

Summary
The persistence of a pool of latently HIV-1-infected cells despite combination Anti-Retroviral Therapy (cART) treatment is the major roadblock for a cure. The BAF (mammalian SWI/SNF) chromatin remodeling complex is involved in establishing and maintaining viral latency, making it an attractive drug target for HIV-1 latency reversal. Here we report a high throughput screen for inhibitors of BAF-mediated transcription in cells and the subsequent identification of a 12-membered macrolactam. This compound binds ARID1A-specific BAF complexes, prevents nucleosomal positioning, and relieves transcriptional repression of HIV-1. Through this mechanism, these compounds are able to reverse HIV-1 latency in an in vitro T cell line, an ex vivo primary cell model of HIV-1 latency, and in patient CD4+ T cells without toxicity or T cell activation. These macrolactams represent a class of latency reversal agents with unique mechanism of action, and can be combined with other LRAs to improve reservoir targeting.
Graphical Abstract
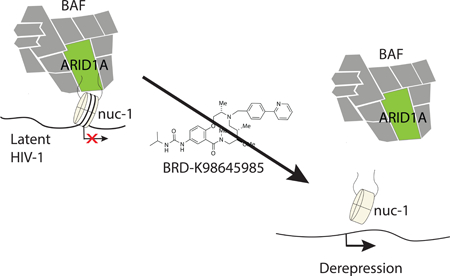
eTOC Blurb
The BAF (SWI/SNF) chromatin remodeling complex is involved in repressing HIV-1 transcription in latently infected T cells. Using high throughput screening, we identified a macrolactam that inhibits ARID1A-containing BAF complexes to reverse HIV-1 latency without T cell activation or toxicity.
Introduction
Since the discovery of HIV-1 as the causative agent of AIDS in 1983 (Barré-Sinoussi et al., 1983), enormous progress has been made in treating HIV-1 infections and prolonging the lifespan of HIV-1 infected individuals. State of the art treatment is a cocktail of drugs acting on different viral targets, known as combination Anti-Retroviral Therapy (c-ART). c-ART is extremely effective at suppressing HIV-1 to undetectable levels, preventing progression to AIDS; however, treatment must be maintained for life and as of yet, HIV-1 eradication is not achievable (Deeks et al., 2013; Maartens et al., 2014). Despite being highly efficient in stopping active viral replication, anti-retroviral drugs do not target latently infected cells that harbor replication competent but transcriptionally silent proviruses. Latently infected cells persist in the body for life and, not being targeted by either c-ART or immune cells, they constitute the viral reservoir (Chun et al., 1997; Finzi et al., 1997; 1999). When these cells are activated, transcription from latent HIV-1 provirus is induced and in the absence of c-ART, viral replication rebounds (Chun et al., 2010; Dahabieh et al., 2015; De Crignis and Mahmoudi, 2017; Ruelas and Greene, 2013; Siliciano et al., 2003).
Currently, there are two major non-redundant strategies to eliminate this population of latently infected cells in HIV-1-infected individuals (Churchill et al., 2016; Cillo and Mellors, 2016; Margolis, 2017; Siliciano and Siliciano, 2016). The first approach is harnessing the immune system to eliminate latently infected cells (Barouch and Deeks, 2014; Brockman et al., 2015; Martrus and Altfeld, 2016; Perreau et al., 2017; Trautmann, 2016); the second, also known as the “shock and kill” strategy, is aimed at inducing HIV-1 transcription in latently infected cells such that all cells harboring replication competent virus can be targeted by the immune system (Deeks, 2012; Margolis and Archin, 2017; Margolis et al., 2016; Rasmussen et al., 2016).
HIV-1 latency is established and maintained through complex genetic and epigenetic mechanisms that create a specific repressive chromatin configuration at the viral promoter or 5’-LTR (Verdin, 1991; Verdin et al., 1993). Active HIV-1 transcription is driven by Tat and its multiple activating co-factor complexes, while HIV-1 latency is driven through epigenetic regulators that maintain increased nucleosome occupancy at the 5’-LTR (Kumar et al., 2015; Mbonye and Karn, 2014; Turner and Margolis, 2017; Van Lint et al., 2013). Histone deacetylases (HDACs) play a prominent role in the repressive chromatin environment that drives HIV-1 latency and as such, HDAC inhibitors are able to reverse HIV-1 latency in in vitro and ex-vivo models (Archin et al., 2014; 2012; Conrad and Ott, 2016; De Crignis and Mahmoudi, 2017; Rasmussen et al., 2013; Sheridan et al., 1997; Van Lint et al., 1996; Wei et al., 2014; Wightman et al., 2013). Results from clinical trials, however, indicate that the HDAC inhibitors tested are unable to significantly reduce the frequency of latently infected cells (Elliott et al., 2014; Rasmussen et al., 2013; Spivak and Planelles, 2016; Søgaard et al., 2015) (Delagrèverie et al., 2016). Among the alternate epigenetic targets being investigated for reversing HIV-1 latency, one potential candidate is the mammalian SWI/SNF chromatin remodeling complex, BAF, which has been shown to contribute to HIV-1 transcriptional repression (Boese et al., 2009; Rafati et al., 2011; Van Duyne et al., 2011). BAF complexes are multisubunit ATP-dependent chromatin remodelers known for their roles in development and cancer (Ho and Crabtree, 2010; Hodges et al., 2016; Pulice and Kadoch, 2017). In latent cells harboring HIV-1 proviruses, BAF complexes are required for maintaining increased nucleosome density immediately downstream of the HIV-1 transcription start site (Rafati et al., 2011). During latency reversal, the closely related PBAF complex, which shares many of the same subunits, replaces BAF and directly or indirectly promotes removal of the repressive Nuc-1, activating HIV-1 transcription (Agbottah et al., 2006; Easley et al., 2010; Mahmoudi et al., 2006; Rafati et al., 2011; Tréand et al., 2006; Van Duyne et al., 2011). Consistent with the pivotal role of the BAF complex in HIV-1 latency, a recent report demonstrated that the latency reversal activity of BRD4 bromodomain inhibitors is due to the requirement for a short BRD4 isoform that recruits BAF to the HIV-1 5’ LTR (Conrad et al., 2017).
Inhibitors specifically targeting the ARID1A subunit-containing BAF complex (but not PBAF) would be invaluable as HIV-1 latency reversal agents. We recently reported a medium throughput screen using qRT-PCR to identify compounds that alter the transcription of BAF target genes in mouse embryonic stem cells (ESCs) (Dykhuizen et al., 2012), and several compounds identified from this screen displayed an ability to reverse HIV-1 latency; however, many of these compounds have known targets besides BAF, raising the possibility for toxic off-target effects (Stoszko et al., 2016). To identify specific and nontoxic small molecule inhibitors of the BAF complex, we developed a high throughput assay for screening large libraries of diverse small molecules in ESCs. From a screen of almost 350,000 compounds, we identified a 12-membered macrolactam scaffold with low toxicity in cells and the ability to regulate a panel of BAF target genes. These macrolactams reverse HIV-1 latency in several relevant in vitro cell line and primary cell models of HIV-1 latency. In addition, they enhance the activity of other clinically used latency reversal agents targeting HDACs and PKC. Target identification experiments implicate ARID1A-containing BAF complexes as the primary target, and the compounds act to reverse HIV-1 latency by reducing repressive nucleosome occupancy at the 5’ LTR.
Results
Development and confirmation of a luciferase reporter cell line
In murine ESCs the BAF complex is essential for maintaining repression of certain polycomb repressive complex 1 subunit genes including Bmi1 and Ring1a while activating genes involved in maintaining the pluripotent state, such as Fgf4 (Dykhuizen et al., 2012; Ho et al., 2009). For high throughput screening of BAF inhibitors, we developed a knock-in luciferase reporter of BAF transcriptional repression. We used homologous recombination to construct a mESC line with firefly luciferase inserted at exon 1 of Bmi1. Southern blotting confirmed successful recombination of the original targeting vector, as well as subsequent Cre-mediated excision of the Neo selection marker (Figure 1A). To confirm that the reporter line was a reliable indicator of esBAF-mediated repression of Bmi1, we knocked down the gene encoding the BAF ATPase BRG1 using lentiviral shRNA. Three days after infection, a stable four-fold increase in luciferase levels was observed when corrected for cell number, indicating successful de-repression of Bmi1 (Figure 1B). In an effort to simplify the requirements for high throughput screening, we performed the Brg1 knockdown without correcting for cell number and found that the reporter line still displays robust three-fold induction of luciferase 72 hours after infection with shBrg1-expressing lentivirus (Figure 1B).
Figure 1: High throughput screen for inhibitors of BAF-mediated transcriptional repression.

A. The generation of a luciferase knock-in at the Bmi1 locus using homologous recombination in mouse embryonic stem cells and validation using Southern blot analysis. B. Validation of the Bmi1-luciferase reporter cell line using lentiviral-mediated knockdown of Brg1 either with (above) or without (below) normalizing by cell number. C. The robustness of the screen was determined using positive control compound 63. D. The five hits identified from high throughput screening efforts. See also Figure S1.
High throughput Assay Development:
The knock-in ESC line was generated on mouse embryonic fibroblasts (MEFs) as a feeder layer, which is not compatible with high throughput screening. Therefore, we first made the cell line feeder free by passaging at high density five times on gelatin. While we previously developed methods to gelatin coat 384-well plates (Dykhuizen et al., 2012), this was not feasible for this scale of screening effort. Instead we identified the Corning High Bind® surface to support normal ESC morphology and normal alkaline phosphatase levels (indicative of maintained pluripotency) compared to gelatin coated plates (Figure S1A). The assay was optimized in 384-well format at the Broad Institute Probe Development Center (BIPDEC). The coefficient of variance (CV) for firefly luciferase reading across a 384-well plate was 4.5% and using a non-specific hit identified in the pilot qRT-PCR screen (compound 63) as our positive control (Dykhuizen et al., 2012), we calculated a Z-factor of 0.6, indicating a robust screen (Figure 1C).
High Throughput Screen:
We screened 347,670 compounds in duplicate (Figure S1B for illustration of reproducibility). The compounds included the MLPCN validation set of natural products, known bioactives, commercially available compound libraries, and compound libraries designed by scientists at the Broad Institute (PubChem assay entry AID 602393 for full description of library). We defined hits as compounds with luciferase inductions that were at least 3 standard deviations above the mean, which corresponded to approximately 40% of the maximal activity observed with Brg1 knockdown. We identified 7048 hits (hit rate of 2%), which is high, but not unexpected from cell-based luciferase assays that tend to identify nonspecific luciferase stabilizers (Auld et al., 2008). Hits identified in >5 luciferase screens or >10% of luciferase screens on PubMed were eliminated, as were compounds containing functional groups with known reactivity, including a-chloroketones, imines, and nitro groups. This resulted in a refined hit list of 1157 compounds for a hit rate of 0.33%, more in line with the expected hit rate for a robust screen (see Figure S1C for summary of the screening tree). We rescreened the 1157 compounds at eight doses in the cell-based luciferase assay as a confirmatory assay along with a counter-screen for viability. We confirmed 548 hits with EC50 < 10 µM and toxicity EC50 > 30 µM. We then treated cells with these hits at a single dose (30 µM) in the qRT-PCR screen previously reported (Dykhuizen et al., 2012). From this secondary assay, we found five compounds that increase Bmi1 at least six-fold, Ring1 at least two-fold and decreased Fgf4 at least five-fold (Figure 1D). STK177924, a known non-specific pan-assay interference (PAINS) scaffold, was eliminated (Figure 1D) (Baell and Holloway, 2010). We next used chemoinformatics to investigate the SAR of the thiophene (MLS003122856) from the primary screen and developed a small library of analogs to explore additional SAR for linker attachment (Figure S1D). The biotin-linked compound on solid support failed to enrich subunits of the BAF complex from lysates (Figure S1E), and in addition, this compound had only moderate HIV latency reversal activity (Figure S1F). Therefore, while the thiophene may have interesting biological activity worth investigating, there is substantial evidence that it doesn’t directly target the BAF complex or phenocopy the effects of BAF deletion in latent HIV-1 infected T cells.
Broad DOS library macrocycle SAR:
The three remaining hits shared a similar macrocyclic scaffold with only slight variations of substituents off macrocycle aniline: n-propyl amide (BRD-K83694683), n-propyl urea (BRD-K21001652) and isopropyl urea (BRD-K98645985). Re-evaluation of hit BRD-K98645985 in the luciferase assay provided an EC50 of approximately 2.37 ± 0.98 µM (Figure 2A). In addition, we observed a 5-fold increase in Bmi1, 2.6-fold increase in Ring1, and 3.3-fold decrease in Fgf4 upon treatment with 30 µM of compound, a profile that closely mimics Brg1 KO (Figure 2B). We observe no toxicity to ESCs up to 30 µM (Figure 2C), or to HepG2, HEK293T and A549 cells (Figure S2A), increasing enthusiasm for this scaffold. The three hits containing this scaffold are members of the DOS library synthesized using head – to –tail scaffold design (Fitzgerald et al., 2012). The primary screen contained 3618 compounds containing the same 12-membered macrolactam scaffold. Substituents off the scaffold vary at two positions (R1 off the core aniline and R2 off the core secondary amine) and the stereochemistry varies at three positions along the ring, C2, C5, and C6. While all stereoisomers with varying stereochemistry at C2, C5, and C6 were included for BRD-K98645985, BRD-K83694683 and BRD-K21001652, all of the top hits have the same stereochemical configuration. Taking a closer look at the primary screen scores for the eight stereoisomers of BRD-K98645985 the stereoisomer S,S,R showed significantly increased activity over the other stereoisomers, supporting a specific target (Figure 2D). A similar profile was observed for BRD-K83694683 and BRD-K21001652 (Figure S2B). To further define the SAR from initial screen data we also looked at varied substituents at two positions, R1 and R2 (Figure 2E). The most common building block used at R1 was n-propyl amide, which was included in 218 macrolactam library members. For these 218 library members R2 groups included aliphatic groups, aromatic groups, and sulfonamides, but only compounds with 2-phenyl pyridine moiety at R2 were hits in the primary screen. To ensure that the preference for 2-phenyl pyridine isn’t due to nonspecific interactions, we also looked at the 75 compounds in the initial compound library that contained the 2-phenyl pyridine moiety and confirmed that only the DOS macrolactams were hits. Substituting phenyl for 2-phenyl pyridine at R2 increased the cLogP from 3.5 to 4.2, which in conjunction with a lowered efflux ratio, could indicate that the increased activity is due to improved cell permeability (Over et al., 2016). There were 26 compounds that contained the 2-phenyl pyridine at position R2 and varying substituents at R1 and all were hits to some degree in the primary screen. This likely indicates that the R1 position is more permissible to variation than the R2 position; however, the variations at R1 in the screen were minimal, including only simple aliphatic groups connected via amides, ureas and carbamates.
Figure 2: 12-membered macrolactams are inhibitors of BAF-mediated transcription.

A. The EC50 was measured for the best screen hit BRD-K98645985 after 24 h compound treatment with the Bmi1-luciferase reporter cell line. Each concentration was dosed in triplicate. B. The fold change of the transcription of three BAF target genes was calculated using qRT-PCR after 18 h BRD-K98645985 treatment (30 µM) or Brg1 knockdown compared to untreated cells. C. Viability measurements in wild type ESCs were performed after 72 h of compound or DMSO treatment using CellTiter-Glo®. D. The structure activity relationship of the eight stereoisomers of BRD- K98645985 based on initial luciferase induction from the primary screen. E. The structure activity relationship of the 3618 macrolactam library members based on initial luciferase induction from the primary screen. See also Figure S2.
Solution Phase Macrolactam Library:
Based on this initial SAR data, we synthesized a 30-membered library to further investigate substitutions at the R1 and R2 positions (Figure 3). For this library, we developed a solution phase synthesis based on the previously published solid phase synthesis of the scaffold with minor alterations (Figure S3) (Fitzgerald et al., 2012). For the 30 library members, we performed the luciferase screen in dose and also calculated the fold change in Bmi-1, Fgf4, and Ring1 at a single dose (30 µM) using qRT-PCR (Figure 3, Figure S3). We found high correlation between activity in the two assay formats. We varied substituents at the R1 position and confirmed that small aliphatic substituents linked via amide, urea and carbamates are tolerated (Figure 3A). Interestingly, a small aromatic ring is tolerated at R1, while a squarate linkage is not. Importantly, it is revealed that the free aniline is almost as equally potent as the parent compound and that no substituent is actually required at the R1 position. At the R2 position, 2-benzyl pyridine is still the best substituent, although 3-benzyl pyridine and 4-benzyl pyridine are tolerated (Figure 3B). Not surprisingly, smaller substituents containing a single aromatic ring are not active, but interestingly, the closely related 2-benyzoyl pyridine and amide phenyl pyridine are also not active, indicating that the benzyl pyridine may be involved in a specific binding mechanism, and not simply functioning to increase cell permeability. The last thing we looked at is the position of the R2 substituent off the macrocycle aromatic. We found no tolerance for moving the phenyl pyridine from the para to the meta position (Figure S3D). We measured solubility and neither this value nor the cLogP values for library members strongly correlated with activity (Table S1). Additionally, the compounds all displayed low toxicity towards cell lines with the exception of a few, which showed slight toxicity, possibly correlated to low solubility (Table S1).
Figure 3: The structure activity relationship between members of a solution phase 12-membered macrolactam analog library:

A solution phase library of 30 analogs was synthesized and tested to further explore structure activity relationship for compounds with variations at A. the aniline (R1), and B. the secondary amine (R2). Activity was defined as the EC50 in the luciferase reporter screen and as the fold transcriptional change of three BAF targets (Bmi1, Ring1, Fgf4) at a single compound concentration (30 µM) determined using qRT-PCR. n = 3. Data presented as mean ± S.D. NA = no activity. See also Figure S3 and Table S1.
Latency Reversal:
Using a primary cell model of HIV-1 latency established in ex vivo infected human CD4 + T cells (Lassen et al., 2012), we measured HIV-1 reversal activity of six compounds from the follow up library: three with high activity in the Bmi1 luciferase reporter assays (BRD-K98645985, BRD-K25923209, BRD-K80443127), one with moderate activity (BRD-K17257309), and two with low/no activity (BRD-K13648511, BRD-K04244835). We found remarkable correlation between the ability to induce Bmi1 transcription in ESCs and the ability to increase transcription of latent HIV-1 in a concentration dependent manner (Figure 4A). The activity of three potent compounds (BRD-K98645985, BRD-K25923209, BRD-K80443127) was similar in this assay (Figure 4A); however, since BRD-K80443127 displayed slightly higher activity at low concentrations, we continued with this carbamate analog for further characterization. The activation was confirmed in a second ex vivo model of HIV-1 latency (Figure S4A) (Bosque and Planelles, 2009) and the differential expression of two BAF target genes, namely c-MYC and p21, upon treatment with BRD-K80443127 was consistent with BAF inhibition (Figure 4B) (Bin Guan et al., 2011; Cheng et al., 1999; Pham et al., 2010; Shi et al., 2013). The compounds show excellent activity even in the low concentration range of 1– 10 µM, and importantly, the compounds show no toxicity to T cells at these concentrations (Figure 4C, Figure S4B). Unwanted general immune activation reduces the clinical applicability of LRAs (DeChristopher et al., 2012; Korin et al., 2002); therefore, to investigate whether inhibitor treatment results in stimulation of T cells, we treated human primary CD4+ T cells isolated from two healthy donors with BRD-K80443127 for 24 and 72 hours, followed by detection of activation markers CD25 and CD69. As expected, PMA/Ionomycin treatment resulted in activation of T cells while BRD-K80443127 treatment had no effect on CD25 and CD69 expression (Figure S4C). We also found that the compounds can be used in conjunction with other HIV latency reversal agents, such as HDAC inhibitors (SAHA, VPA and romidepsin) or PKC modulators (prostratin and bryostatin), to boost activity compared to single agent treatments (Figure 4D). The activities were primarily additive, with slight synergy when compared across multiple drug concentrations (Figure S4D) (Laird et al., 2015). To confirm whether inhibitors could reverse HIV-1 latency in T-cells obtained from HIV-1 infected patients, we treated CD4+ T cells from three aviremic patients with BRD-K80443127, prostratin, BRD-K80443127 and prostratin, and αCD3/αCD28 magnetic beads as a positive control (Figure 4E). Patient 1 CD4 T cells responded to all treatments, while interestingly, double treatment with BRD-K80443127 and prostratin resulted in 63% latency reversal compared to positive control αCD3/αCD28 (Figure 4E). Cells from patient 2 and 3 did not result in a significant increase in cell associated HIV-1 POL after treatment with BRD-K80443127 alone; however, when co-treated with the PKC agonist prostratin, BRD-K80443127 showed significant increase in POL copies (Figure 4E). As biomarkers can be clinically useful as surrogate measures of compound activity, we examined expression levels of BAF target genes p21 and C-MYC in the three patients and we observed significant decrease of p21 and C-MYC transcripts levels, confirming BRDK80443127 activity in these cells.
Figure 4: 12-membered macrolactams reactivate latent HIV-1 in primary model systems of HIV-1 latency and patient samples with limited toxicity or T-cell activation.

A. A panel of six macrolactams with varying EC50 values from the Bmi1-luciferase assay were tested in an ex vivo model of HIV-1 latency using primary CD4+ T cells from healthy donors (Lassen et al., 2012). Each point represents a single experiment using T cells from at least two different healthy donors. Luciferase levels are normalized with total protein levels. Error bars represent mean ± S.D. Asterisks indicate the level significance compared to untreated cells using student’s T test (* p< 0.05 ** p< 0.01, *** p< 0.001, **** p< 0.0001). B. mRNA expression levels of two BAF target genes were determined after treatment of CD4+ T cells isolated from 3 healthy donors with BRD-K80443127. Bars represent the average ± SD, Asterisks indicate the level significance compared to untreated cells using student’s T test (* p< 0.05 ** p< 0.01, *** p< 0.001, **** p< 0.0001). C. The number of apoptotic human primary CD4+ T cells in the presence of macrolactams was measured using Annexin V staining and flow cytometry analysis. Data presented as mean ± S.D. of experiments performed on cells isolated from 6 healthy donors. D. Latency reversal activity of BRD-K80443127 in combination with known LRAs was assessed in the ex-vivo model of HIV-1 latency. BRD-K80443127 was used at a concentration of 5 µM alone or in combination with known LRAs at a single dose. Luciferase levels are normalized with total protein levels. Data presented as mean ± S.D. of experiments performed in duplicate using cells from two healthy donors. Asterisks indicate the level significance compared to untreated cells using student’s T test (* p< 0.05 ** p< 0.01, *** p< 0.001, **** p< 0.0001). E. Cell associated HIV Pol mRNA levels were quantitated in CD4+ T cells obtained from three c-ART treated virologically suppressed HIV-1 infected patients after ex vivo treatment with BRD-K80443127 (10 µM), Prostratin (200 nM) or αCD3/αCD28 dynabeads as indicated in triplicate. Bars represent average of treatments in triplicate ± SD, asterisks indicate the level of significance using one-way ANOVA followed by Tukey test (p< 0.05). mRNA expression levels of biomarker genes of BAF activity, c-MYC and p-21 were also quantitated in the patient CD4+ T cells after treatment with DMSO or BRD-K80443127 (10 µM). See also Figure S4.
Validation of BAF-mediated transcriptional effects:
To define how well the BAF inhibitors mimic the transcriptional profile of BAF deletion we performed RNA-Seq analysis on mESCs treated with 30 µM BRD-K98645985 or DMSO for 18h. We found 3534 differentially regulated genes upon compound treatment (1.5-fold change, P<0.05), with 1518 up and 1916 down upon compound treatment. Comparing the gene overlap with published RNA-Seq data from Brg1 knockout in mES cells (King and Klose, 2017) leads to significant (2.3-fold enrichment over predicted, p = 1.47 × 10−33) overlap of gene expression (Figure 5A). This high degree in overlap indicates that BRD-K98645985 targets similar pathways regulated by BRG1 in ES cells. The incomplete overlap could be due to off-target effects, phenotypic difference between acute inhibition and genetic deletion, compound targeting a subset of BRG1 function, heterogeneity of different ES cell lines, different experimental conditions, and/or different analysis conditions. Pathway analysis of the overlapping genes shows significant enrichment of genes related to neuronal development and morphogenesis, consistent with the loss of pluripotency and a propensity for neuronal differentiation reported for Arid1a knockout ESCs (Gao et al., 2008). We then performed RNA-Seq analysis with Arid1a knockout ESCs and identified 1141 differentially regulated genes (1.5-fold change, P < 0.05). A higher percentage of the ARID1A-regulated genes overlapped with compound treated cells (2.7-fold enrichment over predicted, p = 6.55 × 10−58) than BRG1-regulated genes. In addition, the canonical pathways identified for the differential genes from each dataset were the most similar between the Arid1a KO cells and BRD-K98645985 treated cells. This, in conjunction with parallel studies with screen hits in the context of ARID1A mutant cancer cell lines and ATR synergy (Chory et al. Submitted), led us to hypothesize ARID1A as the primary target of the macrolactams. Still, the incomplete overlap of gene expression between Arid1a KO cells and BRD-K98645985 treatment raises the possibility that BRD-K98645985 modulates Arid1a function in a manner different from genetic deletion, and/or that it has additional biological targets.
Figure 5: 12-membered Macrolactams are inhibitors of ARID1A-containing BAF complexes.

A. Differential gene expression of mESCs treated with 30 µM of BRD-K98645985 for 18 h was compared to published differential gene expression in Brg1 KO mESCs (King and Klose, 2017) to determine overlapping gene sets. Data was acquired from RNA-Seq analyses. B. The luciferase induction upon treatment with macrolactams with propyl amide (BRD-K83694683), butyl amide (CAM2–64) and biotin-hexylamide (CAM2–56) appended off the aniline was determined using the BMI1-luciferase reporter cell line. C. Pulldowns were performed from ESC lysates pretreated with DMSO or 200 µM BRD- K25923209 using biotin or CAM2–56 prebound to streptavidin resin. Protein enrichment was determined using immunoblot analysis. D. Protein stabilization by BRD-K25923209 was determined using CETSA in mESCs. The stabilization of ARID1A, PBRM1 and LAMINB1 was detected using immunoblot analysis of soluble proteins after incubation in a temperature gradient. E. Sequential salt extractions were performed on ESC nuclei. The chromatin was washed with increasing concentrations of salt containing DMSO or BRD-K98645985 (30 µM) and the elution of ARID1A and PBRM1 were analyzed using immunoblot analysis. The percent of protein elution was calculated across all five washes using ImageJ for ARID1A and PBRM1. n = 3. F. J-Lat 11.1 cells were treated with increasing concentrations of BRD-K80443127 and reactivation was quantitated at 48 h post treatment. Percent GFP positive cells (left axis, green bars), which corresponds to the level of HIV-1 activation, and cell viability (right axis, transparent bars) were both evaluated by flow cytometry. n = 3. G. Levels of nucleosome occupancy at the HIV-1 5’-LTR region following treatment with BRD-K80443127, CAPE, and PMA were analyzed using FAIRE assay. n = 3. Data are presented as mean ± S.D. Asterisks indicate the level significance compared to untreated cells using student’s T test (* p< 0.05 ** p< 0.01, *** p< 0.001, **** p< 0.0001). See also Figure S5.
Validation of ARID1A as a target:
Based on the SAR we hypothesized that the R1 position would be amenable to linker attachment for protein target identification; however, the synthesis of analog CAM 2–64 with a single methylene addition compared to BRD-K83694683 resulted in a significant decrease in activity (Figure 5B). Further modification with a longer linker attached to biotin in CAM 2–56 further reduced activity in a cell-based system (Figure 5B). While CAM 2–56 may suffer from poor cell permeability due to the addition of biotin, it is unlikely that the pharmacokinetics of CAM 2–64 is significantly altered due to the addition of a single methylene onto BRD-K83694683, implying that the binding site doesn’t tolerate additional bulk at R1. Nevertheless, since we observed some activity of the biotin-linked compound CAM 2–56, we used it in combination with streptavidin support for affinity purification. In accordance with reduced binding affinity of these linkered macrolactams, we saw only moderate enrichment of ARID1A from ES cell lysates (Figure 5C, Figure S5A); however, we did observe a complete reduction of ARID1A enrichment upon preincubation of lysates with 200 µM of soluble BRD-K25923209, indicating selective binding (Figure 5C, Figure S5A). In addition, we observed only non-specific enrichment of PBAF subunit PBRM1 and loading control LaminB1. In agreement with the compounds targeting the ARID1A-containing BAF complexes, we observed a decrease in enrichment for BAF155 and BAF47, which are subunits of both BAF and PBAF. While these experiments indicate direct binding to ARID1A-containing BAF complexes, they are complicated by the decrease in affinity upon linking the compound to solid support. To circumvent the need for a derivatized scaffold for target ID, we turned to CETSA, a cell-based technique (Savitski et al., 2014). CETSA is based on the principle that the stability of a protein will be increased upon binding to a small molecule ligand, which can be visualized using immunoblot analysis after incubation of cells across a temperature gradient followed by removal of the insoluble, denatured proteins. Since ARID1A is a dedicated member of the large, 1–2 MDa SWI/SNF complex, changes in protein stability from binding a small molecule would be difficult to discern; however, we did observe a small but reproducible increase in ARID1A stability, but not PBRM1 or LaminB1 stability, upon compound treatment, providing further preliminary evidence that the compound binds ARID1A-containing complexes (Figure 5D). The known enzymatic activity of the BAF chromatin remodeling complex is DNA-stimulated ATP hydrolysis via the BRG1 subunit; however, we observed only slight inhibition of the ATPase activity at high concentrations of compound (Figure S5B) indicating that this is not its primary mode of action. Similarly, we performed glycerol gradients to assess whether compounds affect BAF complex formation and did not observe any significant disruption to BAF complex integrity upon compound treatment (Figure S5C). When we used a sequential salt extraction to investigate the possibility of the compounds inhibiting ARID1A association with chromatin, we saw a significant and reproducible shift in ARID1A elution but not PBRM1 elution upon treatment with compound (Figure 5E). Although these shifts are small in nature, they are very reproducible between experiments and consistent with affinity shifts previously observed upon deletion of a single BAF complex subunit or mutation of a single chromatin binding domain within large BAF complexes (Porter and Dykhuizen, 2017).
Mechanism of action in HIV latency reversal:
To examine the mechanism for how these BAF inhibitors might be inhibiting ARID1A-mediated repression, we used the J-Lat T-cell line models of HIV-1 latency, in which activation of transcription from the latent provirus results in GFP synthesis. We confirmed concentration dependent latency reversal upon treatment with BRD-K98645985, although at higher concentrations than in primary cells (Figure 5F, Figure S5D)). We investigated how nucleosome occupancy at the 5’-LTR changes upon treatment with BRD-K80443127 in J-Lat 11.1 using the formaldehyde assisted isolation of regulatory elements (FAIRE) assay, which is a measure of chromatin accessibility (Giresi et al., 2007). Overnight compound treatment resulted in increased chromatin accessibility at the Nuc-1 position of the 5’ LTR (Figure 5G) mimicking the effect elicited by the siRNA knockdown of ARID1A and treatment with BAF inhibitors identified in our previous studies (Rafati et al., 2011; Stoszko et al., 2016).
Discussion:
While the mammalian SWI/SNF chromatin remodeling complex is often referred to as a single protein complex, it is actually a heterogeneous assembly of closely related protein complexes with different biochemical and biological functions (Hargreaves and Crabtree, 2011; Wu et al., 2009). Several of these subcomplexes have been determined to be misregulated in disease, implicating subunits of SWI/SNF complexes as potential drug targets (Hohmann and Vakoc, 2014; Schiaffino-Ortega et al., 2014). A significant challenge for drug development is that general inhibition of SWI/SNF chromatin remodeling function may have undesirable toxicity as many, if not most, cell types require some form of SWI/SNF chromatin remodeling for basic viability (Dykhuizen et al., 2013; Hohmann and Vakoc, 2014). Further complicating matters, it is still unclear how the biochemical functions of individual subunits of chromatin remodeling complexes are related to desired phenotypes, making it difficult to design and implement biochemical screening programs. To circumvent these issues, we developed a robust high throughput phenotypic screen designed to identify small molecules that inhibit BAF-mediated transcription without affecting cellular viability. From this screen, we have identified a 12-membered macrolactam with low toxicity and the ability to inhibit the transcriptional activities of the BAF complex. These compounds have significant promise as an HIV-1 latency reversal agent, particularly because of their potential for clinical use in combination therapy with other currently available LRAs. Indeed, we show that treatment with the structurally similar macrolactams, BRD-K98645985, BRD-K25923209, and BRD-K80443127, triggered HIV transcription in ex vivo infected primary CD4+T cells harboring latent HIV and potentiated the effect of other latency reversal agents when used in combination. In CD4+ T cells isolated from c-ART treated virologically suppressed HIV-1 infected patients, a significant increase in cell associated HIV mRNA was observed after ex vivo treatment with BRD-K80443127 alone in one patient, while in all three patients, BRD-K80443127 treatment lead to significant potentiation of prostratin activity. The mechanism of action appears to be that of de-repression, or inhibition of the HIV-1 LTR-bound repressive BAF complex. Our observed modest and variable effects in latency reversal by BRD-K80443127 alone is consistent with this notion and points to the need for larger patient cohorts in order to test the activity of this compound with robust across patient statistics. Importantly, in line with the mechanism of action of BRD-K80443127 as an LTR de-repressor, co-treatment with the PKC agonist prostratin, which is a bona fide activator of HIV transcription resulted in significant increase in cell associated HIV RNA in all patients. This is in line with recent findings that ARID1A degraders from our previous studies (Stoszko et al., 2016) act to increase transcriptional noise (frequency or burst) at the HIV-1 LTR promoter (Megaridis et al., 2018). This points to the potential for this class of compounds for inclusion in combinatorial therapy with other drugs targeting different steps in HIV-1 transcription.
Target identification and mechanistic work indicates that this inhibitor binds the ARID1A-containing BAF complexes and prevents ARID1A function at the 5’ LTR of HIV-1, although the exact mechanism of compound action is still to be resolved. The SAR from this study indicates that further variation of substituents at the R1 and R2 positions on the macrolactam scaffold will likely not improve potency much, but optimization at other unexplored positions around the macrolactam ring could increase potency and also facilitate derivatization for more in-depth target identification. Deciphering how this class of compounds inhibits ARID1A activity will be critical for further optimization of these inhibitors as HIV-1 latency reversal agents targeting the BAF complex. In addition, we have found that BAFi synergizes with ATR inhibitors for the killing of specific types of cancer, particularly those with high mutational backgrounds (Chory et al. submitted). Here again, a detailed understanding of the exact mechanism of actions for these inhibitors will be necessary for pharmacologic and therapeutic optimization as well as a tool to understand the mechanism of action of ARID1A-containing BAF complexes in polycomb eviction and resolution of facultative heterochromatin (Kadoch et al., 2016; Miller et al., 2017; Stanton et al., 2016).
STAR Methods:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Emily Dykhuizen (edykhui@purdue.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Line Authentification:
Cell lines were obtained directly from ATCC or NIH AIDS reagent program and used at less than 15 passages.
Cell Culture Conditions:
E14 ESC and Bmi-luc ESC culture:
Mouse male ESCs were cultured in ESC media: (DMEM (Gibco), 15% ES tested FBS (Applied Stem Cell), 1% HEPES (Gibco), 1% Sodium pyruvate (Gibco), 1% Pen/Strep (Invitrogen), 1% Glutamine (Invitrogen), 1% non-essential amino acids (Gibco) and 0.1% Lif-condition media from Cos-Lif cells. Media was changed daily. After 72 h, the cells were split with 0.25% Trypsin-EDTA (Gibco) and plated at the same density on tissue culture plates that had been treated with 0.1% gelatin in water (Millipore) for 30 minutes and removed.
Arid1af/f:CreERT2 ESCs:
Mouse ESCs were cultured in Knockout™ DMEM (Thermo Fisher Sci #10829018) supplemented with 15% ESC-Sure FBS serum (Applied Stem Cell #ASM-5007) and Knockout™ Serum Replacement (Thermo Fisher Sci #10828028), 2 mM L-glutamine (Gibco #35050061), 10 mM HEPES (Gibco #15630080), 1 mM sodium pyruvate (Gibco #11360070), 100 U/mL penicillin/streptomycin (Gibco #15140122), 0.1 mM non-essential amino acids (Gibco #11140050), 0.1 mM beta-mercaptoethanol (Gibco 21985023) and leukemia inhibitory factor (LIF). ESCs were maintained on gamma-irradiated mouse embryonic fibroblast (MEF) feeders at 37°C, 5% CO2 with daily media changes and passaged every other day.
HEK293T Cell Culture:
Human female HEK293T cells were cultured in (DMEM (Gibco), 10% FBS (Omega), 1% Sodium pyruvate (Gibco), 1% Pen/Strep (Invitrogen). After 72 h, the cells were split 1:4 with 0.25% Trypsin-EDTA (Gibco).
A549 Cell Culture:
Human male A549 cells were cultured in (DMEM (Gibco), 10% FBS (Omega), 1% Sodium pyruvate (Gibco), 1% Pen/Strep (Invitrogen). After 72 h, the cells were split 1:4 with 0.25% Trypsin-EDTA (Gibco).
HepG2 Cell Culture:
Human male HepG2 cells were cultured in (DMEM (Gibco), 10% FBS (Omega), 1% Sodium pyruvate (Gibco), 1% Pen/Strep (Invitrogen). After 72 h, the cells were split 1:4 with 0.25% Trypsin-EDTA (Gibco) and replated.
Jurkat Cell Culture:
Human male J-Lat A2 and J-Lat 11.1 cells were cultured in RPMI-1640 medium (Sigma Aldrich) supplemented with 10% FBS and 100 µg/ml penicillin-streptomycin (Sigma Aldrich) at 37 °C in a humidified 95% air-5% CO2 atmosphere. After 72 h, the cells were diluted to a concentration of 2 × 105 cells/mL with fresh media.
Primary human CD4+ T cells:
Primary human CD4+ T cells from either healthy donors or HIV+ patients were obtained via either blood donations (buffy coats) or leukapheresis respectively. PBMCs were isolated by Ficoll gradient followed by isolation (negative selection) of CD4+ T cells by RosseteSep kit (Stem Cell Technologies) or by negative selection with EasySep (Stem Cell Technologies) from healthy donors or HIV+ patients respectively. CD4+ T cells were cultured at a density of 1–1.5 × 106/ml in RPMI-1640 medium supplemented with 7% FBS and 100µg/ml penicillin-streptomycin at 37 °C in a humidified 95% air-5% CO2 atmosphere before treatment with compounds or incubation in presence of αCD3/αCD28 beads.
Subject details:
This study was conducted in accordance with the ethical principles of the Declaration of Helsinki. The patients involved in the study provided signed informed consent and the study protocol was approved by The Netherlands Medical Ethics Committee (MEC-2012–583). Three otherwise healthy adult HIV-1 infected patients on stable c-ART with effective viral suppression <50 c/mL were recruited at the Erasmus MC HIV outpatient clinic. Patient Inclusion criteria: 1. Age >18 years, 2. Confirmed HIV-1 infection. 3. Plasma HIV RNA viral load <50 copies per ml. 4. on cART. Patient Exclusion criteria: 1. Inability to place 2.5 cm venous catheter, 2. Previous use of any known latency reversal agents. 3. Pregnancy 4. Major comorbidities including anemia, defined as a hemoglobin level of <6.0 mmol/L (women) or <6.5 mmol/L (men), cardiovascular disease, hepatitis B/C co-infection, severe psychiatric disorder, active drug use.
METHOD DETAIL
Bmi1-luciferase reporter cell line:
Low passage mESCs derived from 129 mice (20 million, p10) were electroporated with 40 µg of a linearized construct consisting of 2 kb homology upstream of the Bmi1 locus, firefly luciferase at exon 1 of Bmi1 followed by loxP neo and a 6 kb 3’ homology arm with thymidine kinase outside the homology arms. The cells were plated on 10 gelatin treated plates (60 mm) of irradiated neo resistant MEFs and selected with G418 and ganciclovir for 5 days. 384 colonies were selected, trypsinized and replated in gelatin treated 24-well plates for expansion. The cells were split and DNA was isolated for digestions with EcoRI or BamHI. We confirmed the successful homologous recombination in 7 out 384 colonies using Southern blot analysis at both the 5’ (EcoRI) and 3’ (BamHI) end (see Table S2 for probe and primer sequences). We deleted the neomycin cassette using transfected Cre recombinase and confirmed the excision at all clones using a second round of Southern blot analysis at the 3’ end (BamHI).
Lentiviral Infection
HEK293T cells were transfected with lentiviral constructs along with lentiviral packaging vectors pMD2.G and psPAX2. After 48 h, supernatants were collected and virus isolated using ultracentrifugation at 20,000 r.p.m. for 2 h. Viral pellets were re-suspended in PBS and used to infect cell lines. Cells were selected with puromycin and harvested 72 h after infection.
Compound Treatment:
10,000 cells in 30 µL ESC media were plated in white 384 well CellBind plates. The cells were cultured in a 37 °C, 5% CO2 incubator for 24 h and 100nL/well of 0.75mM of positive control (Pubchem SID: 85814977) or 100 nL/well of 3.75 mM compound library (primary screen) was added via pin transfer into plates. There was no effect on assay readout at DMSO concentrations up to 0.5%. The cells were cultured in a 37 °C, 5% CO2 incubator for 24 h for luciferase reporter assay and 18 h for qRT-PCR assay.
Luciferase assay:
(PubChem AID 602393 primary, PubChem AID 651717 confirmatory.) The assay plates were removed from the incubator and equilibrates for 10 minutes to room temperature. Promega SteadyGlo® solution (10 µL/well) was added to each well of the assay plates. The assay plates were mixed at 1000 rpm for 15 seconds and then incubated for 10 minutes at room temperature. The luciferase levels were read on a Perkin Elmer Envision in Ultra Sensitive Luminescence mode. Signals remained stable up to 2 hours.
qRT-PCR secondary screen (PubChem AID 743180, 743177, 743176)
The qRT-PCR screen was performed as published (Dykhuizen et al., 2012). In brief, 5,000 ES cells were plated on gelatin-coated 384-well tissue culture plates and cultured in a 37C, 5% CO2 incubator. 24 h later, hit compounds in 100 nL DMSO were treated at eight different doses. 18h after compound treatment cells were washed two times with 100 µL PBS and all excess PBS was removed by centrifuging the plates upside down at 1000 rpm. The Ambion ® Cells-to-Ct kit was used to generate cDNA. In brief, the cells were lysed in the plate in 10 µL lysis buffer containing DNase for 10 minutes and quenched with 1 µL lysis stop buffer. The lysate (2 µL) was added to 5 µL RT reaction buffer (2X), 2.5 µL nuclease-free water and 0.5 µL Reverse transcriptase (20X) and incubated at 37 °C for 60 minutes and 95 °C for 1 minute to generate 10 µL of cDNA. cDNA (1 µL) was used in each 5 µL qPCR reaction with Roche master mix and TaqMan probes (Applied Biosystems) for Bmi1-FAM (Mm00776122_gH), Ring1a-FAM (Mm01278940_m1/4331182), or Fgf4-FAM (Mm00438916_g1/4351372) alongside actin-VIC (4352341E) for a loading control. The qPCR was run accordingly: 95 °C for 10 minutes Then 55 cycles of: 95 °C for 10 seconds followed by 60 °C for 30 seconds. The fold increase in transcription was calculated using the ∆∆CT method (Livak and Schmittgen, 2001).
Viability Assays (PubChem AID: 743188, 743189, 743190, 1053139, 1053140, 1053141)
Cell culture: HepG2, A549 and HEK293 cells were propagated to 95% confluence in DMEM containing 10% FBS 1% Pen/Strep, 1% L-Glutamine. Cells were plated at 2000 per well in 40 µL media in white tissue culture treated 384-well plates and incubated at 5% CO2; 95% humidity, 37 ºC for 24 hours. Compound (100 nL) was added to wells using a pin tool (CyBi Well) alongside 100 nL cytotoxic compounds, mitoxantrone (final concentration of 10 µM) as a positive control. The cells were incubated for 72 hours at 37 ºC, 95% humidity 5% CO2. Plates were removed from the incubator, equilibrated for 15 minutes to room temperature; and 20 µL 50% Promega CellTiterGlo (diluted 1:1 with PBS, pH 7.4) was added. The plates were read on Perkin-Elmer EnVision with standard luminescence settings for 0.1 sec per well.
qRT-PCR screen confirmation and DOS analog Library (SYBR)
50,000 mESCs were plated on gelatin coated 24-well plates. After 24 hours, the cells were treated with 30 µM of compound and incubated at 5% CO2; 95% humidity, 37 °C for 18 hours. RNA was isolated using Trizol® and cDNA was synthesized from 1 µg RNA using Superscript III Reverse Transcriptase with Oligo(dT)12–18 primers (Thermo) and diluted 10x with water. 1 µL of this cDNA mixture was used for qPCR with 2x SYBR (Roche) and the following primers: Bmi1: Forward: TACCATGAATGGAACCAGCA; reverse: AAAGGAAGCAAACTGGACGA, Ring1a: Forward: CCTGGACATGCTGAAGAACA; reverse: TCCCGGCTAGGGTAGATTTT, FGF4: Forward: GGGTGTGGTGAGCATCTTCGGA; reverse: GGTATGCGTAGGACTCGTAGGGC, Gapdh: Forward: TGCACCACCAACTGCTTAG; reverse: GGATGCAGGGATGATGTTT.
Chemical Synthesis of DOS analog library:
The synthesis of the original DOS library was performed according to the Head–to-Tail strategy for combinatorial synthesis of multiple scaffold simultaneously (Fitzgerald et al., 2012). For the synthesis of the 30 macrolactam library members in solution the backbone (compound 2141–017) was synthesized according to published procedures in the scheme in (Figure S3). The synthesis of the six representative compounds used in HIV latency experiments (Figure 4, Figure S3) from this backbone are outlined below:

Nitro Reduction (2141–018b):
To macrocycle (2141–017) (.675 g, 1.550 mmol) dissolved in MeOH (Volume: 15.50 ml) was added tin(II) chloride dihydrate (3.50 g, 15.50 mmol). The reaction mixture was stirred at room temperature for 2 days or until LC/MS indicates complete conversion. The residue was dissolved in EtOAc and washed with 2 M aq. KOH (2x). The combined aqueous layers were washed with EtOAc (2x) and the resulting organic layers were washed with brine, dried over MgSO4, filtered, and concentrated. The crude aniline (2141–018b) was used without purification. Note: The workup as described above produces a lot of precipitate/emulsion. This can be overcome by extensive washing or by quenching with 1 volume 1 M NaOH and filtration over celite prior to workup. (M+H)+ calculated = 406.24 (M+H)+ measured (LC/MS) = 405.98
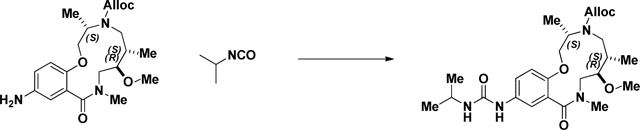
Acylation of the Aniline (2141–018):
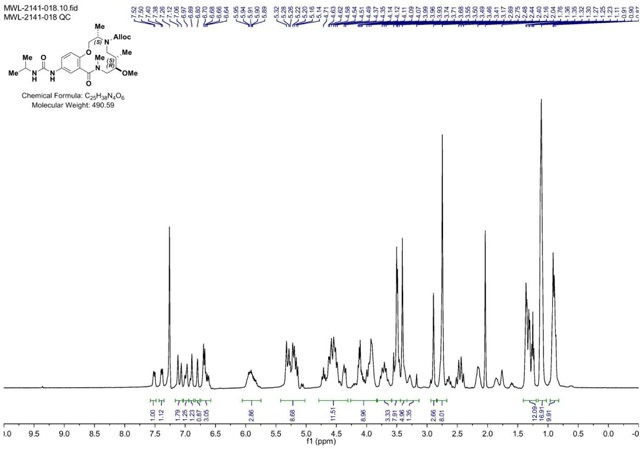
Isopropyl isocyanate (0.174 ml, 1.767 mmol) was added dropwise to a stirring solution of crude aniline (2141–018b) (.597g, 1.472 mmol) in CH2Cl2 (Volume: 7.36 ml) and reaction was stirred at rt overnight. LC/MS showed conversion into the desired product. The solvent was evaporated and the residue was purified via ISCO (0.5–9% MeOH in CH2Cl2, 18 min); Collected fractions 40–52 to afford the product as a white solid foam. 644 mg (89% over 2 steps) (M+H)+ calculated = 491.29 (M+H)+ measured (LC/MS) = 492.73
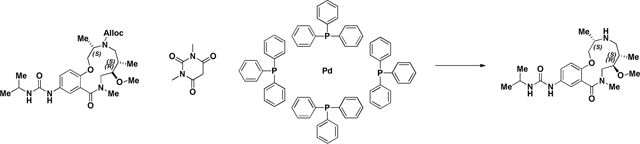
Alloc deprotection (2141–022):
A 100 mL round-bottomed flask was charged with a solution of Urea (2141–018) (.625g, 1.274 mmol) in EtOH/CH2Cl2 (2:1). 1,3-Dimethylbarbituricacid (0.298 g, 1.911 mmol) was added in one portion at room temperature, followed by Tetrakis(triphenylphosphine)palladium(0) (0.147 g, 0.127 mmol). The mixture was stirred at 40 °C for ~16 h (overnight) or until LC/MS demonstrate d conversion into a product with the same mass. The solvents were removed in vacuo, and the crude residue was dissolved in CH2Cl2 and passed through a plug of acidic resin (5 equiv relative to SM) rinsing with CH2Cl2 (~3 column volumes). The amine was then eluted with 1M NH3 in MeOH to afford sufficiently clean material from next step. (M+H)+ calculated = 407.27 (M+H)+ measured (LC/MS) = 407.56
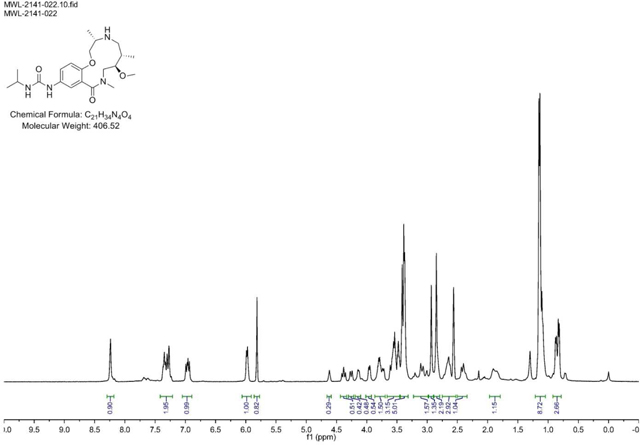
BRD-K13648511:
Benzaldehyde (0.090 ml, 0.886 mmol) was added to a DMF (Volume: 2.95 ml) solution of crude amine (2141–022) (.12g, 0.295 mmol) at rt. Acetic acid (0.017 ml, 0.295 mmol) was added and the mixture was stirred for 30 min before sodium triacetoxyhydroborate (0.250 g, 1.181 mmol) was added. The resulting mixture was stirred at rt overnight. LCMS indicates complete SM consumption. Saturated aqueous sodium bicarbonate solution was slowly added until gas evolution ceased. The reaction mixture was diluted with EtOAc and the layers were separated. The organic layer was washed with brine, dried with MgSO4, filtered, and concentrated. The residue was purified via ISCO (SiO2, 1–12% MeOH in CH2Cl2, 20 min, 254 nm); Collected fractions 47–54 to afford the product as a white solid in 23% yield (34 mg) over 2 steps. (M+H)+ calculated = 497.3123, (M+H)+ average (3 ESI replicates) = 497.3131 ± 1.81
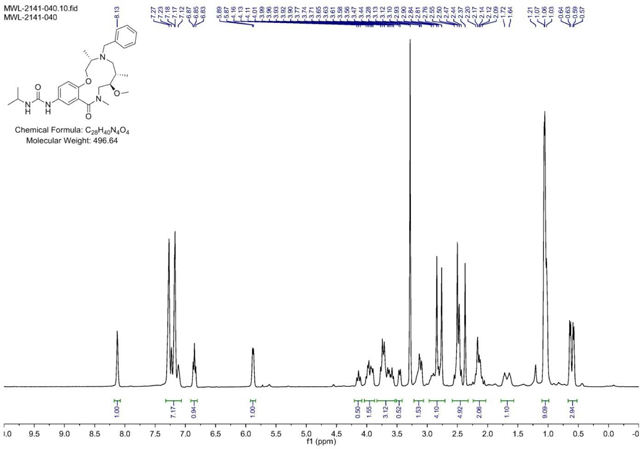
BRD-K17257309:
4-(pyridin-3-yl)benzaldehyde (151 mg, 0.827 mmol) was added to a DMF (Volume: 1378 µl, Density: 0.944 g/ml) solution of Crude amine (2141–022) (112mg, 0.276 mmol) at rt. Acetic acid (15.77 µl, 0.276 mmol) was added and the mixture was stirred for 30 min before sodium triacetoxyhydroborate (234 mg, 1.102 mmol) was added and the mixture was stirred at rt overnight or until LC/MS indicates conversion into product. Saturated aq sodium bicarbonate solution was slowly added until gas evolution ceased. The reaction mixture was diluted with EtOAc and the layers were separated. The aqueous phase was extracted with EtOAc (3x). The combined organic layers were washed with brine, dried with MgSO4, filtered, and concentrated. The residue was purified via ISCO (0.5–7% MeOH in CH2Cl2, 13 min) to afford the product as a yellow solid in 24% yield (38 mg) over 2 steps. (M+H)+ calculated = 574.3388 (M+H)+ average (3 ESI replicates) = 5.74.3394 ± 0.85
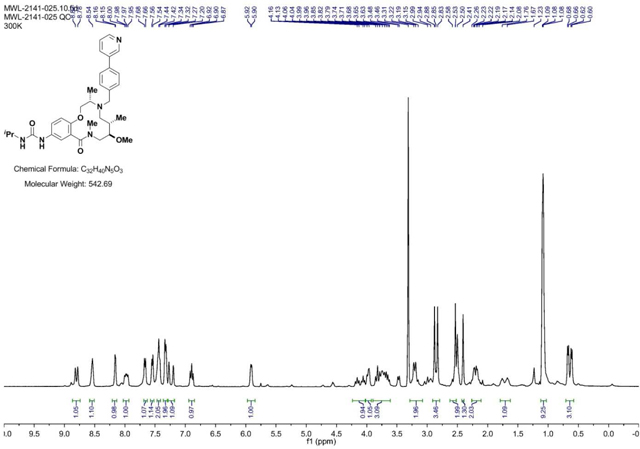
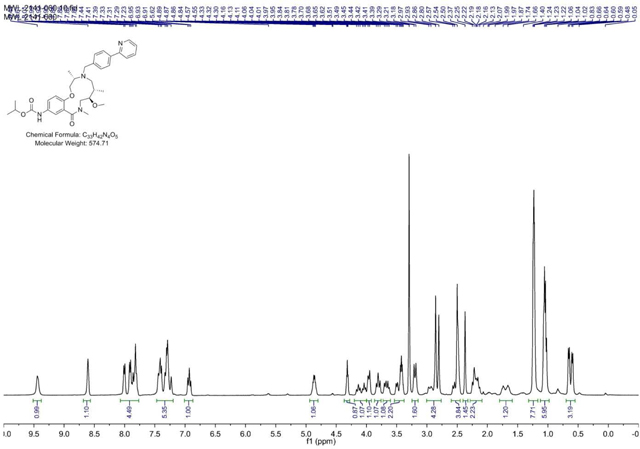
BRD-K98645985:
4-(Pyridin-2-yl)benzaldehyde (199 mg, 1.085 mmol) was added to a DMF (Volume: 1808 µl) solution of crude amine (2141–022) (147mg, 0.362 mmol) at room temperature. Acetic acid (21.72 mg, 0.362 mmol) was added and the mixture was stirred for 30 min before NaBH(OAc)3 (307 mg, 1.446 mmol) was added and the mixture was stirred at rt overnight. LC/MS indicated conversion into the desired product. Saturated aqueous sodium bicarbonate solution was slowly added until gas evolution ceased. The reaction mixture was diluted with EtOAc and the layers were separated. The aqueous phase was extracted with EtOAc (3x). The combined organic layers were washed with brine, dried with MgSO4, filtered, and concentrated. The residue was purified via ISCO (0.5–7% MeOH in CH2Cl2, 13 min); Collected fractions 66–73 to afford the product as a brown solid in 26% yield (54 mg) over 2 steps. (M+H)+ calculated = 574.3388 (M+H)+ average (3 ESI replicates) = 5.74.3395 ± 1.36.
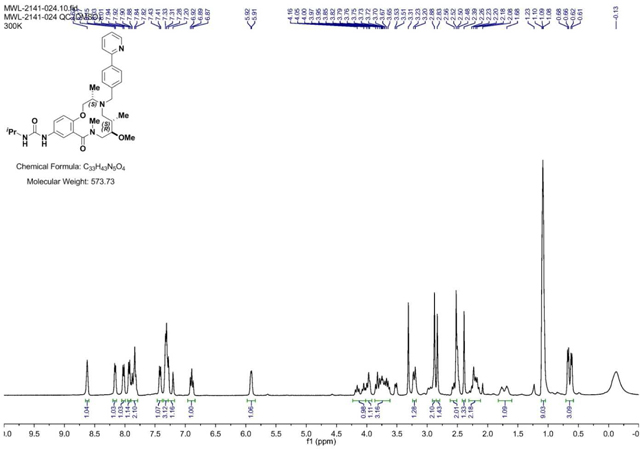
Alloc Deprotection (2141–039a):
A round-bottomed flask was charged with a solution of macrocycle (2141–017) (.675 g, 1.550 mmol) in EtOH/ CH2Cl2 (2:1). 1,3-Dimethylbarbituricacid (0.363 g, 2.325 mmol) was added in one portion at room temperature, followed by Pd(PPh3)4 (0.179 g, 0.155 mmol). The mixture was stirred under ambient conditions for ~16 h (overnight). The reaction was monitored by LC/MS and demonstrated complete starting material consumption and the presence of the desired mass. The reaction mixture was then passed over a silica plug eluting with 15% MeOH in CH2Cl2 (with 2% triethylamine). The filtrate was concentrated and used in the next step without purification (used theoretical yield). (M+H)+ calculated = 352.19 (M+H)+ measured (LC/MS) = 351.90
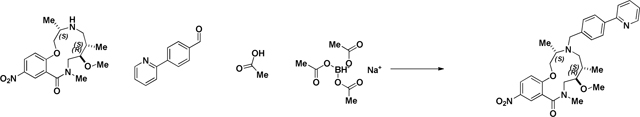
Reductive Amination (2141–039b):
4-(2-Pyridinyl)-benzaldehyde (852 mg, 4.65 mmol, 3 eq) was added to a DMF (Volume: 7755 µl) solution of crude amine (2141–039a) (545 mg, 1.551 mmol, 1 eq) at room temperature. Acetic acid (89 µl, 1.551 mmol, 1 eq) was added and the mixture was stirred for 30 min before sodium triacetoxyhydroborate (1315 mg, 6.20 mmol, 4 eq) was added. The mixture was stirred at room temperature for 2 days when LC/MS indicated complete conversion into product (presence of SM by LCMS). Saturated aqueous sodium bicarbonate solution was slowly added until gas evolution ceased. The reaction mixture was diluted with EtOAc and the layers were separated. The aqueous layer was extracted with EtOAc (3x). The organic layer was washed with brine, dried with MgSO4, filtered, and concentrated. Material was taken forward without further purification attempts. (M+H)+ calculated = 519.29 (M+H)+ measured (LC/MS) = 518.91.
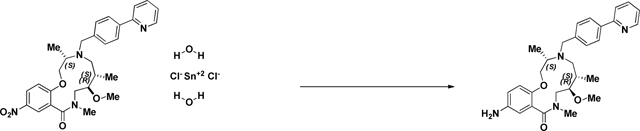
BRD-K25923209:
To Macrocycle 2141–039b (.804g, 1.550 mmol) dissolved in MeOH (Volume: 15.50 ml) was added tin(II) chloride dihydrate (3.50 g, 15.50 mmol). The reaction mixture was stirred at 40 °C for 24h until LC/MS indicated complete consumption of starting material (nitro) and presence of desired mass. Upon completion, the reaction mixture was concentrated and the resulting residue was dissolved in EtOAc and washed with 2 M aq. KOH (2x). The combined aqueous layers were washed with EtOAc (4X). The resulting organic layers were washed with brine, saturated aqueous NaHCO3, water, and brine, dried over MgSO4, filtered, and concentrated. The crude aniline was sufficiently pure to use in the capping step and therefore was used without purification.
Note: The workup as described above produces a lot of precipitate/emulsion. This can be overcome by extensive washing or alternately, the reaction can be quenched with 1 volume of 1 M NaOH, stirred with celite for 10 minutes, and filtered prior to workup to yield 33.8% (17.2 mg) over 3 steps. (M+H)+ calculated = 489.286 (M+H)+ average (3 ESI replicates) = 489.2866 ± 0.92.
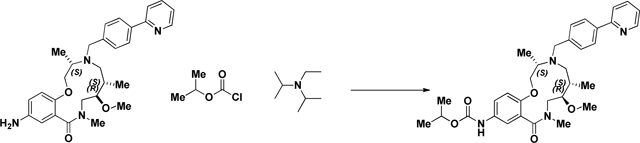
BRD-K80443127:
A screwtop vial was charged with aniline (BRD-K25923209) (43.3mg, 0.089 mmol) and CH2Cl2 (Volume: 886 µl, Density: 1.325 g/ml). Isopropyl chloroformate (115 µl, 0.115 mmol) and DIEA (46.4 µl, 0.266 mmol) were added dropwise and reaction stirred under ambient conditions for 1 h. LC/MS showed complete sm consumption. The reaction mixture was loaded directly onto SiO2 and purified via ISCO (1–12% MeOH in CH2Cl2, 20 min); Collected fractions 40–44 to afford the product as a yellow oil in 33.8% (17.2 mg) yield. (M+H)+ calculated = 575.3228 (M+H)+ average (3 ESI replicates) = 575.3235 ± 1.32.
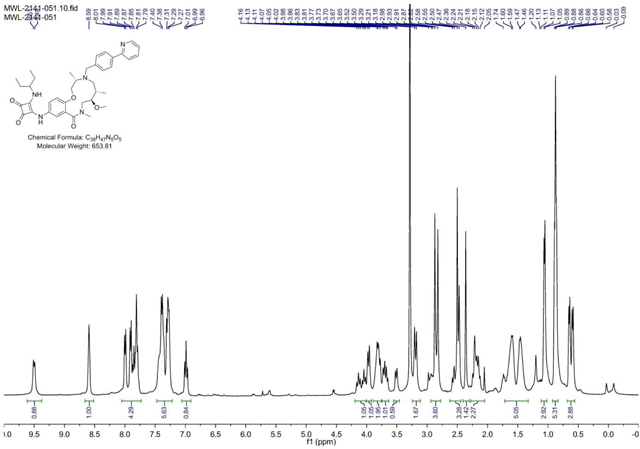
BRD-K04244835:
A 2.5mL microwave vial was charged with aniline (BRD-K25923209) (40 mg, 0.082 mmol) and EtOH (Volume: 819 µl, Density: 0.81 g/ml). Squaramide (CRE-III-001) (20.75 mg, 0.098 mmol), DIEA (42.8 µl, 0.246 mmol), and DMAP (2.000 mg, 0.016 mmol) were added in sequence and the vial was sealed. The resulting solution was stirred at 85 °C overnight or until LC/MS indicated complete starting material consumption. The reaction mixture was loaded directly onto SiO2 and purified via ISCO (1–12% MeOH in CH2Cl2, 20 min); fractions 50–56 were collected to afford the product as a yellow solid in 48.6% (26 mg) yield. (M+H)+ calculated = 654.365 (M+H)+ average (3 ESI replicates) = 654.3661 ± 1.64.
Nuclear Isolation:
Cells were trypsinized and washed 1x with PBS. Cells were suspended in Buffer A (25 mM HEPES pH 7.6, 5 mM MgCl2, 25 mM KCl, 0.05 mM EDTA, 10% glycerol, 0.1% NP-40) and cell membranes were lysed by passing cells through a 22-gauge needle 10 times and incubation on ice for 5 minutes. Nuclei were pelleted by centrifugation at 400 × g for 5 minutes.
Cell Lysis:
Pellets of whole cells or isolated nuclei were lysed with lysis buffer (50 mM Tris-HCl pH 8.0, 300 mM NaCl, 0.1% NP-40, protease inhibitors) by rotating at 4 °C for 30 minutes. The lysate was cleared by centrifugation (20,000 × g) for 5 minutes and transferred to a fresh tube.
ATPase Assay:
Adapted from (Bultman et al., 2005). Each sample utilized 70 µg nuclear ESC lysates in 50 µL cell lysis buffer. The nuclear lysates were incubated with 1 µL anti-Brg1 antibody (ab110641) and 5 µL protein A dynabeads (Thermo) for 4 h and washed 1 × with lysis buffer and 1 × with wash buffer (10 mM Tris-HCl pH 7.5, 50 mM NaCl, 5 mM MgCl2, protease inhibitor). After washing, 20 µL reaction buffer (10 mM Tris pH 7.5, 50 mM mM NaCl, 5 mM MgCl2, 20% glycerol, 1 mg/ml BSA, 20 µM ATP, 20 nM plasmid DNA, 1 µCi gamma-32P ATP, protease inhibitors, 1 µL DMSO or compound to 250 µM final concentration) was added to the beads. The reaction was incubated with shaking (900 rpm) at 37 °C. After 30 minutes the reaction mixture (1 µL) was spotted on PEI cellulose TLC plates (Sorbtech) and the TLC was run in 0.5M LiCl, 1M formic acid. The plates were dried and developed using phosphoimager technology. The percent conversion from starting material to product was determined using Image J software and normalized to the no enzyme control.
Immunoblot Analysis:
Total protein was denatured for 10 min at 95 °C, separated on a 4–12% SDS-polyacrylamide gel, and transferred to a PVDF membrane (Immobilon FL, EMD Millipore, Billerica, MA). The membrane was blocked with 5% bovine serum albumin (VWR, Batavia, IL) in PBS containing 0.1% Tween-20 (PBST) for 30 mins at room temperature and then incubated in primary antibodies overnight at 4°C. The primary antibodies used were directed against ARID1A (Santa Cruz Biotechnology Inc., Dallas, TX; sc-32761), PBRM1 (Bethyl Laboratories, Montgomery, TX; A301–591A), BAF155 (Santa Cruz Biotechnology Inc., Dallas, TX; Sc-32763), BAF47 (Santa Cruz Biotechnology Inc., Dallas, TX; Sc-166165), LAMIN B1 (Santa Cruz Biotechnology Inc., Dallas, TX; Sc-377000). The primary antibodies were detected by incubating the membranes in goat-anti-rabbit or goat-anti-mouse secondary antibodies (LI-COR Biotechnology, Lincoln, NE) conjugated to IRDye 800CW or IRDye 680 respectively for 1 h at room temperature, and the signals were visualized using Odyssey Clx imager (LI-COR Biotechnology, Lincoln, NE).
Glycerol Gradients:
20 million mESCs were plates on gelatin coated 150 mm tissue culture plates and incubated for 24 h. Cells were treated with 30 µM BRD-K25923209 or DMSO for 24 h. Nuclear lysate from ~50 million cells (~0.6– 1 mg nuclear protein) was layered on top of a 10–30% glycerol gradient (10 mL) in HEMG buffer (25 mM Hepes 7.6, 0.1 mM EDTA, 12.5 mM MgCl2, 100 mM KCl). The protein was separated by the gradients through ultracentrifugation in Beckman rotor SW41 at 40,000 RPM for 16 h. Twenty 500 µL fractions were removed sequentially from the top of the gradients and 50 µL of each fraction was used for immunoblot analysis.
Sequential Salt Extractions:
Sequential salt extractions were performed as published 80, with the following modifications: mESCs were treated for 2 h with 30 µM BRD-K25923209 or DMSO. Nuclei were isolated into two tubes (5 million each) and the salt extractions were performed with 30 µM of BRD- K25923209 or DMSO in each salt wash.
Biotin Pull Downs:
Compound 2–57 (200 µM in 200 µL PBS) was pre-bound to 20 µL high capacity streptavidin beads (Solulink) for 30 minutes. The beads were washed 1x with lysis buffer and incubated with cell lysates preincubated with DMSO or 200 µM of BRD-K25923209 for 2 h. The beads were washed once with 1 mL lysis buffer and boiled in SDS loading buffer for immunoblot analysis.
CETSA protocol:
CETSA was performed as published (Jafari et al., 2014) with the following modifications: mESCs were treated with 50 µM of compound BRD-K25923209 or DMSO for 1 hour. The cells were trypsinized, washed 1 × with PBS and 2 million cells of each condition were resuspended in 1 mL PBS. Cells (100 µL) were transferred into eight wells of a strip tube and put in a temperature gradient PCR block for 3 minutes, followed by incubation at room temperature for 3 minutes. The cells were lysed with 2 × freeze-thaw cycles on a 25 °C heat block and the cell lysis was transferred to microcentrifuge tubes and centrifuged at 15,000 × g for 20 minutes at 4 ºC. The soluble protein (90 µL) was removed from pellets and added to SDS loading buffer for immunoblot analysis.
RNA-Seq ECS with BAFi:
Mouse ESE14 cells were plated at a density of 2.5 × 105 in a gelatin coated 6-well plate and in a 37 °C, 5% CO2 incubator for 24 h. RNA was purified from ESCs treated with BAF inhibitor BRD-K98645985 or a DMSO mock treatment for 16h. RNA was extracted using TRIzol reagent (Life Technologies Corporation, Grand Island, NY) according to the manufacturer’s instructions and cleaned up using RNeasy Mini Kit (Qiagen Inc., Valencia, CA). RNA libraries were prepared for sequencing using standard Illumina protocols. Sequencing was performed using an Illumina HiSeq 2500. Reads were 100 bp and paired-ended. Reads were trimmed using Trimmomatic and aligned to the mm10 reference genome using STAR. Gene expression levels were computed using htseq-count using default parameters. Differential expression analysis was performed with DESeq2 using default parameters (Anders and Huber, 2010). Processed and unprocessed data is deposited in GEO GSE113627.
RNA-Seq Arid1a f/f ESCs:
Arid1af/f mice were a kind gift from Terry Magnuson (UNC School of Medicine) (Chandler et al., 2015). Arid1af/f mice were bred to Actin:CreERT2 mice to obtain Arid1af/+;ActinCreERT2, which were subsequently interbred with Arid1af/f mice. Timed matings were set up between Arid1af/f;ActinCreERT2 and Arid1af/f and oviducts were flushed at day 3.5. ESCs were derived as described (Ho et al., 2011). Arid1af/f:CreERT2 ESCs were treated with either ethanol or 1 µM 4-hydroxytamoxifen for 24 hours then passaged. RNA was collected 72 hours after treatment. RNA was isolated using Quick-RNA Miniprep Kit (Zymo Research). RNA-Seq libraries were prepared using either Illumina TruSeq RNA Library Prep Kit v2 or Illumina TruSeq Stranded mRNA Kit following the manufacturer’s instructions. Sequencing was performed using an Illumina HiSeq 2500. Reads were 50 bp and single-ended. Fastq files were evaluated for quality using FastQC and trimmed using Trimmomatic. Trimmed sequences were mapped to the mm10 reference genome using HISAT2 in single-end mode with default parameters. The average counts per million (cpm) was calculated for each sample condition using custom R scripts, and differentially expressed genes were identified using the edgeR package in Bioconductor (Robinson et al., 2010). A false discovery threshold of 0.05 was imposed using the toptags function of edgeR. Processed and unprocessed data is deposited in GEO GSE113872
Cell line models of HIV-1 latency:
J-Lat A2 (LTR-Tat-IRES-GFP Cells) (Jordan et al., 2001) and J-Lat 11.1 (integrated full-length HIV-1 genome mutated in env gene and harboring GFP in place of Nef)(Jordan et al., 2003)) cells were cultured in RPMI-1640 medium (Sigma Aldrich) supplemented with 10% FBS and 100 µg/ml penicillin-streptomycin at 37 °C in a humidified 95% air-5% CO2 atmosphere. Cells were treated with compounds or DMSO for 48 h, followed by quantitation of GFP positive cells using flow cytometry. Data was normalized as a fold increase over DMSO treated control. Data are presented as mean of at least 3 independent experiments ± SD.
Ex Vivo HIV latency model (Lassen et al., 2012):
Viral pseudotyped particles were obtained by co-transfecting HXB2 Env together with the HIV-1 backbone plasmid (pNL4.3.Luc.R-E-) into HEK 293 T cells using PEI (Polyethylenimine) transfection reagent. At 48 h and 72 h post-transfection, the pseudovirus-containing supernatant was collected, filtered through a 0.45 µm filter, aliquoted, and stored at − 80 °C. Primary CD4 + T cells were isolated from buffy coats from healthy donors by Ficoll gradient followed by density-based negative selection of CD4+ T cells with RosetteSep kit (StemCells Technologies). Twenty-four hours after isolation, cells were spin-infected as described, with minor modifications (Lassen et al., 2012; Stoszko et al., 2016). CD4+ T cells were spin-infected at 1200 g for 2 h with the HBX2 Env pseudotyped pNL4.3-Luc virus. Eighteen hours after spin-infection cells were washed and cultured in growth media supplemented with 5 µM saquinavir mesylate. After three days, latently infected cells were treated with BAFi’s or left untreated for 24 h in the presence of 30 µM raltegravir, followed by luciferase assay (Promega). Data was normalized as a fold increase over untreated control. Synergy was calculated using Bliss score formula (BLISS, 1939): Sexp =[1 − (1 – A)× (1− B)], where Sexp. is the expected percentage of cells reactivated after combinatorial treatment in absence of synergism and A and B correspond to the percentage of cells reactivated by the single treatments. Combination was considered synergistic if the observed effect of combined treatments was significantly higher than calculated value (Sexp.), and is indicated with an S in the figure.
Ex Vivo HIV latency model (Bosque-Planelles)(Bosque and Planelles, 2009):
Primary CD4 + T cells were isolated from buffy coats from healthy donors by Ficoll gradient followed by density-based negative selection of CD4+ T cells with RosetteSep kit (StemCells Technologies). Twenty four hours after isolation primary CD4+ T cells were cultured in the presence of 10 ng/ml TGF-β (Sigma-Aldrich), 1 µg/ml α-IL-4 (PeproTech) and αCD3/CD28 dynabeads (Life Technologies) at the cell:bead ratio 1:1 for 3 days. αCD3/CD28 dynabeads were removed, cells washed and cultured for 4 days in growth media supplemented with 30 IU/ml rIL-2 (Roche) Then cells were washed and subjected to spininfection (90 min, 1200 g) and incubated over-night. Next day cells were washed and re-suspended in growth media supplemented with 30 IU/ml rIL-2 and Saquinavir Mesylate (5 µM). Seven days post-infection cells were treated with BRD-K80443127 in increasing concentrations or with PMA/Ionomycin in the presence of Raltegravir (30 µM). After 24 hours of stimulation cells were collected and subjected to the luciferase assay, RLU was normalized to the total protein content.
Biomarkers:
CD4+ T cells were isolated from three healthy donors and treated in duplicate with 3 µM BRD-K80443127 or control DMSO for 16 hours. Cells were lysed with TRIreagent (Sigma) and total RNA was isolated with Total RNA Zol-Out (A&A Biotechnology) kit and cDNA was synthetized using random primers and Superscript II Reverse Transcriptase (Life Technologies). Real-time PCR was performed using GoTaq qPCR Master Mix (Promega) on CFX Connect Real-Time PCR Detection System thermocycler (BioRad) using following conditions: 95 °C for 3 min, followed by 40 cycles of 95 °C for 10 sec and 60 °C for 30 sec. Products quality was assessed by theirs melting curve analysis. Relative expression of target genes was normalized to β−2-microglobulin and calculated using Livak-Schmittgen method (Livak and Schmittgen, 2001). Primers: (p21: For-AGCAGAGGAAGACCATGTGGAC, Rev-TTTCGACCCTGAGAGTCTCCAG. cMYC: For- AAGCCACAGCATACATCC, Rev-GCACAAGAGTTCCGTAGC. B2M: For- AGCGTACTCCAAAGATTCAGGTT, Rev-ATGATGCTGCTTACATGTCTCGAT)
HIV latency reversal from patient samples:
All patients were older than 18 years, c-ART treated for at least 3 years, and their viral loads were below 50 copies/ml for more than 12 months with no blips in the past two years. CD4+ T cells from aviremic HIV+ patients were isolated as described previously (Stoszko et al., 2016) with minor modifications. Briefly, frozen PBMCs were cultured in RPMI medium over-night to recover. Then next day CD4+ T cells were isolated twice (enriched CD4+ T cells were subjected to a second round of CD4+ T cell enrichment) and left for 6hrs to recover. Three million cells were treated with DMSO, 10 µM BRD-K80443127, 200 nM Prostratin, 10 µM BRD-K80443127 and 200 nM prostratin, and αCD3/CD28 magnetic beads (at cell:bead ration 1:1) as a positive control in triplicate. After 24 hours cells were lysed in TRIreagent (Sigma), total RNA was isolated with Total RNA Zol-Out (A&A Biotechnology) kit and cDNA was synthetized using random primers and Superscript II Reverse Transcriptase (Life Technologies). Detection of cellular associated pol RNA was performed as described previously (Pol: For GGTTTATTACAGGGACAGCAGAGA, Rev-ACCTGCCATCTGTTTTCCATA) (Stoszko et al., 2016). This study was conducted in accordance with the ethical principles of the Declaration of Helsinki. HIV-1 infected patient volunteers were informed and provided signed consent to participiate in the study. The study protocol was approved by The Netherlands Medical Ethics Committee (MEC-2012–583). cDNA generated from control and 10 µM BRD-K80443127 treated samples was used to assess expression of BAF target genes biomarker genes– p21 and C-MYC. Primers: (p21: For-AGCAGAGGAAGACCATGTGGAC, Rev- TTTCGACCCTGAGAGTCTCCAG. cMYC: For-AAGCCACAGCATACATCC, Rev- GCACAAGAGTTCCGTAGC. B2M: For-AGCGTACTCCAAAGATTCAGGTT, Rev- ATGATGCTGCTTACATGTCTCGAT)
Activation markers, apoptosis and viability of primary CD4+ T cells.
Activation markers, namely CD25 and CD69 were analyzed as described previously (Stoszko et al., 2016). Briefly, CD4+ T cells were treated with DMSO, compound, or PMA/Ionomycin for 24 and 72 hours. Cells were collected, washed with PBS and stained for 30 min at 4 °C with α-CD25-APC (17–0259-42, eBioscience) and α-CD69-FITC (11–0699-42, eBioscience). Following two washes with PBS, cells were fixed with 1% HCHO at 4 °C and analyzed by flow cytometry with Becton Dickinson Fortessa instrument. To determine percent of apoptotic cells after treatment cells were stimulated for 24 and 72 hours and stained with anti-AnnexinV-PE (BD Biosciences, cat. 556454) in the presence of 2.5 mM CaCl2 for 20 min at 4 °C. Cells were analyzed by Becton Dickinson Fortessa flow cytometer. Data represents the average of six experiments performed on cells from different healthy donors. Viability of ex-vivo infected primary CD4+ cells was determined by flow cytometry on the basis of forward versus side scatter analysis.
FAIRE:
FAIRE experiment was performed as described, with minor modifications (Stoszko et al., 2016). Eighteen hours prior to analysis, J-Lat 11.1 cells were treated with BAFi’s where indicated. Cells were fixed for 10 min by adding formaldehyde to a final concentration of 1% at room temperature. Twenty million cells were used per FAIRE experiment. The reaction was quenched with 125 mM glycine. Cross-linked cells were washed with PBS followed by washes with buffer B and buffer C. For sonication, cells were re-suspended in ChIP incubation buffer and chromatin was sheared by sonication to an apparent length of ~ 200–400 bp (corresponding to ~ 100– 200 bp of free DNA) using a BioRuptor sonicator (Cosmo Bio Co., Ltd) with 20 times 60-s pulses at maximum setting at 4 °C. Sonicated chromatin was once phenol:chloroform:isoamyl alcohol (24:24:1) extracted, washed with chloroform:isoamylalcohol (24:1) and ethanol precipitated. Isolated DNA was subjected to Sybergreen qPCR cycles with specific primers (Nuc-0: For-ATCTACCACACACAAGGCTAC, rev-GTACTAACTTGAAGCACCATCC; HSS: for-AAGTTTGACAGCCTCCTAGC, rev-CACACCTCCCTGGAAAGTC; Nuc-1: for-TTTGCCTGTACTGGGTCTCTCTGG, rev-CACAACAGACGGGCACACACT) with a CFX Connect Real-Time PCR Detection System (BioRad) and GoTaq qPCR Mastermix (Promega).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details can be found in the Figure legends. Bar graphs are plotted as mean ± S.D. Statistical significance was calculated using Prism 7. Asterisks indicate the level of significance using student’s T test (* p< 0.05 ** p< 0.01, *** p< 0.001, **** p< 0.0001).
Supplementary Material
Document S1: Figures S1-S5
Table S1: Summary of biological activities and physical properties of the 31-member solution phase library of macrolactams. Related to Figure 3.
Table S2: List of primers for Southern blot probes (related to Figure 1), qRT-PCR (related to Figure 2, 3 and 4) and FAIRE (related to Figure 5).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-ARID1A | Santa Cruz | Cat# Sc-32761 |
| Mouse monoclonal anti-BAF155 | Santa Cruz | Cat# Sc-32763 |
| Mouse monoclonal anti-BAF47 | Santa Cruz | Cat# Sc-166165 |
| Mouse monoclonal anti-LAMIN1B | Santa Cruz | Cat# Sc-377000 |
| Rabbit polyclonal anti-PBRM1 | Bethyl | Cat# A301-591A |
| α-CD25-APC | eBioscience | Cat# 17-0259-42 |
| α-CD69-FITC | eBioscience | Cat# 11-0699-42 |
| anti-AnnexinV-PE | BD Biosciences | Cat# 556454 |
| Biological Samples | ||
| Healthy adult T cells | Sanquin blood bank, Rotterdam, The Netherlands | NVT0080 |
| HIV-1 infected patient T cells | Erasmus Medical Center, approved by The Netherlands Medical Ethics Committee | (MEC-2012-583) |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SAHA | Bioconnect | Cat# S1047 |
| VPA | Sigma | Cat# P4543-10G |
| PMA/Iono | Sigma | Cat# P1585- |
| 10MG/I9657 | ||
| Prostatin | Sigma | Cat# P0077-1MG |
| Bryostatin | Santa Cruz | Cat# sc-201407 |
| Romidepsin | Sigma | Cat# SML1175-1MG |
| Saquinavir Mesylate | Centre for AIDS Reagents, Euripred | Cat# ARP983 |
| Raltegravir | Centre for AIDS Reagents, Euripred | Cat# ARP980 |
| Compound 68 | Dykhuizen et al., 2012 | N/A |
| Critical Commercial Assays | ||
| Steady Glo® | Promega | Cat# E2510 |
| Cell-titer Glo® | Promega | Cat# G7570 |
| Dual Glo® | Promega | Cat# E2920 |
| Cells to Ct™ | Thermo Fisher | Cat# 4399002 |
| Deposited Data | ||
| Raw and analyzed RNA-Seq data Arid1a KO | This paper | GEO: GSE113872 |
| Raw and analyzed RNA-Seq data Compound treated cells | This paper | GEO: GSE113627 |
| Experimental Models: Cell Lines | ||
| E14ES cells | ATCC | Cat# CRL-1821 RRID: CVCL_C320 |
| Bmi-luc reporter ES cell line | This paper | N/A |
| HEK293T | ATCC | Cat# CRL-3216 |
| A549 lung adenocarcinoma cell line | ATCC | Cat# CCL-185 |
| HepG2 hepatocarcinoma cell line | ATCC | Cat# HB-8065 |
| JLat Tat-GFP Cells (A2) | NIH AIDS Reagent Program | Cat# 9854 |
| JLAT11.1 | Kind gift from Dr Eric Verdin Jordan et al., EMBO, 200x3 | N/A |
| Oligonucleotides | ||
| Southern probes (see Table S2) | This paper | N/A |
| Primers for qRT-PCR (see Table S2) | Dykhuizen et al., 2012 | N/A |
| Primers for FAIRE (see Table S2) | This paper | N/A |
| Taqman Probe mouse Bmi1-FAM | Applied Biosystem | Cat# Mm00776122_gH |
| Taqman Probe mouse Ring1a-FAM | Applied Biosystem | Cat# Mm01278940_m1/4 331182 |
| Taqman Probe mouse Fgf4-FAM | Applied Biosystem | Cat# Mm00438916_g1/43 51372 |
| Taqman Probe mouse actin-VIC | Applied Biosystem | Cat# 4352341E |
| Recombinant DNA | ||
| Bmi1-Luciferase knock-in targeting construct | This paper | N/A |
| pNL4.3.Luc.R-E | Centre for AIDS Reagents | Cat# 3418 |
| HIV-1 HXB2-Env expression vector | Centre for AIDS Reagents | Cat# 1069 |
| PLKO.1 lentiviral vector containing shRNA to mouse Brg1: hairpin sequence: CCGGCG-CCCGACACATTATTGAGAACTCGAGTTCT-CAATAATGTGTCGGGCGTTTTTG targeted to CGCCCGACACATTATTGAGAA | Dharmacon | Clone ID: TRCN0000071386 Cat# RMM3981-201797880 |
| Software and Algorithms | ||
| DESeq | Anders and Huber 2010 | http://bioconductor.org/packages/release/bioc/html/DESeq.html |
| EdgeR | Robinson et al. 2010 | http://bioconductor.org/packages/release/bioc/html/edgeR.html |
| Other | ||
| PubChem Assay ID | This paper | AID 602436 |
Significance:
The BAF (SWI/SNF) chromatin remodeling complex has long been an attractive target for drug development; however, the heterogenous nature of BAF complexes, along with undefined biochemical functions for disease-related subunits has made the development of small molecule screening platforms particularly challenging. Here, we have developed a reporter cell line of BAF-mediated transcriptional repression and identified a macrolactam inhibitor of the BAF chromatin remodeling complex using high throughput screening. This optimized class of compounds compounds activates the expression of BAF-repressed genes in embryonic stem cells and are similarly able to activate transcription in in vitro cell line models of HIV-1 latency and in primary human CD4+ T cells harboring latent HIV-1. Importantly, these compounds do not display T cell toxicity or T cell activation, which is associated with many latency reversal agents. Target identification and phenotypic analysis points to the inhibition of ARID1A-containing BAF complexes, which are selectively involved in maintaining HIV-1 latency. This study validates the strategy of targeting individual BAF subcomplexes involved in disease and identifies a macrolactam scaffold developed using diversity oriented synthesis. This class of compounds is a useful starting point for more potent and selective BAF inhibitors, which can be used in combination with other latency reversal agents to activate HIV-1 transcription and eliminate the latently infected cell population.
Highlights.
HTS identifies a macrolactam that inhibits BAF transcriptional repression.
The macrolactams are likely targeting specific ARID1A-containing BAF complexes.
The macrolactams reverse HIV-1 latency and are non-toxic to T-cells.
The macrolactams can be combined with other latency reversal agents.
Acknowledgements:
We would like to thank: Nicola Tolliday, and Stuart Schreiber at the Broad Institute for their continued support, Helen Bodmer for technical assistance, Naoki Hosen for assistance with the Bmi1-luciferase knock-in cell line, and Emma Chory for helpful discussion. Arid1af/f mice were a gift from Terry Magnuson (UNC School of Medicine). HIV-1 molecular clone pNL4.3.Luc.R-E-, HIV-1 HXB2-Env expression vector, Saquinavir Mesylate and Raltegravir were provided by the Centre for AIDS Reagents, NIBSC. HIV-1 molecular clone pNL4.3.Luc.R-E- and HIV-1 HXB2-Env expression vector were donated by Dr. Nathaniel Landau and Drs Kathleen Page and Dan Littman, respectively. This work was directly supported by NIH grant DA032469 to G.R.C. and E.C.D, American Cancer Society ACS 121535-PF-11–145-01-DMC to E.C.D., and the Showalter Trust to E.C.D. E.C.D. is supported by The V Foundation for Cancer Research (V2014–004 and D2016–030) the NIH (CA207532), and the DoD (W81XWH-17–1-0267). Opinions, interpretations, conclusions and recommendations are those of the author and are not necessarily endorsed by the Department of Defense. G.R.C. is supported by NIH grants (CA163915, NS046789), CIRM RB4–05886, SFARI and the Howard Hughes Medical Institute. T.M. is supported by the European Research Council Programme (FP/2007–2013) / ERC STG 337116 Trxn-PURGE, Dutch AIDS Fonds grant 2014021and ErasmusMC mRACE.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: Gerald R. Crabtree is a co-founder of Foghorn Therapeutics.
DATA AND SOFTWARE AVAILABILITY
RNA-Seq datasets are deposited in GEO with accession number GSE113872 for Arid1af/f:CreERT2 ESCs and GSE113627 for BRD-K98645985 treated ESCs.
ADDITIONAL RESOURCES
A full description of the high throughput screen and associated bioassays can be found on PubChem under assay number 602436.
References:
- Agbottah E, Deng L, Dannenberg LO, Pumfery A, and Kashanchi F (2006). Effect of SWI/SNF chromatin remodeling complex on HIV-1 Tat activated transcription. Retrovirology 3, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archin NM, Bateson R, Tripathy MK, Crooks AM, Yang KH, Dahl NP, Kearney MF, Anderson EM, Coffin JM, Strain MC, et al. (2014). HIV-1 Expression Within Resting CD4+ T Cells After Multiple Doses of Vorinostat. The Journal of Infectious Diseases 210, 728–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, Parker DC, Anderson EM, Kearney MF, Strain MC, et al. (2012). Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 487, 482–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auld DS, Thorne N, Nguyen D-T, and Inglese J (2008). A Specific Mechanism for Nonspecific Activation in Reporter-Gene Assays. ACS Chem. Biol 3, 463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell JB, and Holloway GA (2010). New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- Barouch DH, and Deeks SG (2014). Immunologic strategies for HIV-1 remission and eradication. Science 345, 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barré-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, Dauguet C, Axler-Blin C, Vézinet-Brun F, Rouzioux C, et al. (1983). Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 220, 868–871. [DOI] [PubMed] [Google Scholar]
- Guan Bin, Wang T-L, and Shih I-M (2011). ARID1A, a Factor That Promotes Formation of SWI/SNF-Mediated Chromatin Remodeling, Is a Tumor Suppressor in Gynecologic Cancers. Cancer Research 71, 6718–6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLISS CI (1939). THE TOXICITY OF POISONS APPLIED JOINTLY1. Annals of Applied Biology 26, 585–615. [Google Scholar]
- Boese A, Sommer P, Holzer D, Maier R, and Nehrbass U (2009). Integrase interactor 1 (Ini1/hSNF5) is a repressor of basal human immunodeficiency virus type 1 promoter activity. - PubMed - NCBI. J. Gen. Virol 90, 2503–2512. [DOI] [PubMed] [Google Scholar]
- Bosque A, and Planelles V (2009). Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 113, 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockman MA, Jones RB, and Brumme ZL (2015). Challenges and Opportunities for T-Cell-Mediated Strategies to Eliminate HIV Reservoirs. Frontiers in Immunology 6, 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultman S, Gebuhr T, and Magnuson T (2005). A Brg1 mutation that uncouples ATPase activity from chromatin remodeling reveals an essential role for SWI/SNF-related complexes in beta-globin expression and erythroid development. Gene Dev 19, 2849–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler RL, Damrauer JS, Raab JR, Schisler JC, Wilkerson MD, Didion JP, Starmer J, Serber D, Yee D, Xiong J, et al. (2015). Coexistent ARID1A–PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signalling. Nat Commun 6, 6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SWG, Davies KP, Yung E, Beltran RJ, Yu J, and Kalpana GV (1999). c-MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nat Genet 22, 102–105. [DOI] [PubMed] [Google Scholar]
- Chory EJ, Kirkland JG, Chang C-Y, D’Andrea VD, Dykhuizen EC, Crabtree GR. A Novel SWI/SNF Chromatin Remodeling Complex Inhibitor Synergizes with ATR Inhibition in Cancer. Submitted.
- Chun T-W, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn TC, et al. (1997). Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. - PubMed - NCBI. Nature 387, 183–188. [DOI] [PubMed] [Google Scholar]
- Chun T-W, Justement JS, Murray D, Hallahan CW, Maenza J, Collier AC, Sheth PM, Kaul R, Ostrowski M, Moir S, et al. (2010). Rebound of plasma viremia following cessation of antiretroviral therapy despite profoundly low levels of Hiv reservoir: implications for eradication. Aids 24, 2803–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill MJ, Deeks SG, Margolis DM, Siliciano RF, and Swanstrom R (2016). HIV reservoirs: what, where and how to target them. Nature Reviews Microbiology 14, 55–60. [DOI] [PubMed] [Google Scholar]
- Cillo AR, and Mellors JW (2016). Which therapeutic strategy will achieve a cure for HIV-1? Current Opinion in Virology 18, 14–19. [DOI] [PubMed] [Google Scholar]
- Conrad RJ, and Ott M (2016). Therapeutics Targeting Protein Acetylation Perturb Latency of Human Viruses. ACS Chem. Biol acschembio.5b00999. [DOI] [PMC free article] [PubMed]
- Conrad RJ, Fozouni P, Thomas S, Sy H, Zhang Q, Zhou M-M, and Ott M (2017). The Short Isoform of BRD4 Promotes HIV-1 Latency by Engaging Repressive SWI/SNF Chromatin-Remodeling Complexes. Mol Cell 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahabieh M, Battivelli E, and Verdin E (2015). Understanding HIV Latency: The Road to an HIV Cure. Annual Review of Medicine 66, 407–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Crignis E, and Mahmoudi T (2017). The Multifaceted Contributions of Chromatin to HIV-1 Integration, Transcription, and Latency. International Review of Cell and Molecular Biology 328, 197–252. [DOI] [PubMed] [Google Scholar]
- DeChristopher BA, Loy BA, Marsden MD, Schrier AJ, Zack JA, and Wender PA (2012). Designed, Synthetically Accessible Bryostatin Analogues Potently Induce Activation of Latent HIV Reservoirs in vitro. Nature Chem 4, 705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks SG (2012). HIV: Shock and kill. Nature 487, 439–440. [DOI] [PubMed] [Google Scholar]
- Deeks SG, Lewin SR, and Havlir DV (2013). The end of AIDS: HIV infection as a chronic disease. - PubMed - NCBI. The Lancet 382, 1525–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delagrèverie HM, Delaugerre C, Lewin SR, Deeks SG, and Li JZ (2016). Ongoing Clinical Trials of Human Immunodeficiency Virus Latency-Reversing and Immunomodulatory Agents. Open Forum Infect Dis 3, ofw189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykhuizen EC, Carmody LC, Tolliday N, Crabtree GR, and Palmer MAJ (2012). Screening for Inhibitors of an Essential Chromatin Remodeler in Mouse Embryonic Stem Cells by Monitoring Transcriptional Regulation. Journal of Biomolecular Screening 17, 1221–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykhuizen EC, Hargreaves DC, Miller EL, Cui K, Korshunov A, Kool M, Pfister S, Cho Y-J, Zhao K, and Crabtree GR (2013). BAF complexes facilitate decatenation of DNA by topoisomerase IIα. Nature 497, 624–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easley R, Carpio L, Dannenberg L, Choi S, Alani D, Van Duyne R, Guendel I, Klase Z, Agbottah E, Kehn-Hall K, et al. (2010). Transcription through the HIV-1 nucleosomes: Effects of the PBAF complex in Tat activated transcription. Virology 405, 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott JH, Wightman F, Solomon A, Ghneim K, Ahlers J, Cameron MJ, Smith MZ, Spelman T, McMahon J, Velayudham P, et al. (2014). Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog 10, e1004473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, et al. (1997). Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278, 1295–1300. [DOI] [PubMed] [Google Scholar]
- Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, Smith K, Lisziewicz J, Lori F, Flexner C, et al. (1999). Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med 5, 512–517. [DOI] [PubMed] [Google Scholar]
- Fitzgerald ME, Mulrooney CA, Duvall JR, Wei J, Suh B-C, Akella LB, Vrcic A, and Marcaurelle LA (2012). Build/Couple/Pair Strategy for the Synthesis of Stereochemically Diverse Macrolactams via Head-to-Tail Cyclization. ACS Comb. Sci 14, 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Tate P, Hu P, Tjian R, Skarnes WC, and Wang Z (2008). ES cell pluripotency and germ-layer formation require the SWI/SNF chromatin remodeling component BAF250a. Proceedings of the National Academy of Sciences 105, 6656–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giresi PG, Kim J, McDaniell RM, Iyer VR, and Lieb JD (2007). FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Research 17, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves DC, and Crabtree GR (2011). ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res 21, 396–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, and Crabtree GR (2010). Chromatin remodelling during development. Nature 463, 474–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, Jothi R, Ronan JL, Cui K, Zhao K, and Crabtree GR (2009). An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proceedings of the National Academy of Sciences 106, 5187–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, Miller EL, Ronan JL, Ho WQ, Jothi R, and Crabtree GR (2011). esBAF facilitates pluripotency by conditioning the genome for LIF/STAT3 signalling and by regulating polycomb function. Nat Cell Biol 13, 903–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges C, Kirkland JG, and Crabtree GR (2016). The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb Perspect Med 6, a026930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AF, and Vakoc CR (2014). A rationale to target the SWI/SNF complex for cancer therapy. Trends Genet 30, 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundbäck T, Nordlund P, and Molina DM (2014). The cellular thermal shift assay for evaluating drug target interactions in cells. Nature Protocols 9, 2100–2122. [DOI] [PubMed] [Google Scholar]
- Jordan A, Bisgrove D, and Verdin E (2003). HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. Embo J 22, 1868–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan A, Defechereux P, and Verdin E (2001). The site of HIV-1 integration in the human genome determines basal transcriptional activity and response to Tat transactivation. The EMBO Journal 20, 1726–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, Williams RT, Calarco JP, Miller EL, Weber CM, Braun SMG, Pulice JL, Chory EJ, and Crabtree GR (2016). Dynamics of BAF-Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat Genet [DOI] [PMC free article] [PubMed]
- King HW, and Klose RJ (2017). The pioneer factor OCT4 requires the chromatin remodeller BRG1 to support gene regulatory element function in mouse embryonic stem cells. eLife Sciences 6, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korin YD, Brooks DG, Brown S, Korotzer A, and Zack JA (2002). Effects of Prostratin on T-Cell Activation and Human Immunodeficiency Virus Latency. J Virol 76, 8118–8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Darcis G, Van Lint C, and Herbein G (2015). Epigenetic control of HIV-1 post integration latency: implications for therapy. Clin Epigenetics 7, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird GM, Bullen CK, Rosenbloom DIS, Martin AR, Hill AL, Durand CM, Siliciano JD, and Siliciano RF (2015). Ex vivo analysis identifies effective HIV-1 latency–reversing drug combinations. Journal of Clinical Investigation 125, 1901–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassen KG, Hebbeler AM, Bhattacharyya D, Lobritz MA, and Greene WC (2012). A Flexible Model of HIV-1 Latency Permitting Evaluation of Many Primary CD4 T-Cell Reservoirs. PLoS ONE 7, e30176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2− ∆∆CT method. Methods [DOI] [PubMed]
- Maartens G, Celum C, and Lewin SR (2014). HIV infection: epidemiology, pathogenesis, treatment, and prevention. The Lancet 384, 258–271. [DOI] [PubMed] [Google Scholar]
- Mahmoudi T, Parra M, Vries RGJ, Kauder SE, Verrijzer CP, Ott M, and Verdin E (2006). The SWI/SNF chromatin-remodeling complex is a cofactor for Tat transactivation of the HIV promoter. J Biol Chem 281, 19960–19968. [DOI] [PubMed] [Google Scholar]
- Margolis DM (2017). Towards an HIV Cure: a View of a Developing Field. The Journal of Infectious Diseases 215, S109–S110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis DM, and Archin NM (2017). Proviral Latency, Persistent Human Immunodeficiency Virus Infection, and the Development of Latency Reversing Agents. - PubMed - NCBI. The Journal of Infectious Diseases 215, S111–S118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis DM, Garcia JV, Hazuda DJ, and Haynes BF (2016). Latency reversal and viral clearance to cure HIV-1. Science 353, aaf6517–aaf6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martrus G, and Altfeld M (2016). Immunological strategies to target HIV persistence. - PubMed - NCBI. Current Opinion in HIV and AIDS 11, 402–408. [DOI] [PubMed] [Google Scholar]
- Mbonye U, and Karn J (2014). Transcriptional control of HIV latency: cellular signaling pathways, epigenetics, happenstance and the hope for a cure. - PubMed - NCBI. Virology 454–455, 328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megaridis MR, Lu Y, Tevonian EN, Junger KM, Moy JM, Bohn-Wippert K, and Dar RD (2018). Fine-tuning of noise in gene expression with nucleosome remodeling. APL Bioengineering 2, 026106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EL, Hargreaves DC, Kadoch C, Chang C-Y, Calarco JP, Hodges C, Buenrostro JD, Cui K, Greenleaf WJ, Zhao K, et al. (2017). TOP2 synergizes with BAF chromatin remodeling for both resolution and formation of facultative heterochromatin. Nat Struct Mol Biol 24, 344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Over B, Matsson P, Tyrchan C, Artursson P, Doak BC, Foley MA, Hilgendorf C, Johnston SE, Lee MD, Lewis RJ, et al. (2016). Structural and conformational determinants of macrocycle cell permeability. Nat Chem Biol 12, 1065–1074. [DOI] [PubMed] [Google Scholar]
- Perreau M, Banga R, and Pantaleo G (2017). Targeted Immune Interventions for an HIV-1 Cure. - PubMed - NCBI. Trends Mol Med [DOI] [PubMed]
- Pham LV, Tamayo AT, Li C, Bueso-Ramos C, and Ford RJ (2010). An epigenetic chromatin remodeling role for NFATc1 in transcriptional regulation of growth and survival genes in diffuse large B-cell lymphomas. Blood 116, 3899–3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter EG, and Dykhuizen EC (2017). Individual Bromodomains of Polybromo-1 Contribute to Chromatin Association and Tumor Suppression in Clear Cell Renal Carcinoma. J Biol Chem 292, 2601–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulice JL, and Kadoch C (2017). Composition and Function of Mammalian SWI/SNF Chromatin Remodeling Complexes in Human Disease. Cold Spring Harb. Symp. Quant. Biol 031021. [DOI] [PubMed]
- Rafati H, Parra M, Hakre S, Moshkin Y, Verdin E, and Mahmoudi T (2011). Repressive LTR Nucleosome Positioning by the BAF Complex Is Required for HIV Latency. Plos Biol 9, e1001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen TA, Søgaard OS, Brinkmann C, Wightman F, Lewin SR, Melchjorsen J, Dinarello C, Østergaard L, and Tolstrup M (2013). Comparison of HDAC inhibitors in clinical development: Effect on HIV production in latently infected cells and T-cell activation. Human Vaccines & Immunotherapeutics 9, 993–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen TA, Tolstrup M, and Søgaard OS (2016). Reversal of Latency as Part of a Cure for HIV-1. - PubMed - NCBI. Trends in Microbiology 24, 90–97. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruelas DS, and Greene WC (2013). An Integrated Overview of HIV-1 Latency. Cell 155, 519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski MM, Reinhard FBM, Franken H, Werner T, Savitski MF, Eberhard D, Molina DM, Jafari R, Dovega RB, Klaeger S, et al. (2014). Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346, 1255784–1255784. [DOI] [PubMed] [Google Scholar]
- Schiaffino-Ortega S, Balinas C, Cuadros M, and Medina PP (2014). SWI/SNF proteins as targets in cancer therapy. J Hematol Oncol 7, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan PL, Mayall TP, Verdin E, and Jones KA (1997). Histone acetyltransferases regulate HIV-1 enhancer activity in vitro. Gene Dev 11, 3327–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Whyte WA, Zepeda-Mendoza CJ, Milazzo JP, Shen C, Roe J-S, Minder JL, Mercan F, Wang E, Eckersley-Maslin MA, et al. (2013). Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Gene Dev 27, 2648–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliciano JD, and Siliciano RF (2016). Recent developments in the effort to cure HIV infection: going beyond N = 1. Journal of Clinical Investigation 126, 409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, Kovacs C, Gange SJ, and Siliciano RF (2003). Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med 9, 727–728. [DOI] [PubMed] [Google Scholar]
- Spivak AM, and Planelles V (2016). HIV-1 Eradication: Early Trials (and Tribulations). Trends Mol Med 22, 10–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton BZ, Hodges C, Calarco JP, Braun SMG, Ku WL, Kadoch C, Zhao K, and Crabtree GR (2016). Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin. Nat Genet [DOI] [PMC free article] [PubMed]
- Stoszko M, De Crignis E, Rokx C, Khalid MM, Lungu C, Palstra R-J, Kan TW, Boucher C, Verbon A, Dykhuizen EC, et al. (2016). Small Molecule Inhibitors of BAF; A Promising Family of Compounds in HIV-1 Latency Reversal. EBioMedicine 3, 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Søgaard OS, Graversen ME, Leth S, Olesen R, Brinkmann CR, Nissen SK, Kjaer AS, Schleimann MH, Denton PW, Hey-Cunningham WJ, et al. (2015). The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS Pathog 11, e1005142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautmann L (2016). Kill. Current Opinion in HIV and AIDS 11, 409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tréand C, Chéné, du I, Brès V, Kiernan R, Benarous R, Benkirane M, and Emiliani S (2006). Requirement for SWI/SNF chromatin-remodeling complex in Tat-mediated activation of the HIV-1 promoter. Embo J 25, 1690–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner A-MW, and Margolis DM (2017). Chromatin Regulation and the Histone Code in HIV Latency. Yale J Biol Med 90, 229–243. [PMC free article] [PubMed] [Google Scholar]
- Van Duyne R, Guendel I, Shafagati N, Kehn-Hall K, Easley R, Klase Z, Nekhai S, Tyagi M, and Kashanchi F (2011). Varying modulation of HTLV-1 LTR activity by BAF complexes. Retrovirology 8, A180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, Ott M, and Verdin E (1996). Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. Embo J 15, 1112–1120. [PMC free article] [PubMed] [Google Scholar]
- Van Lint C, Bouchat S, and Marcello A (2013). HIV-1 transcription and latency: an update. Retrovirology 10, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E (1991). DNase I-hypersensitive sites are associated with both long terminal repeats and with the intragenic enhancer of integrated human immunodeficiency virus type 1. J Virol 65, 6790–6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E, Paras P, and Van Lint C (1993). Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. Embo J 12, 3249–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei DG, Chiang V, Fyne E, Balakrishnan M, Barnes T, Graupe M, Hesselgesser J, Irrinki A, Murry JP, Stepan G, et al. (2014). Histone Deacetylase Inhibitor Romidepsin Induces HIV Expression in CD4 T Cells from Patients on Suppressive Antiretroviral Therapy at Concentrations Achieved by Clinical Dosing. PLoS Pathog 10, e1004071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wightman F, Lu HK, Solomon AE, Saleh S, Harman AN, Cunningham AL, Gray L, Churchill M, Cameron PU, Dear AE, et al. (2013). Entinostat is a histone deacetylase inhibitor selective for class 1 histone deacetylases and activates HIV production from latently infected primary T cells. Aids 27, 2853–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JI, Lessard J, and Crabtree GR (2009). Understanding the Words of Chromatin Regulation. Cell 136, 200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1: Figures S1-S5
Table S1: Summary of biological activities and physical properties of the 31-member solution phase library of macrolactams. Related to Figure 3.
Table S2: List of primers for Southern blot probes (related to Figure 1), qRT-PCR (related to Figure 2, 3 and 4) and FAIRE (related to Figure 5).