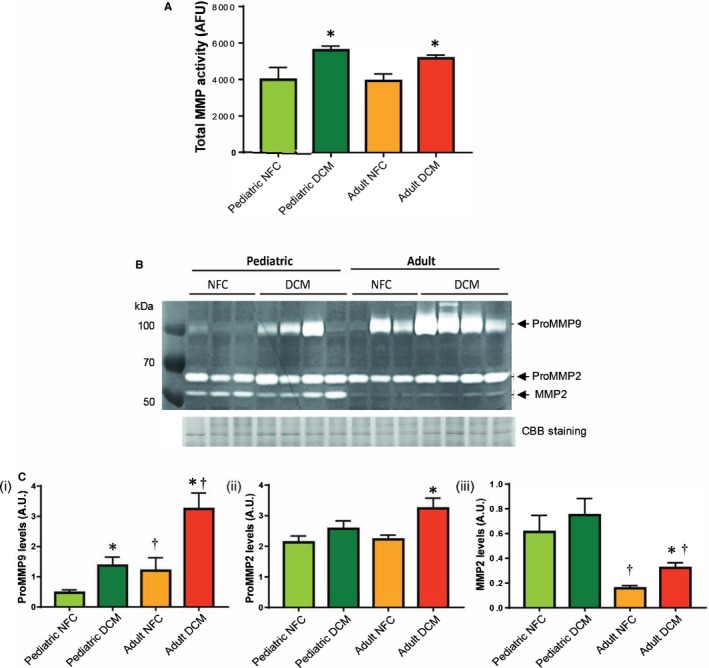
Proteolytic activities of matrix metalloproteases (MMPs) in control and failing hearts of pediatric and adult populations. A, Total MMP activity, measured using a fluorescent‐based in vitro activity assay, in the indicated groups. B, A Representative gelatin zymogram showing MMP‐9 and MMP‐2 levels in nonfailing control (NFC) and dilated cardiomyopathy (DCM) hearts from pediatric and adult groups. Coomassie blue (CBB)–stained SDS‐PAGE was used as the loading control. C, Quantification of gelatinolytic activities for (i) proMMP9, (ii) proMMP2, and (iii) MMP2. n=12 to 16 per group. Data represent mean±SEM. *P<0.05 compared with the corresponding control, † P<0.05 compared with the corresponding pediatric group. AU indicates arbitrary units.
TIMPs in Pediatric and Adult Failing Hearts
The in vivo activity of the metalloproteinases (MMPs, ADAMs, ADAMTSs) is negatively regulated by their endogenous inhibitors, TIMPs. In assessing the TIMP levels, we found higher TIMP1 levels in the control adult group compared with the pediatric group, but comparable levels between the failing hearts (Figure 8A, 8Bi). TIMP2 increased significantly in pediatric and adult failing hearts compared with their corresponding control group (Figure 8A, 8Bii). TIMP3 decreased to comparable levels in adult and pediatric failing hearts (Figure 8A, 8Biii), and TIMP4 levels remained comparable among all groups (Figure 8A, 8Biv). Overall, TIMPs exhibit a similar pattern of alteration in failing hearts regardless of age group.
Figure 8.
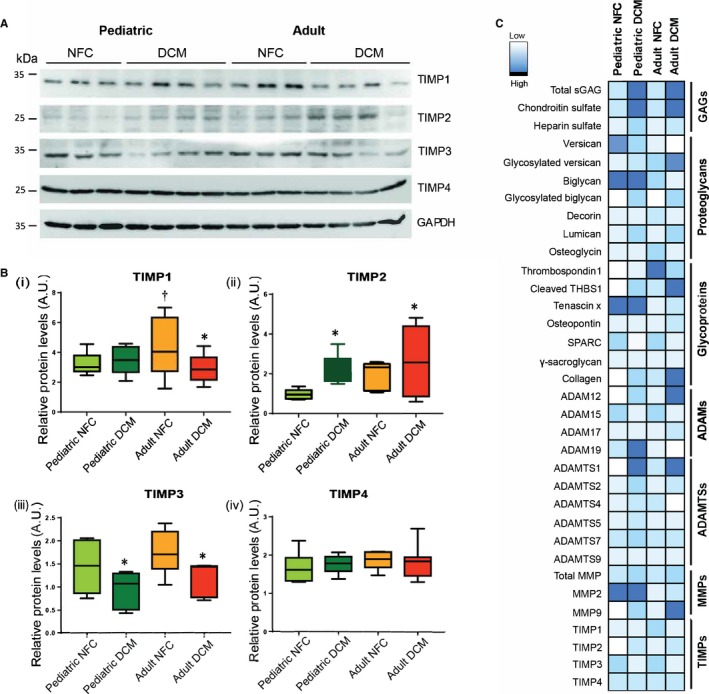
Altered tissue inhibitors of metalloprotease (TIMP) levels in nonfailing control (NFC) and dilated cardiomyopathy (DCM) hearts from pediatric and adult populations. Representative Western blots (A) and averaged protein quantification (B) for TIMP1, TIMP2, TIMP3, and TIMP4. n=12 to 16 per group. Data represent median±interquartile range. (C) A collective array showing a summary of protein expression levels for the molecules analyzed in this study in pediatric and adult NFC and DCM hearts. The color scale indicates the relative expression level. *P<0.05 compared with the corresponding control, † P<0.05 compared with the corresponding pediatric group. ADAMS indicates A disintegrin and metalloproteinases; ADAMTSs, a disintegrin and metalloproteinase with thrombospondin motifs; AU, arbitrary units; MMPs, matrix metalloproteases; sGAGs, sulfated glycosaminoglycans; SPARC, secreted protein acidic and cysteine‐rich; THBS1, thrombosondin‐1.
A summary of protein expression levels for proteoglycans, glycoproteins, ADAMs, ADAMTSs, MMPs, and TIMPs in pediatric and adult NFC and DCM failing hearts is presented using a collective array (Figure 8C).
Discussion
Adverse myocardial remodeling is a hallmark of heart disease and ultimately results in HF in children and adults. Despite great advancement in understanding the role of ECM remodeling and its contribution to heart disease development and progression,16 the impact of age at the time of HF on this process is poorly understood. In this study we performed a comprehensive analysis of the ECM proteins including elements of the fibrillar network (pertaining to fibrosis) and the nonfibrillar ECM, proteoglycans (and their glycosaminoglycan side branches), glycoproteins, and other matricellular proteins in failing hearts from pediatric and adult patients with nonischemic DCM.
Proteoglycans are composed of a core protein and a number of linear glycosaminoglycan chains, the unbranched polysaccharides (Figure S1). Heparan sulfate proteoglycans and hyaluronic acid have been studied in vascular diseases,19, 20 immune response, and inflammation.21, 22 However, the role of proteoglycans and glycosaminoglycans in cardiac remodeling related to HF, particularly in the pediatric population, has not been detailed. This study was designed based on the strong interaction that was detected by STRING analysis between the families of proteoglycans and glycoproteins, ADAMTSs, ADAMs, MMPs, and TIMPs (Figure 1A). Our data indicate a unique and age‐dependent pattern of fibrillar ECM remodeling in juvenile compared with adult failing hearts, with more severe fibrosis in adults than in children with DCM. We propose that this could be caused by a differentially modified glycosaminoglycan binding affinity for TGF‐β1, the predominant profibrotic cytokine, in the pediatric versus adult DCM hearts. Since the total glycosaminoglycan levels were increased similarly in both failing groups, but glycosaminoglycan binding affinity for TGF‐β was reduced to a greater extent in adult compared with pediatric DCM, we postulate that increased bioavailability of TGF‐β1 exists in adult failing hearts, with enhanced activation of the associated downstream signaling pathway (p‐Smad2/3) and a more pronounced fibrosis in these adult DCM compared with pediatric DCM hearts. The more pronounced fibrosis in adult failing hearts has been previously observed,23 and here we demonstrate a mechanism that could explain this effect. Further, control and DCM pediatric and adult hearts exhibited differential alterations in protein expression of the nonfibrillar ECM proteins and their regulatory enzymes (Figure 8C), supporting the notion that remodeling may progress differently in pediatric hearts. The relevance of this observation lies in the fact that pediatric DCM may not always constitute an end stage of HF. Up to 20% of cases of pediatric DCM have been reported to show echocardiographic normalization over time,24 which suggests that a pathway towards reverse remodeling may still be available in juvenile DCM hearts.
Our analysis of proteoglycans and glycoproteins revealed enhanced glycosylation of biglycan and versican in DCM hearts of both age strata, which is consistent with the comparable rise in glycosaminoglycan content in both. Lumican, thrombospondin, and tenascin‐X, however, showed age‐dependent alterations. Since tenascin‐X is important in the assembly of elastin fibers and hence in maintaining the flexibility of myocardial tissue, its downregulation has been proposed to be protective in the setting of cardiac remodeling.25 We speculate that the markedly higher tenascin‐X levels seen in pediatric control compared with adult control hearts might represent an age‐dependent advantage towards myocardial compliance in the young. The observed accumulation of chondroitin sulfate glycosaminoglycans in adult DCM hearts has been reported to contribute to inflammation and fibrosis.26 We found a similar increase in chondroitin sulfate levels in pediatric and adult DCM hearts, but also observed substantially more fibrosis in adult DCM than pediatric DCM hearts. Therefore, it is plausible that a direct correlation between fibrosis and glycosaminoglycan content may not be consistent across the spectrum of diseased myocardium and that the age of the patient may be at least as important.
TGF‐β1 is a prominent profibrotic cytokine, and its activation and activity can be modulated at many levels and by different factors. TGF‐β is secreted as a latent complex in which the mature dimer remains noncovalently bound to its prodomain, latency‐associated peptide, and is sequestered within the ECM where it remains inactive. Activation of TGF‐β1 requires its release from the ECM (often proteolytically by MMP2, MMP9, or MT1‐MMP),9 and then the release of the 25 kDa TGF‐β1 dimer from the latency‐associated peptide. This can occur through the interaction of the latent complex (latency‐associated peptide–TGF‐β) with integrins,9 glycoproteins such as thrombospondin‐127 or tenascin‐x.28 TGF‐β1 has also been reported to regulate the expression of small leucine‐rich proteoglycans29 and to suppress the expression and release of lumican in cancer cells.30 Inversely, small leucine‐rich proteoglycans such as lumican can negatively impact TGF‐β function by binding to this cytokine and limiting its ability to activate its receptors.31, 32 Consistent with this notion, we found an inverse relationship between the changes in lumican levels (greater in pediatric DCM versus adult DCM) and TGF‐β activity (greater in adult DCM versus pediatric DCM).
Finally, MMPs are primarily known for degrading ECM proteins during physiological and pathological turnover. In addition to MMPs, other metalloproteinases (ADAMs and ADAMTS) contribute to remodeling of the ECM through proteolytic processing of its nonfibrillar components.33 We previously reported that total proteolytic activity of metalloproteases is elevated in failing adult DCM hearts.12 In this study, we report that the alterations in protein levels of key proteases appear to be modulated by the age of the patient. Meanwhile, TIMPs, the endogenous inhibitors for metalloproteinases, show the same pattern in pediatric and adult hearts, and only TIMP3 is decreased in failing hearts. TIMP3 is a potent inhibitor of ADAMTSs, including ADAMTS4 and ADAMTS5,34 as well as ADAMs and MMPs.18 Therefore, the increased levels of cleaved thrombospondin‐1 in the DCM hearts may indicate the elevated activity of ADAMTSs in the face of reduced TIMP3 in the failing hearts of both age groups.
Limitations
The specimens used in this study were procured from explanted failing hearts at an advanced stage of the disease. Therefore, information regarding the molecular/cellular events during the early stages of disease progression remains unknown. The control adult hearts used in this study were procured from brain‐dead donors a few days after injury, and the control pediatric specimens were from congenitally diseased hearts, which may have experienced altered hemodynamics that could have also caused remodeling. However, these hearts lacked the features of remodeling that are seen in DCM, and therefore constitute reasonable control groups. As such, we have referred to these groups as “controls” rather than “normal” or “healthy.”
Conclusions
This study reveals that although pediatric and adult DCM hearts are similar at the end stage of HF, their ECM components (fibrillar and nonfibrillar) appear to have remodeled in a different fashion. We have determined that the reduced binding affinity of the glycosaminoglycans for TGF‐β1 in the diseased adult myocardium gives rise to increased bioavailability of TGF‐β1 and activation of its profibrotic downstream signaling pathway. Moreover, protein expression of proteoglycans, glycoproteins, and the enzymes responsible for their turnover and ECM remodeling are altered differently in pediatric versus adult failing hearts. These findings raise the possibility that manipulation of the remodeling process may differ in hearts of younger patients.
Sources of Funding
This study was funded by a Canadian Institutes of Health Research project grant to Kassiri (PJT‐153328) and a Women and Children's Health Research Institute innovation grant to Oudit. Zhang is supported by the Chinese Scholarship Council.
Disclosures
None.
Supporting information
Table S1. Clinical Characteristics of Pediatric and Adult Patients With DCM
Figure S1. Proteoglycans and glycosaminoglycans in healthy and failing hearts.
(J Am Heart Assoc. 2018;7:e010427 DOI: 10.1161/JAHA.118.010427)
References
- 1. Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, Messere J, Cox GF, Lurie PR, Hsu D, Canter C, Wilkinson JD, Lipshultz SE. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296:1867–1876. [DOI] [PubMed] [Google Scholar]
- 2. Kirk R, Edwards LB, Kucheryavaya AY, Aurora P, Christie JD, Dobbels F, Rahmel AO, Stehlik J, Hertz MI. The Registry of the International Society for Heart and Lung Transplantation: thirteenth official pediatric heart transplantation report––2010. J Heart Lung Transplant. 2010;29:1119–1128. [DOI] [PubMed] [Google Scholar]
- 3. Kirk R, Dipchand AI, Rosenthal DN, Addonizio L, Burch M, Chrisant M, Dubin A, Everitt M, Gajarski R, Mertens L, Miyamoto S, Morales D, Pahl E, Shaddy R, Towbin J, Weintraub R. The International Society for Heart and Lung Transplantation Guidelines for the management of pediatric heart failure: executive summary [Corrected]. J Heart Lung Transplant. 2014;33:888–909. [DOI] [PubMed] [Google Scholar]
- 4. Kantor PF, Abraham JR, Dipchand AI, Benson LN, Redington AN. The impact of changing medical therapy on transplantation‐free survival in pediatric dilated cardiomyopathy. J Am Coll Cardiol. 2010;55:1377–1384. [DOI] [PubMed] [Google Scholar]
- 5. Shaddy RE, Boucek MM, Hsu DT, Boucek RJ, Canter CE, Mahony L, Ross RD, Pahl E, Blume ED, Dodd DA, Rosenthal DN, Burr J, LaSalle B, Holubkov R, Lukas MA, Tani LY; Pediatric Carvedilol Study G . Carvedilol for children and adolescents with heart failure: a randomized controlled trial. JAMA. 2007;298:1171–1179. [DOI] [PubMed] [Google Scholar]
- 6. Rossano JW, Shaddy RE. Heart failure in children: etiology and treatment. J Pediatr. 2014;165:228–233. [DOI] [PubMed] [Google Scholar]
- 7. Fan D, Creemers EE, Kassiri Z. Matrix as an interstitial transport system. Circ Res. 2014;114:889–902. [DOI] [PubMed] [Google Scholar]
- 8. Moore L, Fan D, Basu R, Kandalam V, Kassiri Z. Tissue inhibitor of metalloproteinases (TIMPs) in heart failure. Heart Fail Rev. 2012;17:693–706. [DOI] [PubMed] [Google Scholar]
- 9. Takawale A, Sakamuri SS, Kassiri Z. Extracellular matrix communication and turnover in cardiac physiology and pathology. Comprehensive Physiology. 2015;5:687–719. [DOI] [PubMed] [Google Scholar]
- 10. Takawale A, Zhang P, Azad A, Wang W, Wang X, Murray AG, Kassiri Z. Myocardial overexpression of TIMP3 following myocardial infarction exerts beneficial effects through promoting angiogenesis and suppressing early proteolysis. Am J Physiol Heart Circ Physiol. 2017:313:H224–H236. [DOI] [PubMed] [Google Scholar]
- 11. Vanhoutte D, Heymans S. TIMPs and cardiac remodeling: ‘Embracing the MMP‐independent‐side of the family’. J Mol Cell Cardiol. 2010;48:445–453. [DOI] [PubMed] [Google Scholar]
- 12. Sakamuri SS, Takawale A, Basu R, Fedak PW, Freed D, Sergi C, Oudit GY, Kassiri Z. Differential impact of mechanical unloading on structural and nonstructural components of the extracellular matrix in advanced human heart failure. Transl Res. 2016;172:30–44. [DOI] [PubMed] [Google Scholar]
- 13. Huynh MB, Morin C, Carpentier G, Garcia‐Filipe S, Talhas‐Perret S, Barbier‐Chassefiere V, van Kuppevelt TH, Martelly I, Albanese P, Papy‐Garcia D. Age‐related changes in rat myocardium involve altered capacities of glycosaminoglycans to potentiate growth factor functions and heparan sulfate‐altered sulfation. The Journal of biological chemistry. 2012;287:11363–11373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac Fibrosis. The Fibroblast Awakens. 2016;118:1021–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Segura AM, Frazier OH, Buja LM. Fibrosis and heart failure. Heart Fail Rev. 2014;19:173–185. [DOI] [PubMed] [Google Scholar]
- 16. Spinale FG, Frangogiannis NG, Hinz B, Holmes JW, Kassiri Z, Lindsey ML. Crossing into the next frontier of cardiac extracellular matrix research. Circ Res. 2016;119:1040–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stanton H, Melrose J, Little CB, Fosang AJ. Proteoglycan degradation by the ADAMTS family of proteinases. Biochim Biophys Acta. 2011;1812:1616–1629. [DOI] [PubMed] [Google Scholar]
- 18. Zhang P, Shen M, Fernandez‐Patron C, Kassiri Z. ADAMs family and relatives in cardiovascular physiology and pathology. J Mol Cell Cardiol. 2016;93:186–199. [DOI] [PubMed] [Google Scholar]
- 19. Gonzales JC, Gordts PL, Foley EM, Esko JD. Apolipoproteins E and AV mediate lipoprotein clearance by hepatic proteoglycans. J Clin Invest. 2013;123:2742–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Osterholm C, Folkersen L, Lengquist M, Ponten F, Renne T, Li J, Hedin U. Increased expression of heparanase in symptomatic carotid atherosclerosis. Atherosclerosis. 2013;226:67–73. [DOI] [PubMed] [Google Scholar]
- 21. Strand ME, Aronsen JM, Braathen B, Sjaastad I, Kvaloy H, Tonnessen T, Christensen G, Lunde IG. Shedding of syndecan‐4 promotes immune cell recruitment and mitigates cardiac dysfunction after lipopolysaccharide challenge in mice. J Mol Cell Cardiol. 2015;88:133–144. [DOI] [PubMed] [Google Scholar]
- 22. Jiang D, Liang J, Campanella GS, Guo R, Yu S, Xie T, Liu N, Jung Y, Homer R, Meltzer EB, Li Y, Tager AM, Goetinck PF, Luster AD, Noble PW. Inhibition of pulmonary fibrosis in mice by CXCL10 requires glycosaminoglycan binding and syndecan‐4. J Clin Invest. 2010;120:2049–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Woulfe KC, Siomos AK, Nguyen H, SooHoo M, Galambos C, Stauffer BL, Sucharov C, Miyamoto S. Fibrosis and fibrotic gene expression in pediatric and adult patients with idiopathic dilated cardiomyopathy. J Card Fail. 2017;23:314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Everitt MD, Sleeper LA, Lu M, Canter CE, Pahl E, Wilkinson JD, Addonizio LJ, Towbin JA, Rossano J, Singh RK, Lamour J, Webber SA, Colan SD, Margossian R, Kantor PF, Jefferies JL, Lipshultz SE; Pediatric Cardiomyopathy Registry Investigators . Recovery of echocardiographic function in children with idiopathic dilated cardiomyopathy: results from the pediatric cardiomyopathy registry. J Am Coll Cardiol. 2014;63:1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Petersen JW, Douglas JY. Tenascin‐X, collagen, and Ehlers‐Danlos syndrome: tenascin‐X gene defects can protect against adverse cardiovascular events. Med Hypotheses. 2013;81:443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhao RR, Ackers‐Johnson M, Stenzig J, Chen C, Ding T, Zhou Y, Wang P, Ng SL, Li PY, Teo G, Rudd PM, Fawcett JW, Foo RSY. Targeting chondroitin sulfate glycosaminoglycans to treat cardiac fibrosis in pathological remodeling. Circulation. 2018;137:2497–2513. [DOI] [PubMed] [Google Scholar]
- 27. Crawford SE, Stellmach V, Murphy‐Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, Boivin GP, Bouck N. Thrombospondin‐1 is a major activator of TGF‐beta1 in vivo. Cell. 1998;93:1159–1170. [DOI] [PubMed] [Google Scholar]
- 28. Alcaraz LB, Exposito JY, Chuvin N, Pommier RM, Cluzel C, Martel S, Sentis S, Bartholin L, Lethias C, Valcourt U. Tenascin‐X promotes epithelial‐to‐mesenchymal transition by activating latent TGF‐beta. J Cell Biol. 2014;205:409–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bassols A, Massague J. Transforming growth factor beta regulates the expression and structure of extracellular matrix chondroitin/dermatan sulfate proteoglycans. J Biol Chem. 1988;263:3039–3045. [PubMed] [Google Scholar]
- 30. Kang Y, Roife D, Lee Y, Lv H, Suzuki R, Ling J, Rios Perez MV, Li X, Dai B, Pratt M, Truty MJ, Chatterjee D, Wang H, Thomas RM, Wang Y, Koay EJ, Chiao PJ, Katz MH, Fleming JB. Transforming growth factor‐beta limits secretion of lumican by activated stellate cells within primary pancreatic adenocarcinoma tumors. Clin Cancer Res. 2016;22:4934–4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yamaguchi Y, Mann DM, Ruoslahti E. Negative regulation of transforming growth factor‐beta by the proteoglycan decorin. Nature. 1990;346:281–284. [DOI] [PubMed] [Google Scholar]
- 32. Nikitovic D, Katonis P, Tsatsakis A, Karamanos NK, Tzanakakis GN. Lumican, a small leucine‐rich proteoglycan. IUBMB Life. 2008;60:818–823. [DOI] [PubMed] [Google Scholar]
- 33. Chute M, Jana S, Kassiri Z. Disintegrin and Metalloproteinases (ADAMs and ADAM‐TSs), the emerging family of proteases in heart physiology and pathology. Current Opinion in Physiology. 2018;1:34–45. [Google Scholar]
- 34. Kashiwagi M, Tortorella M, Nagase H, Brew K. TIMP‐3 is a potent inhibitor of aggrecanase 1 (ADAM‐TS4) and aggrecanase 2 (ADAM‐TS5). J Biol Chem. 2001;276:12501–12504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Clinical Characteristics of Pediatric and Adult Patients With DCM
Figure S1. Proteoglycans and glycosaminoglycans in healthy and failing hearts.