

Abstract
Mice Jacking Cu/Zn-superoxide dismutase (Sod1−/− or sod1KO mice) show high levels of oxidative stress/damage and a 30% decrease in lifespan. The Sod1KO mice also show many phenotypes of accelerated aging with the loss of muscle mass and function being one of the most prominent aging phenotypes. Using various genetic models targeting the expression of Cu/Zn-superoxide dismutase to specific tissues, we evaluated the role of motor neurons and skeletal muscle in the accelerated loss of muscle mass and function in Sod1KO mice. Our data are consistent with the sarcopenia in Sod1KO mice arising through a two-hit mechanism involving both motor neurons and skeletal muscle. Sarcopenia is initiated in motor neurons leading to a disruption of neuromuscular junctions that results in mitochondrial dysfunction and increased generation of reactive oxygen species (ROS) in skeletal muscle. The mitochondrial ROS generated in muscle feedback on the neuromuscular junctions propagating more disruption of neuromuscular junctions and more ROS production by muscle resulting in a vicious cycle that eventually leads to disaggregation of neuromuscular junctions, denervation, and loss of muscle fibers.
1. Introduction
Aging in humans is accompanied by a 30–40% reduction in skeletal muscle mass/function and is the primary cause of frailty and loss of independence in the elderly [17]. This phenomenon, called sarcopenia, is universal to all mammalian species and is observed even in the absence of disease. The loss of muscle mass involves both loss of muscle fibers and atrophy of the remaining fibers. Aging in skeletal muscle is associated with an increased number of fibers per motor unit, fiber type grouping, and atrophy of fibers, especially Type IIB glycolytic fibers. These changes in the structural organization of muscle are consistent with a cycle of continual denervation and innervation [8]. In addition, changes in motor neurons have also been reported during aging that could contribute to muscle atrophy, including loss of motor neurons and alterations in axonal sprouting [2]. While the etiology of sarcopenia is poorly understood, several potential contributing factors have been identified, including loss of innervation, mitochondrial dysfunction, and oxidative stress [3].
Our groups initially began studying the role oxidative stress/ damage played in sarcopenia because it was generally accepted at the time that the age-related increase in oxidative damage/stress played a major role in aging and the pathologies (e.g., loss of muscle mass/ function) associated with aging [15]. To directly test the role of oxidative stress in sarcopenia, we studied the effect of altering the antioxidant defense system on the age-related decline in muscle mass. If oxidative stress played a role in sarcopenia, we rationalized that compromising the antioxidant defense system would lead to increased oxidative stress and a more rapid progression of sarcopenia. Table 1 lists the mouse models we studied with deficiencies in various components of the antioxidant defense system. Of the five models studied, only mice in which Cu/Zn-superoxide dismutase (Cu/ZnSOD) was completely deleted (Sod1KO) showed an accelerated loss of muscle mass.
Table 1.
Mouse models deficient in antioxidant enzymes that were studied with respect to sarcopenia.
| Genotype | Name of gene product | Expression | ΔMuscle mass |
|---|---|---|---|
| Sod1 +/− | Cu/Zn-Superoxide Dismutase | ~ 50% | No Change |
| Sod1−/− | Cu/Zn-Superoxide Dismutase | Not Detectable | ~40–50% Decrease |
| Sod2+/− | Mn-Superoxide Dismutase | ~ 50% | No Change |
| Gpx1−/− | Glutathione Peroxidase 1 | Not Detectable | No Change |
| Gpx4+/− | Glutathione Peroxidase 4 | ~ 50% | No Change |
The genotypes of the mice that were used study the effect of oxidative stress on muscle mass are listed with the expression of each of genes that were knocked out. The change in muscle mass for each genotypes is compared to WT mice at 10–12 month of age.
2. Sod1KO mouse model of accelerated sarcopenia
Cu/ZnSOD is the primary antioxidant enzyme responsible for removing superoxide anions in the cytosol and mitochondria and protecting cells from oxidative stress/damage. The Sod1KO mice, which lack Cu/ZnSOD in all tissues, show a dramatic increase in oxidative damage in all tissues studied, including muscle and neuronal tissue, and a 30% decrease in lifespan [10,12]. The Sod1KO mice also exhibit several signs of accelerated aging, especially muscle atrophy and weakness [1]. As shown in Fig. 1, the Sod1KO mice show a significant loss of muscle mass (gastrocnemius) by 8–12 months of age, which mimics changes seen in 25- to 30-month-old wild type (WT) mice [10,4,7]. A significant decrease in the mass of all muscle types is observed in Sod1KO mice except for soleus [10]. The Sod1KO mice also show a decline in muscle function as measured by isometric contractile force generated in situ by the gastrocnemius [4,7]. The lower forces generated by the muscle of Sod1KO mice are not simply a consequence of smaller muscle mass. When normalized to muscle cross-sectional area, the specific force was still significantly lower for Sod1KO mice compared to WT mice. By comparing force generation by nerve and direct muscle stimulation, Larkin et al. [7] showed that muscle weakness in Sod1KO mice was due in part to functionally denervated fibers, i.e., force generated by direct muscle stimulation was significantly greater than the force generated by nerve stimulation. The decrease in contractile force in the muscle of Sod1KO mice was paralleled by a decline in grip strength [1], rota-rod performance [10], voluntary wheel running [10], and treadmill endurance [4]. In addition, the Sod1KO mice exhibit most of the changes in function observed in frail humans [1].
Fig. 1. Comparison of muscle atrophy in WT and Sod1KO mice as they age.
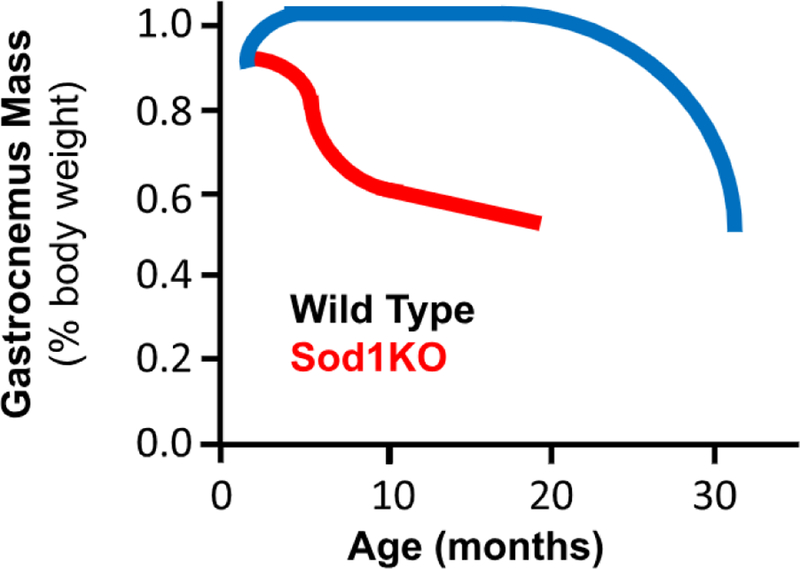
The mass of the gastrocnemius is shown relative to the lifespan of WT (blue) and Sod1KO (red) mice. The figure represents a composite of data from Muller et al. [10], Jang et al. [4]; and Larkin et al. [7].
Because innervation of the muscle by motor neurons is essential for maintaining muscle size, fiber type, structure, and function, we compared the structure and function of neuromuscular junctions (NMJs) in the Sod1KO to young and old WT mice. As shown in Fig. 2, dramatic changes were observed in the structure of the NMJs in Sod1KO mice at 18 months of age, which were comparable to the changes we observed in WT mice at 30 months of age. These changes include deterioration of the NMJ, acetylcholine receptor (AChR) cluster fragmentation, and the degeneration and retraction of motor neurons [4,5]. Interestingly, dietary restriction, which increases the lifespan of mice, including Sod1KO mice [19], attenuated the loss of muscle mass/function and NMJ degeneration in Sod1KO mice [6]. Thus, the changes in skeletal muscle and motor neurons not only mimic sarcopenia but other motor neuron diseases as well, e.g., muscular dystrophy and amyotrophic lateral sclerosis (ALS).
Fig. 2. Neuromuscular junction degeneration in WT and Sod1 KO mice.
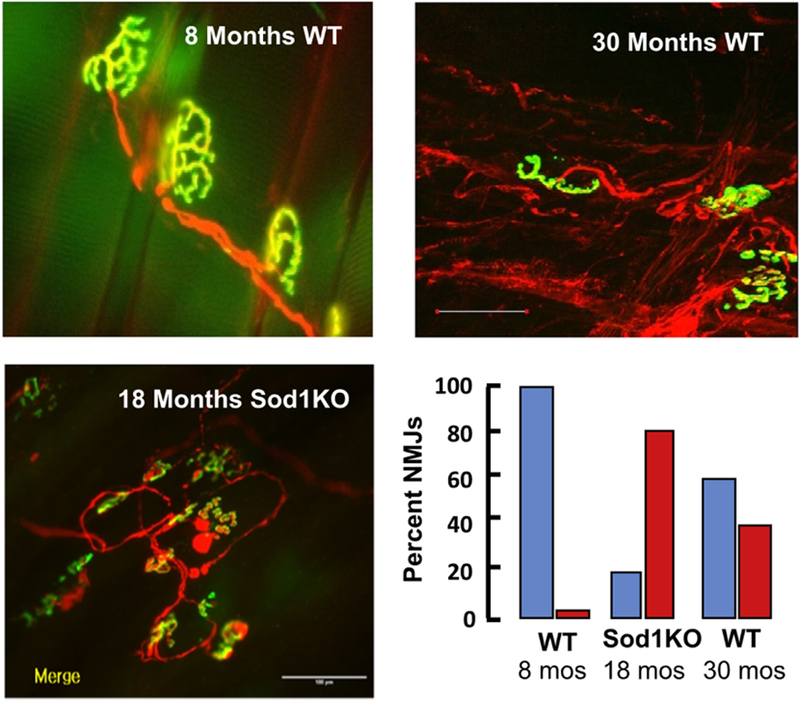
The photographs show the NMJ immunofluorescence for gastrocnemius of 8- and 30-month-old WT mice and 18-month-old Sod1KO mice. The graph shows the percent of NMJs that are normal (blue bars) and fragmented (red bars). Data are taken from Larkin et al. [7] and Jang et al. [5,6].
Our first insight into a possible molecular mechanism involved in muscle atrophy came from our studies in several mouse models of sarcopenia. As shown in Fig. 3, the loss of muscle mass is associated with a dramatic increase in the production of reactive oxygen species (ROS) by isolated muscle mitochondria, e.g., with age [11,9] and in Sod1KO mice [10,11] and an ALS mouse model [11]. As shown in Fig. 3, we found that mitochondrial ROS (mtROS) production by muscle is increased approximately 30% in 30-month-old WT mice, more than 50% in 20-month-old Sod1KO mice, and approximately 75% in muscle from symptomatic ALS mice. We believe these changes in mtROS generation arise from denervation, because sciatic nerve transection resulted in a greater than 30-fold increase in muscle mtROS [11]. Furthermore, we found that dietary restriction of Sod1KO mice dramatically reduced mtROS production by skeletal muscle and attenuated muscle atrophy [6]. Thus, there is a strong association between mtROS generation by muscle, muscle atrophy, and NMJ denervation, suggesting that mtROS produced by muscle plays a key role in sarcopenia. The mitochondria isolated from the muscles of Sod1KO mice also show decreased ATP generation and oxygen consumption and enhanced mitochondria-mediated apoptosis [4].
Fig. 3. ROS generation by muscle mitochondria in three models of sarcopenia.
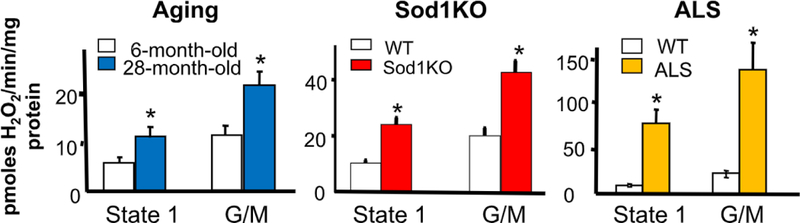
The generation of hydrogen peroxide by isolated mitochondria from the gastrocnemius is show for young and old mice, Sod1KO mice (20 month old) and ALS mice, (G93A). These data are taken from Muller et al. [10,11], and the * indicates those values that are significantly different from the WT controls.
In summary, Sod1KO mice mimic changes that occur with age in WT mice, e.g., a loss of motor function and contractility, a decline in nerve conduction and number of motor units, partial denervation, degeneration of NMJs, increased mtROS generation and mitochondrial dysfunction, increased oxidative damage and content of heat shock proteins in muscle [1,18]. Therefore, the Sod1KO allows one to study in less than a year the process of sarcopenia, which takes over two years to occur with normal aging in WT mice. In addition, by genetically manipulating the expression of Sod1, it is possible to study the mechanism of sarcopenia, e.g., whether changes proximal or distal to neuromuscular synapses play a role in sarcopenia. Below we describe the effect of the following genetic manipulations on sarcopenia: expressing Sod1 in neurons of Sod1KO mice (nTg/Sod1KO) and knocking out Sod1 in muscle (mSod1KO) or neurons (nSod1KO). Fig. 4 compares the effect of these three manipulations on muscle mass and function to Sod1KO mice.
Fig. 4. Effect of genetic manipulations on the mass and function of muscle.
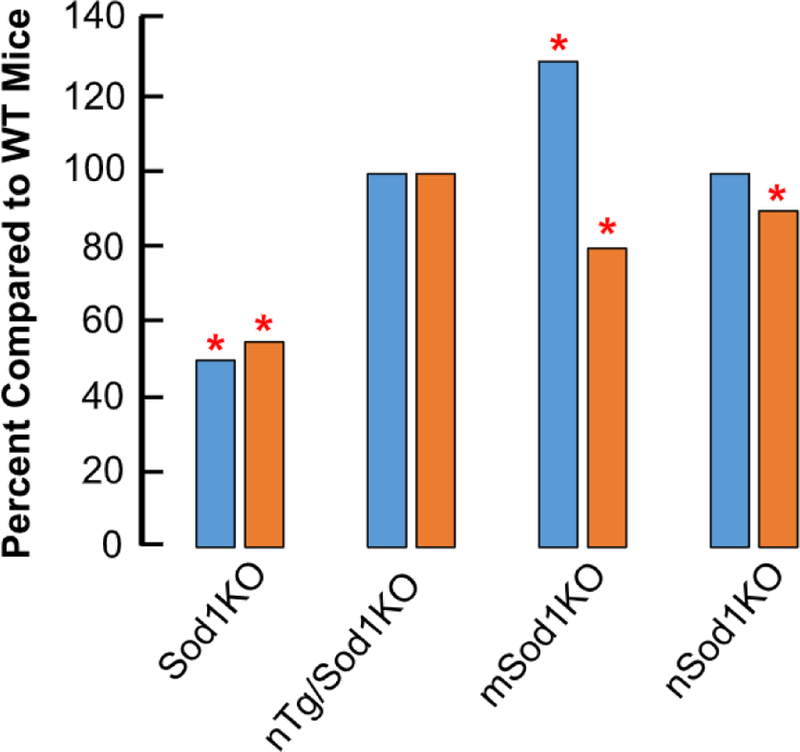
The mass (blue) and function (maximum isometric specific force, gold) for gastrocnemius is shown at 8–10 months of age for four genetic mouse models compared to WT mice. The * indicates those values that are significantly different than WT mice. Data are taken from Larkin et al. [7] for Sod1KO mice; Sakellariou et al. [13] for nTg/Sod1KO mice; Zhang et al. [19] for mSod1KO mice, and Sataranataranjan et al. [16] for nSod1 KO mice.
3. Sod1KO mice that express Sod1 in neuronal tissues (nTg/Sod1KO mice)
To study the role motor neurons play in the accelerated loss of muscle mass/function in Sod1 mice, we generated Sod1KO mice that expressed Cu/ZnSOD only in neurons using a transgene containing the 4.5 kb rat synapsin 1 promoter fused to the human SOD1 gene. When we crossed these transgenic mice to Sod1KO mice, we generated mice that expressed Cu/ZnSOD only in neurons (nTg/Sod1KO mice). The expression of Cu/ZnSOD was not detectable in skeletal muscle, as well as other tissues; however, Cu/ZnSOD expression (~20% of wild type levels) was observed in neuronal tissues [13]. As shown in Fig. 4, these mice do not show the accelerated loss of muscle mass or function that is observed in Sod1KO mice. In addition, the structure and function of NMJs are maintained in the nTg/Sod1KO mice. Interestingly, the nTg/Sod1KO mice show no increase in mtROS production or increased oxidative damage in muscle even though Cu/ZnSOD is completely absent. These data demonstrate the importance of the motor neurons in sarcopenia. Currently, it appears that impaired redox signaling rather than oxidative damage in peripheral nerves is key in initiating sarcopenia observed Sod1KO mice [14].
4. Mice that do not express Sod1 in muscle (mSod1KO mice)
Our groups generated the first Sod1flox/flox mice [19], and these mice have been crossed to the muscle-specific Acta 1-cre-recombinase transgenic mice to generate skeletal muscle-specific Sod1 knockout mice (mSod1KO mice). The Cu/ZnSOD levels in the skeletal muscle of mSod1KO mice was less than 5% of the WT levels; however, there was no change in Cu/ZnSOD levels in brain or other tissues. The decrease in Cu/ZnSOD was observed in all muscle types tested. As shown in Fig. 4, the mSod1 KO mice actually show an increase (~ 30%) in muscle mass; however, the specific force generating capacity of the gastrocnemius from the mSod1KO mice was reduced 20%. These data suggest that the muscle atrophy and loss of function are occurring by different mechanisms. However, there was no difference in muscle function as measured by rota-rod or treadmill performance. Again, no increase in mtROS generation or measures of oxidative damage was observed in the muscles from the mSod1KO mice even though the level of Cu/ZnSOD in skeletal muscle is negligible. More recently, Sakellariou et al. [14] reported that the mSod1KO mice did not show any evidence of NMJ degeneration.
5. Mice that do not express Sod1 in neuronal tissue (nSod1KO mice)
We also crossed the Sod1flox/flox mice to transgenic mice expressing the neuron-specific Cre-recombinase, nestin-cre to generate mice deficient in Cu/ZnSOD in neuronal tissue [16]. Cu/ZnSOD protein levels were undetectable in the brains of nSod1KO mice, and Cu/ZnSOD activity is barely detectable (< 5% of the activity of WT mice). We also observed that the level/activity of Cu/ZnSOD was reduced 90–95% in other neuronal tissues, e.g., sciatic nerve and spinal cord. On the other hand, the levels and activity of Cu/ZnSOD in the skeletal muscle of the nSod1KO mice was similar to WT mice. The expression of Cu/ZnSOD was similar in nSod1KO and WT mice in all muscle types, e.g., gastrocnemius, quadriceps, soleus, etc.
Based on our data showing that expression of Sod1 in neurons of Sod1KO mice rescued the sarcopenia phenotype of Sod1KO mice, we were surprised to find that the nSod1KO mice had little effect on muscle mass [16]. The mass of the gastrocnemius and extensor digitorum longus was similar for the nSod1 KO and WT mice. A slight, but significant decrease in the mass of the soleus (< 10%) and quadriceps (14%) was observed in the nSod1KO mice. A slight (~10%) but significant decrease in force was observed in the gastrocnemius and extensor digitorum longus but not the soleus of nSod1KO mice. This decrease in force is much less than the 40% decrease observed in Sod1KO mice [4,7]. NMJ morphology was altered and the expression of genes associated with denervation were increased in the nSod1KO mice [16]; however, there was no evidence of NMJ fragmentation in the nSod1 KO mice, which is seen as in Sod1KO mice (Fig. 2). Thus, while deleting Sod1 in neuronal tissue has an effect on NMJ structure/function, it is much less severe than we find in Sod1KO mice and does not lead to an increase in mtROS production by muscle or evidence of denervation that occurs in nSod1 KO mice.
6. Two-hit model of sarcopenia in Sod1KO mice
Based on our data with the three genetic models, we propose that muscle atrophy in Sod1KO mice occurs through a ‘two-hit’ mechanism involving both motor neurons and skeletal muscle as shown in Fig. 5. The ‘first-hit’ occurs when redox homeostasis is compromised in motor neurons resulting in NMJ dysfunction, which triggers increased mtROS production in compromised muscle (e.g., muscle fibers with a reduced antioxidant defense system). We propose that the ‘second-hit’ occurs when the increased mtROS generated in skeletal muscle, triggers a retrograde response inducing further damage to NMJs, which leads to increased NMJ dysfunction. The increased NMJ dysfunction, in turn, increases mtROS generation in muscle, resulting in a vicious cycle that ultimately results in NMJ fragmentation, denervation, loss of muscle fibers, and sarcopenia.
Fig. 5. Proposed two-hit model describing the molecular mechanism leading to loss of muscle mass and function in Sod1KO mice.
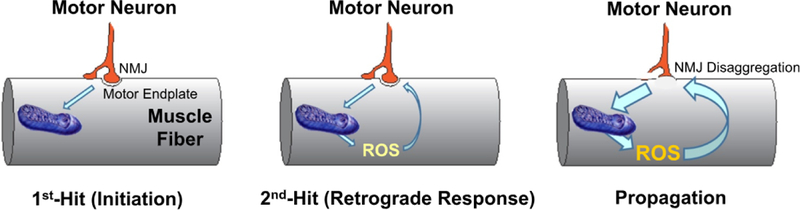
The oxidized redox state in motor neurons initiates disruption of NMJs leading to mitochondrial dysfunction and increased ROS production. The mtROS generated in muscle feeds-back on the NMJs propagating more NMJ dysfunction and more mtROS production resulting in a vicious cycle that eventually leads to NMJ disaggregation, denervation, and loss of muscle fibers.
Preventing the initial motor neuron defect, as in the nTg/Sod1KO mouse, prevents mtROS generation in skeletal muscle as well as the cascade of events leading to sarcopenia even though the muscle is compromised. The mSod1KO mice show that without the initial motor neuron dysfunction there is no trigger to increase mtROS production by muscle over that observed in WT mice and, therefore, no NMJ fragmentation, These data also suggest that the dysfunction in motor neurons/NMJ is necessary to initiate the induction of mtROS in skeletal muscle. Finally, the nSod1KO mice show little or no muscle atrophy because the initial NMJ changes in structure/function cannot trigger mitochondria dysfunction and mtROS generation in a ‘healthy’, uncompromised muscle fiber with a robust antioxidant defense system. Therefore, the nSod1KO mice lack the second-hit, which would lead to the vicious cycle resulting in NMJ fragmentation and denervation. Because the accelerated sarcopenia phenotype observed in Sod1KO mice closely mimics sarcopenia observed in normal, WT mice, we believe it is likely that sarcopenia that occurs with age occurs through a similar two-hit mechanism.
Acknowledgements
The data presented were obtained by our groups over the past fifteen years as part of program project grants (AG020591, AG AG051442) from the National Institute on Aging.
References
- [1].Deepa SS, Bhaskaran S, Espinoza S, Brooks SV, McArdle A, Jackson MJ, Van Remmen H, Richardson A, A new mouse model of frailty: the Cu/Zn superoxide dismutase knockout mouse, Geroscience 39 (2) (2017) 187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fagg GE, Scheff SW, Cotman CW, Axonal sprouting at the neuromuscular junction of adult and aged rats, Exp. Neurol 74 (1981) 847–854. [DOI] [PubMed] [Google Scholar]
- [3].Fulle S, Protasi F, Di Tano G, Pietrangelo T, Beltramin A, Boncompagni S, Vecchiet L, Fanò G, The contribution of reactive oxygen species to sarcopenia and muscle ageing, Exp. Gerontol 39 (2004) 17–24. [DOI] [PubMed] [Google Scholar]
- [4].Jang YC, Lustgarten MS, Liu Y, Muller FL, Bhattacharya A, Liang H, Salmon AB, Brooks SV, Larkin L, Hayworth CR, Richardson A, Van Remmen H, Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration, FASEB J 24 (2010) 1376–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jang YC, Van Remmen H, Age-associated alterations of the neuromuscular junction, Exp. Gerontol 46 (2011) 193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jang YC, Liu Y, Hayworth CR, Bhattacharya A, Lustgarten MS, Muller FL, Chaudhuri A, Qi W, Li Y, Huang JY, Verdin E, Richardson A, Van Remmen H, Dietary restriction attenuates age-associated muscle atrophy by lowering oxidative stress in mice even in complete absence of CuZnSOD, Aging Cell 11 (2012) 77782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Larkin LM, Davis CS, Sims-Robinson C, Kostrominova TY, Van Remmen H, Richardson A, Feldman EL, Brooks SV, Skeletal muscle weakness due to deficiency of Cu, Zn superoxide dismutase is associated with loss of functional innervation, Am. J. Physiol. Regul. lntegr. Comp. Physiol 301 (2011) Rl40Rl407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Larsson L, Edstrom L, Effects of age on enzyme-histochemical fibre spectra and contractile properties of fast- and slow-twitch skeletal muscles in the rat, J. Neurol. Sci 76 (1986) 69–89. [DOI] [PubMed] [Google Scholar]
- [9].Mansouri A, Muller FL, Liu Y, Ng R, Faulkner J, Hamilton M, Richardson A, Huang TT, Epstein CJ, Van Remmen H, Alterations in mitochondrial function, hydrogen peroxide release and oxidative damage in mouse hind-limb skeletal muscle during aging, Mech. Ageing Dev 127 (2006) 298–306. [DOI] [PubMed] [Google Scholar]
- [10].Muller FL, Song W, Liu Y, Chaudhuri A, Pieke-Dahl S, Strong R, Huang TT, Epstein CJ, Roberts LJ 2nd, Csete M,Faulkner JA, Van Remmen H, Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy, Free Radie. Biol. Med 40 (2006) 1993–2004. [DOI] [PubMed] [Google Scholar]
- [11].Muller FL, Song W, Jang YC, Liu Y, Sabia M, Richardson A, Van Remmen H, Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production, Am.J. Physiol.-Reg. Integr 293 (2007) Rl 159–Rl 168. [DOI] [PubMed] [Google Scholar]
- [12].Perez V i., Bokov A, Van Remmen H, Mele J, Ran Q, Ikeno Y, Richardson A, Is the oxidative stress theory of aging dead? Biochim. Biophys. Acta 1790 (2009) 1005–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sakellariou GK, McDonagh B, Porter H, Giakoumaki II, Earl KE, Nye GA, Vasilaki A, Brooks SV,Richardson A, Van Remmen H, McArdle A, Jackson MJ, Comparison of whole body SODlknockout with muscle-specific SODl knockout mice reveals a role for nerve redox signaling in regulation of degenerative pathways in skeletal muscle, Antioxid. Redox Signal 28 (2014) 275–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sakellariou GK, Davis CS, Shi Y, Ivannikov MV, Zhang Y, Vasilaki A, Macleod GT, Richardson A, Van Remmen H, Jackson MJ, McArdle A, Brooks SV, Neuron-specific expression of CuZnSOD prevents the loss of muscle mass and function that occurs in homozygous CuZnSOD-knockout mice, FASEB J 28 (2018) 1666–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Salmon AB, Richardson A, Pérez VI, Update on the oxidative stress theory of aging: does oxidative stress play a role in aging or healthy aging? Free Radie. Biol. Med 48 (5) (2010) 642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sataranataranjan K, Qaisar R, Davis C, Sakellariou GK, Vasilaki A, Zhang Y, Liu Y, Bhaskaran s., McArdle A, Jackson M, Brooks SV, Richardson A, Van Remmen H, Neuron specific reduction in CuZnSOD is not sufficient to initiate a full sarcopenia phenotype, Redox Biol 5 (2015) 14148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Roubenoff R, Hughes VA, Sarcopenia: current concepts, J. Gerontol. A Biol. Sci. Med. Sci 55 (2000) M716–M724. [DOI] [PubMed] [Google Scholar]
- [18].Vasilaki A, Richardson A, Van Remmen H, Brooks SV, Larkin L, McArdle A, Jackson MJ, Role of nerve-muscle interactions and reactive oxygen species in regulation of muscle proteostasis with ageing, J. Physiol 595 (20) (2017) 6409–6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhang Y, Davis C, Sakellariou GK, Shi Y, Kayani AC, Pulliam D, Bhattacharya A, Richardson A, Jackson MJ, McArdle A, Brooks SV, Van Remmen H, CuZnSOD gene deletion targeted to skeletal muscle leads to loss of contractile force but does not cause muscle atrophy in adult mice, FASEB J 27 (2013) 3536–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]