

Abstract
Peritoneal response to various kinds of injury involves loss of peritoneal mesothelial cells (PMC), danger signalling, epithelial-mesenchymal transition and mesothelial-mesenchymal transition (MMT). Encapsulating peritoneal sclerosis (EPS), endometriosis (EM) and peritoneal metastasis (PM) are all characterized by hypoxia and formation of a vascularized connective tissue stroma mediated by vascular endothelial growth factor (VEGF). Transforming growth factor-β1 (TGF-β1) is constitutively expressed by the PMC and plays a major role in the maintenance of a transformed, inflammatory micro-environment in PM, but also in EPS and EM. Persistently high levels of TGF-β1 or stimulation by inflammatory cytokines (interleukin-6 (IL-6)) induce peritoneal MMT, adhesion formation and fibrosis. TGF-β1 enhances hypoxia inducible factor-1α expression, which drives cell growth, extracellular matrix production and cell migration. Disruption of the peritoneal glycocalyx and exposure of the basement membrane release low molecular weight hyaluronan, which initiates a cascade of pro-inflammatory mediators, including peritoneal cytokines (TNF-α, IL-1, IL-6, prostaglandins), growth factors (TGF-α, TGF-β, platelet-derived growth factor, VEGF, epidermal growth factor) and the fibrin/coagulation cascade (thrombin, Tissue factor, plasminogen activator inhibitor [PAI]-1/2). Chronic inflammation and cellular transformation are mediated by damage-associated molecular patterns, pattern recognition receptors, AGE-RAGE, extracellular lactate, pro-inflammatory cytokines, reactive oxygen species, increased glycolysis, metabolomic reprogramming and cancer-associated fibroblasts. The pathogenesis of EPS, EM and PM shows similarities to the cellular transformation and stromal recruitment of wound healing.
Keywords: CAPD, DAMPs, endometriosis, EPS, HIF, hyaluronan, peritoneal metastasis, TGF-β1, VEGF
Introduction
Thirty years ago, Harold Dvorak published a seminal paper entitled “Tumors: Wounds that do not heal”. The novelty was based on two findings, namely, the discovery of vascular permeability factor (VPF, subsequently renamed as vascular endothelial growth factor [VEGF]) as a tumour product and the recognition that the chronic vascular hyperpermeability (CVH) induced by VPF/VEGF likely accounted for the fibrin deposited in solid tumours and in early stages of wound healing [1].
Dvorak made the crucial observation that both wounds and tumours are hypoxic. When cells experience shortage of oxygen, they produce a family of transcription factors named Hypoxia-inducible factors (HIF). HIF is instrumental for the survival of cells under hypoxia or physiological stress. HIF induces angiogenesis by stimulating the outsprouting of new vessels from existing vessels, designed to deliver oxygen and nutrients to the growing (or healing) tissue. Overexpression of HIF-1α was observed in peritoneal dialysis patients [2], in women with endometriosis (EM) [3] and in patients with peritoneal metastasis (PM) [4]. Both HIF-1α and HIF-1β activate the transcription of the gene encoding VEGF [5]. By binding to its receptor, VEGF stimulates neoangiogenesis. VEGF has multiple interdependent pathways which contribute to chronic inflammation, organ fibrosis and carcinogenesis [6, 7]. These mechanisms are involved in the peritoneal response to injury, particularly epithelial-mesenchymal transition (EMT) and mesothelial-mesenchymal transition (MMT).
Tissue regeneration is regulated by members of the transforming growth factor-β (TGF-β) superfamily, which also regulate cell fate and differentiation in embryonic development. During embryonic development, TGF-β can induce EMT. TGF-β signalling can either inhibit or foster tumour growth [5]. The creation of a vascularized connective tissue stroma in healing wounds and in EM is mediated by VEGF. VEGF was also found to promote PM and ascites formation [7, 8, 9].
The recognition of tissue injury and the subsequent inflammatory response is fundamental to the resolution of cellular damage and successful healing. If the initial injury is severe or repetitive, a wound is formed which becomes chronic. This review will highlight the similarities in the pathophysiology of encapsulating peritoneal sclerosis (EPS), EM and PM. The contribution of TGF-β superfamily, HIF and VEGF in inflammation, cellular proliferation, stromal recruitment and angiogenesis in these diseases will be reviewed.
Mechanisms of peritoneal injury
Intact peritoneal mesothelial cells (PMC) express epithelial-like markers and phenotype. They maintain peritoneal homeostasis by controlling fluid and electrolyte transport, initiation and resolution of inflammation, leucocyte migration, fibrinolysis and cellular signalling. The peritoneal mesothelium produces a glycocalyx comprised of surfactants, phospholipids and glycosaminoglycans (GAGs) which provides a slippery protective surface on which the viscera slide. The major component of the glycocalyx is high molecular weight (1,000–2,000 kDa) hyaluronan (HMW-HA), a linear, non-sulphated GAG which covers the mesothelial microvilli [10]. Hyaluronan (HA) is a polymer composed of repeating disaccharides of β-(1, 4) glucuronic acid and β-(1, 3) N-acetyl glucosamine [11].
Disruption of HA is pro-inflammatory
Native HMW-HA has unique viscoelastic, hygrostatic, rheologic, anti-oxidant, anti-inflammatory and anti-angiogenic properties [10]. Disruption of the glycocalyx or damage to the PMC releases low molecular weight hyaluronan (LMW-HA), which initiates a cascade of pro-inflammatory mediators [12]. Such mediators include peritoneal cytokines (TNF-α, interleukin [IL]-1, IL-6, prostaglandins), growth factors (TGF-α, TGF-β, platelet-derived growth factor [PDGF], VEGF, epidermal growth factor [EGF]) and the fibrin/coagulation cascade (thrombin, Tissue factor, PAI-1/2) This is part of the damage-associated molecular patterns (DAMPs) response to cellular injury [13]. HA interacts with CD44 (cluster of differentiation cell surface HA binding protein), Hyaluronan Mediated Motility Receptor (RHAMM) and Toll-like receptor (TLR)-4 on mesothelial and cancer cells, which can promote both EMT and MMT [12, 14, 15]. When activated or injured, PMCs remodel the extracellular matrix (ECM) or invade the basement membrane (BM) by the production of matrix metalloproteases (MMP-2/MMP-9) and degradation of type IV collagen [10]. This may initiate attempted healing of injured peritoneum but, by the same mechanisms, enhance the growth of transcoelomic carcinoma. For example, active CD44 facilitates MMP-9 on the surface of cancer cells, which degrades the ECM and thereby activates latent TGF-β1 and promotes angiogenesis and tumour invasion. TGF-β1 also promotes transformation of fibroblasts to myofibroblasts via CD44v6 signalling [16].
Mesothelial-mesenchymal transition
MMT is a feature of chronic inflammation of the peritoneum, particularly in EPS. MMT in EPS involves loss of peritoneal mesothelium, impaired fibrinolysis, submesothelial myofibroblast proliferation and fibrous collagen and advanced glycation end products (AGE) deposition in the submesothelial layer. Vascular effects include neoangiogenesis, medial hyalinization and sclerosis of the peritoneal vasculature. The submesothelial myofibroblasts appear to be derived from PMC which have been transformed. MMT is typified by loss of PMC epithelial markers such as intercellular adhesion molecule (ICAM-1), calretinin, CA-125 and E-cadherin (CDH1), with increased expression of mesenchymal markers including N-cadherin, SNAIL, fibronectin, vimentin, fibroblast-specific protein-1, Collagen-1, MMP-2, and α-smooth muscle actin (α-SMA) [11, 15, 17]. Acquisition of a mesenchymal phenotype is a non-linear five-dimensional process, controlled by a different transcription factor for each axis involving:
-
–
Increased cellular polarity with fibroblast shape
-
–
BM remodelling
-
–
Loss of cell–cell adhesion
-
–
Apical constriction
-
–
Cell migration [18].
MMT and EMT are induced by AGE-RAGE interaction, HIF, TGF-β and repression of E-cadherin. E-cadherin transcriptional repressors include the zinc finger proteins Snail and Slug, zinc finger E-box binding homeobox (ZEB1, ZEB2) and twist basic helix loop helix transcription factor 1 (TWIST1), SIP1, FOXC1, FOXC2. Induction of EMT by these different EMT‐Transcriptional factors (EMT-TFs) may drive epithelial and mesothelial cells into different regions in the five-dimension landscape, leading to variable phenotypical behaviour and epithelial/mesenchymal plasticity [18]. Both the TGF-β1-induced SMAD-dependent and SMAD-independent pathways converge on the activation of Snail, which is a strong inducer of EMT in PMC [19]. Mammalian SMAD is the homologue of Drosophila protein MAD (Mothers Against Decapentaplegic) and the Caenorhabditis elegans protein SMA (Small body size). The SMAD-independent, non-canonical TGF-β pathway also induces long non-coding RNA activated by TGF-β (lncRNA-ATB). LncRNA-ATB increases ZEB1 and ZEB2 mRNA and protein levels through competitively inhibiting miR 200s, resulting in EMT [20]. Other cytokines and growth factors involved in MMT/EMT include hepatocyte growth factor (HGF), PDGF and IL-1β [15]. The Wnt/β-catenin signalling pathway which promotes Human epidermal growth factor-2 (HER2) activity, EMT and cell invasion is linked to TGF-β1 by TWIST [21].
Chronic inflammation, TGF-β1 and IL-6
TGF-β1 was first discovered in 1983 when it was shown to stimulate the growth of cultured rat fibroblasts, and is the master cytokine in liver fibrogenesis [22]. In the meantime, the TGF-β superfamily is recognized as a fundamental mediator in the development of EPS, organ fibrosis, EM and PM. This superfamily includes at least 40 structurally and functionally related proteins, such as bone morphogenetic proteins (BMPs), activins, inhibins, anti-Mullerian hormone, growth differentiation factors (GDFs), and glial-derived neurotrophic factors [22]. TGF-β1 is regarded as a principle factor in the transformation of PMCs into a mesenchymal phenotype in both in vivo and in vitro studies. The mammalian TGF-β family members (TGF-β1, -β2 and -β3) are induced and activated in a variety of fibrotic diseases [23].
Persistently high levels of TGF-β1 or stimulation by inflammatory cytokines (IL-6) induce peritoneal MMT, adhesion formation and fibrosis [24, 25] (Figure 1). Recently a murine continuous ambulatory peritoneal dialysis (CAPD) model showed exposure to TGF-β1 alone directed naïve CD4+T cell precursors towards the Treg lineage, whereas the combination of TGF-β1 and IL-6 caused progenitors to adopt a Th17 signature. Peritoneal Treg cells produce IL-10, which is an anti-inflammatory cytokine. TGF-β1 was strongly upregulated in the peritoneal cavity of cd69 -/- mice exposed to peritoneal dialysis fluid (PDF). The lack of CD69 expression on Treg cells caused Th17 cell expansion, the release of the pro-inflammatory cytokine IL-17 and peritoneal fibrosis. PDF-induced fibrosis was blocked by anti-IL-17 antibodies in cd69 -/- mice and enhanced by neutralizing anti-CD69 antibody in wild-type mice [26]. HIF-1α is also involved in the inflammatory process via the retinoic acid-related orphan receptor-γt to drive Th17 differentiation. Th17 cells undergo clonal expansion and switch to glycolysis to maintain ATP production. This Warburg-like metabolic reprogramming occurs independently of oxygen levels and is controlled by an mammalian target of rapamycin (mTOR)-dependent nutrient-sensing pathway [27]. TNF receptor-associated factor (TRAF6) regulates both canonical TGF-β1/SMAD pathways via cytokines, and SMAD-independent pathways involving TGF-β-activated kinase-1 (TAK1)/mitogen-activating protein kinase kinase kinase (MAP3K)/p38. IL-1α and TNF-α cause Activin A secretion via TAK-1/p38/NFκB signalling. The resulting activin A signals in an autocrine way via ALK/SMAD2,3/AKT to promote myofibroblast formation. Activin A levels were respectively increased by 1,152% and 459% after treatment with IL-1α and TNF-α [28]. Thus peritoneal sclerosis and MMT proceed as a result of continued inflammatory cytokine stimulation of TGF-β1 release and exaggerated DAMP signalling.
Figure 1:
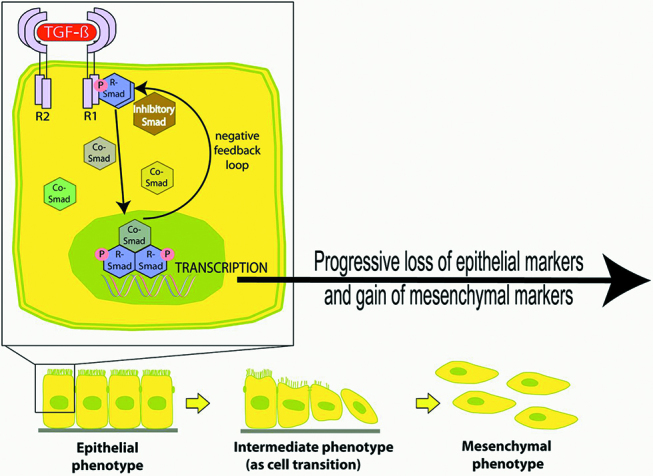
TGF signalling and peritoneal MMT.
HA size regulates inflammation and TGF-β1
The molecular size of HA polymers released after cellular injury appears to be a discriminating factor in the initiation and also resolution of tissue inflammation and MMT [10]. Medium size HA fragments (100–250 kDa) promote cell migration and cytokine production. LMW-HA (6–50 kDA) significantly increases TLR-4 expression compared to medium-to-HMW-HA (100–1,800 kDA). Upregulation of the TLR4–Myeloid Differentiation Factor 88 (MYD88) axis results in the down-regulation of BAMBI, a pseudoreceptor which normally antagonises TGF-β receptor activation. Very small MW HA fragments (four saccharides) promote chemotaxis. Small HA oligomers (<6 kDA) upregulate TGF-β1 and TNF-α by 35-fold compared to native HA, and impair cellular reparation after injury, promoting continued inflammation [10]. Myofibroblast differentiation in response to TGF-β1 is dependent on, and facilitated by, HA interactions with CD44 and EGFR. This has downstream effects on extracellular signal-regulated kinase (ERK) phosphorylation and calcium/calmodulin kinase II activation [29].
TGF-β role in chronic inflammation
Chronic inflammatory conditions include retroperitoneal fibrosis, hepatic fibrosis, idiopathic pulmonary fibrosis, systemic sclerosis (CREST syndrome), Crohn’s disease, radiation enteritis, collagenous colitis, EPS and others [19]. Such conditions share a common pathophysiology of pro-inflammatory mediators, TGF-β overexpression and myofibroblast generation. TGF-β1 is constitutively expressed by most cells, including fibroblasts, mesothelial and epithelial cells in the peritoneum and gastrointestinal tract [30, 31]. TGF-β1 has a pro-fibrogenic activity and can transform rat mesothelial cells, mouse mesangial cells and human smooth muscle cells into myofibroblasts [32]. These myofibroblasts have the ability to produce collagen and α-SMA and contract the ECM resulting in wound contraction or tissue fibrosis [19].
Continued platelet activation, deposition of ECM and impaired fibrinolysis also potentiate chronic wounds. Thrombin activation of platelets triggers the release of TGF-β1 from the α-granules of platelets, and thrombin activity can also release latent TGF-β1 from the ECM [33]. The synthesis of different collagen type I polypeptides is determined by two separate pathways: the TGF-β1 protein activation pathway and the SMAD signalling pathway [34].
Activation of TGF-β1
The inactive pro-TGF-β complex contains TGF-β and the latency-associated peptides (LAP). It is connected to the ECM by the latent-TGF-β-binding proteins 1/2 (LTBP 1/2). Thus the ECM is a sequestrant reservoir for latent TGF-β1, which acts as an extracellular sensor and transducer of mechanical forces, oxidative stress or tissue damage [34]. Active extracellular TGF-β1 is released from the pro-TGF-β complex under proteolytic activity from plasmin, MMP-2 and MMP-9 or thrombospondin (TSP-1) [30]. Activation can also occur by mild acids (lactate), integrin αV-β6, reactive oxygen species (ROS), angiotensin II, low-density lipoproteins (LDL), glucose, thromboxane A2, calpain, cathepsin D, chymase, elastase, endoglycosidase F, kallikrein, or neuraminidase [31, 35]. The released TGF-β1 then acts on its type I (TβR-I) and type II (TβR-II) serine/threonine kinase cell surface receptors. TGF-β1 normally inhibits epithelial cell cycling and promotes apoptosis, and as such is regarded as a tumour suppressor in the early stages of carcinoma. TGF-β1 also promotes epithelial to mesenchymal transition, which is a transient stage in the response to injury and acute wound healing. However, dysregulation of TGF-β1 signalling occurs in sustained or severe peritoneal injury or peritoneal malignancy, which contributes to progressive EMT/MMT, angiogenesis and cellular proliferation [31].
TGF-β1 and SMAD signalling pathway
TGF-β1 interacts as a ligand with type 1 receptors (TβRI and Activin-Like-Kinase 1) and type 2 receptors (TβRII). After binding of a TGF-β dimer to dimers of type 1 and type 2 receptors, a stable complex is formed and Receptor-regulated SMAD (R-SMAD) is phosphorylated by the kinase of the receptor. SMAD proteins transduce signals from the cell membrane to the nuclear DNA. Some of the SMAD-binding transcription factors in the nucleus are relevant for the function of TGF-β superfamily members in cell growth and migration [5]. In endothelial cells, SMAD2/3 signalling inhibits cellular proliferation and migration. In contrast, SMAD1, SMAD5 and SMAD8 signalling promotes these cellular processes.
The type 1 receptor Activin like kinase (ALK5) is the main ALK expressed in epithelial cells and is commonly referred as TβRI [36]. TGFβ/activin pathways usually signal through SMAD2, SMAD3; and BMP/GDF pathways via SMAD1, SMAD5 and SMAD8. However, TGF-β1 may signal via SMAD1/5 in some cells, including endothelial, fibroblast, immortalized epithelial, adenoma and carcinoma cell lines [37]. Prior to being activated, SMADs are anchored to the cell membrane by SMAD Anchor for Receptor Activation which brings the SMADs close to the TGF receptor kinases [38]. TGF-β1 first binds to TβR-II, and then recruitment of TβR-I ensues. Both receptors form the TβR heteromeric complex in which TβR-II phosphorylates and activates TβR-I. The active TβR-I then phosphorylates SMAD2/3 (R-SMADS). This results in retention of p-SMAD2/3 in the cytoplasm and association with SMAD4 (co-SMAD). The resulting hetero-oligomeric complex is translocated to the nucleus and activates SMAD-dependent transcriptional activity. These include DNA sequence-specific binding sites Activating Transcription Factor-2 and SMAD Binding Element (SBE), which regulate gene expression [39,40, 41, 42] (Figure 1). TGF-β1 transcriptional factors include c-jun, p300/CPB, c-myc, cyclin D1, cyclin-dependent kinase 4, cyclin dependent kinase (CDK)-interacting protein 1 (p21cip), cyclin-dependent kinase inhibitor 1B (p27 Kip1), cyclin-dependent kinase 4 inhibitor B/multiple tumour suppressor 2 (p15 INK4B), retinoblastoma protein (Rb), integrins, E-cadherin, collagen and other DNA-binding proteins [41]. Tumour suppressor p53 (TP53) is a cofactor for SMAD-mediated transcription, including the full activation of p21cip. Both p21cip and p15 INK4B, when translated, interfere with cyclin-CDK interactions, which inhibits CDK activation. p15 INK4B also prevents phosphorylation of Rb, resulting in suppression of release of the transcription factor E2F. E2F controls transition from G1 to S phase in cell cycling. TGF-β1 causes further cell cycle arrest by transcriptional repression of c-Myc proto-oncogene and upregulation of apoptosis factor Bim [43]. Loss of function, mutations or lack of expression of either TβR receptor or their downstream effectors (including TP53, Rb, CDKN1A, or the CDKN2A/B locus) leads to failure of growth arrest, which occurs in advanced epithelial cancers [43, 44].
Regulation of TGF-β1 signalling
SMAD6 and SMAD7 are inhibitory SMADs which provide negative feedback control of TGF-β1 family signalling activity. SMAD6 preferentially blocks BMP signalling [45]. SMAD7 competes for TβR-I binding and inhibits phosphorylation of SMAD2 or SMAD3 [46]. SMAD7 decreases the stability and activity of TβR-I by inducing receptor degradation. SMAD7 competes with SMAD4 to associate with R-SMADs and recruits the E3 ubiquitin ligases (including SMAD ubiquitin regulatory factors (SMURF1, SMURF2, PRAJA, WWP1, NEDD4L)), to R-SMADs for polyubiquination and proteosomal degradation [31, [47]. SMAD7 also blocks the interaction of the hetero-oligomeric SMAD complex with its nuclear targets [45]. Active TGF-β1 not only induces SMAD7 transcription but also promotes the degradation of SMAD7 by activating the SMAD ubiquitination regulatory factor-2 (SMURF2)/arkadia-mediated ubiquitin–proteasome degradation pathway. This involves the formation of a SMURF2/SMAD7 complex, which targets TβR-I ubiquitylation-mediated degradation. SMAD and TβR-I ubiquitination is reversed by deubiquitination enzymes, which include ubiquitin-specific proteases (USPs), ubiquitin C-terminal hydrolases and ovarian tumour proteases [48]. Three examples of deubiquitinating enzymes for TβR-I are ubiquitin-specific peptidase-4 (USP4), −11 (USP11) and −15 (USP15), all of which antagonize the effect of SMAD7 and strongly induce TGF-β1 signalling [49]. The latent form of TGF-β1 can protect against renal fibrosis and inflammation by upregulating SMAD7 [50]. Expression of SMAD7 by active TGF-β1 is defective in chronic fibrosing conditions including EPS, related to interactions between AP-1, Sp1, SMAD proteins and counteregulation by nuclear factor-κB (NF-κB) [51]. Increased levels of SMURF2 and decreased SMAD7 promote the TGF-β1-induced repression of E-cadherin and upregulation of vimentin, thus promoting EMT/MMT [49]. HSP90 increases TGF-β1 signalling by binding to TβR-I and TβR-II, which protects these receptors from SMURF2/SMAD7-mediated ubiquitylation [37]. TRAF6-mediated cleavage of TβR-I inhibits normal SMAD3 pathways of TGF-β1 signalling. The intracellular retention of the cleaved TβR-I fragment and its nuclear translocation promotes angiogenesis and cellular invasion by Snail and Jag1 gene expression [45].
TGF-β1 and non-SMAD signalling pathways
The SMAD 2/3 independent, non-canonical pathways are directly activated by the TGF superfamily members, including TGF-β1 and Activin [44, 52]. These pathways include NF-κB, Mitogen-activated protein kinases (MAPK) pathway components (ras, raf, ERK1/2, p38 MAPK, JNK), the phosphatidylinositol-3 kinase (PI3K):AKT:mTOR signalling axis, and cadherin junction regulators RhoA and RAC [53]. PI3K binds to and is activated by TβR-I, leading to increased ECM production [49]. AKT also enhances TGF-induced SMAD signalling by increasing SMURF2 and USP4, which worsens peritoneal fibrosis in murine models of EPS [49]. PI3K/AKT contributes to HIF-1α stabilization by provoking expression of heat shock proteins [54]. TAK-1 is a serine/threonine kinase which belongs to the MAP3K family. TAK1 functions in response to TRAF6 and TGF-β receptor activation, and promotes downstream activation of JNK/c-Jun [55]. c-Jun transcriptional target genes, including receptor for activated C kinase1 (RACK1) and cyclin D1, lead to promotion of EMT [56]. Another non-canonical pathway is JAK interactions with TβR-I to activate STAT3. Phosphorylated STAT3 is required for much of the TGF-β target genes in hepatic stellate cells, with SMAD3/STAT3 being crucial in TGF-β hepatic fibrosis [57]. c-Src is the mammalian proto-oncogene homologue of the Rous sarcoma virus transforming protein v-src. The protein product of c-Src is Src, a non-receptor tyrosine kinase required for phosphorylation of TβR-II, prior to TGF-β-mediated p38 MAPK activation and cell invasion [58, 59]. The integrin-linked kinase (ILK) mediates TGF-β1-induced EMT through Snail and Slug, which is independent of the SMAD signalling pathway [60]. TGF-β signalling via focal adhesion kinase (FAK) is involved in myofibroblast differentiation and remodelling of connective tissue.
TGF-β1 causes fibrosis by three main mechanisms:
-
–
Myofibroblast differentiation via EMT/MMT/endothelial mesenchymal transition (Endo-MT).
-
–
ECM production through canonical (SMAD 2/3-dependent) and non-canonical (non-SMAD dependent) pathways.
-
–
Excessive accumulation of ECM by suppression of MMP and promotion of tissue inhibitor of metalloprotease (TIMP), connective tissue growth factor (CTGF) and PAI [22].
TGF-β, ROS and Redox fibrosis
ROS are formed by mitochondrial metabolism, inflammation, oxidative stress, ischaemia/reperfusion injury or exposure to ionizing radiation, ultraviolet light, chemotherapy or asbestos [34]. ROS activate expression of TGF-β1, and activated TGF-β1 also increases ROS production.
Activation of TGF-β1 by ROS appears to be direct, as release of recombinant latent TGF-β1 can occur in the absence of cellular machinery by ionizing radiation or by metal ion-catalysed ascorbate reactions. Activation of TGF-β by ROS only occurs with TGF-β1 and not TGF-β2 or TGF-β3. This is related to ROS oxidation of the LAP-beta1 protein at methionine253 [35]. The selective activation of TGF-β1 suggests specific DAMPs signalling for different latent TGF-β family members. Increased ROS production by activated TGF-β1 is mainly due to induction of NADPH oxidases (NOX). TGF-β1-induced NOX 4 expression is associated with idiopathic pulmonary fibrosis and hepatic fibrosis. TGF-β1 upregulates NOX4 expression and the NOX4-dependent generation of hydrogen peroxide, which is required for TGF-β1-induced myofibroblast differentiation, ECM production and contractility [35]. Furthermore, TGF-β1 decreases antioxidant enzyme activity including Superoxide Dismutase 1 (SOD1), SOD2, Glutathione (GSH) peroxidase, GSH and Catalase (CTL). These are particularly involved in the suppression of Fenton reaction generation of hydroxyl radical, hydrogen peroxide and superoxide [35]. The persistent activation of LTGF-β1 by oxidative stress and TGF-β1-mediated nett persistence of ROS may represent a ROS/TGF-β1 positive feedback loop, leading to amplification of cellular damage, carcinogenesis and fibrosis [34]. Such disturbance in ROS generation and scavenging is referred to as “redox-fibrosis” [35].
ROS, TGF-β and platelets
Platelets contain 40–100 times the levels of TGF-β1 than other, non-neoplastic human cells. When released, 99% of the platelet-derived TGF-β1 is LTGF-β1 complexed with LAP. This is activated in a similar way to ECM TGF-β1 by exposure to integrins, MMP, plasmin or ROS. ROS and glucose degradation product (GDP)-related damage to the peritoneum is characterized by increased levels of 8-iso-prostaglandin F2α (8-iso-PGF2α) [61, 62, 63, 64]. 8-iso-PGF2α is produced from arachidonic acid via non-enzymatic lipid peroxidation, catalysed by oxygen-free radical action on cell membranes and LDL. It is independent of cyclo-oxygenases (COX) and thus unaffected by COX inhibitors such as aspirin and indobufen. High glucose levels and protein glycation enhance LDL oxidation by metal ions, and these reactions also produce AGEs [62]. 8-iso-PGF2α causes irreversible platelet aggregation in the presence of collagen, ADP, arachidonic acid, and PGH2/TXA2 analogues that fail to aggregate platelets when acting alone [62]. Activated platelets release TGF-β1, VEGF, PDGF, fibroblast growth factor, EGF, HGF, and insulin-like growth factor (IGF) from their cytoplasmic α-granules. This leads to increased migration of endothelial cells, neoangiogenesis, vascular permeability, fibrosis and MMT and EMT in the peritoneum [65]. Other agents that activate platelets include glycoprotein Ib alpha, sialyl Lewis x/sialyl Lewis a, Necl-5, integrins, TSP-1, high mobility group protein B1 (HMGB1) and podoplanin/Aggrus [65].
Hypoxia induces HIF-1α generation
In most human tissues under physiological conditions, the concentration of molecular oxygen lies between 10 and 30 μM. Under these normoxic conditions, a member of the HIF-α prolyl hydroxylase family (PHD) hydroxylates one or both of the critical proline residues (pro402 or pro564) in HIF-1α chains [5]. HIF is a protein dimer, in which one of three different α chains combines with one or two β chains. Among HIFαs, HIF-1α and HIF-2α are the best characterized. Hydroxylation of specific HIF-α proline residues increases the affinity of HIF-α peptide for the von Hippel-Lindau protein (pVHL)-elonginB-elonginC (VBC) complex by at least three orders of magnitude [66, 67]. Polyubiquinated HIF-α is degraded by the 26S proteasome. Thus, under normoxic conditions, there are usually minimal HIF-α cellular levels due to rapid metabolism of HIF-1α and HIF-2α by ubiquitin-proteasomal pathways [68].
Hypoxic conditions
Because tumours often have a hypoxic central “core” with inadequate diffusion of nutrients and a vascularized periphery, HIF generation is enhanced, which drives growth and proliferation of the cancer cells and the transformed stroma [69]. Under hypoxic conditions, the α chain is no longer hydroxylated, HIF-1α is stabilized and HIF-1α cellular level increases. When stabilized, HIF-α is transported to the cell nucleus where it combines with HIF-β to form the heterodimeric HIF. HIF activates the hypoxia response element (HRE) via transcriptional coactivator complexes, including p300/CBP and steroid receptor coactivator (SRC). The HRE has over 100 validated downstream transcription targets [70]. (Table 1) (Figure 2)
Table 1:
Target genes of HIF.
| Function | Transcription target | Reference |
|---|---|---|
| Angiogenesis | VEGFA, VEGFR-1, PAI-1 production | [5, 81] |
| Erythropoiesis | EPO | [68] |
| Glucose metabolism | Hexokinase II (HK2), Phosphofructokinase-1, glucose 6 phosphate dehydrogenase (G6PD), Aldolase A (ALDOA), α-enolase (ENO1), phosphoglycerate kinase 1 (PGK1), pyruvate kinase M2(PKM2), pyruvate dehydrogenase kinase 1 (PDK1), Lactose dehydrogenase (LDHA) and SLC2A1 | [71] |
| Cell growth | EGF, IGF-1, Nip3, Cyclin D/Cdk4 | |
| Iron absorption and transport | Transferrin, ceruloplasmin, heme-oxygenase-1, divalent metal transporter-1, duodenal cytochrome b, ferroportin | [68] |
| Drug resistance | Multidrug resistance transporter P-glycoprotein | [70] |
| Extracellular matrix production | Procollagen prolyl hydroxylase α1, CTGF | [35] |
| Hormonal regulation | Leptin | [74] |
| Cell migration | c-Met, CXCR4, carbonic anhydrase IX (CAIX) | [27, 35] |
| Cell fate | RAGE expression | [27, 66, 68] |
| Epithelial-mesenchymal transition | Snail, Twist and Zeb1 | [71] |
| Recognition of PAMPs and DAMPs | TLR2/4 | [27] |
| Hypoxia-induced autophagy | HIF-1α-STAT3-Src axis | [58] |
| Immune response | Programmed death-ligand 1 (PDL-1), Th17 differentiation | [27] |
| Lactate shuttling | Monocarboxylate transporter 4 (MCT 4) | [71] |
| Cytokine production | Interleukin 1β (IL-1β) | [27] |
Figure 2:

Target genes of HIF.
HIF target genes affect glucose metabolism
The metazoan HIF pathway has evolved to sense cellular hypoxia, but also detect failure of oxidative phosphorylation and ATP production, the presence of mitochondrial oxidative stress, heat shock or molecular damage, loss of redox potential or iron deficiency. The HRE provides adaptive capability via transcriptional response pathways, including switching from mitochondrial respiration to anaerobic glycolysis as an alternative energy source. ATP yield of oxidative phosphorylation is almost 20 times higher with oxidative than with anaerobic glycolysis. HIF regulates the transcription of almost all the glycolytic enzymes and membrane glucose transporters (GLUTs). The upregulation of glycolytic enzymes by HIF and EMT-TF is also directly involved in EMT [71].
The “glycolytic switch” is mediated by HIF and Snail. Snail inhibits mitochondrial respiration by repression of cytochrome C oxidase via wnt/Snail signalling. Snail directly targets Fructose-1,6-bisphosphatase (FBP1), the rate limiting enzyme in gluconeogenesis. FBP1 repression also occurs via Zeb1. Loss of FBP1 decreases mitochondrial oxygen consumption and increases glucose uptake, glycolysis and lactate generation. Twist has been shown to upregulate LDHA, PKM2, HK2 and G6-PD in Twist-overexpressing cancer cells [71]. Loss of Rb or E2F-1 function leads to increased PDK4 expression and decreased glucose oxidation [72].
Active transport of glucose into cells occurs via the glucose transporter (SLC2A1/GLUT), which is dependent on ATP and thiamine for energy. Hexokinase II and Phosphofructokinase drive flux of glucose down the hexokinase pathway. G6-PD is important for ribose and NADPH production via the Pentose Phosphate Pathway, which is utilized by rapidly proliferating cells to increase their biomass synthesis [73]. Aldolase A (ALDOA) catalyses the reversible conversion of fructose-1,6-bisphosphate to glyceraldehyde 3-phosphate and dihydroxyacetone phosphate. Pyruvate kinase (PK) converts phosphoenolpyruvate (PEP) and ADP to pyruvate and ATP [71] in the final rate limiting step of glycolysis [74]. Lactate dehydrogenase A (LDHA) converts pyruvate to lactate, and pyruvate dehydrogenase kinase 1 (PDHK1) inhibits the conversion of pyruvate to acetyl co-enzyme A by PDH. Acetyl Co-A is required for normal functioning of the Krebs’ cycle in mitochondria. PDHK1 causes build-up of the Krebs’ cycle intermediates citrate, fumarate or succinate, leading to further normoxic stabilization of HIF-1α [27]. Increased glucose transport and glycolytic enzymes, together with inhibition of mitochondrial oxidative phosphorylation, contribute to lactate production and the Warburg effect [75, 76]. Oncogenic growth signalling pathways such as PI-3K, Ras and Wnt are also involved in normoxic stabilization of HIF-1α and the metabolic reprogramming of cancer cells [73]. Leptin is another target of HRE and also promotes cancer cell EMT, activates the PI3K/AKT signalling pathway and upregulates PKM2 expression [74].
Role of glycolytic enzymes in EMT
Apart from their role in rapid generation of ATP and lactate, the glycolytic enzymes have also been found to promote EMT and carcinogenesis. These include ALDOA, ENO1, PKM2, LDHA and PDK1. ALDOA upregulates N-cadherin and vimentin and downregulates E-cadherin [71]. Over expression of ENO1 is associated with tumour progression and poor prognosis in pancreatic cancer, cholangiocarcinoma, neuroendocrine tumours, neuroblastoma, prostate cancer, thyroid carcinoma, hepatocellular carcinoma (HCC) and breast cancer. ENO1 has been shown to promote cell proliferation, cycle progression, cancer migration and invasion [77]. ENO1 expression is upregulated by HIF under hypoxia. It is associated with increased plasmin and Ki67 activity and inversely correlated with p53 expression in pancreatic cancer tissues. This was reflected in larger tumour size, lymph node involvement and a poorer prognosis (HR=2.469; 95% CI: 1.348–4.522; p=0.003) in pancreatic cancer patients with high pancreatic ENO1 levels. High expression of ENO1 was found on IHC in 47% of pancreatic cancer tissue specimens [78].
PKM2 is not normally found in adult human cells, but is overexpressed in HCC and oesophageal cancer [71]. PKM2 translocates to the nucleus under the effect of ERK and EGF, and acts as a protein kinase via interactions with Src, β catenin and C-myc. Activated β catenin increases cyclin D1 and the expression of SNAIL and vimentin, and decreases expression of E-cadherin. EMT is also mediated by PKM2 interacting with TGF-β-induced factor homeobox 2, which recruits histone deacetylase 3 to the promoter of E-cadherin and represses its expression [71]. PKM2 phosphorylates STAT3 using PEP as the phosphate donor, a process independent of JaK2 and Src pathways. p-STAT3 activates downstream EMT genes involved in the migration and motility of many cancer types. STAT3 is usually activated by inflammatory cytokines such as IL-6, and phosphorylation by PKM2 provides a plausible pathway for constitutive STAT3 activation in cancers [79]. C-Myc promotes the expression of heterogeneous nuclear ribonucleoproteins which favours alternative splicing of PK to generate PKM2 isoform in tumour cells. PKM2 has low activity in the production of pyruvate from PEP, as compared to the high activity of native PK. Thus PKM2 directs glucose metabolism towards lactate production rather than mitochondrial oxidative phosphorylation [80].
PKM2 is a direct target gene of HRE, and HIF-1 controls the expression of PKM2. PKM2 is hydroxylated at Pro403 and Pro408 by PHD3, which enhances the interaction of PKM2 with HIF-1α. PKM2 also enhances HIF-1α occupancy and histone acetyltransferase p300 recruitment, which may prevent negative regulation by FIH-1. PKM2 promotes the HIF activated HRE targets in glycolysis including genes that mediate increased glucose uptake (GLUT1), increased lactate production (LDHA), decreased oxidative metabolism (PDK1), and also the HRE target genes for increased angiogenesis (VEGF) [81]. The activation of HIF-1α by PKM2 and downstream transcription of PKM2 by the HRE represents a positive feedback loop that cancer cells utilize to enhance glycolysis, lactate production, cellular proliferation, angiogenesis, EMT and the Warburg effect. (Figure 2)
TGF-β1 and HIF‐1α are interdependent
HIF-1α expression is increased 2.2-fold by TGF-β1 alone under normoxic conditions. Under hypoxic conditions, TGF-β1 enhances hypoxia-stimulated HIF-1α expression 1.4-fold above that seen with hypoxia alone [82]. TGF‐β1 decreases the expression of HIF PHD2 via a SMAD2/3‐dependent mechanism, resulting in HIF‐1α stabilization and up‐regulation of collagen I and PAI expression in renal epithelial cells [83]. Both SMAD3 and HIF-1α are interdependent contributors to TGF-β1-stimulated collagen expression under cellular normoxia. Inhibiting HIF-1α decreases TGF-β1-induced SBE-Luc activation, collagen I mRNA expression and promoter activity. Disruption of SMAD3 signalling decreases normoxic, TGF-β-stimulated, HIF-1α-mediated induction of HRE activity [82]. HIF-1α protein stability is also regulated through an O2-independent pathway involving the interaction of HSP90 and RACK1 [84].
EMT of renal tubular epithelial cells into myofibroblasts and tubulointerstitial fibrosis is related to the downstream targets of HRE, stromal cell-derived factor-1 also named CXCL12 and its receptor CXCR4 [35]. Fibrosis is correlated with expression of HRE target genes for ECM formation including PAI-1, TIMP-1 and CTGF [35]. Late TGF-β1-induced deposition of ECM contributes to hypoxia in the peritoneal submesothelial fibroproliferative zone. This induces a secondary hypoxic response of HIF-1α expression and further angiogenesis (Figure 2). The mTOR inhibitor rapamycin blocks these late hypoxia/VEGF-induced angiogenic effects, but does not influence the direct TGFβ-mediated fibrosis and peritoneal VEGF angiogenesis in EPS [85].
Pathophysiology of EPS
In this section, we will focus on the effect of non-physiologic conditions on the peritoneum, including peritoneal dialysis and intraperitoneal chemotherapy (IPC) solutions. EPS is a chronic fibrosing condition of the peritoneum with sclerosis, calcification, thickening or encapsulation of the small intestine [86]. EPS was first described by Owtschinnikow in 1907 as “peritonitis chronica fibrosa incapsulata” [87]. EPS is classified as primary or secondary. Secondary EPS is most commonly caused by CAPD. It has also been described after major abdominal surgery, generalized peritonitis, peritoneal malignancy, IPC, EM, peritoneal lavage with sclerosing antiseptics such as chlorhexidine gluconate, β-blocker treatment (practolol/propranolol), Familial Mediterranean Fever, protein S deficiency, abdominal tuberculosis, autoimmune diseases or sarcoidosis [87, 88, 89]. Simple peritoneal sclerosis tends to affect the parietal peritoneum without intestinal encapsulation, and is common in CAPD [90]. It is unclear whether EPS is related solely to progression of simple sclerosis [91]. However, it was shown in a mouse model that MMT and peritoneal fibrosis progress in direct proportion to the duration of CAPD [17] (Figure 3).
Figure 3:

Progression of peritoneal fibrosis mediated by TGF.
Symptoms and signs of EPS include bloody ascites, anorexia, nausea, diarrhoea and abdominal pain. Early EPS can be associated with inflammatory features of elevated CRP, fever, malaise, anaemia and hypoalbuminaemia. Progression of intestinal encapsulation leads to constipation, abdominal mass, weight loss, severe malnutrition and intermittent subacute bowel obstruction. Diagnosis is based on clinical presentation, radiological imaging, peritoneal biopsy and analysis of peritoneal dialysate fluid [92].
EPS in CAPD – the genesis of a wound
The frequency of EPS in CAPD is directly related to the duration of peritoneal dialysis. A 1998 Australian multicentre, retrospective, observational cohort study of 7,374 patients reported the EPS risk with time was 1.9%, 6.4%, 10.8% and 19.4% after 2, 5, 6 and 8 years of CAPD [93]. The subsequent 2010 Binational Australasian Registry of 7,618 patients found the EPS risk with time had fallen to 0.3%, 0.8% and 3.9% after 3, 5 and 8 years of CAPD [92]. This may be related to changes in PDF biocompatibility and dialysis practices in this time period. However, the EPS risk varies greatly between different countries, ranging from an incident rate/1,000 patient years of 1.8 in Australia and New Zealand (1995–2007) to 13.6 in Scotland (2000–2007). The mortality of EPS is substantial (25–56%), and in some case series up to 60% of patients may die within 4 months of diagnosis [87, 94].
The use of acidic, hyperosmolar, hyperglycaemic, bio-incompatible dialysis fluid generates the initial damage to the peritoneum in CAPD. This is mediated in part by uraemia, lactate, GDP and AGEs [95, 96]. AGEs are formed by the Maillard reaction, a process of irreversible non-enzymatic glycation and oxidation of proteins, nucleic acids, and lipids [97, 98]. Further damage to the peritoneum promotes the development of EPS. This “second-hit” injury includes recurrent infectious peritonitis (Pseudomonas, S. aureus or fungal spp.), endotoxins (LPS) or recurrent haemoperitoneum in CAPD [17]. Similarly, when the peritoneum is repeatedly or severely injured in other disease processes such as PM, IPC, EM or chemical peritonitis, EPS may also result. EPS is typified by progressive loss and MMT of PMC, inflammatory cell infiltrate with persistent secretion of cytokines, growth factors, and chemokines, and a complex feedback network of chronic peritoneal inflammation.
The signalling pathways involved in EPS include TGF-β1 (and TGF-β1 receptors), integrin-linked kinases (ILK), TRAF6, IL-6, NF-κB, AGE-RAGE, HRE, Src, tyrosine kinase receptors, protein kinase C (AKT), glycogen synthase 3, Wnt/β-catenin, platelet activation and VEGF [37].
Indicators of EPS: peritoneal cytokeratin markers and cytokine levels
Indicators of EPS in CAPD include failure of PD ultrafiltration [99], declining small solute clearance, decreased glucose reabsorption (D2/D0), higher dialysate-to-plasma urea ratio (D/Purea) [87, 100, 101], low levels of CA-125 and high levels of cytokines IL-1β, IL-6, IL-8, HGF, PDGF, TGF-β and VEGF in the dialysate [91, 102, 103]. Abrahams et al. [104] reported increased gene expression of CCN2 (35-fold), TGF-β1(24-fold) and VEGF (77-fold) in the peritoneal membrane of EPS patients compared to CAPD patients without EPS. Sampimon et al. [105] analysed the time course of low CA-125 and elevated IL-6 preceding the diagnosis of EPS. In CAPD patients with ultrafiltration failure, a dialysate appearance rate of CA-125 less than 33 U/min and IL-6 greater than 350 pg/min had a sensitivity of 70% and a specificity of 89% for the diagnosis of EPS. The disappearance of CA-125 from CAP dialysate is related to the mesenchymal transformation of PMC (MMT) and loss of epithelial cytokeratin characteristics [95]. Levels of IL-6 in dialysate efflux reflect the continuing peritoneal inflammatory response and predict the loss of individual peritoneal membrane solute transport function [103].
Role of TGF-β in EPS
Bio-incompatible dialysis fluid, reactive carbonyl species/GDPs (e.g. formaldehyde, glyoxal, methylglyoxal, 3-deoxyglucosone) and formation of AGEs stimulate production of TGF-β1 [106, 107]. Both hyperglycaemic PDF and cellular damage/necrosis cause release of the nuclear protein HMGB1 also named amphoterin. HMGB1 normally resides in the nucleus and regulates DNA replication, transcription, recombination and repair. When released into the extracellular space, HMGB1 also acts as a DAMPs signalling ligand. This is dependent on HMGB1 binding to pattern recognition receptors (PRR), including TLR-2, TLR-4 and RAGE. There is cross ligand binding of different PRRs by endogenous “alarmin” molecules including AGEs, HMGB1, macrophage-1 antigen (MAC-1) and calcium binding protein (S-100), as well as exogenous LPS from gram negative bacteria. Microbial-derived ligands such as LPS are called pathogen-associated molecular patterns (PAMPs). Both DAMPs and PAMPs can synergize in the synthesis and secretion of pro-inflammatory cytokines such as IL-1β in CAPD peritonitis [108]. Activation of RAGE and TLR-4 strongly upregulates MAPK/NF-κB and protein kinase C (AKT) signalling pathways, and CTGF and TGF-β release during peritoneal inflammation [98].
NF-κB is a transcription factor which is kept inert by its cytoplasmic inhibitor protein, I-κB. Degradation of IκB and the subsequent nuclear translocation of NF-κB results from cell/PRR/RAGE stimulation by a variety of agents, examples of which include HMGB1, AGEs, DAMPS, PAMPs, oxidized LDL, ROS, inflammatory cytokines, mitogens, angiotensin II, HSP60, BMP2/4, erythropoietin receptor and HA [109, 110]. HMGB1 stimulates MCs to secrete monocyte chemoattractant protein 1 (MCP-1) and IL-8, which causes recruitment of leukocytes to the peritoneum. In addition to MCP-1 and IL-8, endothelial cells also secrete TNF-α in response to HMGB1 [111]. Release of TGF-β1 promotes peritoneal angiogenesis via MMT of PMC, myofibroblast differentiation and VEGF release, and also by the expansion of hypoxic submesothelial ECM/tissue expressing HIF-1α. Normal peritoneum does not contain myofibroblasts [19]. Lineage tracing shows myofibroblasts can be derived from mesothelial cells (MMT), epithelial cells (EMT), resident pericytes (fibroblast to myofibroblast transition), endo-MT, Th17 lymphocytes or CD34+ bone marrow-derived cells. In situ MMT may also contribute to submesothelial myofibroblast differentiation via cellular and ECM signalling [112].
Blocking the cell surface receptor for AGE (RAGE) with monoclonal antibodies (anti-RAGE Ab) reduced TGF-β1-related peritoneal MMT, interstitial fibrosis and vascular sclerosis in a murine uraemia model. Peritoneal AGE accumulation in uraemia results from reactive carbonyl compounds derived from either carbohydrates or lipids. Heat sterilization of glucose containing dialysis fluid may contribute to GDPs and AGE formation. GDPs have strong oxidant properties and can be directly cytotoxic to mesothelial cells or cause AGE accumulation [113].
Progression of EPS
Podoplanin-positive myofibroblasts derived from PMC MMT in a methylglyoxal-induced murine peritoneal injury model may reflect the progression of fibrosis in human CAPD [114]. In patients with EPS, peritoneal podoplanin is expressed by activated calretinin-positive mesothelial cells, lymphatic endothelial cells, and α-SMA-positive myofibroblasts. The level of podoplanin expression in peritoneal biopsies can be useful in distinguishing simple peritoneal fibrosis from EPS [90]. Peritoneal fibrosis often accelerates after renal transplantation, as calcineurin (CN) inhibitors such as Tacrolimus and Cyclosporin upregulate TGF-β1 [91]. Use of m-TOR inhibitors such as Everolimus instead of CN inhibitors after renal transplantation may reduce the risk of EPS [95]. Histological changes in hyalinizing vasculopathy and EPS, and the use of diagnostic peritoneal biopsy have been recently reviewed in this journal [89].
Pathophysiology of PM
TGF-β1 is a potent inducer of apoptosis and suppressor of proliferation and immortalization in normal epithelial cells [36]. However, TGF-β1 can lose this cytostatic effect and promote EMT, cancer-associated fibroblasts (CAFs) and continued production of ECM. This is referred to as the TGF-β paradox, which is a feature of the tumour micro-environment and is related to multiple factors [37, 39, 53]. (Table 2, Figure 4)
Table 2:
TGF-β1 paradox.
| Action | Mechanism | Ref. |
|---|---|---|
| Loss of TGF tumour suppression | TP53/Rb mutations leading to aberrant TGF-β1 signalling | [43] |
| Loss of TGF tumour suppression | TGF mutations (40–50% for SMAD4, 3–10% for TGFBRII), overexpression of TGF β1,2,3 and Src in pancreatic ductal cancer | [115] |
| HIF promotion | TGF-β normoxic stabilization of HIF/production of SNAIL | [82] |
| VEGF | Association of HIF-1α and SMAD3 cooperatively activating the VEGF promoter. | [116] |
| Src activation | Activation of Src by 17β-estradiol and enhanced TGF-β1/SMAD signalling and cyclin D | [117] |
| Glycolysis | HIF-induced glycolytic switch. | [68, 70] |
| Lactate | Lactate production from the Warburg effect and shuttling of lactate via MCT4 and CAFs. | [75, 118] |
| EMT | Glycolytic enzyme induced EMT. | [71] |
| Stromal recruitment | Recruitment of tumour-associated macrophages (TAM), tumour associated neutrophils (TANS), Th17 and CAFs by TGF-β1 in the TME. | [119, 120, 136] |
| USP4 | Stabilization of vimentin by USP4 in gastric cancer and associated down-regulation of miR-320a. Overexpression of USP4 is found in colorectal, ovarian and lung adenocarcinomas. | [121] |
| HSP90 | Stabilization of TβR-I and TβR-II receptors by HSP90. | [37] |
| RAGE | Persistent RAGE activation by its ligands and feed forward expression of RAGE via RAGE-NF-κB signalling. | [97, 122] |
| TRAF | TRAF6 activation of SMAD/non-SMAD signalling and Redox fibrosis. | [61] |
| ILK | ILK activity-dependent formation of the ILK/Rictor complex, which is required for TGF-β1-mediated EMT. This occurs in epithelial cancers but not in normal epithelial cells. | [60] |
| IL-17 | Immune evasion due to TGF‐β1 secreted from regulatory T cells (Tregs) up‐regulating IL‐17rb through downstream SMAD2/3/4 signalling. | [123] |
| Activin | TGF-β-induced activin pathway activation selectively enhancing cancer cell migration and not TGF-β-induced growth suppression. Both tumoural TGF-β1 and activin expression in human colorectal cancer=shorter survival (median OS 11.5 months vs. 67 months, p ≤ 0.05) | [44, 52] |
| CAFS | TβR-I down-regulation in cancer cells and continued TGF-β1 response in CAFs. Excessive TGF-β1 in the TME due to CAF-TGF-β1 feed forward autocrine effect, and tumour cell-platelet aggregates. | [136] |
| Trogocytosis | Trogocytosis and release of platelet-derived TGF-β1. Activated platelets coat tumour cells with a cell-fibrin-platelet aggregate shield, protecting them from shear forces and host immune attack by natural killer T lymphocytes. Transference of MHC class I proteins and glucocorticoid-induced tumour necrosis factor receptor-related (GITR) ligand from platelets to the tumour cells (trogocytosis) also assists in evasion from host immunosurveillance. Trogocytosis disrupts the recognition of tumour cell missing self, resulting in impaired cytotoxicity and IFN-g production by NK cells. | [124, 125, 126] |
| Platelet-derived TGF-β1 | Platelet-derived TGF-β1, released upon tumour cell–platelet interaction, also inhibits NK-cell mediated immunosurveillance through down-regulation of the activating NK receptor NKG2D. Formation of ovarian cancer spheres, chemoattraction, cancer cell migration, EMT and stem cell markers is increased by platelets. This enhances both haematogenous and transcoelomic metastasis. | [124, 127] |
Figure 4:

The TGF paradox.
Upregulation of TGF-β1 signalling and HIF expression occurs in colon, esophageal, gastric, hepatocellular and pancreatic carcinoma. This is associated with tumour progression, metastasis, angiogenesis and a poor prognosis [128, 129, 130]. For example, elevated TGF-β1 signalling and increased p-SMAD2/3 staining was found in 16% of metastatic colorectal cancers. This was associated with the worst outcome, no response to chemotherapy (cisplatin, oxaliplatin or 5–FU), and a mesenchymal phenotype [131]. Increased lncRNA-ATB production via autocrine TGF-β/ZEB/miR-200 signalling was also shown to be an independent risk factor for invasion, lymph node metastasis, distant metastasis, higher tumour stage and poorer disease free survival, recurrence free survival and overall survival in gastric cancer, CRC and HCC. High levels of lncRNA-ATB were also associated with transtuzumab resistance in breast cancer patients [132, 133, 134].
The initial adhesion of carcinoma cells to the mesothelium is mediated by “cross talk” involving ligand-receptor pairs, including α5β1 integrins–fibronectin, αVβ3 integrins–vitronectin (VN), and CD44–hyaluronic acid. Disruption of the mesothelial glycocalyx and HA oligomer release by ROS, physical trauma, peritoneal desiccation, hypoxia or reperfusion injury enhances carcinoma cell adhesion, inflammatory cytokine release, and peritoneal MMT [12, 15]. The non-aggressive ovarian carcinoma cell line (FOC3) phenotype could be transformed to that of metastasizing ovarian cancer (MFOC3) by exposure to IL-1β. Endogenous IL-1β production by MFOC3 cells stimulated mesothelial cell surface β1 integrin expression and enhanced cancer cell adhesion via a paracrine IL-1β/β1 integrin axis [135].
Inflammatory cytokines IL-1β and TNF‐α act synergistically with TGF‐β1 in upregulating VEGF and IL‐6 production in MCs. Normal murine peritoneum which has been activated by TGF‐β1 is more receptive to subsequent intraperitoneal ovarian cancer cell inoculation and tumour growth. Even the calretinin positive staining peritoneum distant to malignant implants in metastatic ovarian cancer patients showed nuclear expression of pSMAD3. This indicated that the TGF‐β1 pathway had already been widely activated in the peritoneal cavity [136]. Autocrine/paracrine pathways of tumour cell IL-1β/β1 integrin, TGF‐β1/TGF-β1R and VEGF/VEGR ligand binding also promote peritoneal inflammation, cancer cell adhesion and MMT. PMC are transformed into CAFs [137] which promote cell migration, ECM invasion, MMP-2 and −9 release, cleavage of ECM proteins, release of ECM latent VEGF and TGF-β1 and further carcinoma cell invasion. Ovarian and gastric cancer xenografts grow more rapidly when co-injected with omental MCs than with cancer cells alone [138, 139]. Such recruitment and transformation of PMCs into CAFs by cancer cells results in a neovascularized stroma, which is the advancing front of PM and ascites formation [137].
Interestingly, the VHL gene is known a tumour-suppressor gene. Inactivating germline mutations of the gene predisposes affected individuals to the development of several hereditary tumours. Familial VHL syndrome is related to loss of VHL protein, failure of ubiquitylation of HIF-α and promotion of vascular, neuroendocrine and renal cell tumours. This has similarities to the normoxic stabilization of HIF-α by TGF-β1 which enables downstream VEGF release in PM [140].
The Warburg effect may, in part, explain the cancer cachexia syndrome seen in patients with advanced peritoneal metastatic disease. Weight loss can be exacerbated by peritoneal carbonyl stress or continued IL-1 release, causing anorexia and food avoidance. Cancer cachexia and age-related sarcopenia may also be related to TGF-β1 stimulation of myostatin, activin A and activin B, which results in profound muscle wasting [28, 141].
Common pathophysiological features in EPS, EM and PM
For the clinician, diseases such as EPS, EM and PM may appear unrelated pathological entities. The unprepared reader may not suspect that their underlying pathophysiology is indeed quite similar. The pretext for understanding the pathophysiology of these diseases is to realize that damage signalling and wound healing mechanisms are involved in the peritoneal response to various kinds of injury, including EMT and MMT.
EPS is a chronic fibrosing condition of the peritoneum with sclerosis, calcification, thickening or encapsulation of the small intestine [86]. EPS is most commonly due to repeated injury of the peritoneum caused by CAPD. On histology, EPS is typified by progressive loss and MMT of PMC, inflammatory cell infiltrate with persistent secretion of cytokines, stromal growth factors, and chemokines, and a complex feedback network of chronic peritoneal inflammation [17].
EM, a chronic estrogen-dependent gynecological disease, occurs in 10–15% of reproductive-age women. During the proliferative phase of the menstrual cycle, endometrial cells proliferate under the influence of 17β-estradiol, which activates telomerase activity [142, 143]. In EM, this stem cell-like self-renewal ability is enhanced, together with estrogen effects on VEGF release. 17β-Estradiol production is increased due to aromatase activity in EM [142], which drives EMT via HGF, and TNF-α and IL-6 release from peritoneal macrophages [144]. Markers of EMT including overexpression of SLUG, SNAIL, TWIST1 and repression of E-cadherin (CDH1) are found in ectopic compared to eutopic endometrium. E-cadherin-negative, N-cadherin-positive endometriotic epithelial cells show invasive growth in vitro. Increased expression of MYC, Cyclin D1 and Ki67 genes in EM reflects higher levels of proliferation and loss of cell cycle regulation [144, 145]. EM may share common features with type 1 ovarian cancer, including replicative advantage, evasion of apoptosis, genetic changes and tumour suppressor gene mutations [143]. Type 1 ovarian carcinoma (OC) includes EM-associated carcinoma (Clear cell ovarian cancer (CCC)) and endometrioid ovarian tumour, and has different genomic profiles to the more common Type 2 or high grade serous ovarian cancer (HGSC) [146].
Loss of heterozygosity (LOH) or somatic loss of function mutations in tumour suppressor genes including phosphatase and tensin homolog (PTEN), TP53, p16Ink4, human MutL homolog 1 (hMLH1) and B cell lymphoma-2 (BCL-2) have been reported in both EM and type 1 OC. Other genes that may be affected by LOH include the estrogen receptor, progesterone receptor, Apolipoprotein A2, and SOD genes [142, 147]. Endometrioid ovarian cancer has frequent mutations in PTEN, catenin β1 (CTNNB1), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit α (PIK3CA) and AT-rich interaction domain 1A (ARID1A). PIK3CA and ARID1A are also commonly mutated in CCC together with hepatocyte nuclear factor-1β (HNF-1β) overexpression [146]. PTEN is an important suppressor of the PI3K pathway, and loss of PTEN was found in 0% of normal endometrium, 20.6% of EM cysts, 0–5% of CCC and 14–31% of endometrioid ovarian tumours [148]. Loss of PTEN and K-ras mutations appear to act synergistically in the progression from EM to type 1 ovarian cancer [143, 149]. Methylation of human androgen receptor gene (HUMARA) and phosphoglycerate kinase 1 (PGK-1) genes on the X chromosome are used as markers for monoclonal EM associated ovarian cancers arising from EM. This supports the concept of the EM-type 1 OC sequence [142, 150].
The presence of ectopic menstrual blood and high levels of free iron ions in endometriotic cysts leads to the production of ROS through the Fenton reaction. ROS and IL-1 are also released from activated tissue-associated macrophages. This may contribute to release of PGF2α into the peritoneal micro-environment, which promotes pelvic pain and oncogene and inflammatory cytokine activation [142]. NF-ĸB and AP molecular pathway activation by ROS are mediated by convergence of the molecular signals received from activation of receptor activator of NF-ĸB and TGF-β1 receptors [142]. Down-regulation of miR-33b in EM compared to normal uterine endometrium was found to enhance endometrial proliferation and VEGF and MMP-9 release [151]. Thus EM lesions grow by EMT, BM invasion, TGF-β1-induced transformation and stromal recruitment, in a similar way to PM [151].
Peritoneal metastases are a deadly mode of transcoelomic tumour spread in gastrointestinal and gynecological cancer. In order to survive, PM induce the vascularized connective tissue stroma that they require. The formation of a vascularized connective tissue stroma is mediated by VEGF. VEGF increases vascular permeability of endothelial cells with a potency 50,000 times that of histamine, allowing egress of plasma and platelets into the extravascular space and exposure to the ECM. VEGF release in PM thus initiates a chain of events which resemble that of wounds, including CVH, fluid extravasation, platelet activation, fibrin deposition, chemotaxis and inflammatory cytokines [7].
TGF-β1 induces a Warburg effect by switching cellular energy production from mitochondrial phosphorylation to cytosolic glycolysis in EM and peritoneal malignancies, resulting in excess lactic acid production and stabilization of HIF-α [27]. The lactate appears to be derived from transformed PMC adjacent to endometriotic lesions, metastatic carcinoma or tumour-associated macrophages (TAM). Lactate is pumped out of cells into the extracellular space by monocarboxylate 4, which contributes to extracellular acidosis in the coelomic micro-environment [46, 71]. TGF-β1 is released from the latency-associated peptide (LAP) and LTBP when exposed to factors such as ROS, plasmin or acid [22]. Increased lactate can activate latent TGF-β1, leading to a positive feedback loop of increasing TGF-β1 and lactate. This causes further mesenchymal transformation of PMC, promoting EM cell invasion or progression of peritoneal metastases [75]. Extracellular acidosis aids lamellipodia formation, genome instability, cysteine protease, MMP and glycosidase breakdown of the ECM, tumour invasion, angiogenesis, drug resistance, and immune evasion [71]. Thus the transformed PMC create an acidic, pseudohypoxic micro-environment which is conducive to continued inflammation, fibrosis and tumourogenesis [71, 152].
Conclusions
The peritoneal response to various kinds of injury involves loss of PMC, activation of PRRs, EMT and MMT. EPS, EM and PM are all characterized by hypoxia and formation of a vascularized connective tissue stroma mediated by VEGF. TGF-β1 is constitutively expressed by the PMC and plays a major role in the maintenance of a transformed, inflammatory coelomic micro-environment in PM, but also in EPS and EM. Persistently high levels of TGF-β1 or stimulation by inflammatory cytokines (IL-6) induce peritoneal MMT, adhesion formation and fibrosis. TGF-β1 enhances hypoxia-stimulated HIF-1α expression, which drives cell growth, ECM production and cell migration. Disruption of the peritoneal glycocalyx and exposure of the BM release LMW-HA and ECM components, initiating a cascade of pro-inflammatory mediators, including peritoneal cytokines (TNF-α, IL-1, IL-6, prostaglandins), growth factors (TGF-α, TGF-β, PDGF, VEGF, EGF) and the fibrin/coagulation cascade (thrombin, Tissue factor, PAI-1/2). Chronic inflammation and cellular transformation are mediated by DAMPs, PRR, AGE-RAGE, extracellular lactate, pro-inflammatory cytokines, ROS, increased glycolysis, TGF-β1 release, metabolomic reprogramming, TAM and CAFs. The pathogenesis and chronic progression of EPS, EM and PM have common pathways, related to dysregulated mechanisms of wound healing, just as Harold Dvorak originally postulated in 1986.
Footnotes
Author contributions: The author has accepted responsibility for the entire content of this submitted manuscript and approved submission.
Research funding: None declared.
Employment or leadership: None declared.
Honorarium: None declared.
Competing interests: None declared.
References
- 1.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–59. [DOI] [PubMed]
- 2.Morishita Y, Ookawara S, Hirahara I, Muto S, Nagata D. HIF-1α mediates hypoxia-induced epithelial-mesenchymal transition in peritoneal mesothelial cells. Ren Failure. 2016;38:282–89. [DOI] [PubMed]
- 3.Wu MH, Chen KF, Lin SC, Lgu CW, Tsai SJ. Aberrant expression of leptin in human endometriotic stromal cells is induced by elevated levels of hypoxia inducible factor-1alpha. Am J Pathol. 2007;170:590–98. [DOI] [PMC free article] [PubMed]
- 4.Varghese S, Burness M, Xu H, Beresnev T, Pingpank J, Alexander HR. Site-specific gene expression profiles and novel molecular prognostic factors in patients with lower gastrointestinal adenocarcinoma diffusely metastatic to liver or peritoneum. Ann Surg Oncol. 2007;14:3460–71. [DOI] [PubMed]
- 5.Wagener C, Stocking C, Müller O. Cancer signaling: from molecular biology to targeted therapies. Wiley-VCH, Weinheim. 2017:183–204.
- 6.Ceelen W, Pattyn P, Mareel M. Surgery, wound healing, and metastasis: recent insights and clinical implications. Crit Rev Oncol Hematol. 2014;89:16–26. [DOI] [PubMed]
- 7.Dvorak HF. Tumors: wounds that do not heal-Redux. Cancer Immunol Res. 2015;3:1–11. [DOI] [PMC free article] [PubMed]
- 8.Byun JS, Gardner K. Wounds that will not heal: pervasive cellular reprogramming in cancer. Am J Pathol. 2013;182:1055–64. [DOI] [PMC free article] [PubMed]
- 9.Flier JS, Underhill LH, Dvorak HF. Tumors: wounds that do not heal. N Engl J Med. 1986;315:1650–59. [DOI] [PubMed]
- 10.D’Agostino A, Stellavato A, Corsuto L, Diana P, Filosa R, La Gatta A, et al. Is molecular size a discriminating factor in hyaluronan interaction with human cells?. Carbohydr Polym. 2017;157:21–30. [DOI] [PubMed]
- 11.Kawanishi K. Diverse properties of the mesothelial cells in health and disease. Pleura Peritoneum. 2016;1:79–89. [DOI] [PMC free article] [PubMed]
- 12.Matsuzaki S, Vernis L, Bonnin M, Houlle C, Fournet-Fayard A, Rosano G, et al. Effects of low intraperitoneal pressure and a warmed, humidified carbon dioxide gas in laparoscopic surgery: a randomized clinical trial. Sci Rep. 2017;7:11287. [DOI] [PMC free article] [PubMed]
- 13.Bertheloot D, Latz E. HMGB1, IL-1α, IL-33 and S100 proteins: dual-function alarmins. Cell Mol Immunol. 2017;14:43–64. [DOI] [PMC free article] [PubMed]
- 14.Tjhay F, Motohara T, Tayama S, Narantuya D, Fujimoto K, Guo J, et al. CD44 variant 6 is correlated with peritoneal dissemination and poor prognosis in patients with advanced epithelial ovarian cancer. Cancer Science. 2015;106:1421–28. [DOI] [PMC free article] [PubMed]
- 15.Wilson R. Changes in the coelomic microclimate during carbon dioxide laparoscopy: morphological and functional implications. Pleura Peritoneum. 2017;2:17–31. [DOI] [PMC free article] [PubMed]
- 16.Misra S, Hascall VC, Markwald RR, Ghatak S. Interactions between hyaluronan and its receptors (CD44, RHAMM) regulate the activities of inflammation and cancer. Front Immunol. 2015;6:201. [DOI] [PMC free article] [PubMed]
- 17.Loureiro J, Sandoval P, delPeso G, Gónzalez-Mateo G, Fernández-Millara V, Santamaria B, et al. Tamoxifen ameliorates peritoneal membrane damage by blocking mesothelial to mesenchymal transition in peritoneal dialysis. PLoS One. 2013;23:e61165. [DOI] [PMC free article] [PubMed]
- 18.Jolly MK, Ware KE, Gilja S, Somarelli JA, Levine H. EMT and MET: necessary or permissive for metastasis?. Molecular Oncology. 2017;11:755–69. [DOI] [PMC free article] [PubMed]
- 19.Strippoli R, Moreno-Vicente R, Battistelli C, Cicchini C, Noce V, Amicone L, et al. Molecular mechanisms underlying peritoneal EMT and fibrosis. Stem Cells Int. 2016;2016:3543678. [DOI] [PMC free article] [PubMed]
- 20.Li W, Kang Y. A new Lnc in metastasis: long non-coding RNA mediates the pro-metastatic functions of TGF-β. Cancer Cell. 2014;25(5):557–59. [DOI] [PMC free article] [PubMed]
- 21.Wu Y, Tran T, Dwabe S, Sarkissyan M, Kim J, Nava M, et al. A83-01 inhibits TGF-β-induced upregulation of Wnt3 and epithelial to mesenchymal transition in HER2-overexpressing breast cancer cells. Breast Cancer Res Treat. 2017;163:449–60. [DOI] [PMC free article] [PubMed] [Retracted]
- 22.Xu F, Liu C, Zhou D, Zhang L. TGF-β/SMAD pathway and its regulation in hepatic fibrosis. J Histochem Cytochem. 2016;64:157–67. [DOI] [PMC free article] [PubMed]
- 23.Govinden R, Bhoola KD. Genealogy, expression, and cellular function of transforming growth factor-beta. Pharmacol Ther. 2003;98:257–65. [DOI] [PubMed]
- 24.Jin X, Ren S, Macarak E, Rosenbloom J. Pathobiological mechanisms of peritoneal adhesions: the mesenchymal transition of rat peritoneal mesothelial cells induced by TGF-β1 and IL-6 requires activation of Erk1/2 and SMAD2 linker region phosphorylation. Matrix Biol. 2016;51:55–64. [DOI] [PubMed]
- 25.Falk P, Angenete E, Bergström M, Ivarsson ML. TGF-beta1 promotes transition of mesothelial cells into fibroblast phenotype in response to peritoneal injury in a cell culture model. Int J Surg. 2013;11:977–82. [DOI] [PubMed]
- 26.Liappas G, González-Mateo GT, Sánchez-Díaz R, Lazcano JJ, Lasarte S, Matesanz-Marín A, et al. Immune-regulatory molecule CD69 controls peritoneal fibrosis. J Am Soc Nephrol. 2016;27:3561–76. [DOI] [PMC free article] [PubMed]
- 27.Corcoran SE, O’Neill LA. HIF-1α and metabolic reprogramming in inflammation. J Clin Invest. 2016;126:3699–707. [DOI] [PMC free article] [PubMed]
- 28.Trendelenburg AU, Meyer A, Jacobi C, Feige JN, Glass DJ. TAK-1/p38/nNFκB signaling inhibits myoblast differentiation by increasing levels of Activin A. Skeletal Muscle. 2012;2:3. [DOI] [PMC free article] [PubMed]
- 29.Midgley AC, Rogers M, Hallett MB, Clayton A, Bowen T, Phillips AO, et al. Transforming growth factor-β1 (TGF-β1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts. J Biol Chem. 2013;288:14824–38. [DOI] [PMC free article] [PubMed]
- 30.Pohlers D, Brenmoehl J, Löffler I, Müller CK, Leipner C, Schultze-Mosgau S, et al. TGF-beta and fibrosis in different organs – molecular pathway imprints. Biochim Biophys Acta. 2009;1792:746–56. [DOI] [PubMed]
- 31.Zarzynska JM. Two faces of TGF-beta1 in breast cancer. Mediators Inflamm. 2014;2014:141747. [DOI] [PMC free article] [PubMed]
- 32.Li YS, Ni SY, Meng Y, Shi XL, Zhao XW, Luo HH, et al. Angiotensin II facilitates fibrogenic effect of TGF-β1 through enhancing the down-regulation of BAMBI caused by LPS: a new pro-fibrotic mechanism of angiotensin II. Chen Y Ed PLoS One. 2013;8(10):e76289. [DOI] [PMC free article] [PubMed]
- 33.Alexander ET, Minton AR, Peters MC, van Ryn J, Gilmour SK. Thrombin inhibition and cisplatin block tumor progression in ovarian cancer by alleviating the immunosuppressive microenvironment. Oncotarget. 2016;7:85291–305. [DOI] [PMC free article] [PubMed]
- 34.Andrarawewa KL, Kirshner J, Mott JD, Barcellos-Hoff MH. TGF β: roles in DNA responses. Cancer treatment and therapy. In: Jakowlew SB, editor. From transforming growth factor β in cancer therapy (Vol. II). New York: Human Press, 2010:321–31.
- 35.Richter K, Kietzmann T. Reactive oxygen species and fibrosis: further evidence of a significant liaison. Cell Tissue Res. 2016;365:591–605. [DOI] [PMC free article] [PubMed]
- 36.Lebrun JJ. The dual role of TGF-β in human cancer: from tumor suppression to cancer metastasis. ISRN Mol Biol. 2012;381428:1–28. [DOI] [PMC free article] [PubMed]
- 37.Moustakas A, Heldin CH. The regulation of TGFβ signal transduction. Development. 2009;136:3699–714. [DOI] [PubMed]
- 38.Otten J, Bokemeyer C, Fiedler W. TGF-beta superfamily receptors-targets for antiangiogenic therapy?. J Oncol. 2010:317068. [DOI] [PMC free article] [PubMed]
- 39.Alsina-Sanchís E, Figueras A, Lahiguera A, Gil-Martín M, Pardo B, Piulats JM, et al. TGFβ controls ovarian cancer cell proliferation. Int J Mol Sci. 2017;18:1658. [DOI] [PMC free article] [PubMed]
- 40.Gupta DK, Singh N, Sahu DK. TGF-β mediated crosstalk between malignant hepatocyte and tumor microenvironment in hepatocellular carcinoma. Cancer Growth Metastasis. 2014;7:1–8. [DOI] [PMC free article] [PubMed]
- 41.Chruścik A, Gopalan V, Lam AK. The clinical and biological roles of transforming growth factor beta in colon cancer stem cells: a systematic review. Eur J Cell Biol. 2018;97:15–22. [DOI] [PubMed]
- 42.Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci USA. 2009;106:13445–50. [DOI] [PMC free article] [PubMed]
- 43.Eser PÖ, Jänne PA. TGFβ pathway inhibition in the treatment of non-small cell lung cancer. Pharmacol Ther. 2017 Nov 10. pii: S0163-7258(17)30288-7. DOI: 10.1016/j.pharmthera.2017.11.004. [Epub ahead of print] [DOI] [PubMed]
- 44.Staudacher JJ, Bauer J, Jana A, Tian J, Carroll T, Mancinelli G, et al. Activin signaling is an essential component of the TGF-β induced pro-metastatic phenotype in colorectal cancer. Sci Rep. 2017;7:5569. [DOI] [PMC free article] [PubMed]
- 45.Hata A, Chen YG TGF-beta signaling from receptors to Smads. Cold Spring Harb 2016 Perspect Biol 8: a022061. [DOI] [PMC free article] [PubMed]
- 46.Young VJ, Ahmad SF, Duncan WC, Horne AW. The role of TGF-β in the pathophysiology of peritoneal endometriosis. Hum Reprod Update. 2017;23(5):548–59. [DOI] [PubMed]
- 47.Yan X, Liao H, Cheng M, Shi X, Lin X, Feng XH, et al. Smad7 protein interacts with Receptor-regulated Smads (R-Smads) to inhibit Transforming Growth Factor-β (TGF-β)/Smad Signaling. J Biol Chem. 2016;291:382–92. [DOI] [PMC free article] [PubMed]
- 48.Zhang J, Zhang X, Xie F, Zhang Z, van Dam H, Zhang L, et al. The regulation of TGF-β/SMAD signaling by protein deubiquitination. Protein Cell. 2014;5:503–17. [DOI] [PMC free article] [PubMed]
- 49.Xiao L, Peng X, Liu F, Tang C, Hu C, Xu X, et al. AKT regulation of mesothelial-to-mesenchymal transition in peritoneal dialysis is modulated by Smurf2 and deubiquitinating enzyme USP4. BMC Cell Biol. 2015;16:7. [DOI] [PMC free article] [PubMed]
- 50.Meng XM, Tang PM, Li J, Lan HY. TGF-β/Smad signaling in renal fibrosis. Front Physiol. 2015;6:82. [DOI] [PMC free article] [PubMed]
- 51.Guo H, Leung JC, Lam MF, Chan LY, Tsang AW, Lan HY, et al. Smad7 transgene attenuates peritoneal fibrosis in uremic rats treated with peritoneal dialysis. J Am Soc Nephrol. 2007;18:2689–703. [DOI] [PubMed]
- 52.Basu M, Bhattacharya R, Ray U, Mukhopadhyay S, Chatterjee U, Roy SS. Invasion of ovarian cancer cells is induced by PITX2-mediated activation of TGF-beta and Activin-A. Mol Cancer. 2015;14:162. [DOI] [PMC free article] [PubMed]
- 53.Lee YH, Schiemann WP. Chemotherapeutic targeting of the transforming growth factor-β pathway in breast cancers. Breast Cancer Manage. 2014;3:73–85. [DOI] [PMC free article] [PubMed]
- 54.Zhou J, Schmid T, Frank R, Brüne B. PI3K/Akt is required for heat shock proteins to protect hypoxia-inducible factor 1alpha from pVHL-independent degradation. J Biol Chem. 2004;279:13506–13. [DOI] [PubMed]
- 55.Ren K, Mo ZC, Liu X, Tang ZL, Jiang Y, Peng XS, et al. TGF-beta down-regulates apolipoprotein M expression through the TAK-1-JNK-c-Jun pathway in HepG2 Cells. Lipids. 2017;52:109–17. [DOI] [PubMed]
- 56.Zhang S, Che D, Yang F, Chi C, Meng H, Shen J, et al. Tumor-associated macrophages promote tumor metastasis via the TGF-β/SOX9 axis in non-small cell lung cancer. Oncotarget. 2017;8(59):99801–15. [DOI] [PMC free article] [PubMed]
- 57.Zhang YE. Mechanistic insight into contextual TGF-β signaling. Curr Opin Cell Biol. 2017 Nov 14;51:1–7. DOI: 10.1016/j.ceb.2017.10.001. [DOI] [PMC free article] [PubMed]
- 58.Bartscht T, Rosien B, Rades D, Kaufmann R, Biersack H, Lehnert H, et al. Dasatinib blocks transcriptional and promigratory responses to transforming growth factor-beta in pancreatic adenocarcinoma cells through inhibition of Smad signalling: implications for in vivo mode of action. Mol Cancer. 2015;14:199. [DOI] [PMC free article] [PubMed]
- 59.Rahman MS, Akhtar N, Jamil HM, Banik RS, Asaduzzaman SM. TGF-β/BMP signaling and other molecular events: regulation of osteoblastogenesis and bone formation. Bone Res. 2015;3:15005. [DOI] [PMC free article] [PubMed]
- 60.Serrano I, McDonald PC, Lock FE, Dedhar S. Role of the integrin-linked kinase (ILK)/Rictor complex in TGF beta-1-induced epithelial-mesenchymal transition (EMT). Oncogene. 2013;32:50–60. [DOI] [PubMed]
- 61.Ahamed J, Laurence J. Role of platelet-derived transforming growth factor-β1 and reactive oxygen species in radiation-induced organ fibrosis. Antioxid Redox Signal. 2017;27:977–88. [DOI] [PMC free article] [PubMed]
- 62.Davi G, Ciabattoni G, Consoli A, Mezzetti A, Falco A, Santarone S, et al. In vivo formation of 8-iso-prostaglandin F2α and platelet activation in diabetes mellitus: effects of improved metabolic control and vitamin E supplementation. Circulation. 1999;99:224–29. [DOI] [PubMed]
- 63.Souza AM, Wang CC, Chu CY, Lam PM, Rogers MS. The effect of intra-abdominal pressure on the generation of 8-iso prostaglandin F2 during laparoscopy in rabbits. Hum Reprod. 2003;18:2181–88. [DOI] [PubMed]
- 64.Terawaki H, Hayashi Y, Zhu WJ, Matsuyama Y, Terada T, Kabayama S, et al. Transperitoneal administration of dissolved hydrogen for peritoneal dialysis patients: a novel approach to suppress oxidative stress in the peritoneal cavity. Med Gas Res. 2013;3:14. [DOI] [PMC free article] [PubMed]
- 65.Takemoto A, Okitaka M, Takagi S, Takami M, Sato S, Nishio M, et al. A critical role of platelet TGF-β release in podoplanin-mediated tumour invasion and metastasis. Sci Rep. 2017;7:42186. [DOI] [PMC free article] [PubMed]
- 66.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–54. [DOI] [PubMed]
- 67.Karuppagounder SS, Ratan RR. Hypoxia-inducible factor prolyl hydroxylase inhibition: robust new target or another big bust for stroke therapeutics?. J Cereb Blood Flow Metab. 2012;32:1347–61. [DOI] [PMC free article] [PubMed]
- 68.Haase VH. HIF-prolyl hydroxylases as therapeutic targets in erythropoiesis and iron metabolism. Hemodialysis Int. 2017;21:S110–S124. [DOI] [PMC free article] [PubMed]
- 69.Lamichhane SP, Arya N, Kohler E, Xiang S, Christensen J, Shastri VP. Recapitulating epithelial tumor microenvironment in vitro using three dimensional tri-culture of human epithelial, endothelial, and mesenchymal cells. BMC Cancer. 2016;16:581. [DOI] [PMC free article] [PubMed]
- 70.Slemc L, Kunej T. Transcription factor HIF1A: downstream targets, associated pathways, polymorphic hypoxia response element (HRE) sites, and initiative for standardization of reporting in scientific literature. Tumour Biol. 2016;37:14851–61. [DOI] [PubMed]
- 71.Huang R, Zong X. Aberrant cancer metabolism in epithelial–mesenchymal transition and cancer metastasis: mechanisms in cancer progression. Crit Rev Oncol Hematol. 2017;115:13–22. [DOI] [PubMed]
- 72.Clem BF, Chesney J. Molecular pathways: regulation of metabolism by RB. Clin Cancer Res. 2012;18:6096–100. [DOI] [PubMed]
- 73.Lee G, Won HS, Lee YM, Choi JW, Oh TI, Jang JH, et al. Oxidative dimerization of PHD2 is responsible for its inactivation and contributes to metabolic reprogramming via HIF-1α activation. Sci Rep. 2016;6:18928. [DOI] [PMC free article] [PubMed]
- 74.Wei L, Li K, Pang X, Guo B, Su M, Huang Y, et al. Leptin promotes epithelial-mesenchymal transition of breast cancer via the upregulation of pyruvate kinase M2. J Exp Clin Cancer Res. 2016;35:166. [DOI] [PMC free article] [PubMed]
- 75.Young VJ, Brown JK, Maybin J, Saunders PT, Duncan WC, Horne AW. Transforming growth factor-β induced Warburg-like metabolic reprogramming may underpin the development of peritoneal endometriosis. J Clin Endocrinol Metab. 2014;99:3450–59. [DOI] [PMC free article] [PubMed]
- 76.Nass N, Brömme HJ, Hartig R, Korkmaz S, Sel S, Hirche F, et al. Differential response to α-oxoaldehydes in tamoxifen resistant MCF-7 breast cancer cells. PLoS One. 2014;9:e101473. [DOI] [PMC free article] [PubMed]
- 77.Fu QF, Liu Y, Fan Y, Hua SN, Qu HY, Dong SW, et al. Alpha-enolase promotes cell glycolysis, growth, migration, and invasion in non-small cell lung cancer through FAK-mediated PI3K/AKT pathway. J Hematol Oncol. 2015;8:22. [DOI] [PMC free article] [PubMed]
- 78.Sun L, Guo C, Cao J, Burnett J, Yang Z, Ran Y, et al. Over-expression of alpha-enolase as a prognostic biomarker in patients with pancreatic cancer. Int J Med Sci. 2017;14:655–61. [DOI] [PMC free article] [PubMed]
- 79.Gao X, Wang H, Jenny JY, Liu X, Liu ZR. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell. 2012;45:598–609. [DOI] [PMC free article] [PubMed]
- 80.He X, Du S, Lei T, Li X, Liu Y, Wang H, et al. PKM2 in carcinogenesis and oncotherapy. Oncotarget. 2017;8:110656–70. [DOI] [PMC free article] [PubMed]
- 81.Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–44. [DOI] [PMC free article] [PubMed]
- 82.Basu RK, Hubchak S, Hayashida T, Runyan CE, Schumacker PT, Schnaper HW. Interdependence of HIF-1α and TGF-β/Smad3 signaling in normoxic and hypoxic renal epithelial cell collagen expression. Am J Physiol Renal Physiol. 2011;300:F898–905. [DOI] [PMC free article] [PubMed]
- 83.Liu M, Ning X, Li R, Yang Z, Yang X, Sun S, et al. Signalling pathways involved in hypoxia-induced renal fibrosis. J Cell Mol Med. 2017;21:1248–59. [DOI] [PMC free article] [PubMed]
- 84.Liu YV, Hubbi ME, Pan F, McDonald KR, Mansharamani M, Cole RN, et al. Calcineurin promotes hypoxia-inducible factor 1alpha expression by dephosphorylating RACK1 and blocking RACK1 dimerization. J Biol Chem. 2007;282:37064–73. [DOI] [PMC free article] [PubMed]
- 85.Sekiguchi Y, Zhang J, Patterson S, Liu L, Hamada C, Tomino Y, et al. Rapamycin inhibits transforming growth factor β-induced peritoneal angiogenesis by blocking the secondary hypoxic response. J Cell Mol Med. 2012;16:1934–45. [DOI] [PMC free article] [PubMed]
- 86.Kawaguchi Y, Saito A, Kawanishi H, Nakayama M, Miyazaki M, Nakamoto H, et al. Recommendations on the management of encapsulating peritoneal sclerosis in Japan, 2005: diagnosis, predictive markers, treatment, and preventive measures. Perit Dial Int. 2005;25(Suppl. 4):S83–S95. [PubMed]
- 87.Oran E, Seyit H, Besleyici C, Ünsal A, Alıs H. Encapsulating peritoneal sclerosis as a late complication of peritoneal dialysis. Ann Med Surg. 2015;4:205–07. [DOI] [PMC free article] [PubMed]
- 88.Kawanishi H. Surgical and medical treatments of encapsulation peritoneal sclerosis. Contrib Nephrol. 2012;177:38–47. [DOI] [PubMed]
- 89.Kawanishi K, Honda K, Hamada C. Recommendations for pathological diagnosis on biopsy samples from peritoneal dialysis patients. Pleura Peritoneum. 2017;2:3–15. [DOI] [PMC free article] [PubMed]
- 90.Braun N, Fritz P, Ulmer C, Latus J, Kimmel M, Biegger D, et al. Histological criteria for encapsulating peritoneal sclerosis – a standardized approach. PLoS One. 2012;7:e48647. [DOI] [PMC free article] [PubMed]
- 91.Moinuddin Z, Summers A, Van Dellen D, Augustine T, Herrick SE. Encapsulating peritoneal sclerosis – a rare but devastating peritoneal disease. Front Physiol. 2014;5:470. [DOI] [PMC free article] [PubMed]
- 92.Brown EA, Bargman J, van Biesen W, Chang MY, Finkelstein FO, Hurst H, et al. Length of time on peritoneal dialysis and encapsulating peritoneal sclerosis – position paper for ISPD: 2017 update. Perit Dial Int. 2017;37:362–74. [DOI] [PubMed]
- 93.Rigby RJ, Hawley CM. Sclerosing peritonitis: the experience in Australia. Nephrol Dial Transplant. 1998;13:154–59. [DOI] [PubMed]
- 94.Brown MC, Simpson K, Kerssens JJ, Mactier R. Encapsulating peritoneal sclerosis in the new millennium: a national cohort study. Clin J Am Soc Nephrol. 2009;4:1222–29. [DOI] [PMC free article] [PubMed]
- 95.Cornelis T, Oreopoulos DG. Update on potential medical treatments for encapsulating peritoneal sclerosis; human and experimental data. Int Urol Nephrol. 2011;43:147–56. [DOI] [PMC free article] [PubMed]
- 96.Mortier S, DeVriese AS, VandeVoorde J, Schaub TP, Passlick-Deetjen J, Lameire NH. Hemodynamic effects of peritoneal dialysis solutions on the rat peritoneal membrane: role of acidity, buffer choice, glucose concentration, and glucose degradation products. J Am Soc Nephrol. 2002;13:480–89. [DOI] [PubMed]
- 97.Lin L, Park S, Lakatta EG. RAGE signaling in inflammation and arterial aging. Front Biosci (Landmark Ed). 2009;14:1403–13. [DOI] [PMC free article] [PubMed]
- 98.Byun K, Yoo Y, Son M, Lee J, Jeong GB, Park YM, et al. Advanced glycation end-products produced systemically and by macrophages: a common contributor to inflammation and degenerative diseases. Pharmacol Ther. 2017;177:44–55. [DOI] [PubMed]
- 99.Paudel K, Fan SL. Is there an end in sight for encapsulating peritoneal sclerosis?. Perit Dial Int. 2014;34(6):576–78. [DOI] [PMC free article] [PubMed]
- 100.Lin F, Wu X, Zhang H, You X, Zhang Z, Shao R, et al. A microrna screen to identify regulators of peritoneal fibrosis in a rat model of peritoneal dialysis. BMC Nephrol. 2015;16:48. [DOI] [PMC free article] [PubMed]
- 101.De Sousa-Amorim E, Del Peso G, Bajo MA, Alvarez L, Ossorio M, Gil F, et al. Can EPS development be avoided with early interventions? The potential role of tamoxifen–a single-center study. Perit Dial Int. 2014;34:582–93. [DOI] [PMC free article] [PubMed]
- 102.Zweers MM, Struijk DG, Smit W, Krediet RT. Vascular endothelial growth factor in peritoneal dialysis: a longitudinal follow-up. J Lab Clin Med. 2001;137:125–32. [DOI] [PubMed]
- 103.Schmitt CP, Aufricht C. Is there such a thing as biocompatible peritoneal dialysis fluid?. Pediatr Nephrol. 2017;32:1835–43. [DOI] [PMC free article] [PubMed]
- 104.Abrahams AC, Habib SM, Dendooven A, Riser BL, Van Der Veer JW, Toorop RJ, et al. Patients with encapsulating peritoneal sclerosis have increased peritoneal expression of connective tissue growth factor (CCN2), transforming growth factor-β1, and vascular endothelial growth factor. PLoS One. 2014;9:e112050. [DOI] [PMC free article] [PubMed]
- 105.Sampimon DE, Korte MR, Barreto DL, Vlijm A, de Waart DR, Struijk DG, et al. Early diagnostic markers for encapsulating peritoneal sclerosis – a case control study. Perit Dial Int. 2010;30:163–69. [DOI] [PubMed]
- 106.Semchyshyn HM. Reactive carbonyl species in vivo: generation and dual biological effects. Sci World J. 2014;2014:417842. [DOI] [PMC free article] [PubMed]
- 107.Flessner MF. The transport barrier in intraperitoneal therapy. Am J Physiol Renal Physiol. 2005;288:F433–42. [DOI] [PubMed]
- 108.Saïd-Sadier N, Ojcius D. Alarmins, inflammasomes and immunity. Biomed J. 2012;35:437–49. [DOI] [PMC free article] [PubMed]
- 109.El-Far AH. The role of receptors for advanced glycation end product in pancreatic carcinogenesis. Pancreat Disord Ther. 2016;6:166.
- 110.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–66. [DOI] [PubMed]
- 111.Chu Y, Wang Y, Zheng Z, Lin Y, He R, Liu J, et al. Proinflammatory effect of high glucose concentrations on HMrSV5 cells via the autocrine effect of HMGB1. Front Physiol. 2017;8:762. [DOI] [PMC free article] [PubMed]
- 112.Padwal M, Margetts PJ. Experimental systems to study the origin of the myofibroblast in peritoneal fibrosis. Kidney Res Clin Pract. 2016;35:133–41. [DOI] [PMC free article] [PubMed]
- 113.De Vriese AS, Tilton RG, Mortier S, Lameire NH. Myofibroblast transdifferentiation of mesothelial cells is mediated by RAGE and contributes to peritoneal fibrosis in uraemia. Nephrol Dial Transplant. 2006;21:2549–55. [DOI] [PubMed]
- 114.Hirahara I, Sato H, Imai T, Onishi A, Morishita Y, Muto S, et al. Methylglyoxal induced basophilic spindle cells with podoplanin at the surface of peritoneum in rat peritoneal dialysis model. Biomed Res Int. 2015;2015:289751. [DOI] [PMC free article] [PubMed]
- 115.Ungefroren H, Sebens S, Groth S, Gieseler F, Fändrich F. Differential roles of Src in transforming growth factor-ß regulation of growth arrest, epithelial-to-mesenchymal transition and cell migration in pancreatic ductal adenocarcinoma cells. Int J Oncol. 2011;38(3):797–805. [DOI] [PubMed]
- 116.Liu W, Shen SM, Zhao XY, Chen GQ. Targeted genes and interacting proteins of hypoxia inducible factor-1. Int J Biochem Mol Biol. 2012;3(2):165–78. [PMC free article] [PubMed]
- 117.Wang X, Deng H, Zou F, Fu Z, Chen Y, Wang Z, et al. ER-α36-mediated gastric cancer cell proliferation via the c-Src pathway. Oncol Lett. 2013 Aug;6(2):329–35. [DOI] [PMC free article] [PubMed]
- 118.Mao L, Li Y, Zhao J, Li Q, Yang B, Wang Y, et al. Transforming growth factor-β1 contributes to oxaliplatin resistance in colorectal cancer via epithelial to mesenchymal transition. Oncology Letters. 2017;14:647–54. [DOI] [PMC free article] [PubMed]
- 119.Brechbuhl HM, Finlay-Schultz J, Yamamoto TM, Gillen AE, Cittelly DM, Tan AC, et al. Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen. Clin Cancer Res. 2017;23(7):1710–21. [DOI] [PMC free article] [PubMed]
- 120.Singel KL, Segal BH. Neutrophils in the tumor microenvironment: trying to heal the wound that cannot heal. Immunol Rev. 2016;273(1):329–43. [DOI] [PMC free article] [PubMed]
- 121.Zhu Y, Zhang Y, Sui Z, Zhang Y, Liu M, Tang H. USP14 de-ubiquitinates vimentin and miR-320a modulates USP14 and vimentin to contribute to malignancy in gastric cancer cells. Oncotarget. 2017;8(30):48725–36. [DOI] [PMC free article] [PubMed]
- 122.Bongarzone S, Savickas V, Luzi F, Gee AD. Targeting the receptor for advanced glycation endproducts (RAGE): a medicinal chemistry perspective. J Med Chem. 2017;60(17):7213–32. [DOI] [PMC free article] [PubMed]
- 123.Huang S, Wei P, Hwang‐Verslues WW, Kuo W, Jeng Y, Hu C, et al. TGF‐β1 secreted by Tregs in lymph nodes promotes breast cancer malignancy via up‐regulation of IL‐17RB. EMBO Mol Med. 2017;9(12):1660–80. [DOI] [PMC free article] [PubMed]
- 124.Takemoto A, Miyata K, Fujita N. Platelet-activating factor podoplanin: from discovery to drug development. Cancer Metastasis Rev. 2017;36(2):225–34. [DOI] [PMC free article] [PubMed]
- 125.Placke T, Örgel M, Schaller M, Jung G, Rammensee HG, Kopp HG, et al. Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res. 2012;72(2):440–48. [DOI] [PubMed]
- 126.Placke T, Salih HR, Kopp HG. GITR ligand provided by thrombopoietic cells inhibits NK cell antitumor activity. J Immunol. 2012 Jul 1;189(1):154–60. [DOI] [PubMed]
- 127.Erices R, Cubillos S, Aravena R, Santoro F, Marquez M, Orellana R, et al. Diabetic concentrations of metformin inhibit platelet-mediated ovarian cancer cell progression. Oncotarget. 2017;8(13):20865–80. [DOI] [PMC free article] [PubMed]
- 128.Li DP, Fan J, Wu YJ, Xie YF, Zha JM, Zhou XM. MiR-155 up-regulated by TGF-β promotes epithelial-mesenchymal transition, invasion and metastasis of human hepatocellular carcinoma cells in vitro. Am J Transl Res. 2017;9:2956–65. [PMC free article] [PubMed]
- 129.Wigerup C, Påhlman S, Bexell D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol Ther. 2016;164:152–69. [DOI] [PubMed]
- 130.Chen XL, Chen ZQ, Zhu SL, Liu TW, Wen Y, Su YS, et al. Prognostic value of transforming growth factor-beta in patients with colorectal cancer who undergo surgery: a meta-analysis. BMC Cancer. 2017 4;Apr(17):240. [DOI] [PMC free article] [PubMed]
- 131.Salazar R, Roepman P, Willems SM, Brunen D, Kellner U, Midgley RA, et al. Molecular subtyping of colorectal cancer to identify a mesenchymal tumor type that might benefit from TGF-beta pathway inhibition. J Clin Oncol. 2014;32(3 suppl):456–456.
- 132.Saito T, Kurashige J, Nambara S, Komatsu H, Hirata H, Ueda M, et al. A long non-coding RNA activated by transforming growth factor-β is an independent prognostic marker of gastric cancer. Ann Surg Oncol. 2015;22(Suppl 3):S915–22. [DOI] [PubMed]
- 133.Fan YH, Ji CX, Xu B, Fan HY, Cheng ZJ, Zhu XG. Long noncoding RNA activated by TGF-β in human cancers: a meta-analysis. Clin Chim Acta. 2017;468:10–16. [DOI] [PubMed]
- 134.Yang Y, Junjie P, Sanjun C, Ma Y. Long non-coding RNAs in colorectal cancer: progression and future directions. J Cancer. 2017;8(16):3212–25. [DOI] [PMC free article] [PubMed]
- 135.Watanabe T, Hashimoto T, Sugino T, Soeda S, Nishiyama H, Morimura Y, et al. Production of IL1-beta by ovarian cancer cells induces mesothelial cell beta1-integrin expression facilitating peritoneal dissemination. J Ovarian Res. 2012;5:7. [DOI] [PMC free article] [PubMed]
- 136.Rynne-Vidal A, Au-Yeung CL, Jiménez-Heffernan JA, Pérez-Lozano ML, Cremades-Jimeno L, Bárcena C, et al. Mesothelial-to-mesenchymal transition as a possible therapeutic target in peritoneal metastasis of ovarian cancer. J Pathol. 2017;242:140–51. [DOI] [PMC free article] [PubMed]
- 137.Rynne-Vidal A, Jiménez-Heffernan JA, Fernández-Chacón C, López-Cabrera M, Sandoval P. The mesothelial origin of carcinoma associated-fibroblasts in peritoneal metastasis. Cancers (Basel). 2015;7:1994–2011. [DOI] [PMC free article] [PubMed]
- 138.Mikula-Pietrasik J, Sosinska P, Kucinska M, Murias M, Maksin K, Malinska A, et al. Peritoneal mesothelium promotes the progression of ovarian cancer cells in vitro and in a mice xenograft model in vivo. Cancer Lett. 2014;355:310–15. [DOI] [PubMed]
- 139.Tsukada T, Fushida S, Harada S, Yagi Y, Kinoshita J, Oyama K, et al. The role of human peritoneal mesothelial cells in the fibrosis and progression of gastric cancer. Int J Oncol. 2012;41:476–82. [DOI] [PMC free article] [PubMed]
- 140.Crespigio J, Berbel LCL, Dias MA, Berbel RF, Pereira SS, Pignatelli D, et al. Von Hippel-Lindau disease: a single gene, several hereditary tumors. J Endocrinol Invest. 2018;41:21–31. [DOI] [PubMed]
- 141.Chen JL, Walton KL, Hagg A, Colgan TD, Johnson K, Qian H, et al. Specific targeting of TGF-β family ligands demonstrates distinct roles in the regulation of muscle mass in health and disease. Proc Natl Acad Sci U S A. 2017;114:E5266–E5275. [DOI] [PMC free article] [PubMed]
- 142.Pollacco J, Sacco K, Portelli M, Schembri-Wismayer P, Calleja-Agius J. Molecular links between endometriosis and cancer. Gynecol Endocrinol. 2012;28:577–81. [DOI] [PubMed]
- 143.Viganó P, Somigliana E, Chiodo I, Abbiati A, Vercellini P. Molecular mechanisms and biological plausibility underlying the malignant transformation of endometriosis: a critical analysis. Hum Reprod Update. 2006 Jan-Feb;12(1):77–89. [DOI] [PubMed]
- 144.Proestling K, Birner P, Gamperl S, Nirtl N, Marton E, Yerlikaya G, et al. Enhanced epithelial to mesenchymal transition (EMT) and upregulated MYC in ectopic lesions contribute independently to endometriosis. Reprod Biol Endocrinol. 2015;13:75. [DOI] [PMC free article] [PubMed]
- 145.Bischoff FZ, Heard M, Simpson JL. Somatic DNA alterations in endometriosis: high frequency of chromosome 17 and p53 loss in late stage endometriosis. J Reprod Immunol. 2002;55:49–64. [DOI] [PubMed]
- 146.Kawahara N, Ogawa K, Nagayasu M, Kimura M, Sasaki Y, Kobayashi H. Candidate synthetic lethality partners to PARP inhibitors in the treatment of ovarian clear cell cancer. Biomed Rep. 2017;7(5):391–99. [DOI] [PMC free article] [PubMed]
- 147.Shin JC, Ross HL, Elias S, Nguyen DD, Mitchell-Leef D, Simpson JL, et al. Detection of chromosomal aneuploidy in endometriosis by multi-color fluorescence in situ hybridization (FISH). Hum Genet. 1997;100:401–06. [DOI] [PubMed]
- 148.Rojas V, Hirshfield KM, Ganesan S, Rodriguez-Rodriguez L. Molecular characterization of epithelial ovarian cancer: implications for diagnosis and treatment. Wong K-K, ed. Int J Mol Sci. 2016;17(12):2113. [DOI] [PMC free article] [PubMed]
- 149.Otsuka J, Okuda T, Sekizawa A, Amemiya S, Saito H, Okai T, et al. K-ras mutation may promote carcinogenesis of endometriosis leading to ovarian clear cell carcinoma. Med Electron Microsc. 2004 Sep;37(3):188–92. [DOI] [PubMed]
- 150.Chang CM, Yang YP, Chuang JH, Chuang CM, Lin TW, Wang PH, et al. Discovering the deregulated molecular functions involved in malignant transformation of endometriosis to endometriosis-associated ovarian carcinoma using a data-driven, function-based analysis. Int J Mol Sci. 2017;18(11):2345. [DOI] [PMC free article] [PubMed]
- 151.Yang YM, Yang WX. Epithelial-to-mesenchymal transition in the development of endometriosis. Oncotarget. 2017;8:41679–89. [DOI] [PMC free article] [PubMed]
- 152.Worzfeld T, Pogge Von Strandmann E, Huber M, Adhikary T, Wagner U, Reinartz S, et al. The unique molecular and cellular microenvironment of ovarian cancer. Front Oncol. 2017;7:24. [DOI] [PMC free article] [PubMed]