

Summary
Resolution of acute inflammation is governed, in part, by specialized pro-resolving mediators (SPM), including lipoxins, resolvins, protectins and maresins. Among them, resolvin D1 (RvD1) exhibits potent pro-resolving functions via activating human resolvin D1 receptor (DRV1/GPR32). RvD1 is a complex molecule that requires challenging organic synthesis, diminishing its potential as a therapeutic. Therefore, we implemented a high throughput screening of small molecule libraries and identified several chemotypes that activated recombinant DRV1, represented by NCGC00120943 (C1A), NCGC00135472 (C2A), pMPPF and pMPPI. These chemotypes also elicited rapid impedance changes in cells overexpressing recombinant DRV1. With human macrophages, they each stimulated phagocytosis of serum-treated zymosan at concentrations comparable to RvD1, the endogenous DRV1 ligand. Additionally, macrophage phagocytosis of live E. coli was significantly increased by these chemotypes in DRV1-transfected macrophages, compared to mock-transfected cells. Taken together, these chemotypes identified by unbiased screens act as RvD1 mimetics, exhibiting pro-resolving functions via interacting with human DRV1.
Keywords: G protein-coupled receptor, inflammation, resolution, phagocytosis, macrophage
eTOC blurb:
Chiang et al., identified synthetic molecules that activate human DRV1/GPR32 receptor using high throughput screening (>48,000 compounds). DRV1/GPR32 and its ligand Resolvin D1 display pro-resolving actions in inflammation. These molecules act as resolvin D1 mimetics, offering templates to facilitate therapeutic development targeting DRV1 to promote resolution of inflammation.
Graphical Abstract
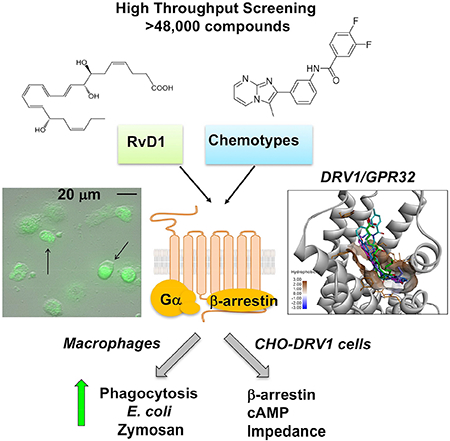
Introduction
Acute inflammation, while protective, when uncontrolled can lead to excessive chronic inflammation, which is recognized today as a major component of many widely occurring diseases (Cotran et al., 1999) as well as infection (Nathan, 2012). Diseases associated with inflammation include cardiovascular disease (Ridker et al., 2017), periodontal disease (Van Dyke and Serhan, 2006), arthritis (Norling and Perretti, 2013), neurodegenerative diseases, pulmonary, renal and metabolic syndrome, to name a few of the organ systems impacted by excessive inflammation and collateral tissue damage (Perretti et al., 2015). Currently, the treatment approaches used for controlling inflammation mostly focus on enzyme inhibitors and receptor antagonists that can block pro-inflammatory and inflammation initiators; while effective, these can turn immunosuppressive for most patients (Serhan, 2014, 2017). Hence, additional approaches are needed to control unwanted excessive inflammation.
The identification of previously unknown resolution phase mediators in inflammation demonstrated that resolution is a biosynthetically active process, and also characterized specialized functions of these potent mediators in the control of resolution of inflammation (Chiang and Serhan, 2017; Serhan, 2014). For example, Resolvin D1 (RvD1; 7S,8R,17S-trihydroxy-4Z,9E,11E,13Z,15E,19Z-docosahexaenoic acid) is a member of the resolvin family (Serhan et al., 2002) belonging to the new superfamily coined specialized proresolving mediators (SPM), which are agonists targeting leukocytes and other cell types. SPM are biosynthesized in the resolution phase with their major defining functions that: a) limit further neutrophil recruitment, b) counterregulate pro-inflammatory mediators, enhance macrophage phagocytosis of apoptotic leukocytes, microbes and cell debris as well as reduce pain (Chiang and fproinflammatory leukotrienes (Fredman et al., 2014). Together Serhan, 2017; Serhan, 2014). Thus, the SPM activate the signaling for resolution of inflammation (Chiang and Serhan, 2017; Serhan, 2014, 2017).
The potent anti-inflammatory and pro-resolving functions of Resolvin D1 (Serhan et al., 2002) have now been confirmed by others and extended to many preclinical disease models including reducing ocular inflammation (Li et al., 2013; Rossi et al., 2015; Settimio et al., 2012), protection from myocardial infarct (Gilbert et al., 2014), reducing pain (Bang et al., 2010; Huang et al., 2011), salivary inflammation as in Sjögren’s syndrome (Leigh et al., 2014; Odusanwo et al., 2012) and airway inflammation, tissue injury (Cox et al., 2015; Eickmeier et al., 2013; Hsiao et al., 2014; Tang et al., 2014), pulmonary infection (Croasdell et al., 2016) and allergic airway responses (Rogerio et al., 2012), as well as reduction of cancer burden, tumor growth and enhancing cancer therapy in mice (Sulciner et al., 2018). Resolvin D1 has also shown to be renal protective (Zhang et al., 2013) and reduces inflammation in adipose tissue and obese fat (Clària et al., 2012), as well as enhances wound healing (Tang et al., 2013).
Resolvin D1 also stimulates macrophage autophagy (Prieto et al., 2015) and has direct actions in the adaptive immune response acting on B cells to reduce IgE production (Kim et al., 2016) and on T cells to regulate their responses (e.g. reducing TNFα production (Chiurchiu et al., 2016). Resolvin D1 regulates the biosynthesis of pro-inflammatory leukotrienes (Fredman et al., 2014). Together, these findings suggest that resolvin D1 or small molecule mimetics have therapeutic potential (Serhan, 2017). The total organic synthesis of resolvins requires multiple steps (for example, see resolvin D4 synthesis as in Winkler et al., 2018), as do their analog mimetics (Tang et al., 2014). To enable industry-scale synthesis, benzo-containing resolvin D1 mimetics were prepared as a means of reducing the number of chiral centers and steps in organic synthesis (Orr et al., 2015). These substitutions, while proving more chemically stable, reduce potency and still require multi-step synthesis and purification.
The receptor for RvD1 is a G protein-coupled receptor (GPCR) identified via library screening and direct ligand binding with 3H-labeled RvD1, denoted as human DRV1/GPR32 (Krishnamoorthy et al., 2010). In order to facilitate clinical development of small molecules that could act as resolvin D1 mimetics and potentially be unique therapeutic classes of SPMs, we screened a diverse collection of small molecules for compounds that stimulate activity of DRV1, the human resolvin D1 receptor.
Results
Identification of small molecule GPR32 agonists from qHTS
A GPR32 PathHunter® β-Arrestin reporting cell line, which measures recruitment of β-arrestin to the DRV1/GPR32 upon agonist activation and generates chemiluminescent signal, was used for the primary high throughput screening (HTS) of DRV1/GPR32 agonists. The assay was miniaturized to a 1536-well microplate format to enable the automated screening of a large collection of small molecules in dose responses, known as quantitative HTS (qHTS) (Inglese et al., 2006). To test robustness of the GPR32 PathHunter® β-Arrestin Assay for HTS, and before proceeding to an automatic screening of a large small molecule collection, the Library of Pharmacologically Active Compounds LOPAC®1280 (Sigma-Aldrich) including 1280 compounds was screened in a qHTS format (Figure 1). The library of compounds were diluted 1:5 in 7 concentrations in interplate manner, with final concentrations in the assay ranging from 76μM to 5nM. From the screen of the LOPAC collection, we identified p-MPPF which, akin to its close analog p-MPPI, is a serotonin receptor 5-HT(1A) antagonist (Kung et al., 1996; Kung et al., 1995). Following the assay validation, a high-throughput screening (HTS) of a diversity collection of 47,065 compounds were tested at two concentrations (76μM and 15μM) using a fully automated robotic screening system.
Figure 1. Compound identification and validation flow chart.

Depicted is the funnel of the assays, filtering, and analyses performed for identification of the most prominent chemotypes of clusters and singletons selected for further studies.
Figure 1 outlines the screening strategy that was pursued to select and validate active compounds identified from the primary HTS. Among them, 220 hits (0.46% of all screened libraries) were selected from the primary HTS based on efficacy >60% in comparison to the basal activity (DMSO-treated cells). These active compounds were re-tested at 7 concentrations as 3-fold serial dilutions at final assay concentrations 76μM –106nM in the same GPR32 PathHunter® β-Arrestin Assay used for the primary HTS. In this screening, 34 compounds (15% of primary cherry-picks) confirmed their activity in a dose response manner. The confirmed compounds were further tested in a panel of assays including GPR32 cAMP HTRF signaling assay, THP1 cytotoxicity assay, and leukotriene B receptor 1 (BLT1) PathHunter® β-Arrestin assay as a counter assay (Figure 1). Finally, 11 hits were selected which were active in GPR32 cAMP HTRF signaling assay, non-cytotoxic on THP1, and inactive in BLT1 counter assay. Structural clustering analysis showed that the active compounds were rather diverse, with 3 large clusters (5 and more members) and a number of small clusters (2–3 members) and singletons. Structural analysis identified two chemotypes, represented by the hits NCGC00120943 (chemotype-1A; C1A) and NCGC00135472 (chemotype-2A; C2A) shown in Figure 2A, as attractive templates for medicinal chemistry-based structure activity relationship studies.
Figure 2. Identification of unique chemotypes via screening of a diverse collection of small molecules.

(A) Chemical structures of known DRV1 ligands and the most promising agonists identified from the screening. (B-D) β-arrestin (solid dots) and cAMP (clear dots) profiling of the most potent representatives: (B) Initial hit pMPPF and its close analog pMPPI, (C) Chemotypes 1 and (D) Chemotypes 2. Both assays were performed in CHO-GPR32 cell line. Cellular basal activity (0%) in β-arrestin assay is vehicle stimulation (DMSO, 0.57%), 100% activity is a 2-fold signal increase above the basal activity. cAMP assay was performed in the presence of forskolin stimulation (8 μM). The 0% activity corresponds to stimulation with forskolin alone, while vehicle stimulation (DMSO, 0.57%) relates to −100%. β-arrestin and cAMP assays were performed two separate times in triplicates (average of 6, with SD).
For further confirmation, these compounds were re-tested at 11 concentrations in 1:3 dilutions from 57 μM to 1 nM (Figure 2B–D). pMPPF increased the signal in GPR32 β-arrestin cells ~30% above the basal cell activity. These data were confirmed in dose-response manner along with testing of its iodinated analog pMPPI, which increased cAMP, but not β-arrestin activities (Fig.2B). Chemotypes 1 (Fig. 2C) and 2 (Fig. 2D) from the screening of the diversity collection increased β-arrestin by approximately 50% over basal in a dose response manner, and inhibited forskolin-induced cAMP signal by about 50% at similar potencies as those measured in the β-arrestin assay. Each agonist (chemotypes 1 and 2) activates DRV1/GPR32 with different affinities and/or efficacies in the β-arrestin and cAMP assays (Table 1 and Fig. 2). For chemotype 1, C1A and C1B gave similar efficacies in β-arrestin activity, and C1A showed higher affinity (EC50 0.93 μM) than C1B (EC50 3.79 μM) for DRV1. For cAMP activity, they have similar EC50, and C1A exhibited higher efficacies (−46.2 %) than C1B (−30.7 %). For chemotype 2, C2A gave higher affinities (i.e. lower EC50) to DRV1 than C2B in both β-arrestin and cAMP activities, as well as higher potencies (i.e. higher efficacies) in β-arrestin activities.
Table 1.
Beta-arrestin and cAMP profiling of selected lead compounds
| EC50, μM | Efficacy, % | |
|---|---|---|
| C1A [NCGC00120943] | ||
| β-arrestin | 0.93 | 47.0 |
| cAMP | 0.38 | −46.2 |
| C1B [NCGC00120919] | ||
| β-arrestin | 3.79 | 43.0 |
| cAMP | 0.32 | −30.7 |
| C2A [NCGC00135472] | ||
| β-arrestin | 0.37 | 60.8 |
| cAMP | 0.05 | −29 |
| C2B [NCGC00134314] | ||
| β-arrestin | 1.17 | 46.6 |
| cAMP | 0.19 | −32.6 |
| pMPPF | ||
| β-arrestin | 1.16 | 20.5 |
| cAMP | n/a | n/a |
| pMPPI | ||
| β-arrestin | n/a | n/a |
| cAMP | 2.5 | 24.4 |
Both β-arrestin and cAMP assays were performed in CHO-GPR32 cell line. In β-arrestin assay, cellular basal activity (0%) is vehicle stimulation (DMSO, 0.57%; 100% activity is a 2-fold signal increase above the basal activity. cAMP assay was performed in the presence of forskolin stimulation (8 μM). Forskolin alone was taken as 0%, while vehicle alone (DMSO, 0.57%) was taken as −100%. EC50: half maximal effective concentration. Efficacy: the highest activity of each compound expressed as % increase or decrease of forskolin stimulation. The numbers are mean values from two separate experiments in triplicates (average of 6 determinations) for both β-arrestin and cAMP assays.
Full chemical names: C1A: chemotype-1A; NCGC00120943 [3-methoxy-N-(5-morpholin-4-ylsulfonyl-1,3,4-thiadiazol-2-yl)benzamide]
C1B: chemotype-1B; NCGC00120919 [3-bromo-N-(5-morpholin-4-ylsulfonyl-1,3,4-thiadiazol-2-yl)benzamide]
C2A: chemotype-2A; NCGC00135472 [3,4-difluoro-N-[3-(3-methylimidazo[1,2-a]pyrimidin-2-yl)phenyl]benzamide]
C2B: chemotype-2B; NCGC00134314 [3-methoxy-N-[3-(3-methylimidazo[1,2-a]pyrimidin-2-yl)phenyl]benzamide]
p-MPPF: 4-Fluoro-N-{2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl}-N-(2-pyridinyl)benzamide
p-MPPI: 4-iodo-N-{2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl}-N-(2-pyridinyl)benzamide
These differences in efficacies and affinities could impact downstream intracellular signals evoked by each ligand. Taken together, C1A is the more potent agonist for DRV1 in chemotype 1 and C2A is the more potent in chemotype 2, based on both β-arrestin and cAMP activities.
Ligand-receptor dependency for activation of human recombinant DRV1/GPR32
To further examine whether these chemotypes directly activate human DRV1/GPR32, we carried out electrical cell substrate impedance sensing (ECIS) to monitor rapid changes in impedance upon ligand binding to receptors (Peters and Scott, 2009). Using CHO cells overexpressing recombinant human DRV1 (CHO-hDRV1), RvD1 (100 nM) time-dependently reduced impedance (Fig. 3A), consistent with our previous findings (Krishnamoorthy et al., 2012). In comparison, at 1μM, pMPPF increased and pMPPI reduced impedance with CHO-DRV1 cells (Fig. 3B&C). Wild-type CHO cell (CHO-WT) did not give significant impedance changes in response to pMPPF or pMPPI (Fig. 3B&C). We next tested compounds from chemotype-1 (C1A and C1B) and chemotype-2 (C2A and C2B). Each compound (100 nM-10 μM) dose-dependently elicited rapid changes in impedance with CHO-DRV1 (Fig. 3D–G). Their rank order potencies were C2A > C1A > C2B > C1B at 100 nM. These results indicate that each chemotype initiated concentration-dependent signals with CHO cells in a DRV1-dependent manner. In addition, these results are consistent with those obtained with β-arrestin and cAMP activities, in that C1A (Chemotype 1) and C2A (Chemotype 2) are the more potent agonists for DRV1 in the chemotype groups (Fig. 2 and Table 1).
Figure 3. ECIS monitoring of ligand-receptor interaction in CHO-DRV1/GPR32 cells.

CHO-DRV1 (A-G) or CHO-WT (B, C) cells were incubated with test compounds or vehicle alone at indicated concentrations. Impedance changes across cell monolayers were recorded every 4 seconds for 10 min. Results are representative tracings from 3 independent experiments.
Identified chemotypes enhance human macrophage phagocytosis
MΦ phagocytosis of apoptotic cells, microbes and debris is a cellular hallmark of tissue resolution of acute inflammation (Cotran et al., 1999). Since RvD1 enhances MΦ phagocytosis of serum-treated zymosan (STZ) via human DRV1/GPR32 (Krishnamoorthy et al., 2010), we next examined whether these compounds shared this pro-resolving action of RvD1. We selected C2A from chemotype-2 and C1A from chemotype-1 for these studies since they were the two most potent ligands activating recombinant human DRV1 (Figs. 2 and 3). Human MΦ were differentiated from peripheral blood monocytes, and phagocytosis of fluorescent-labeled STZ carried out (see Methods). RvD1 (0.1pM-10 nM) dose-dependently increased phagocytosis with human MΦ (Fig. 4A). In comparison, C2A, C1A, pMPPF and pMPPI also enhanced phagocytosis in a dose-dependent manner (Fig. 4A&B). At a concentration as low as 10 pM, RvD1 and pMPPF were the most efficacious, enhancing phagocytosis by ~60%, followed by C1A and C2A, which increased phagocytosis by ~35–40% (Fig. 4C). pMPPI was significantly less potent than pMPPF. At a higher concentration 10 nM, these compounds were essentially equally efficacious (Fig. 4D).
Figure 4. RvD1 and mimetics promote human macrophage phagocytosis of serum-treated zymosan (STZ).

(A-E) Macrophages (50,000 cells/well in 96-well plates) were incubated with RvD1, mimetics or vehicle control at indicated concentrations for 15 min (37°C, pH 7.45), followed by addition of FITC-STZ at 10:1 ratio (STZ:macrophage) to initiate phagocytosis. Results are dose responses analyzed by curve fitting (A,B); comparisons of RvD1 and mimetics at 10 pM (C) and 10 nM (D); comparisons of RvD1 and 5-HT (E). Results are percent increases of phagocytosis above vehicle; mean±SEM from triplicates of 3–4 separate donors. (C) *P<0.05, vs RvD1; #P<0.05, vs vehicle. (E) *P<0.05, RvD1 vs vehicle control; #P<0.05, RvD1 vs 5-HT. (F) CD163 expression on human macrophages; mean±SEM from 3 separate donors. *P<0.05, RvD1 vs vehicle control.
Since both pMPPF and p-MPPI are known serotonin receptor 5-HT(1A) antagonists (Kung et al., 1996; Kung et al., 1995), we tested whether 5-HT impacts macrophage phagocytosis. At 0.1–10 nM, 5-HT did not significantly increase phagocytosis (Fig. 4E), suggesting that the actions of pMPPF and pMPPI in stimulating phagocytosis were not mediated via 5-HT receptors. In addition to stimulating macrophage phagocytosis, we assessed whether these chemotypes can also promote M2 macrophage differentiation, a known pro-resolving function of RvD1 (Titos et al., 2011). Each compound was incubated with human macrophages for 24h, and levels of CD163, an M2 marker, determined using flow cytometry. RvD1 (10 nM) significantly increased CD163 levels on macrophages by 25.7% compared to cells incubated with vehicle alone (Fig 4F). In comparison, p-MPPI, p-MPPF, C2A, C1A increased CD163 levels by 20.0%, 13.7%, 13.3%, 4.7%, respectively, albeit these increases did not reach statistical significance. Thus, these chemotype agonists of DRV1 shared only some of the pro-resolving actions of RvD1, the endogenous DRV1 ligand, namely stimulating macrophage phagocytosis of microbial particles (Figure 4).
Real-time monitoring of phagocytosis of live E. coli with macrophages overexpressing DRV1/GPR32
Next, we investigated whether phagocytosis stimulated by these compounds was DRV1-dependent. To address this, human MΦ were transfected with either human GPR32 or a mock plasmid. Phagocytosis of fluorescent-labeled E. coli was monitored in real time using microscopy (Fig. 5). Fluorescence-labelled live E. coli was added to macrophages to initiate phagocytosis and images taken every 10 min for 60 min. As early as 10 min, C1A (10 nM) slightly increased phagocytosis in mock-transfected cells (~15% increase above vehicle control), and this action was greatly enhanced with hGPR32 transfection, giving >70% increase above vehicle control (Fig. 5A). C2A also gave significant increase of phagocytosis >40% with DRV1-transfected macrophages at 10 min (Fig. 5B), while pMPPF enhanced phagocytosis >50% with these cells at 20–30 min (Fig. 5C). pMPPI-increased phagocytosis in DRV1-transfected macrophages was higher than that in mock-transfected macrophages, from 20–40 min, albeit did not reach statistical significance (Fig. 5D). For direct comparison, RvD1 (10 nM) increased uptake of E. coli with mock-transfected MΦ (30–40% increase above vehicle control), an action further enhanced with hDRV1 transfection (>100% increase) at 20–30 min (Fig. 5E). Together, these results demonstrated that these unique DRV1 ligands enhanced macrophage phagocytosis of live E. coli, each with different kinetics in a DRV1-dependent manner.
Figure. 5. Phagocytosis of live E. coli with macrophages overexpressing DRV1/GPR32: Real-time monitoring.

Human MΦ were transfected with human DRV1 plasmid or mock. MΦ were plated onto 8-well chamber slides (100,000 cells/well in PBS++) 48h after transfection. Imaging was then carried out 24h after re-plating. MΦ were incubated with test compounds (10 nM) or vehicle control for 15 min at 37°C, followed by addition of BacLight Green-labeled E. coli (E. coli: macrophage 50:1) to initiate phagocytosis. Fluorescent images were then recorded every 10 min for 60 min. Three separate experiments were carried out. In each experiment, 4 fields (40X) per condition (per well) were recorded. (A-E) (Left) Results are mean fluorescence of 4 fields/well from one representative experiment. (Right) % increase in phagocytosis above vehicle control in mock or GPR32 transfected MΦ at 10–60 min; mean±SEM from 3 separate donors. *P<0.05, vs Mock.
Binding models of DRV1 agonists
To gain insight into the binding interactions of these identified agonists with DRV1, we docked the three molecules pMPPF, C1A and C2A to the predicted structural model of DRV1. An ensemble docking approach was applied to account for protein flexibility and induced conformational changes upon ligand binding. As shown in Figure 6, The 7-TM binding site of DRV1 consists of a number of aromatic residues such as Tyr101, Tyr117, Phe-112, Tyr-125, His-220, Phe-272, Phe-302, which forms a highly hydrophobic pocket to accommodate ligand binding. RvD1 binds in this pocket in a manner that the molecule forms an aromatic region in the middle by mimicking π-π stacking interactions with surrounding residues Tyr117, Phe-112, Tyr-125, L276, Phe-272 (Fig. 6A). This is well supported by the binding interaction of Benzo-diacetylenic-17R-RvD1-methyl ester (BDA-RvD1) mimetic, which adopts essentially the same binding conformation as RvD1 with a cyclized aromatic ring bound at the bottom of the pocket (Fig. 6B). This docking model also indicate that the identified agonists bind to the orthosteric site of DRV1, sharing overlapping binding sites with RvD1 (Fig. 6C–E). The core of methoxyphenylpiperidine group of pMPPF is inserted into the bottom of the pocket and formed extensive hydrophobic and aromatic stacking interactions (Fig. 6C). On the other side, the pyridine benzamide group is pointed out to the flexible loop region forming close interactions with ECL2, and pMPPI forms a halogen bond with Y101 (Fig. 6F). The agonists C1A and C2A showed similar binding conformations in the hydrophobic pocket with extensive π-π stacking interactions and H-bonding interaction with Y117, which could contribute to the binding affinity and activity of these agonists to the DRV1 receptor (Fig. 6D). Compared to pMPPI, C1A binds deeper in the pocket and forms a H-bond with F221 (Fig. 6G). Therefore, computational docking of the ligands to the predicted structural model of DRV1 suggested overlapping and distinct interactions for each ligand in the binding pockets of this receptor.
Figure 6. The predicted binding model of DRV1 with selected agonists.

(A-D) Ligands including RvD1(blue), BDA-RvD1 (green), MPPF (cyan), NCGC00120943 (magenta)/NCGC00135472 (violet) were docked into the structural model of GPR32. (E) Overlay of predicted binding model of GPR32 with selected agonists RvD1 (blue), BDA-RvD1 (green), pMPPF (cyan), C1A (magenta), C2A (violet). The transmembrane domain of GPR32 is shown in ribbon (grey). Ligand and key residues surrounding the hydrophobic pocket (in surface) are shown in sticks. (F-G) Comparison of binding interactions of pMPPI (cyan; F) and C1A (magenta; G). The loop ECL2 is shown in red color (only showed in part in C1A binding model for clarity). Key residues interacting with ligand are shown in sticks and H-bond and halogen-bond interactions with ligand are shown in dashes.
Discussion
Resolvin D1 exhibits potent pro-resolving actions via activating human resolvin D1 receptor (DRV1/GPR32) (Krishnamoorthy et al., 2010). By screening a large collection of small molecules, we identified several chemotypes that activate human recombinant DRV1, namely C1A, C2A, pMPPF and pMPPI (Figs. 1 and 2). Each activated recombinant DRV1 monitored by β-arrestin activity and cAMP release (Fig. 2). These chemotypes also elicited rapid impedance changes with recombinant DRV1 (Fig. 3), and stimulated pro-resolving function with human macrophages, i.e. phagocytosis, in a DRV1-dependent manner (Figs. 4&5). Docking these agonists into the structural model of DRV1 indicated that they shared with RvD1 the hydrophobic binding pocket formed by seven-transmembrane segments (Fig. 6). These scaffolds and pharmacophores could be used to develop RvD1 mimetics that require fewer steps in organic synthesis.
Among these compounds, some of the agonists display functional responses that are different from RvD1 responses, e.g. the change in impedance and intracellular cAMP. It is likely that each agonist activates DRV1 with different affinities, potencies and/or potentially evokes different second messengers following DRV1 activation. Results in Figure 2 demonstrated that pMPPF, C1A, C1B, C2A and C2B each dose-dependently reduced forskolin-stimulated cAMP release, suggesting that they activated DRV1 coupling to Gαi protein. On the other hand, pMPPI increased forskolin-stimulated cAMP release suggesting that it might activate Gαs coupling. These results are in line with those obtained from ECIS (Fig. 3), which monitors rapid impedance changes upon ligand activation of GPCR. This cell-based and radiolabel-free system can quantitatively determine direct GPCR activation and distinguish Gαs, Gαi and Gαq coupling. Gαi-coupling and Gαs-coupling give rapid increase and decrease of impedance, respectively, while Gαq-coupling shows a transient dip followed by increase of impedance (Peters and Scott, 2009). In CHO cells expressing DRV1, RvD1 and pMPPI reduced impedance, suggesting that they initiated DRV1 coupling with a Gαs protein. The other ligands pMPPF, C1A, C1B, C2A and C2B increased impedance, suggesting that DRV1 couples with a Gαi protein upon ligand activation. The predicted binding models indicated that pMPPI could bind to an allosteric site by forming a halogen bond with Y101 and extensive hydrophobic interactions with ECL2. These interactions with pMPPI may trigger conformational changes in DRV1 that are different from those triggered by other agonists, thus leading to different G protein coupling and downstream intracellular signals (Fig. 6F&6G). GPCRs are known to couple with different G proteins initiated by different ligands and then transmit separate or overlapped downstream signals (Simon et al., 1991; Cooray et al., 2013). In this context, in human macrophages RvD1 increases cAMP, a second messenger for Gαs protein (Lee and Surh, 2013). This is in line with our earlier and present results obtained with CHO cells expressing recombinant human DRV1 (Krishnamoorthy et al., 2012 and Fig. 3). It remains of interest whether the signals evoked by the chemotypes (i.e. cAMP, β-arrestin, impedance) with recombinant DRV1 are also operative in primary human cells. With human macrophages, all these compounds enhanced phagocytosis (Fig. 4). Thus, it is possible that these compounds activate other signaling components relevant to macrophage phagocytosis, which are not fully functional in CHO cells.
Of interest, both pMPPF and p-MPPI are known serotonin receptor 5-HT(1A) antagonists (Kung et al., 1996; Kung et al., 1995). We tested whether 5-HT has direct impact on macrophage phagocytosis at the same concentration range as RvD1, and found that 5-HT did not significantly increase macrophage phagocytosis of STZ (0.1 nM-10 nM; Fig. 4E). In addition, with CHO cells, the pMPPF- and pMPPI-evoked impedance changes were DRV1-dependent since they did not elicit impedance change with CHOWT cells (Fig. 3B and C). Therefore, it is not likely that the intracellular signals evoked by either pMPPF or pMPPI with CHO-DRV1 cells and their functions with macrophages were mediated via 5-HT receptors. Here, pMPPF and pMPPI activated recombinant DRV1 as agonists and stimulated phagocytosis in a DRV1-dependent manner (Figs. 3 and 5), suggesting that these synthetic serotonin mimetics could have previously unappreciated pro-resolving actions via activating DRV1 in vivo. Regarding the specificities of chemotypes 1 and 2, C1A displayed activity in only 5 out of the 636 bioassays deposited in Pubchem, and none of these 5 bioassays were related to GPCR targets. These targets include glucose-6-phosphate dehydrogenase-6-phosphogluconolactonase and microtubule associated protein tau. (https://pubchem.ncbi.nlm.nih.gov/compound/16017615#section=Biological-Test-Results). C2B was evaluated in 89 bioassays with no reported activity (https://pubchem.ncbi.nlm.nih.gov/compound/7080311#section=Top).
Lipid mediators such as eicosanoids exhibit their potent bioactions via activating cell surface GPCR superfamily (Shimizu, 2009; Woodward et al., 2011). Eicosanoids, including prostaglandins, thromboxane and leukotrienes, are short-lived autocoids that are generated in response to stimuli, act locally and then rapidly metabolized. Thus, metabolically stable analogs were developed to either mimic or block their actions via interacting with their respective GPCRs (Shimizu, 2009; Woodward et al., 2011). For example, Iloprost, a synthetic agonist of prostacyclin (PGI) receptor, is currently used for treatment of pulmonary arterial hypertension (Woodward et al., 2011). Along these lines, lipoxin (LX) A4 stable analogs were designed to resist enzymatic inactivation and prolong their bioactions via activating LXA4 receptor (ALX) (Serhan et al., 1995; Sun et al., 2007). DHA-derived SPM are also biosynthesized locally in response to inflammatory stimuli and subject to rapid further metabolism via enzymatic inactivation. RvD1 is locally converted to 8-oxo-RvD1 and 17-oxo-RvD1 by 15-hydroxyprostaglandin dehydrogenase/eicosanoid oxidoreductase. The 8-oxo-RvD1 remained biologically active, while 17-oxo-RvD1 is inactive, demonstrating stereo-selectivity of RvD1’s bioactions (Sun et al., 2009). Metabolically stable analogs that resist enzymatic inactivation were designed and synthesized, e.g. benzo-diacetylenic 17R-RvD1 methyl ester (BDA-RvD1). This analog mimetic shares defining pro-resolving actions with RvD1, including reducing PMN infiltration in vivo, stimulating phagocytosis of STZ and activating recombinant DRV1 (Orr et al., 2015).
In addition to the affinities, potencies and selectivities of mimetics, the binding kinetics is also essential for GPCR drug development (Strasser et al., 2017). Here, each agonist enhanced phagocytosis with different kinetics as monitored using real-time imaging microscope (Fig. 5). It is possibly that each agonist exhibited different kineticsin ligand binding and/or ligand residence time with DRV1. In these context, each agonist might interact with different amino acid residues in the binding sites (e.g. pMPPI forms a halogen bond with Y101 and C1A binds deeper in the pocket forming H-bond with F221; see Fig 6), which could potentially impact ligand residence time, thus leading to their different kinetics in stimulating phagocytosis. Recently, the crystal structures of cannabinoid receptor 1 (CB1) and leukotriene B4 receptor 1 (BLT1) were determined (Hori et al., 2018; Hua et al., 2017). The plasticity of the binding pocket (formed by seven-transmembrane segments) of CB1 appears to be a common feature among certain class A GPCR (Hua et al., 2017). BLT1, ALX (LXA4 receptor), DRV1/GPR32, DRV2/GPR18 (RvD2 receptor) and ERV1/ChemR23 (RvE1 receptor) (reviewed in ref. Chiang and Serhan, 2017) also belong to the class A GPCR. In the present report, RvD1, BDA-RvD1 and the identified chemotypes appeared to locate in a hydrophobic binding pocket of DRV1 formed by seven-transmembrane segments (Fig. 6). Together, the crystal structures and binding models of GPCRs will provide structural insights into ligand-receptor interactions and facilitate the design and development of mimetics of natural ligands that are receptor-specific.
The chemotypes identified herein provide molecular templates for RvD1 mimetics to stimulate pro-resolving functions via activating human DRV1. These specific mimetics could provide useful tools for understanding ligand-receptor interactions for DRV1. These may offer potentially more cost-effective synthesis to facilitate clinical development of therapeutics that stimulate resolution pathways in vivo. The pro-resolution Rv mimetics could be useful to treat inflammation-related diseases and infection as well as protect from collateral tissue injury, reduce cancer burden and resolve pain. Thus, our present results warrant further investigations for clinical development.
Significance
Resolvin D1 (RvD1) exhibits potent pro-resolving functions in the control of resolution of inflammation via activating human resolvin D1 receptor (DRV1/GPR32). Total organic synthesis of RvD1 requires multiple steps that slow its potential in clinical development. Here, in order to facilitate clinical development of small molecules that could act as RvD1 mimetics, we employed a high throughput system to screen several libraries of small molecules (>48,000 compounds) for their activities in stimulating DRV1. From this, we report the identification of unique chemotypes that activate the human recombinant DRV1 and promote macrophage phagocytosis in a DRV1-dependent manner. In addition, docking these chemotypes into the structural model of DRV1 indicated that they shared the binding pocket with RvD1. Thus, the structures identified herein function as RvD1 mimetics that require fewer steps in organic synthesis. Together, they offer molecular templates to facilitate clinical development of therapeutics that target human DRV1 and its role in the resolution of inflammation.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to the Lead Contact, Prof. Charles N. Serhan (cserhan@bwh.harvard.edu)
Experimental Model and Subject Details
Human primary cells
Healthy human peripheral blood mononuclear cells (PBMCs) were isolated from deidentified leukopacks purchased from Children’s Hospital Blood Bank (Boston, MA), Experiments were conducted in accordance with a protocol approved by Partners Human Research Committee (Partners Human Research Committee Protocol No. 1999-P-001297). Since the leukopacks were obtained from the Children’s Hospital Blood Bank and donors were deidentified to ensure patient confidentiality. While sex and age of the donors are known, the information was not available to the authors because they were deidentified and protected. Also, the information on their involvement in previous experiments or if they were drug or test naïve are not available to the authors. Monocytes were isolated and macrophages (MΦ) were differentiated by culturing freshly isolated monocytes in RPMI media supplemented with 10% FBS and recombinant human GM-CSF for 7 days as in (Krishnamoorthy et al., 2010). CHO GPR32 β-arrestin cell line was purchased from DiscoverX, Chinese Hamster Ovary (CHO-K1) cells were purchased from ATCC (ATCC® CCL-61™; Source: female Chinese hamster). Human acute monocyte leukemia (THP-1) cells were purchased from ATCC (ATCC® TIB-202™; Source: one-year old male). All cells were cultured at 37°C at 5 % CO2
Method Details
PathHunter® β-Arrestin Assay
The CHO GPR32 β-arrestin cell line (DiscoverX, #93–0321C2A) was thawed and plated in thawing media containing F12 HAM (Invitrogen, #11765–054), 10% FBS (Invitrogen, #10082), 1x Glutamine, and 1x Pen/Strep onto the 175-mm flask for 24 hours. Afterwards, thawing media was replaced with growth media, which was the thawing media with 800μg/mL Geneticin (Invitrogen, #10131) and 300μg/mL Hygromycin B (Invitrogen, #10687). For the assay, the cells were harvested at ~80% confluent stage with Cell Dissociation buffer (Gibco, #13151), and seeded at 2,500 cells/ 3μL/well in assay media (PathHunter® Cell Plating Reagent, DiscoverX, #93–0563R1B) using MultiDrop Combi dispenser (Thermo Scientific, Logan, UT) into the wells of white solid-bottom tissue culture-treated 1536-well plates (Greiner # 789173), and allowed to attach overnight at 37°C, 95% humidity, 5% CO 2. Next, 23nL/well of compound solutions in DMSO were added with a Pin Tool (Kalypsis, San Diego, CA) to the assay plates. As the basal control (EC0), 23 nL/well of DMSO was used on each plate. The cells were incubated in the presence of compounds or vehicle for 90 minutes at 37°C, 5% CO2. Subsequently, 1.5μL/well of PathHunter® Detection Reagent prepared per the manufacturer’s instructions was added to the assay plates with a BioRAPTR FRD™ dispenser (Beckman Coulter, San Francisco, CA), and cells incubated for 1 hour at ambient temperature prior to the measurement of the luminescent signal with a ViewLux® uHTS Microplate Imager (Perkin Elmer, Waltham, MA).
Cytotoxicity test of selected compounds
THP-1 (ATCC® TIB-202™) cells were seeded into wells of a 1536-well white solid-bottom tissue culture-treated plate (Greiner #789173) at 5,000 cells/4μL/well using a Multidrop Combi dispenser. The wells with media only were used as “no cells” negative control. Right after dispensing the cells, compounds were pin-transferred into the assay plate, 23nL/well as 11-point 1:3 intraplate serial dilution, in the range of [46μM – 0.8nM] as final assay concentration. 23nL/well of DMSO was added as vehicle control. The plates were incubated at 37°C with 5% CO2 under 95% humidity for 24 hours. Prior to adding CellTiter-Glo® Reagent (Promega, #G7570), the plate and reagent were equilibrated to room temperature, then 5μL/well of the reagent was added to the assay plate. The plate was quickly centrifuged (~15 sec) and shaken for 1 minute to induce cell lysis followed by 15-minute incubation at ambient temperature. The signal was read with ViewLux at luminescent settings with 1 second exposure. Relative luminescent units (RLU) for each well were normalized to the median RLUs from DMSO control wells as 100% viability.
cAMP HTRF® assay
PathHunter™ CHO-K1 GPR32 β-arrestin cells were cultured and harvested for the assay using the same protocol as described above. The cells were seeded into wells of a 1536-well plate (Greiner #789173) at 1,800 cells/3μL/well and incubated for 20 hours at 37°C, 5% CO2. Next, cells were stimulated with 1 L/well forskolin (8 μM final concentration, EC80) with Ro 20–1724 (100 μM final concentration) in PBS. Next, 23 nL/well of compounds were added with a Pin Tool transfer as 1:3 serial dilutions in [57μM −1nM] final concentration range. For controls, one column was pinned with DMSO only (EC0) and another with 57 μM forskolin (EC100). The cells were treated with compounds for 30 min at 37°C, 5% CO2, after which the cAMP HiRange HTRF® assay (Cisbio # 62AM6PEJ) was performed. The HTRF reconstituted reagents (cAMP d2 and anti-cAMP Cryptate) were prepared in distinct vials by dilution 1:20 in HTRF conjugate and lysis buffer (supplied with the assay kit) and dispensed onto the assay wells separately, 1μL/well each. The plates were incubated for 30 min at ambient temperature, and then the signal was read with an EnVision plate reader (Perkin Elmer) with filters for excitation as 340nm, emission as 615nm and 665nm. For normalization for any effects in the donor channel, all data were calculated as a ratio 665/615*10,000.
Electrical Cell-Substrate Impedance Sensing System (ECIS)
Ligand-receptor interactions were determined by measuring impedance across CHO cell monolayers using an Electrical Cell-Substrate Impedance Sensing System (ECIS; Applied Biophysics, Troy, NY) (Peters and Scott, 2009), and carried out essentially as in Krishnamoorthy et al. (Krishnamoorthy et al., 2010). Briefly, GPR32 or mock transfected CHO cells were plated at 0.1×106 per well of an 8-well ECIS array (8W10E+). Test compounds were added to the chambers in serum-free medium, and real-time impedance changes were monitored (0–10 min, 37 °C).
Human leukocyte isolation and macrophage differentiation
Human peripheral blood mononuclear cells from deidentified healthy volunteers were purchased from Children’s Hospital Blood Bank (Boston, MA) (Partners Human Research Committee Protocol No. 1999-P-001297). Monocytes were obtained by density-gradient Ficoll-Histopaque isolation (Krishnamoorthy et al., 2010). Macrophages (MΦ) were differentiated by culturing freshly isolated monocytes in RPMI media supplemented with 10% FBS and recombinant human GM-CSF (10 ng/ml, R&D Systems) for 7 days. GM-CSF is a well-established hemopoietic growth factors that differentiate monocyte to macrophages (Lacey et al., 2012) and is routinely used to differentiate peripheral blood monocytes to macrophages. To access the actions of RvD1 and chemotypes on M2 polarization, each compound was incubated with GMCSF-differentiated macrophages at 10 nM for 24 hours. Cells were then harvested and stained with anti-CD163 Ab (Biolegend). CD163 levels were then determined using flow cytometry.
Macrophage phagocytosis of serum-treated zymosan (STZ)
MΦ were plated onto 96-well plates (50,000 cells/well in PBS++), and phagocytosis carried out 24 hours later. Compounds (0.1 pM-10 nM) was incubated with MΦ for 15 min at 37°C, followed by incubation with FITC-label ed zymosan particles at 10:1 ratio (zymosan: MΦ) for 40 min at 37 °C. Plates were gently washed, e xtracellular fluorescence quenched by trypan blue, and phagocytosis determined by measuring total fluorescence (Ex 493/Em 535 nm) using SpectraMax M3 plate reader (Molecular Devices, San Jose, CA).
GPR32 transfection and macrophage phagocytosis of E. coli
For overexpression of GPR32, MΦ (5 × 106 cells in a 10-cm petri dish) were transfected with a mock vector or with expression vector for human GPR32 (5 μg; Origene, Rockville, MD) using Jet-Pei transfection reagent following manufacturer’s instructions (Polyplus-transfection SA, Illkirch, France). For real-time imaging, MΦ were plated onto 8-well chamber slides (50,000 cells/well in PBS++) 48h after transfection. Imaging was then carried out 24h after re-plating. Chamber slides were kept in a Stage Top Incubation system for microscopes equipped with a built-in digital gas mixer and temperature regulator (TOKAI HIT model INUF-K14). Test compounds were added to MΦ (1 nM, 15 min) followed by BacLight Green-labeled E. coli (2.5 × 106 CFU). Images were then acquired every 10 min for 3h (37°C) with Keyence BZ-9000 (BIOREVO) inverted fluorescence phase-contrast microscope (40X objective) equipped with a monochrome/color switching camera using BZ-II Viewer software (Keyence, Itasca, IL, USA). Green fluorescence intensity was quantified using BZ-II Analyzer.
Homology molecular and docking
Homology model of human GPR32 was constructed using the MOE program (Molecular Operating Environment, version 2010.10; Chemical Computing Group Inc.; Quebec, Canada). Sequence of GPR32 was retrieved from the UniProtKB database (access code O75388). Multiple sequence alignment with the agonist-bound (active) structures of GPCRs showed that the transmembrane domain of GPR32 (414–764) shares highest sequence identity with the human chemokine receptor type 1 (25% in TM and 12% in loops). Therefore, the crystal structure of CXCR1 in the active form (PDB entry 2LNL; https://www.rcsb.org/structure/2LNL) was used as a template for homology model generation. Docking of small molecule agonists to the structural model of GPR32 was performed using MOE dock. The flexible docking protocol was used and the docking pose was refined using PBSA score.
Quantification and Statistical analysis
Results are represented as mean or mean± SEM. Statistical analysis was performed using non-parametric Mann Whitney test for 2 group comparisons and oneway analysis of variance (ANOVA) Kruskal-Wallis test for multiple group comparisons with post-hoc analysis using Dunn’s Multiple Comparison test (GraphPad Prism, La Jolla, CA). P<0.05 was considered to be statistically significant.
Data and Software Availability
Data will be available upon request to the Lead contact.
Highlights:
Identification of chemotypes activating human resolvin D1 receptor DRV1 using HTS.
These chemotypes elicited rapid impedance changes in DRV1 overexpressing cells.
Macrophage phagocytosis was increased by the chemotypes in a DRV1-depedent manner.
Ligand-receptor docking model illustrated specific interactions.
Acknowledgements
The authors thank Mary Small for assistance with manuscript preparation. The authors have no additional financial interests. This research in the Serhan laboratory was supported in part by NIH grant R01GM38765 (C.N.S.). Work at NCATS was supported by the NIH Intramural Research Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: The authors declare no competing interests.
References
- Bang S, Yoo S, Yang TJ, Cho H, Kim YG, and Hwang SW (2010). Resolvin D1 attenuates activation of sensory transient receptor potential channels leading to multiple anti-nociception. Br J Pharmacol 161, 707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang N, and Serhan CN (2017). Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors. Mol Aspects Med 58, 114–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiurchiu V, Leuti A, Dalli J, Jacobsson A, Battistini L, Maccarrone M, and Serhan CN (2016). Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci Transl Med 8, 353ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clària J, Dalli J, Yacoubian S, Gao F, and Serhan CN (2012). Resolvin D1 and resolvin D2 govern local inflammatory tone in obese fat. J Immunol 189, 2597–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooray SN, Gobbetti T, Montero-Melendez T, McArthur S, Thompson D, Clark AJ, Flower RJ, and Perretti M (2013). Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc Natl Acad Sci U S A. 110 18232–18237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotran RS, Kumar V, and Collins T, eds. (1999). Robbins Pathologic Basis of Disease, 6th edn (Philadelphia: W.B. Saunders Co.). [Google Scholar]
- Cox R Jr., Phillips O, Fukumoto J, Fukumoto I, Parthasarathy PT, Arias S, Cho Y, Lockey RF, and Kolliputi N (2015). Enhanced Resolution of Hyperoxic Acute Lung Injury as a result of Aspirin Triggered Resolvin D1 Treatment. Am J Respir Cell Mol Biol 53, 422–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croasdell A, Lacy SH, Thatcher TH, Sime PJ, and Phipps RP (2016). Resolvin D1 Dampens Pulmonary Inflammation and Promotes Clearance of Nontypeable Haemophilus influenzae. J Immunol 196, 2742–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickmeier O, Seki H, Haworth O, Hilberath JN, Gao F, Uddin M, Croze RH, Carlo T, Pfeffer MA, and Levy BD (2013). Aspirin-triggered resolvin D1 reduces mucosal inflammation and promotes resolution in a murine model of acute lung injury. Mucosal Immunol 6, 256–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredman G, Ozcan L, Spolitu S, Hellmann J, Spite M, Backs J, and Tabas I (2014). Resolvin D1 limits 5-lipoxygenase nuclear localization and leukotriene B4 synthesis by inhibiting a calcium-activated kinase pathway. Proc Natl Acad Sci U S A 111, 14530–14535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert K, Bernier J, Godbout R, and Rousseau G (2014). Resolvin D1, a metabolite of omega-3 polyunsaturated fatty acid, decreases post-myocardial infarct depression. Mar Drugs 12, 5396–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori T, Okuno T, Hirata K, Yamashita K, Kawano Y, Yamamoto M, Hato M, Nakamura M, Shimizu T, Yokomizo T, et al. (2018). Na(+)-mimicking ligands stabilize the inactive state of leukotriene B4 receptor BLT1. Nat Chem Biol 14, 262–269. [DOI] [PubMed] [Google Scholar]
- Hsiao HM, Thatcher TH, Levy EP, Fulton RA, Owens KM, Phipps RP, and Sime PJ (2014). Resolvin D1 Attenuates Polyinosinic-Polycytidylic Acid-Induced Inflammatory Signaling in Human Airway Epithelial Cells via TAK1. J Immunol 193, 4980–4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua T, Vemuri K, Nikas SP, Laprairie RB, Wu Y, Qu L, Pu M, Korde A, Jiang S, Ho JH, et al. (2017). Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 547, 468–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Wang C-F, Serhan CN, and Strichartz G (2011). Enduring prevention and transient reduction of post-operative pain by intrathecal resolvin D1. Pain 152, 557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, and Austin CP (2006). Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc Natl Acad Sci U S A 103, 11473–11478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Ramon S, Thatcher TH, Woeller CF, Sime PJ, and Phipps RP (2016). Specialized proresolving mediators (SPMs) inhibit human B-cell IgE production. Eur J Immunol 46, 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy S, Recchiuti A, Chiang N, Fredman G, and Serhan CN (2012). Resolvin D1 receptor stereoselectivity and regulation of inflammation and pro-resolving microRNAs. Am J Pathol 180, 2018–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy S, Recchiuti A, Chiang N, Yacoubian S, Lee C-H, Yang R, Petasis NA, and Serhan CN (2010). Resolvin D1 binds human phagocytes with evidence for pro-resolving receptors. Proc Natl Acad Sci USA 107, 1660–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung HF, Stevenson DA, Zhuang ZP, Kung MP, Frederick D, and Hurt SD (1996). New 5-HT1A receptor antagonist: [3H]p-MPPF. Synapse 23, 344–346. [DOI] [PubMed] [Google Scholar]
- Kung MP, Frederick D, Mu M, Zhuang ZP, and Kung HF (1995). 4-(2’-Methoxyphenyl)-1-[2’-(n-2”-pyridinyl)-p-iodobenzamido]-ethyl-piperazine ([125I]p-MPPI) as a new selective radioligand of serotonin-1A sites in rat brain: in vitro binding and autoradiographic studies. J Pharmacol Exp Ther 272, 429–437. [PubMed] [Google Scholar]
- Lacey DC, Achuthan A, Fleetwood AJ, Dinh H, Roiniotis J, Scholz GM, Chang MW, Beckman SK, Cook AD, and Hamilton JA (2012). Defining GM-CSF- and macrophage-CSF-dependent macrophage responses by in vitro models. J Immunol 188, 5752–5765. [DOI] [PubMed] [Google Scholar]
- Lee HN, and Surh YJ (2013). Resolvin D1-mediated NOX2 inactivation rescues macrophages undertaking efferocytosis from oxidative stress-induced apoptosis. Biochem Pharmacol 86, 759–769. [DOI] [PubMed] [Google Scholar]
- Leigh NJ, Nelson JW, Mellas RE, Aguirre A, and Baker OJ (2014). Expression of resolvin D1 biosynthetic pathways in salivary epithelium. J Dent Res 93, 300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Hodges RR, Jiao J, Carozza RB, Shatos MA, Chiang N, Serhan CN, and Dartt DA (2013). Resolvin D1 and aspirin-triggered resolvin D1 regulate histamine-stimulated conjunctival goblet cell secretion. Mucosal Immunol 6, 1119–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C (2012). Fresh approaches to anti-infective therapies. Sci Transl Med 4, 140sr142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norling LV, and Perretti M (2013). The role of omega-3 derived resolvins in arthritis. Curr Opin Pharmacol 13, 476–481. [DOI] [PubMed] [Google Scholar]
- Odusanwo O, Chinthamani S, McCall A, Duffey ME, and Baker OJ (2012). Resolvin D1 prevents TNF-alpha-mediated disruption of salivary epithelial formation. Am J Physiol Cell Physiol 302, C1331–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr SK, Colas RA, Dalli J, Chiang N, and Serhan CN (2015). Proresolving actions of a new resolvin D1 analog mimetic qualifies as an immunoresolvent. Am J Physiol Lung Cell Mol Physiol 308, L904–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perretti M, Leroy X, Bland EJ, and Montero-Melendez T (2015). Resolution Pharmacology: Opportunities for Therapeutic Innovation in Inflammation. Trends Pharmacol Sci 36, 737–755. [DOI] [PubMed] [Google Scholar]
- Peters MF, and Scott CW (2009). Evaluating cellular impedance assays for detection of GPCR pleiotropic signaling and functional selectivity. J Biomol Screen 14, 246–255. [DOI] [PubMed] [Google Scholar]
- Prieto P, Rosales-Mendoza CE, Terron V, Toledano V, Cuadrado A, Lopez-Collazo E, Bannenberg G, Martin-Sanz P, Fernandez-Velasco M, and Bosca L (2015). Activation of autophagy in macrophages by pro-resolving lipid mediators. Autophagy 11, 1729–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. (2017). Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 377, 1119–1131. [DOI] [PubMed] [Google Scholar]
- Rogerio AP, Haworth O, Croze R, Oh SF, Uddin M, Carlo T, Pfeffer MA, Priluck R, Serhan CN, and Levy BD (2012). Resolvin D1 and aspirin-triggered resolvin D1 promote resolution of allergic airways responses. J Immunol 189, 1983–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi S, Di Filippo C, Gesualdo C, Potenza N, Russo A, Trotta MC, Zippo MV, Maisto R, Ferraraccio F, Simonelli F, et al. (2015). Protection from Endotoxic Uveitis by Intravitreal Resolvin D1: Involvement of Lymphocytes, miRNAs, Ubiquitin-Proteasome, and M1/M2 Macrophages. Mediators Inflamm 2015, 149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN (2014). Pro-resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN (2017). Treating inflammation and infection in the 21st century: new hints from decoding resolution mediators and mechanisms. FASEB J 31, 1273–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, and Moussignac R-L (2002). Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter pro-inflammation signals. J Exp Med 196, 1025–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Maddox JF, Petasis NA, Akritopoulou-Zanze I, Papayianni A, Brady HR, Colgan SP, and Madara JL (1995). Design of lipoxin A4 stable analogs that block transmigration and adhesion of human neutrophils. Biochemistry 34, 14609–14615. [DOI] [PubMed] [Google Scholar]
- Settimio R, Clara DF, Franca F, Francesca S, and Michele D (2012). Resolvin D1 reduces the immunoinflammatory response of the rat eye following uveitis. Mediators Inflamm 2012, 318621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T (2009). Lipid mediators in health and disease: enzymes and receptors as therapeutic targets for the regulation of immunity and inflammation. Annu Rev Pharmacol Toxicol 49, 123–150. [DOI] [PubMed] [Google Scholar]
- Simon MI, Strathmann MP, and Gautam N (1991). Diversity of G proteins in signal transduction. Science 252, 802–808. [DOI] [PubMed] [Google Scholar]
- Strasser A, Wittmann HJ, and Seifert R (2017). Binding Kinetics and Pathways of Ligands to GPCRs. Trends Pharmacol Sci 38, 717–732. [DOI] [PubMed] [Google Scholar]
- Sulciner ML, Serhan CN, Gilligan MM, Mudge DK, Chang J, Gartung A, Lehner KA, Bielenberg DR, Schmidt B, Dalli J, et al. (2018). Resolvins suppress tumor growth and enhance cancer therapy. J Exp Med 215, 115–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y-P, Oh SF, Uddin J, Yang R, Gotlinger K, Campbell E, Colgan SP, Petasis NA, and Serhan CN (2007). Resolvin D1 and its aspirin-triggered 17R epimer: stereochemical assignments, anti-inflammatory properties and enzymatic inactivation. J Biol Chem 282, 9323–9334. [DOI] [PubMed] [Google Scholar]
- Sun Y-P, Tjonahen E, Keledjian R, Zhu M, Yang R, Recchiuti A, Pillai PS, Petasis NA, and Serhan CN (2009). Anti-inflammatory and pro-resolving properties of benzo-lipoxin A4 analogs. Prostaglandins Leukot Essent Fatty Acids 81, 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Liu Y, Yan C, Petasis NA, Serhan CN, and Gao H (2014). Protective actions of aspirin-triggered (17R) resolvin D1 and its analogue, 17R-hydroxy-19-parafluorophenoxy-resolvin D1 methyl ester, in C5a-dependent IgG immune complex-induced inflammation and lung injury. J Immunol 193, 3769–3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Zhang MJ, Hellmann J, Kosuri M, Bhatnagar A, and Spite M (2013). Proresolution Therapy for the Treatment of Delayed Healing of Diabetic Wounds. Diabetes 62, 618–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titos E, Rius B, González-Périz A, López-Vicario C, Morán-Salvador E, Martínez-Clemente M, Arroyo V, and Clària J (2011). Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. J Immunol 187, 5408–5418. [DOI] [PubMed] [Google Scholar]
- Van Dyke TE, and Serhan CN (2006). A novel approach to resolving inflammation. Sci Am Oral and Whole Body Health, 42–45. [Google Scholar]
- Winkler JW, Libreros S, De La Rosa X, Sansbury BE, Norris PC, Chiang N, Fichtner D, Keyes GS, Wourms N, Spite M, et al. (2018). Structural insights into Resolvin D4 actions and further metabolites via a new total organic synthesis and validation. J Leukoc Biol doi: 10.1002/JLB.3MI0617-254R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward DF, Jones RL, and Narumiya S (2011). International Union of Basic and Clinical Pharmacology. LXXXIII: classification of prostanoid receptors, updating 15 years of progress. Pharmacol Rev 63, 471–538. [DOI] [PubMed] [Google Scholar]
- Zhang X, Qu X, Sun YB, Caruana G, Bertram JF, Nikolic-Paterson DJ, and Li J (2013). Resolvin D1 protects podocytes in adriamycin-induced nephropathy through modulation of 14-3-3beta acetylation. PLoS One 8, e67471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be available upon request to the Lead contact.