

SUMMARY
There is a considerable need to identify those individuals with prostate cancer who have indolent disease. We propose that genes that control adult stem cell homeostasis in organs with slow turnover, such as the prostate, control cancer fate. One such gene, KLF4, overexpressed in murine prostate stem cells, regulates their homeostasis, blocks malignant transformation, and controls the self-renewal of tumor-initiating cells. KLF4 loss induces the molecular features of aggressive cancer and converts PIN lesions to invasive sarcomatoid carcinomas; its re-expression in vivo reverses this process. Bioinformatic analysis links these changes to human cancer. KLF4 and its downstream targets make up a gene signature that identifies indolent tumors and predicts recurrence-free survival. This approach may improve prognosis and identify therapeutic targets for advanced cancer.
Graphical Abstract
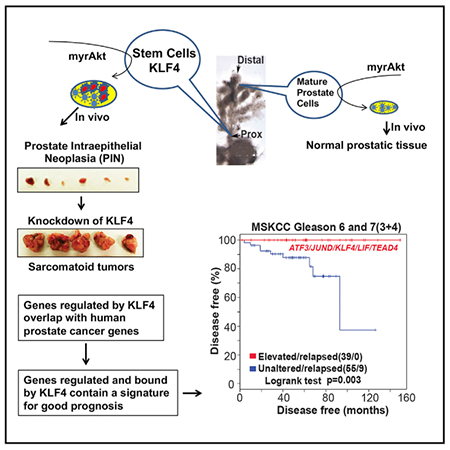
In Brief
Available criteria for segregating prostate cancer patients into those requiring therapeutic intervention and those who can be followed are inadequate. Xiong et al. show that KLF4 and its downstream targets make up a gene signature that identifies indolent tumors. This approach may improve prognosis and identify therapeutic targets for advanced cancer.
INTRODUCTION
Prostate cancer affects ~160,000 men annually in the US and causes ~27,000 deaths (Pentyala et al., 2016), with the majority of patients presenting with an intermediate Gleason score (Gleason 7). Segregation of this group for appropriate treatments is notoriously difficult and in need of more reliable criteria. Our goal is 2-fold: to find biologically relevant molecular signatures that will identify those individuals whose tumors are indolent and who can be spared from unnecessary treatment, while also finding molecules that are responsible for malignant progression for use as potential targets of therapy for aggressive cancers. While most reports propose that the expression of “stem cell genes” in tumor cells correlates with more aggressive cancers (Merlos-Suárez et al., 2011; Smith et al., 2015), the study of genes conferring indolence is limited (Irshad et al., 2013).
The proximal region of prostatic ducts is highly enriched in adult prostate stem cells (APSCs) (Burger et al., 2005, 2009; Tsujimura et al., 2002; Xin et al., 2005). Relevant to our present hypothesis is the fact that although these APSCs are endowed with high proliferative potential, they exist in a predominantly quiescent state. However, when they are challenged, these APSCs are able to fully reconstitute prostatic tissue in vivo (Goto et al., 2006). We postulated that among the genes that are responsible for this behavior of APSCs may be some that if expressed in prostate cancer could restrict its growth and progression. To identify these genes, we compared the molecular signatures of APSC-containing populations to those of mature prostatic cells and found that KLF4 was one of the genes that was overexpressed in APSCs (Blum et al., 2009). KLF4 can either activate or repress transcription, and depending on the cellular context, it can function either as an oncogene or a tumor suppressor (Rowland et al., 2005; Rowland and Peeper, 2006; Shi et al., 2014; Tetreault et al., 2013). In different types of human tumors, the decreased expression of KLF4 has been shown (Rowland and Peeper, 2006; Shi et al., 2014; Tetreault et al., 2013) to have diverse effects, a challenge that is further complicated by evidence showing opposite functions of KLF4 in the same tumor type (Wei et al., 2016; Yan et al., 2016).
Although KLF4 has been shown to serve as a tumor suppressor in prostate cancer (Liu et al., 2012; Wang et al., 2010), its function during prostate cancer initiation and progression has not been elucidated. This deficiency prompted our comprehensive analysis of the ability of KLF4 to antagonize the transformation of APSC by activated Akt and its effect on fully transformed APSCs. Our results show that KLF4 inhibits the proliferation of normal stem cells and the process of their malignant transformation and that in an activated Akt in vivo model of prostate cancer, its expression attenuates tumor progression and reverses aggressive tumors to a more indolent state. Most important, elevated expression of KLF4 appears to be inextricably linked to indolent human prostate cancer, and its presence identifies, with very high specificity, those patients with a long relapse-free survival. Through RNA sequencing (RNA-seq) and chromatin immunoprecipitation sequencing (ChIP-seq), combined with bioinformatic analysis, we identified KLF4-regulated networks of genes that improved the sensitivity of this stratification. This approach also revealed potential targets for the development of future therapies. This work provides the first proof that a uniquely expressed and functionally relevant gene expressed in APSCs controls the fate of prostate tumors.
RESULTS
Klf4 Inhibits Proliferation of Adult Mouse Prostate Stem Cells
We show that the elevated Klf4 mRNA found in Sca-1high APSCs (Blum et al., 2009) is also present in the proximal stem cell niche in situ (Figure 1A). This region, previously characterized by us and others as highly enriched in APSCs (Burger et al., 2005; Tsujimura et al., 2002; Xin et al., 2005), contains 8.5- and 11-fold more Klf4 mRNA and protein, respectively, than the distal regions of ducts that contain more differentiated cells (Figures 1B–1D). Isolated proximal cells (Burger et al., 2005) were infected with small hairpin Klf4 (shKlf4) (Klf4 mRNA reduced by 90%) (Figure S1A) or scrambled shRNA lentivirus (shControl), and cells (3 × 104) were inoculated in collagen gels (Tsujimura et al., 2002) and ducts counted after 7 days. Reduction of KLF4 yielded 1.6-fold more ducts (p < 0.03; Figure 1E), which were also 1.8-fold larger (p < 0.006; Figure S1B), suggesting that Klf4 affects normal stem cell homeostasis. We also inoculated 5 × 104 shKlf4 or shControl cells in combination with 2.5 × 105 urogenital sinus mesenchymal cells (UGM) subrenal capsule (sub-RC) (Burger et al., 2005; Zong et al., 2015). After 8 weeks in vivo, shKlf4 cells produced 1.4-fold more tissue than shControl cells (p < 0.03; Figure 1F), with 2.3-fold more Ki67+ (proliferating) cells (p < 0.00001; Figures 1G and S1C), indicating that Klf4 impedes the proliferation of stem cells in vitro and in vivo. Moreover, the shKlf4 sub-RC tissue had more CK5+ basal cells, increasing the basal to luminal ratio 2.5-fold (p < 0.01; Figures 1G and S1D). Since basal CK5+ cells are more primitive than luminal cells and may contain the stem cell population (Goldstein et al., 2008; Lawson et al., 2007), these results show that in addition to affecting the proliferation of stem cells in vivo, Klf4 also controls their homeostasis.
Figure 1. Klf4 Inhibits Proliferation of Adult Mouse Prostate Stem Cells.
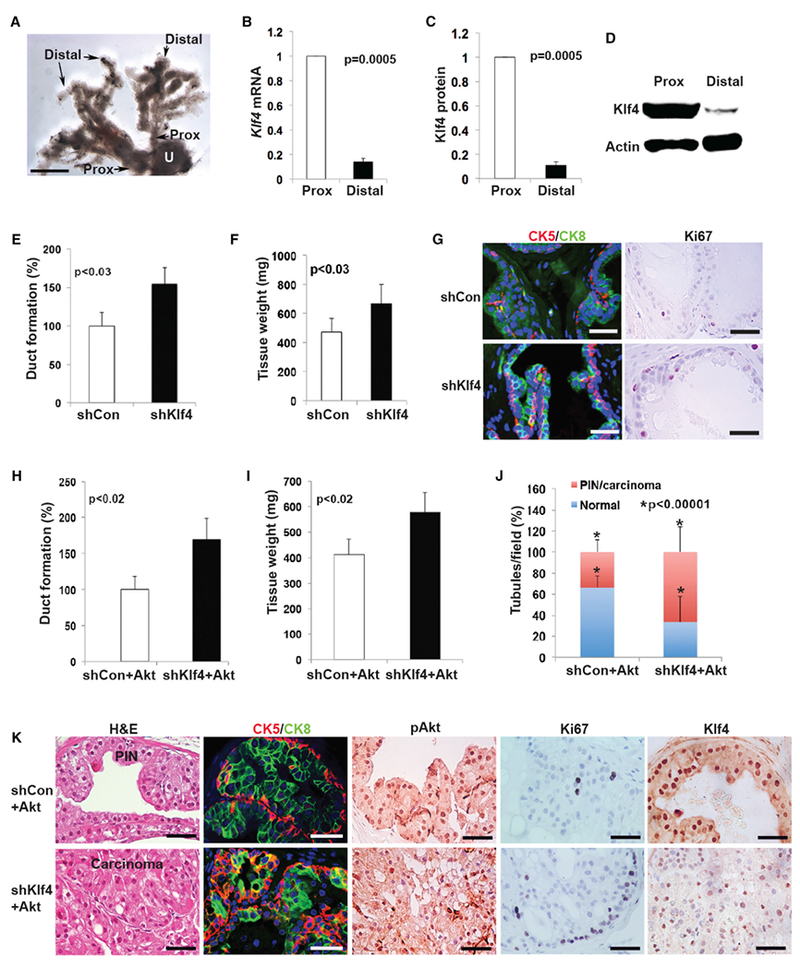
(A) Morphology of the prostatic ductal system of the dorsal prostate showing proximal (Prox) and distal regions. U, urethra. Scale bar, 0.5 mm.
(B and C) The relative expression levels of Klf4 (B) mRNA and (C) protein in the proximal stem cell niche and the distal ductal region. Experiments were repeated 2 times. Data represent means ± SDs. p = 0.0005.
(D) Western blot indicating Klf4 expression in proximal and distal ducts.
(E) Duct-forming ability of proximal cells (3 × 104) in collagen gels infected with shCon or shKlf4 lentivirus. Experiments were repeated at least 3 times. Data represent means ± SDs. p < 0.03.
(F) Proximal cells were infected with shCon or shKlf4 lentivirus and inoculated (5 × 104) sub-RC with UGM (2 × 105). Experiments were repeated at least 3 times. Data represent means ± SDs. p < 0.03.
(G) Prostatic tissue generated by shCon or shKlf4 cells was examined for CK5, CK8, and Ki67 expression. Scale bars, 35 μm.
(H) Proximal cells were infected with Akt lentiviruses combined with control (shCon + Akt) or shKlf4 (shKlf4 + Akt) lentiviruses and seeded (3 × 104) in collagen gels. Experiments were repeated at least 3 times. Data represent means ± SDs. p < 0.02.
(I) Proximal cells were infected with shCon + Akt or shKlf4 + Akt lentiviruses and inoculated sub-RC (5 × 104) with UGM (2 × 105). Experiments were repeated 3 times. Data represent means ± SDs. p < 0.02.
(J) PIN and carcinoma-containing tubules in shCon + Akt or shKlf4 + Akt grafts were determined in all fields in 3 sections from each graft (3 grafts per condition). p < 0.00001.
(K) Grafts from shCon + Akt and shKlf4 + Akt cells were examined by H&E and for expression of CK5, CK8, pAkt, Ki67, and Klf4. Scale bars, 30 μm.
Klf4 Inhibits Malignant Transformation of Stem Cells
We performed fluorescence-activated cell sorting (FACS) of the Sca-1high (APSCs), the Sca-1med/low (transit-amplifying cells), and the Sca-1neg (differentiated cells) (Burger et al., 2005) and infected each subset with a GFP-lentiviral-myrAkt1 or a control construct (Xin et al., 2005) (myrAkt1 was selected as phosphatase and tensin homolog [PTEN], an upstream inhibitor of the Akt pathway, and a frequently inactivated tumor suppressor gene in prostate cancer [Ayala et al., 2004]). All 3 cell subsets had similar levels of phosphorylated Akt (pAkt), but only the Sca-1high cells formed tumors (Figures S1E and S1F), demonstrating that the most primitive cells are the preferential targets of transformation by Akt. APSC-containing primary proximal cells were then infected with a GFP-lentiviral-myrAkt1 construct together with a lentivirus expressing either shKlf4 or a control virus, and their duct-forming ability in collagen gels was tested. After 7 days, the shKlf4-Akt cells formed 70% more ducts than shControl + Akt cells (p < 0.02; Figure 1H) and the shKlf4 + Akt ducts were 3.0-fold larger (p = 0.003; Figure S2A), showing that diminishing the level of Klf4 increases the size of the stem or progenitor cell compartment both in APSCs and in APSCs transformed by activated Akt. We then co-infected primary proximal prostate cells with Akt together with either shKlf4 or an shControl lentivirus and implanted these cells (5 × 104) mixed with 2.5 × 105 UGM sub-RC. The shKlf4-Akt grafts were larger than shControl-Akt (p < 0.02; Figure 1I) and contained large areas of adenocarcinoma, while the smaller shControl-Akt grafts contained mainly prostatic intraepithelial neoplasia (PIN) lesions (Figures 1J, 1K, and S2B) (both had similar pAkt levels [Figure 1K]). The shKlf4-Akt grafts had 14% ± 5% Ki67+ cells compared to only 6% ± 4% in the grafts with intact Klf4 (p < 0.00001; Figure S2C). The basal to luminal cell ratio was 2.5-fold greater in the shKlf4-Akt tumors (p < 0.005; Figures 1K and S2D). Overall, these results indicate that Klf4 regulates the homeostasis of normal prostate stem cells and impedes their malignant transformation by activated Akt.
Klf4 Impedes Tumor Progression
We isolated a prostate stem cell line from an Sca-1high/Akt-derived sub-RC tumor (Figures S1E and S1F). Like the primary parental stem cells, these cells express high levels of Sca-1 and α6 integrin (CD49f) (Figure 2A) and are predominantly basal (~90% CK5+ basal, ~9% CK5+CK8+, and ~1% CK8+ luminal cells). We determined the effect of Klf4 on tumor-initiating cells (TICs) in this cell line by prostatosphere formation in Matrigel and found that the number of prostatospheres formed by shKlf4-Akt-transformed cells was 3.3-fold greater than the number formed by Akt cells with intact Klf4 (p = 0.001; Figure 2B), indicating that the depletion of Klf4 increases self-renewal of TICs. Moreover, the shKlf4-Akt prostatospheres survived at least 6 consecutive passages in Matrigel, while the shControl-Akt cells lost this ability on the second passage (Figure 2C). The shKlf4-Akt prostatospheres initially appeared as large, tight colonies that, with passage, acquired fibroblast-like-looking cells that invaded and spread into the surrounding Matrigel, leading to a reduction in prostatosphere size and a decline in the numbers of spheres with passage (Figure 2C) (Bhagirath et al., 2016). Overexpression of Klf4 in the Akt-transformed cell line reduced the ability of these cells to form prostatospheres (p < 0.006; Figure 2D); the spheres were much smaller and could not be passaged at all. This suggests that increasing the level of Klf4 may significantly reduce TIC content in tumors.
Figure 2. Klf4 Inhibits Proliferation and Self-Renewal of TICs.
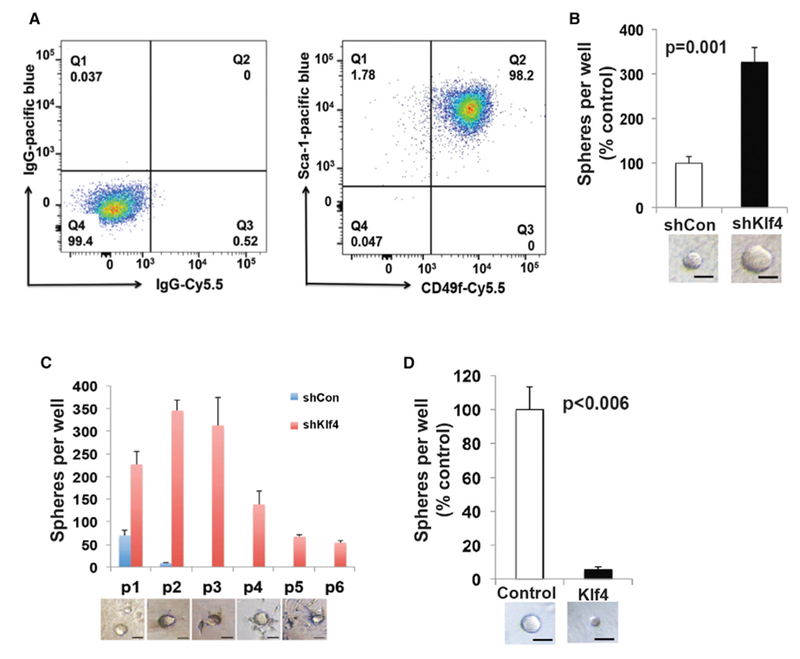
(A) Expression of Sca-1 and CD49f by the Akt cell line (right); cells with control antibodies (left).
(B) Sphere-forming ability of the Akt cell line (3,000 cells/well) in Matrigel after infection with control (shCon) or shKlf4 lentivirus. Scale bars, 30 μm. Experiments were repeated 3 times. Data represent means ± SDs. p = 0.001.
(C) Spheres from shKlf4 and shCon Akt cell line were dissociated, and equal numbers of cells (2,000) were passaged 6 times in triplicate in Matrigel. The sphere number diminished with passage as cells became more migratory and infiltrated the Matrigel (bottom). Scale bars, 30 μm.
(D) Sphere-forming ability of the Akt cell line (3,000 cells/well) in Matrigel after infection with control or Klf4-expressing constructs. Scale bars, 30 μm. Experiments were repeated 3 times. Data represent means ± SDs. p < 0.006.
We next inoculated 2 × 106 cells of the shKlf4-Akt- and shControl-Akt-transformed stem cell lines sub-RC. While the shControl-Akt cells produced barely detectable PIN lesions, the shKlf4-Akt cells produced very large tumors (1.6 ± 0.5 g; Figures 3A and 3B) with sarcomatoid morphology, signs of invasion (Figures 3B and S3A), and high rates of proliferation (69% ± 6% of cells positive for Ki67) (Figures 3C and S3B). The small shControl-Akt PIN lesions were only 15% ± 6% (p < 0.0001) Ki67+ (Figures 3C and S3B). While the PIN lesions had membrane-localized E-cadherin, the sarcomatoid tumors were highly positive for vimentin, with only occasional and nuclear E-cadherin (Figure 3C), a sign of invasiveness and poor prognosis (Chetty et al., 2008). The sarcomatoid morphology and expression of Slug (Figure 3C) suggested that these tumors had undergone epithelial-mesenchymal transition (EMT). A similar conversion to the spindle-shape morphology was observed in the shKlf4 Akt-transformed prostate stem cell line in culture after Klf4 knockdown. The cells lost their epithelial markers such as E-cadherin, ZO-1, CK5 (basal), and CK8 (luminal) while gaining the mesenchymal marker vimentin and expressing proteins associated with EMT, such as Slug, Snail, and Zeb1 (Figures 3D and 3E). This indicates that the loss of Klf4 results in a marked EMT response, confirming published findings (Liu et al., 2012). We conclude that the endogenously expressed Klf4 limits the number of TICs and prevents EMT, two potential mechanisms that control indolence. To determine whether KLF4 had similar effects on EMT in a human stem cell line transformed by Akt, we simultaneously infected WPE cells (Tokar et al., 2010) with AKT and shKLF4 lentivirus and found decreased E-cadherin and increased vimentin (Figure S3C), an indication of EMT as observed in the murine Akt-transformed stem cell line. When KLF4 is re-expressed, E-cadherin is increased and vimentin is decreased, indicating that EMT is reversed (Figure S3C). This indicates that KLF4 also modulates EMT in a human prostate stem cell line.
Figure 3. Decreasing the Levels of Klf4 Induces EMT and Results in Invasive Sarcomatoid Tumors.
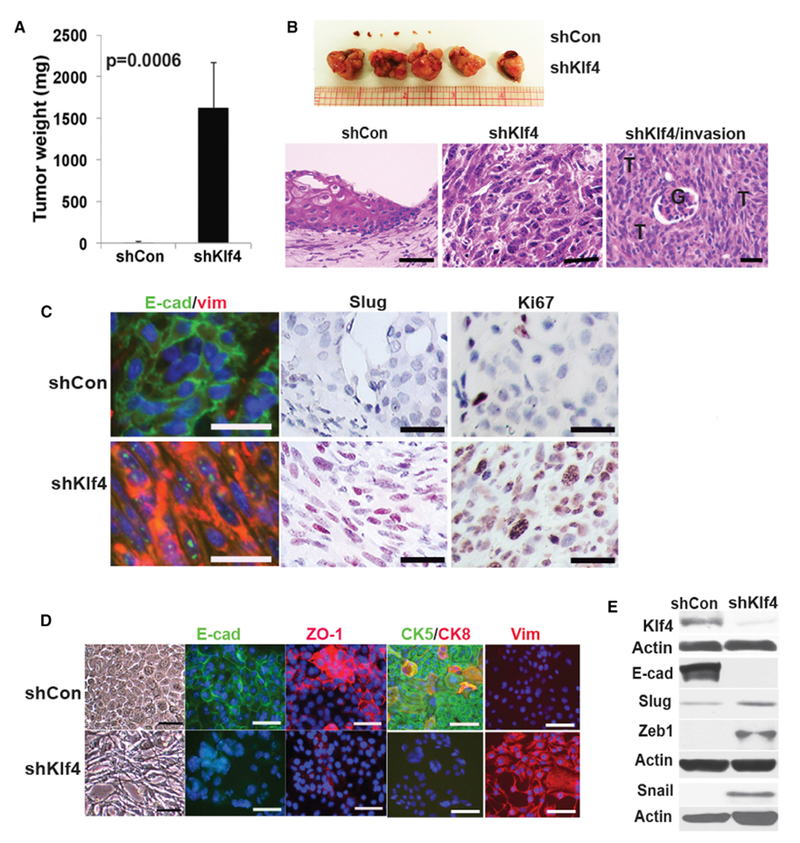
(A) Cells from the Akt cell line were infected with non-silencing control (shCon) orshKlf4 (shKlf4) lentiviruses and implanted (2 × 106) sub-RC. Experiments were repeated at least 3 times. Data represent means ± SDs. p = 0.0006.
(B) Gross tumors after implantation of shCon or shKlf4-infected Akt cell line after 4 weeks. T, tumor; G, glomerulus. Scale bars, 35 μm.
(C) Sub-RC grafts were examined for expression of E-cadherin (E-cad), vimentin (vim), Slug, and Ki67. Scale bars, 25 μm.
(D) Expression of E-cadherin (E-cad), ZO-1, CK5, CK8, and vimentin (vim) in the Akt-transformed cell line after infection with control or shKlf4-expressing lentiviruses. Scale bars, 50 μm.
(E) Western blot of Klf4, E-cadherin (E-cad), Slug, Zeb1, and Snail after infection of the Akt cell line with control or shKlf4-expressing lentiviruses.
See also Figure S3.
Induction of KLF4 in Established Tumors Reverses Their Aggressive Phenotype
We first tested the effects of KLF4 inducibility of the blue fluorescent protein (BFP)-tagged doxycycline (Dox) inducible Tet-on KLF4 system in vitro by adding Dox to the shKlf4-Akt stem cell line using a human KLF4 expressing lentivirus (shKlf4 + KLF4) or an empty vector control lentivirus (shKlf4 + EV). Induction of KLF4 after a 48-hr Dox treatment reduced cell proliferation by 2.3-fold (Figure S3D) and reduced the expression of the EMT markers Slug, Snail, vimentin, and Zeb1 compared with the expression in control shKlf4 + EV cells (Figure 4A).
Figure 4. Re-expression of KLF4 Reverses EMT and Inhibits Tumor Growth.
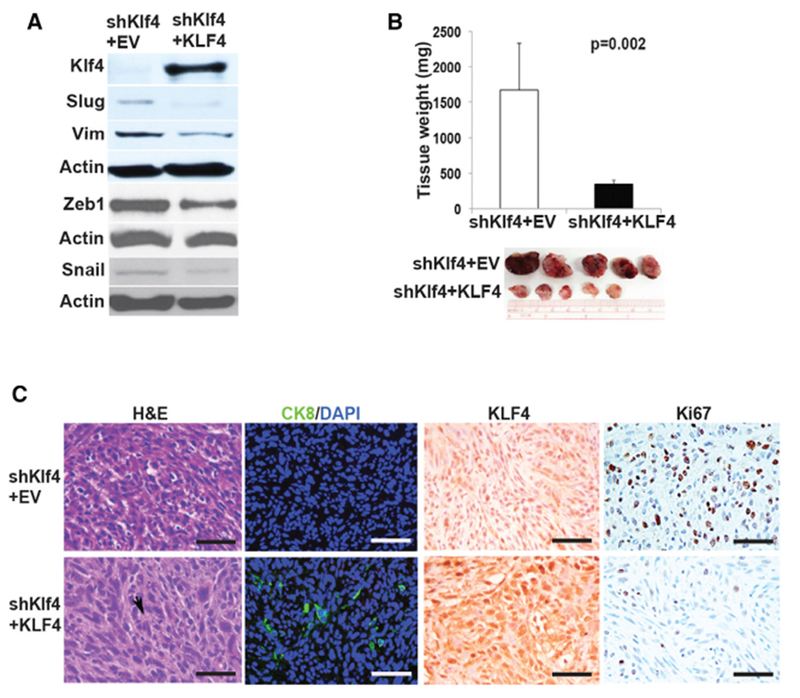
(A) Western blot of KLF4, Slug, vimentin (Vim), Zeb1, and Snail in control cells (shKlf4 + EV) and in cells rescued by expressing KLF4 (shKlf4 + KLF4).
(B) Sub-RC grafts of control (shKlf4 + EV) or KLF4-expressing cells (shKlf4 + KLF4) (1 × 105 cells). Experiments were repeated 3 times. Data represent means ± SDs. p = 0.002.
(C) Sub-RC grafts were examined by H&E (arrowhead indicates duct-like morphology) and for expression of CK8, KLF4, and Ki67. Scale bars, 45 μm.
See also Figure S3.
To determine whether KLF4 can rescue the aggressive sarcomatoid phenotype in vivo, we implanted 1 × 105 Dox inducible shKlf4 + KLF4 cells or control shKlf4 + EV cells sub-RC, maintained the mice without Dox until tumors were established (3 weeks), and administered Dox to all of the mice for 7 days. While Klf4 knockdown (shKlf4 + EV) cells produced large sarcomatoid tumors (1.6 ± 0.6 g), with 47% ± 15% Ki67+ cells (Figures 4B, 4C, and S3E), cells in which KLF4 was induced formed very small (0.3 ± 0.05 g, p = 0.002) tumors, with only 11% ± 7% of cells positive for Ki67 (p < 0.00001; Figures 4B, 4C, and S3E) and with markers of epithelioid differentiation (larger cells with increased cytoplasm and abortive gland formation) and de novo expression of CK8 (Figures 4C and S3F). This indicates that KLF4 can confer a more indolent phenotype even on rapidly growing sarcomatoid tumors.
RNA-Seq Analysis Reveals Novel Gene Signatures Regulated by KLF4
To identify the molecules and pathways responsible for the biological effects of KLF4, we performed RNA-seq analysis after knock down and re-expression of KLF4. We found that Klf4 knockdown significantly (false discovery rate [FDR] <0.01, fold change [FC] >2) altered the expression of 6,250 genes, KLF4 rescue altered 1,436 genes, and 850 genes were altered under both conditions (Figure 5A). As expected from the biological data, transcripts of EMT and of epithelial differentiation were appropriately regulated by the modulation of KLF4 levels (Figure 5B).
Figure 5. RNA-Seq Analysis Reveals Genes and Pathways Targeted by KLF4.
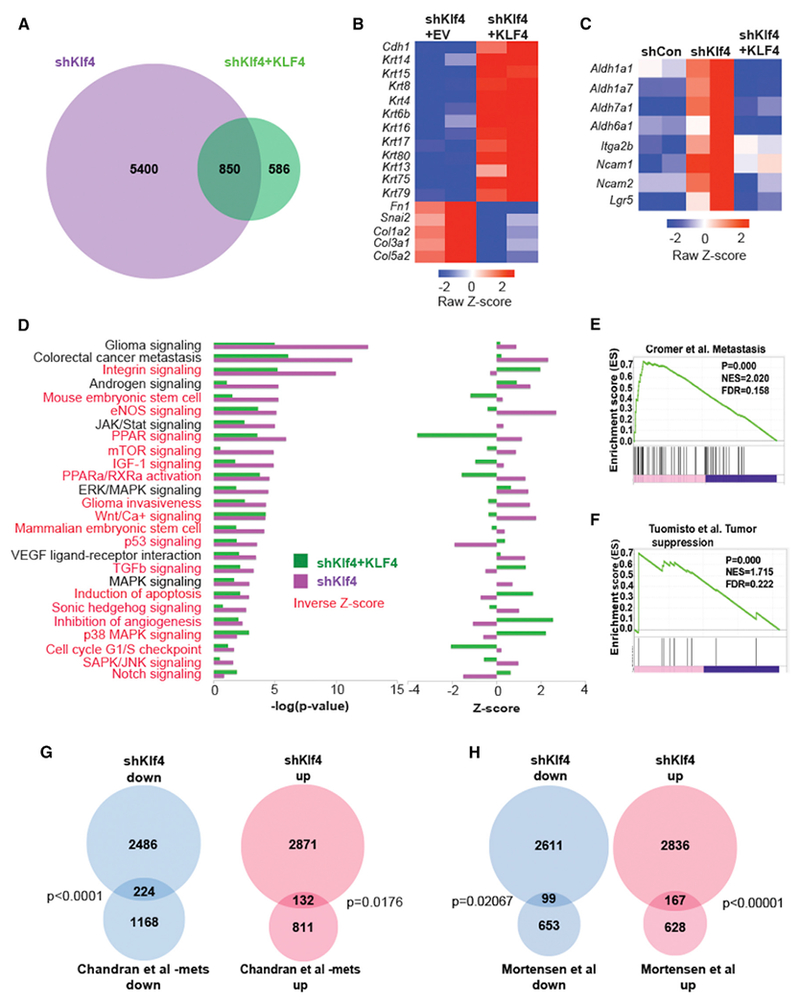
(A) Venn diagram of differentially expressed genes (FDR <0.01, FC >2) in the Akt-transformed cell line after Klf4 knockdown (shKlf4) versus KLF4 re-expression (shKlf4 + KLF4).
(B) Heatmap of EMT genes regulated by decreased (shKlf4 + empty vector [EV]) or increased KLF expression (shKlf4 + KLF4).
(C) Heatmap of stem cell genes regulated by Klf4 knockdown (shKlf4) and KLF4 re-expression.
(D) Ingenuity pathway analysis (canonical pathway) reveals pathways regulated by decreased (shKlf4) or increased (shKlf4 + KLF4) KLF4. Left: −log(p value), right: Z score. Pathways highlighted in red represent those that are inversely regulated by knockdown and re-expression of Klf4 (inverse Z score).
(E) GSEA reveals positive enrichment of genes in a tumor metastasis gene signature after decreased (shKlf4) Klf4 expression.
(F) GSEA reveals positive enrichment of genes in a tumor suppressor gene signature after increased (shKlf4 + KLF4) KLF4 expression.
(G) Overlap of genes after Klf4 knockdown in the Akt-transformed cell line with those expressed in a cohort of human metastatic prostate cancer.
(H) Overlap of genes after Klf4 knockdown in the Akt-transformed cell line with those expressed in a cohort of aggressive human prostate cancer.
Ingenuity pathway analysis (IPA) revealed a significant association between KLF4 and important cancer-related pathways; while downregulation was associated with stemness and with tumor metastasis and EMT pathways (Figures 5C and 5D), its re-expression (shKlf4 + KLF4) reversed these associations (Figure 5D). Reduced KLF4 activated signaling of the glioma, stemness, Wnt/Ca+ and Wnt target peroxisome proliferator-activated receptor (PPAR) pathways, the cell-cycle G1/S checkpoint pathway, the mammalian target of rapamycin (mTOR), insulin growth factor 1 (IGF-1), Sonic hedgehog, and the Jak/Stat, while decreasing the signaling of Notch, transforming growth factor beta (TGF-β), p38 mitogen-activated protein kinase (MAPK), apoptosis, and p53 pathways. With a few exceptions, re-expression of KLF4 reversed the effect of KLF4 knockdown (Figure 5D). The overlap between KLF4-induced pathways and published pathways crucial for tumorigenicity (Gonnissen et al., 2013; Jansson et al., 2005; Valdez et al., 2012) indicates that KLF4 and the pathways it controls represent an important hub of tumor control in both the murine model and human cancer.
A gene set enrichment analysis (GSEA) confirmed this conclusion by showing a correlation between decreased expression of KLF4 and a positive association with a tumor metastasis signature (Figure 5E), a cell migration signature (Figure S4A), and a signature of CDH1 loss (Figure S4B). The analysis also showed an association with a glucose-deprivation gene signature (Figure S4C). Tumor survival and proliferation in low levels of glucose and under hypoxic conditions has been linked with aggressive and drug-resistant tumors that are associated with increased recurrence and metastasis (Cui et al., 2007). In contrast, re-expression of KLF4, in addition to being positively correlated with stem cell quiescence (Figure S4D), is positively correlated with a signature of tumor suppression (Figure 5F) and signatures of depletion of anaplastic lymphoma kinase (ALK) (Figure S4E) and JAK2 (Figure S4F), providing additional support to our original hypothesis of a mechanistic link between stem cell quiescence and prostate cancer indolence.
The overlap between genes regulated by Klf4 in our murine prostate cancer model and those controlling important pathways of human cancer progression compelled further inquiry into the clinical relevance of the Klf4 regulated murine genes. These were compared with two published databases, one specific for metastatic human prostate cancer (Chandran et al., 2007) and one for relapsed aggressive primary tumors (Mortensen et al., 2015). Of the 2,710 genes significantly downregulated (p < 0.01) in the Klf4 knockdown murine model, 224 overlapped with 1,392 genes downregulated in Chandran’s metastatic tumors (Figure 5G; p < 0.0001). Of the 3,003 genes that are significantly upregulated (p < 0.01) after knock down of Klf4, 132 genes overlapped with the 943 upregulated in Chandran’s metastatic tumors (Figure 5G; p = 0.0176; Tables S1 and S2). A similar comparison to Mortenson’s aggressive tumors revealed 99 overlapping downregulated genes after Klf4 knockdown in the murine model (Figure 5H; p < 0.02067) and 167 overlapping genes upregulated after Klf4 knockdown (Figure 5H; p < 0.00001; Tables S3 and S4), strengthening the relevance of our murine model to human prostate cancer.
Klf4 and Genes Bound and Regulated by Klf4 Predict Prognosis of Primary Prostate Cancer
KLF4 is an important transcription factor, with context-dependent functions in normal tissue and tumor tissue (Rowland and Peeper, 2006; Shi et al., 2014). Our results in the murine prostate cancer model implicate Klf4 as a gatekeeper of the indolent state of tumors, and the results of the RNA-seq analysis suggest that this may be the case in human prostate cancer. The first indication that elevated levels of KLF4 may also identify indolent human prostate cancer came from an examination of a publicly available cohort of 131 primary prostate cancer patients (Taylor et al., 2010) that included data on biochemical recurrence-free survival. Of these, none of the 21 patients whose tumors had upregulated KLF4 mRNA levels (Z score >2) relapsed (p = 0.01) (Figure 6A), showing 100% specificity. Elevated levels of KLF4 could stratify patients with low Gleason score (3 + 3 and 3 + 4) tumors, generally considered as having good prognosis, into those who remained recurrence free and those whose cancer could recur despite the favorable pathological prognosis (Figure 6B). Using The Cancer Genome Atlas (TCGA) database (https://gdc.cancer.gov/) of 455 patients with primary prostate cancer, we also confirmed a previous observation (Wang et al., 2010) that an inverse correlation exists between levels of KLF4 and Gleason score; the lowest Gleason score (G6 and G7[3 + 4]) has the highest KLF4 expression, while the higher Gleason scores (G7[4 + 3], p = 0.047; G8 + 9 + 10, p = 0.006) had significantly less KLF4 than tumors with Gleason 6 scores (Figure 6C).
Figure 6. Prognostic Value of KLF4 and Genes Bound by KLF4.
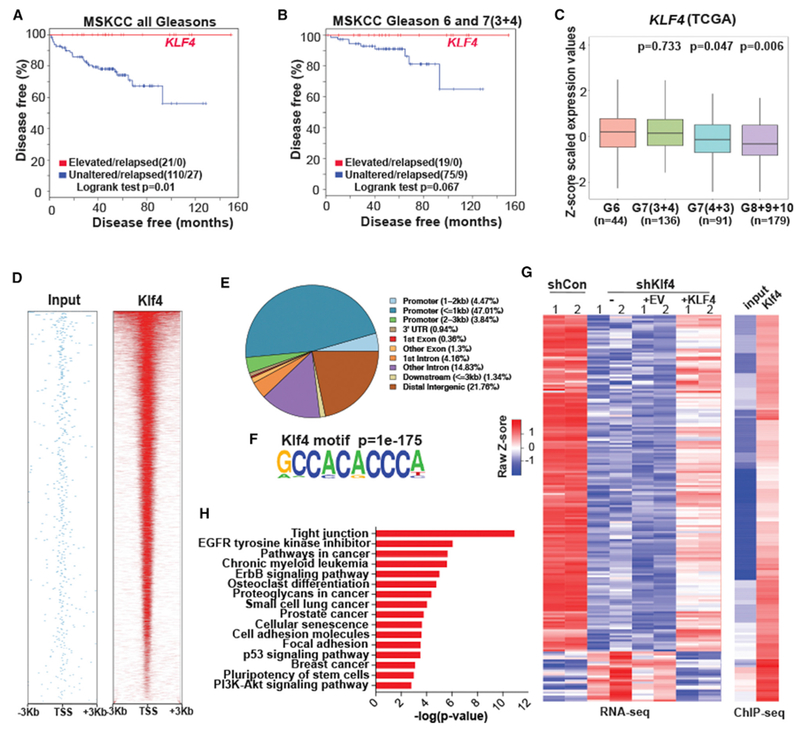
(A) Primary prostate cancer patients whose tumors express increased levels of KLF4 (21 of 131 cases [red line] have no biochemical recurrence for 10 years.
(B) Increased levels of KLF4 predict prolonged survival in primary prostate cancer patients whose tumors have low Gleason scores (3 + 3 and 3 + 4).
(C) Expression of KLF4 in 455 patients with primary prostate cancer from TCGA database as a function of Gleason scores.
(D) Heatmap indicating the chromatin immunoprecipitation sequencing (ChIP-seq) read density within ±3 kb of the TSS region.
(E) Pie chart illustrating the relative distribution of KLF4-bound sequences across the genome.
(F) Motif analyses discovered Klf4 consensus motifs at ChIP-seq peak centers (p = 1e–175).
(G) Heatmap representing 193 direct Klf4 target genes identified by combinatorial ChIP-seq and RNA-seq analyses of shControl, shKlf4, shKlf4 + EV, and shKlf4 + KLF4.
(H) Bar graph illustrating ranked KEGG pathway analysis of 1,528 genes bound by Klf4 and regulated by Klf4 knockdown.
The gene overlap between the murine model and human cancer gleaned from the RNA-seq analysis (Figure 5G and 5H), the unprecedented specificity of KLF4 to predict disease-free survival (Figures 6A and 6B), and the need to improve the sensitivity of prediction prompted the next series of experiments aimed at identifying the KLF4-regulated target genes that are specifically relevant to prostate cancer. To do this, we performed ChIP followed by high-throughput sequencing (ChIP-seq) of the myr-Akt-transformed prostate stem cell line expressing endogenous levels of KLF4. ChIP-seq data revealed a significant enrichment of Klf4 primarily at promoter elements in close proximity (±3 kb) to transcriptional start sites (TSSs) and in intergenic regions (Figures 6D and 6E). Motif analyses discovered a centrally distributed Klf4 consensus motif at the peak summits (p = 1 × 10−175) (Figure 6F), validating the specificity of the ChIP-seq experiments. The bona fide Klf4 gene targets were identified as genes that were also differentially expressed in our RNA-seq data. Of 6,250 genes differentially expressed (FDR <0.1 and FC >2) after Klf4 knockdown, ChIP-seq analysis indicated that 1,528 were bound by Klf4 at the promoter region; when KLF4 was re-expressed, 339 of 1,436 differentially expressed genes (FDR <0.1 and FC >2) were bound by KLF4 at the promoter region. Of the bound genes, 193 overlapped between the 1,528 genes bound by Klf4 after Klf4 knockdown and the 339 genes bound after KLF4 re-expression (Figure 6G; Table S5). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis identified 62 significantly enriched (adjusted p < 0.05) pathway categories (Figures 6H and S5; Table S6) among the 1,528 genes bound by KLF4 at the promoter region after knock down of Klf4. Among the most significantly changed categories were the pathways that regulate tight junctions. As these are regulators of EMT and tumor metastasis (Lamouille et al., 2014), this suggests an important role for Klf4 in these events, supporting the findings of our functional analyses (Figures 3 and 4). Furthermore, consistent with our in vitro and in vivo findings (Figures 3 and 4), we showed that Klf4-bound genes are significantly associated with cancer-related pathways and stem cell pluripotency (Figure 6H; Table S5), suggesting that Klf4 regulates cancer progression and stem cell homeostasis.
We assumed that genes representing the “indolence” signature were likely to be among those that were regulated and bound by KLF4 and that identifying them may result in the ability to stratify a larger proportion of prostate cancer patients with excellent prognosis than that obtained with KLF4 alone (Figures 6A and 6B). We determined that 168 of the 193 target genes that were bound by and inversely regulated by Klf4 were increased, while 25 genes were decreased by KLF4 expression in the Akt cell line (Figure 6G). Interrogation of these 168 target genes indicated that 21 were upregulated (Z score >2) in the Memorial Sloan Kettering Cancer Center (MSKCC) low Gleason score cohort (CBioportal, http://www.cbioportal.org/) (Taylor et al., 2010) and revealed 4 top-ranking signatures (Table S7), including ATF3, JUND, LIF, and TEAD4 (Figure 7A), which, together with KLF4, were able to identify with perfect specificity patients with low Gleason score tumors (GL6 and 7[3 + 4]), 94 patients) (Figure 7B, p = 0.003). These patients are presumed to have a low risk and represent those whose treatment decisions are the most difficult to make. In addition, in the MSKCC (all Gleasons) 131 patient cohort, this 5-gene signature was able to identify, with significantly increased sensitivity (Figure 7C; p = 0.0002), 49 of 104 patients (47% sensitivity and 95% specificity) with prolonged disease-free survival compared with that of KLF4 alone (Figure 6A; p = 0.01, 21 of 104 patients [20%]). These signatures were tested independently in a second prostate cancer cohort (Sboner et al., 2010) that was studied for at least 25 years before the widespread initiation of prostate specific antigen (PSA) screening to monitor disease. The cohort comprises patients with more advanced disease and contains very few non-relapsing patients. As expected in the second cohort (Sboner et al., 2010), while prediction by KLF4 alone does not reach statistical significance (results not shown), the 5-gene signature stratifies patients with low Gleason scores (GL6 and 7 [3 + 4]) (Figure 7D; p = 0.008) and predicts for increased survival of the total cohort (281 patients) (Figure 7E; p = 0.018). Validation of the prognostic power of the 5-gene signature in a second cohort in which sample collection was obtained by transurethral resection of the prostate using paraffin-embedded tissue rather than from prostatectomies (Taylor et al., 2010) indicates the robustness of the ability of this gene signature to stratify prognosis in prostate cancer. Although we only show a 5-gene signature for simplicity here, we extensively tested the performance of gene signatures comprising 2–7 genes with similar results (see Method Details and Figures S6, S7, and S8 for details). An examination of the 5-gene signature (KLF4/ATF3/JUND/LIF/TEAD4) in the TCGA database (https://gdc.cancer.gov/) revealed that the significance of the inverse correlation of this signature with the Gleason score is higher (G8 + 9 + 10, p = 0.0009; Figure 7F) than that of KLF4 alone (p = 0.006; Figure 6C). Multivariate Cox proportional hazard analysis (Grambsch and Therneau, 1994) indicates that our 5-gene signature, combined with the Gleason score, significantly outperforms the Gleason score alone (p = 0.0003). Similarly, when our 5-gene signature is combined with the D’Amico classification (D’Amico et al., 1998), which takes into account the Gleason score, PSA levels, and the clinical T stage, it significantly outperforms the D’Amico classification alone (p = 0.001) (Figure 7G). In addition, the omission of KLF4 from the signature revealed a similar stratification of patients with good prognosis in both cohorts (Figures S9A–S9D), a similar inverse correlation of this signature with high Gleason score in the TCGA database (Figure S9E), and a similar significant independent prognostic value of this gene signature (Figure S9F).
Figure 7. KLF4 and Genes Bound by KLF4 Predict Good Prognosis of Primary Human Prostate Cancer.
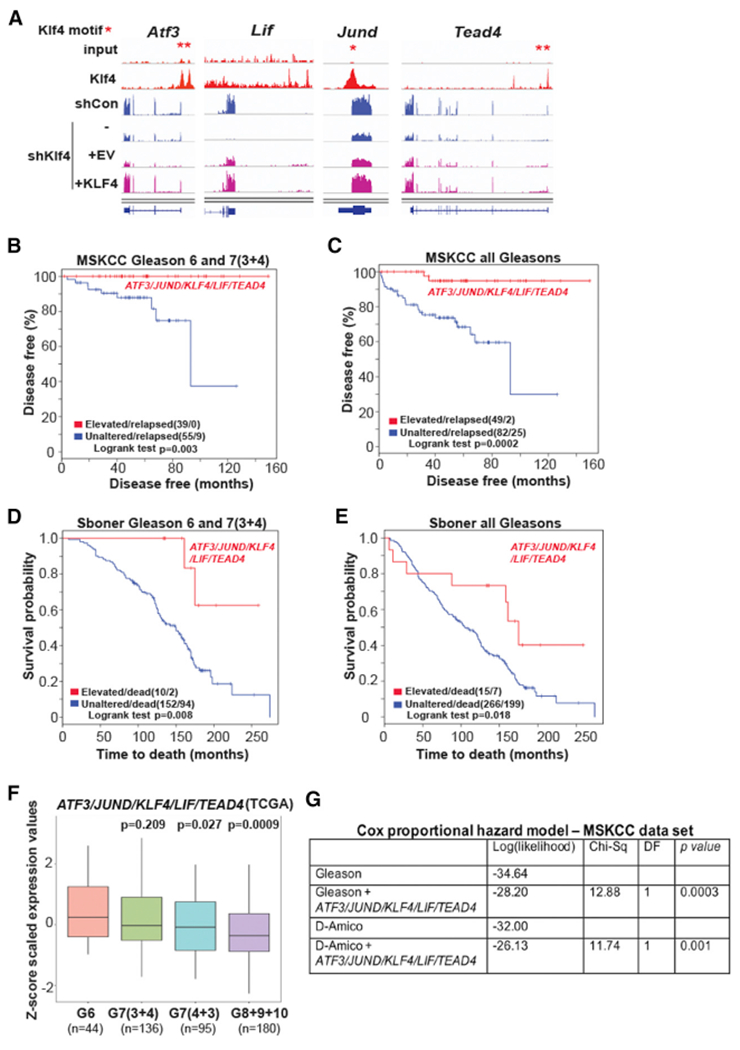
(A) ChIP-seq (red) and RNA-seq (blue and purple) tracks indicate Klf4 enrichment on gene regulatory elements and transcript changes of good prognosis genes (ATF3, JUND, LIF, TEAD4) after Klf4 knockdown and rescue, respectively. shCon and shKlf4 depicted at scale 0–2.63. shKlf4 + EV and shKlf4 + KLF4 depicted at scale 0–0.46. Asterisks depict Klf4 motif locations.
(B) Increased levels of ATF3, JUND, KLF4, LIF, and TEAD4 predict prolonged survival (red line) (p = 0.003) in prostate cancer patients whose tumors have low Gleason scores.
(C) Prostate cancer patients whose tumors express increased levels of ATF3, JUND, KLF4, LIF, and TEAD4 (red line) have a good prognosis (p = 0.0002).
(D) Increased levels of ATF3, JUND, KLF4, LIF, and TEAD4 predict prolonged survival (red line) (p = 0.008) in prostate cancer patients whose tumors have low Gleason scores.
(E) Prostate cancer patients whose tumors express increased levels of ATF3, JUND, KLF4, LIF, and TEAD4 (red line) (p = 0.018) have a good prognosis.
(F) Expression of ATF3, JUND, KLF4, LIF, and TEAD4 in 455 patients with prostate cancer from TCGA database as a function of Gleason scores.
(G) Cox proportional hazard model on Gleason 6 and 7(3 + 4) comparing the performance of ATF3, JUND, KLF4, LIF, and TEAD4 expression with Gleason score alone or with the D’Amico classification. DF, degree of freedom.
DISCUSSION
Results of this study support our hypothesis that APSCs, in a slowly turning-over tissue like the adult prostate, overexpress genes (Blum et al., 2009), some of which control malignant transformation and block tumor progression. We show that in patients with prostate cancer, the expression of KLF4, a significantly overexpressed gene in APSCs, identifies a subpopulation of patients with long-term survival. Incorporating KLF4 expression and the network of genes it controls, once confirmed independently in larger cohorts of patients, may provide a much needed test of prognosis that may spare the overtreatment of patients with prostate cancer. Our approach differs from most genetically based approaches because we focus on a search in tumors for the persistent expression of genes like KLF4 that control the normal homeostasis of prostate stem cells. Tumors expressing such genes are likely to be more indolent because they are still able to maintain a degree of normal tissue regulation. Although we do not yet know why only some prostate cancers continue to express significant amounts of KLF4, the results of our functional studies on the effects of KLF4 in APSCs and in our murine prostate cancer model derived by transforming these cells, as well as the substantial overlap between expression of genes bound by and regulated by KLF4 in this model and genes expressed in human prostate cancer, promise that this approach is likely to result in significant clinical benefit.
APSCs have the highest capacity to differentiate into prostatic ducts in 3-dimensional (3D) cultures, to repopulate prostatic tissue in vivo, and, when infected with constitutively active Akt (myrAkt), to undergo transformation despite containing a relatively high proportion of cells in the G0 stage of the cell cycle (Burger et al., 2005; Tsujimura et al., 2002; Wang et al., 2006; Xin et al., 2005) (Figures S1E and S1F). This suggests a tightly controlled homeostasis. KLF4 has been shown to function as either a tumor suppressor or an oncogene in different tumor types and sometimes even in the same tumor under different conditions (Maddipati and Katz, 2016; Rowland and Peeper, 2006). Ours is the first study that shows that KLF4, by regulating proliferation, possibly by maintaining the population of more slowly cycling APSCs (Figures 1 and S1), maintains tight control of stem cell tumor types’ homeostasis. Can genes and pathways responsible for this control be identified, is their expression and function maintained during malignant transformation, and can they be harnessed for the identification of more indolent forms of prostate cancer or to reverse cancer progression? That primitive prostate cells are the targets of transformation has been described (Lawson et al., 2010; Xin et al., 2005). This fact informed the design of experiments needed to answer the above questions. We developed a murine model of prostate cancer that consisted of a derivative of APSCs transformed by myrAkt. We now show that endogenous Klf4 attenuates the malignant transformation of APSCs by myrAkt and that the malignant potential of Akt-transformed TICs is curtailed (Figures 1, 3, and S2). Ours is the first report implicating Klf4 in this process. We also show that while knock down of Klf4 in Akt-transformed TICs (prostatospheres) increases their proportion (Figure 2) and enables their consecutive passage, TICs that express Klf4 have a very minimal capacity for consecutive passage, a finding that links Klf4 to the inhibition of their self-renewal. This is strong initial evidence in support of our hypothesis that the regulatory function of Klf4 persists in transformed cells and, in the case of TICs, blocks malignant progression. We confirmed these conclusions in in vivo experiments. We (Figure 1) and Xin et al. (2006) showed that myrAkt-transformed primary prostate cells inoculated under the renal capsule form primarily small PIN lesions, but it is our present work that implicates the endogenous Klf4 in keeping the transformed cells in an indolent (PIN) state (Figures 1, 3, and 4). Knock down of Klf4 converts PIN lesions into very large, aggressive, invasive sarcomatoid tumors with features of EMT (Figure 3). A similar induction of EMT and tumor-igenicity that accompanies a loss of Klf4 was noted in a transgenic prostate cancer mouse model (with deletion of TP53 and Pten) (Liu et al., 2012), suggesting that the observation applies to more than one murine model. Most important for the development of future therapies, we show that KLF4 can reverse the invasive phenotype of established sarcomatoid tumors (Figures 4 and S3C–S3E), providing a plausible approach to therapeutic intervention.
Furthering this hypothesis required showing that the profound effect of KLF4 on malignancy, presumably mediated through a network of specific, KLF4-regulated genes, has its counterpart in human cancer, and specifically in prostate cancer. Evidence showing that KLF4 expression is diminished in human prostate cancer metastasis (Liu et al., 2012; Wang et al., 2010) supports this notion. An RNA-seq analysis we performed revealed that knocking down KLF4 induced genes that are part of the tumor metastasis pathways, stem cell pluripotency markers, and, in confirmation of our experimental results, EMT (Figure 3E). Further analysis revealed a substantial overlap between the KLF4-regulated genes in our murine model and those genes that characterize malignancy in cohorts of human prostate cancer (Figures 5G and 5H). Moreover, we show that Klf4 regulates several pathways that are important for tumor progression, including Sonic hedgehog (Shh), Wnt/Ca+, and PPAR pathways (Figure 5D). The Hedgehog (Hh) pathway plays a crucial role in the development and progression of prostate cancer to more aggressive and even therapy-resistant disease states, and the inhibition of Hh signaling has the potential to reduce prostate cancer invasiveness and metastatic potential (Gonnissen et al., 2013). Similarly, Wnt/Ca+ activity, regulated by Klf4 in our model, is increased in prostate cancer cells (Wang et al., 2010), and PPAR activation has been shown to promote prostate cancer cell growth (Forootan et al., 2016). We found a substantial overlap between genes linked to the invasive sarcomatoid tumors induced by Klf4 knockdown (Figures 5G and 5H) and those genes that define metastatic and aggressive human prostate cancer (Chandran et al., 2007; Mortensen et al., 2015), opening a path to identifying new targets for therapy of aggressive cancers and giving support to the relevance of this model to prostate cancer.
Since the Gleason score is among the best available predictors of prostate cancer prognosis, we asked whether KLF4 expression level is correlated with the Gleason score. We examined a cohort of 455 prostate cancer patients for whom Gleason scores and the status of KLF4 expression were known (https://gdc.cancer.gov/). We show (Figure 6C) that KLF4 expression is inversely correlated with the Gleason score; cancers with the lowest Gleason score have a higher expression of KLF4, whereas the level decreases with high Gleason scores. This is important because, for the first time, it links high KLF4 expression with the most indolent prostate cancers in a very large cohort of patients with prostate cancer. A similar correlation was previously described in 3 small datasets from Oncomine (Wang et al., 2010).
A crucial test of our hypothesis was the examination of the prognostic power of KLF4 and a signature of genes we identified that are bound by and regulated by KLF4 in 2 cohorts of prostate cancer patients. In one of the cohorts (Taylor et al., 2010), KLF4 predicted very long recurrence-free survival with perfect specificity but low sensitivity (Figure 6A). That a single overexpressed gene can identify, with perfect specificity, patients with indolent prostate cancer is remarkable, and in spite of its limited sensitivity, it can spare many patients from unnecessary interventions. We sought to improve the sensitivity of the prediction by identifying the bona fide targets of KLF4 using bioinformatic analyses of RNA-seq and ChIP-seq data. Among the network of genes bound by and controlled by KLF4, we identified a signature of 4 additional genes that together with KLF4 (Figures 7A–7C) or separately (Figures S9A and S9B) predict survival with high sensitivity and specificity in the Taylor cohort (Taylor et al., 2010) that significantly increases the sensitivity compared with KLF4 alone. Results from a second cohort (Sboner et al., 2010) validate these findings (Figures 7D, 7E, S9C, and S9D). This signature is much better in predicting survival than KLF4 alone in individuals with cancer with low Gleason scores (Gleason 6 and Gleason 7[3 + 4]) (Figures 7B, 7D, S9A, and S9C); these are considered to be indolent tumors, and treatment decisions for these patients are the hardest to make. The genes identified in this signature (KLF4/ATF3/JUND/LIF/TEAD4) can act in a context-dependent manner to promote or inhibit tumorigenesis (Chen et al., 2012; Pfarr et al., 1994; Qi et al., 2016; Wang et al., 2018). It is evident from our results in human prostate cancer that these genes act to antagonize tumor progression.
Although we have not yet performed functional experiments to examine the possibility that our approach will provide targets for therapy for advanced prostate cancer, we have identified many genes that are inversely regulated by Klf4 and that overlap with genes expressed in human malignant tumors. Potential candidate genes could be Efna3 (a receptor kinase ligand) and Tpx2, which are overexpressed in aggressive prostate cancer (Isayeva et al., 2009; Wang et al., 2009) and predict poor prognosis in colon and gastric cancers (Tomii et al., 2017; Wei et al., 2013) in addition to BGN and ALDH7A1, whose overexpression is associated with a poor prognosis in prostate (Jacobsen et al., 2017; van den Hoogen et al., 2011).
In summary, we show that in cancers whose origins can be traced to organs with slowly turning-over stem cells, such as prostate cancer, identifying genes with specific functions that control the homeostasis of these cells, combined with in vivo experiments in appropriate models and an extensive bioinformatics analysis, yield results that may have profound significance for a patient’s prognosis. Although possibly more laborious than purely genetic approaches, this approach hones from the start on genes whose normal function can be ascertained and then tested during transformation and tumor progression. The advances in bioinformatics make examining the relevance of the findings to human cancer possible. Whether this approach will be applicable to other types of cancers remains to be determined. Our data strongly support the idea that elevated KLF4 can effectively maintain indolence in the murine model of prostate cancer and that the network of genes it controls, when overexpressed, can serve as a prognosticator of long-term survival in human prostate cancer. Although further studies are required to accurately identify all patients with indolent disease, the reassuring trend we present suggests that such a goal may be achievable.
STAR*METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact, Elaine Wilson (elaine.wilson.nyumc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Preparation of Cells, Cell Lines and Cell Culture
Proximal prostate cells were isolated from the proximal region of prostatic ducts of C57BL/6 mice as described (Salm et al., 2005). The proximal region of ventral and dorsolateral prostates of 6-wk-old C57BL/6 mice were removed under a dissecting microscope, minced finely and digested in 0.5% collagenase (Type II from Clostridium histolyticum, Sigma-Aldrich, MO) for 60 min at 37°C, followed by digestion in 0.25% trypsin with 2.21mM EDTA(Cellgro, VA) for 5 min at 37°C. Cells were resuspended in medium (a mixture of basal and luminal epithelial growth media (Salm et al., 2000a) with conditioned medium from a prostatic smooth muscle cell line (Salm et al., 2000b) (40:40:20, vol/vol/vol) (Salm et al., 2005)).
The Akt transformed prostate stem cell line was derived from a sub-RC tumor generated from Sca-1high stem cells isolated from the proximal region of ducts that had been infected with a myr-Akt1 lentiviral construct (obtained from Dr. ON Witte, UCLA, CA). Briefly, primary cells from the tumor were cultured in conditioned medium (see above). Once cultures were established cells were cloned by a limiting dilution assay and the Akt transformed prostate stem cell line was obtained from an epithelial colony derived from a single cell. Cells were cultured in medium as described previously (Salm et al., 2005). UGM cells were prepared as previously described (Goto et al., 2006; Zong et al., 2015). Briefly, UGM was isolated from the urogenital sinus of 18-day-old CD rat embryos after digestion with 0.5% collagenase at 4°C for 90 min. Single UGM cells were obtained by digesting the UGM tissue with trypsin for 10 min at 37°C. Cell proliferation rates were determined after seeding 10,000 cells in 24-well culture plates in triplicate wells. Cells were harvested and counted at 24 hour intervals. The human prostate stem cell line WPE - stem was purchased from ATCC. WPE cells were cultured in Keratinocyte-SFM medium with growth factors (GIBCO, NY) and 10% FBS.
Animal Models
C57BL/6 mice, athymic nude mice and CD rats were housed in a climate-controlled facility and all animal care and procedures were performed in compliance with New York University School of Medicine Institutional Review Board requirements.
METHOD DETAILS
Microdissection of dorsal prostate
Six-week-old C57BL/6 mice were sacrificed, and the urogenital tract was removed en bloc and transferred in Hanks’ balanced salt solution (Mediatech, VA). The dorsal prostate (DP) was removed and dissected under a dissecting microscope in the presence of 0.5% collagenase (1.3 units/mg; Sigma-Aldrich, MO).
Duct formation assay
The ability of cells to form ducts in collagen gels was determined by mixing 30,000 prostate cells with 200 μL collagen (160μl collagen, 20 μL 10× F12 and 20 μL reconstitution buffer) (Tsujimura et al., 2002) and seeding the mixture in triplicate in 8 well chamber slides. Slides were incubated at 37°C for 20 min to allow for the collagen gels to form after which 300 μL conditioned medium (see above) was added to each well. Cells were cultured at 37°C in 5% CO2 incubator and medium was changed every second day. Ducts were counted after 7 to 10 days. For ducts formed by cells infected with shKlf4 and shCon lentivirus, 3μg/ml puromycin (Sigma, MO) was added to the culture medium after 4 days in collagen gels for selection of infected cells. Ducts were counted 3 days after puromycin treatment. Each experiment was repeated 3-4 times.
Prostatosphere assay
Prostatospheres were assayed as described (Lukacs et al., 2010a). In brief, 3,000 Akt transformed prostate stem cells were resuspended in 100 μL of a 60:40 mixture of Matrigel (BD Bioscience, NJ):PrEGM (Lonza, Basel, Switzerland) and plated in triplicate around the rim of a 12-well tissue culture plate. Matrigel was allowed to solidify at 37°C, and 800 μL of PrEGM/well was added. Medium was changed twice weekly and spheres were counted after 7 to 10 days. Each experiment was repeated 3 times. To passage the spheres, wells were treated with 1ml of 1 mg/ml Dispase solution (GIBCO, NJ). Spheres were digested with 0.25% trypsin with 2.21mM EDTA to obtain single cells, and equal numbers of cells (2,000) were seeded in triplicate in Matrigel. The same protocol was used for all 6 passaged generations.
Constructs and viral infection
Constructs expressing constitutively active Akt1 were kindly provided by Dr. ON Witte (UCLA, CA) (Xin et al., 2006). Mouse and human shKlf4 and non-silencing shRNA constructs were purchased from Open Biosystem (Lafayette, CO). The hairpin sequence for mouse shKlf4 is TGAACTCTTCTTTCTCCT. The hair sequence for human shKLF4 is AGAAAAACACATTTGTTCT. The retroviral expression construct of mouse Klf4 (pMXs-Klf4-IP) was purchased from Addgene (www.addgene.org), the empty vector pMXs-IP was generated by removing the Klf4 gene with Not1 (BioLabs, san Diego, CA) from the retroviral sequence. Lentiviral Dox inducible BFP-tagged human KLF4 expressing vector was constructed by modifying the lentiviral vector FUW-tetO-hKLF4 generated by Rudolf Jaenisch (www.addgene.org). This plasmid is under the control of the tetracycline operator and a minimal CMV promoter. The ORF corresponding to human KLF4 was removed by enzymatic digestion with EcoRI (BioLabs, san Diego, CA) and the insert BFP-T2A-hKLF4 (KLF4 overexpression) or BFP-T2A alone (empty vector control, EV) was cloned into the EcoRI sites of the vector backbone FUW-tetO. Lentivirus was produced in 293T cells by co-transfection of plasmids with packaging plasmid Δ8.9 and envelope plasmid VSV-G, viruses were collected and concentrated by centrifugation at 25,000 rpm for 2 hours. Concentrated viruses were reconstituted in prostate cell growth medium. Retroviral particles were generated by transfecting plat E cells with retroviral vectors. Primary prostate proximal cells were isolated from the proximal region of prostatic ducts and cultured in 48 well plates pre-coated with collagen (Biomaterials, Fremont, CA) and were infected with virus (MOI 20) together with 8μg/ml polybrene for 20 hours. Cells were then washed with DMEM and used for in vitro and in vivo experiments. For lentiviral infection of the Akt transformed prostate stem cell line and the WPE human prostate stem cell line, cells were cultured in 10cm dishes and infected with virus for 7 hours, virus was removed and replaced with fresh medium and after 20 hours cells were re-infected as above and incubated for 2 additional days. Puromycin (8 μg/ml) was added to cells for 48 hours and those that survived the selection were processed for experiments. For Dox inducible KLF4 experiments, KLF4 re-expressing cells (shKlf4+KLF4) and control empty vector (shKlf4+EV) cells were treated with Dox (10 ng/ml) for 48 hours before in vitro experiments.
Implantation of cells under renal capsule
Cells isolated from the proximal region of prostatic ducts (30,000-50,000 cells) were combined with 250,000 UGM cells in collagen type I pellets and incubated at 37°C for 16-18 hours prior to implantation under the renal capsule of athymic nude mice (Goto et al., 2006; Zong et al., 2015). Each experiment contained a set of grafts of UGM alone to ensure that tissue growth did not result from contamination of urogenital sinus epithelial cells. Tissues were harvested after 8-10 weeks. Collagen pellets containing cells (1×105-2×106 cells) from the Akt transformed prostate stem cell line were implanted under renal capsule and tumor tissues were harvested after 4 weeks. For experiments using cells infected with the Dox inducible KLF4 construct, KLF4 re-expressing cells (shKlf4+KLF4) and control empty vector (shKlf4+EV) cells were implanted sub-RC. Tumors were allowed to grow for 3 weeks prior to administration of chow containing Dox (200 mg/kg) to all animals (control and KLF4 re-expressing cells) and harvested 7-10 days after initiation of Dox treatment. All experiments were repeated at least 3 times.
Immunohistochemistry
Immunohistochemistry was performed as previously described (Burger et al., 2009; Goto et al., 2006). Briefly, tissue sections were deparaffinized, sections were then heated in a microwave in 10mM sodium citrate pH 6.0 for 10 min for antigen retrieval; endogenous peroxidase activity was blocked with 0.3% hydrogen peroxidase in PBS for 20 min, after which slides were washed and incubated with 5% normal goat serum in PBS containing 1% BSA for 1 hour at room temperature. Proteins were detected by incubating slides with primary antibody overnight at 4°C, followed by incubation with secondary antibody for 1 hour at room temperature. A DAB substrate kit (Vector laboratories, Burlingame, CA) was used for detecting HRP activity. For fluorescent staining, Alexa Fluor 594 or 488 secondary antibodies (Molecular Probes, CA) were used. For the quantitation of CK5+ or Ki67+ cells, 3 sections from each graft (3 grafts per condition) were examined and 8-10 random fields in each section (minimum 600 cells) were analyzed. Data are presented as average percentages per field of view. For determining the numbers of tubules with PIN/carcinoma or normal morphology (Lukacs et al., 2010b), the sections were stained with hematoxylin and eosin (H&E) and all visible fields in 3 sections from each graft (3 grafts per condition) were scored. Data are presented as average percentages per field of view.
Western blot analysis
Protein was extracted from prostate tissue or cultured cells in RIPA buffer supplemented with protease inhibitor cocktail (Roche diagnostics GmbH, Mannheim, Germany) and phosphatase inhibitor (Roche diagnostics GmbH, Mannheim, Germany). Protein concentrations were measured by BCA Protein Assay (Thermo Scientific, NJ). Protein (30-50μg) was loaded on 4%–20% Tris-Glycine gels and blotted onto nitrocellulose membranes. Membranes were blocked with 5% dry milk in Tris-buffered saline for 1hour at room temperature, followed by incubation with primary antibody overnight at 4°C. Membranes were washed with TBST 3 times, incubated with horseradish peroxidase-conjugated secondary antibody for 1hour at room temperature, then washed 3 times with TBST. Immunoreactivity was detected by chemiluminescence as instructed by manufacturer (Thermo Scientific, NJ) for 1min. Membranes were stripped with Restore™ Plus Western Blot Stripping Buffer (Thermo Scientific, NJ) for 10 min at room temperature, then re-probed for mouse β-actin (Sigma, MO) to control for loading consistency. Band intensity was measured using ImageJ software.
FACS analysis and isolation of Sca-1 expressing cells
For the analysis of CD49f and Sca-1 expression in the Akt transformed stem cell line, Cy5.5-CD49f (Biolegend, CA) and pacific blue-Sca-1 antibodies (Biolegend, CA) were used. Cy5.5-rat IgG-2a (Biolegend, CA) and pacific blue-rat IgG-2a (Biolegend, CA) were used for control antibodies. Cells (50,000) were suspended in staining buffer (5% FBS in PBS) and human TruStain FcX™ (Biolegend, CA) was added to block Fc receptors. Cells were incubated at room temperature for 10 min followed by incubation with antibodies or control IgG for 30 min on ice in the dark. Cells were analyzed in the RSLII flow cytometer (BD Sciences, NJ). Proximal prostatic ductal cells were sorted by FACS (Moflo XDP, Beckman Coulter, NJ) into various fractions (Sca-1high, Sca-1med/low and Sca-1neg) according to the mean fluorescence intensity (MFI) of Sca-1 expression by the cells after addition of PE-Sca-1 antibody (BD Sciences, NJ). PE-rat IgG-2a (BD Sciences, NJ) was used as a negative control. Sca-1+ cells with fluorescent intensities in the upper one-third were defined as Sca-1high cells, the remaining Sca-1+ cells served as the Sca-1med/low population while the unlabeled cells served as the Sca-1neg population. Cells (5×104) were implanted with UGM (2×105) sub-renal capsule and grafts examined after 8 weeks.
Quantitative PCR
RNA was isolated using an RNeasy Mini Kit (QIAGEN, CA) as instructed. RNA (500–1000ng) was reverse transcribed using a Reverse Transcriptase (Roche Applied Science, Indianapolis, IN) and relative changes in mouse Klf4 mRNA quantified by real-time qPCR. Primers for Klf4 were: (F) 5′-ATCACGAAGTGGTGAAGTTC-3′ and (R) 5′-TGCTGTAGGAAGCTCATCTC-3′ producing a 176bp product. Primers specific for mActin were: (F) 5′-GTCGACACAGTGGCCATCAGCAGTT-3′ and (R) 5′-TTCCACAGAGCCCTTCTGGTT-3′ producing a 219bp product. Appropriately sized amplicons for all genes of interest were verified by gel electrophoresis and melting curve analyses (data not shown). All gene expression was normalized to mActin expression within each experiment, and relative changes in expression calculated by the 2−ΔΔCT formula. PCR reactions were performed in the iQ5 Real-Time PCR Detection System with iQ SYBR Green Master Mix (Thermo Fisher Scientific, MA). The cycling conditions for all the genes were: 95°C for 4.5 min, 40 cycles of 95°C for 30 s, 60°C for 20 s, and 72°C for 30 s.
RNA-seq data and analysis
The Akt transformed prostate stem cell line was infected with shKlf4 (shKlf4) or non-silencing control (shCon) lentivirus. After 48 hours cells were selected by puromycin (8μg/ml) for 2 days and passaged for 10 days to allow cells to undergo EMT. The infected shCon and shKlf4 cells were collected for RNA extraction. Another cohort of shKlf4 cells was infected with BFP tagged Dox inducible human KLF4 (shKlf4+KLF4) or BFP tagged Dox inducible empty vector (shKlf4+EV) lentivirus. After 3 days, 100ng/ml of Dox was added to cultures of shKlf4+KLF4 and shKlf4+EV cells to induce KLF4 expression while companion cultures (shKlf4+KLF4 and shKlf4+EV cells) received no Dox. Two days later, all shKlf4+KLF4 and shKlf4+EV cells with or without Dox treatment were collected for RNA extraction. Total RNA was extracted using an RNeasy mini kit (QIAGEN, CA). RNA-seq libraries were prepared with the TruSeq sample preparation kit (Illumina, CA).
All the reads were mapped to the mouse reference genome (GRCm38/mm10) using STAR aligner (v2.5.0c). Alignments were guided by a Gene Transfer File (Ensembl GTF version GRCm38.74) and the mean read insert sizes and their standard deviations were calculated using Picard tools (v.1.126) (http://broadinstitute.github.io/picard/). The Read Per Million (RPM) normalized BigWig files were generated using BEDTools (v2.17.0) and bedGraphToBigWig tool (v4). Read count tables were generated using HTSeq (v0.6.0) normalized based on their geometric library size factors using DESeq2 (v3.0) and Deferential Expression (DE) analyses were performed. Differentially expressed genes that were bound by Klf4 (by our ChIP-Seq data) were used for pathway analysis. KEGG pathway analysis and Gene Ontology (GO) analysis was performed using ClusterProfiler R package (v3.0.0). IPA (QIAGEN, CA) was also used for pathway analysis. GSEA (Broad Institute) were performed using the curated gene set (C2) of the Molecular Signature Database (MSigDB) version 4.0 provided by the Broad Institute (https://www.broadinstitute.org/gsea/doc/GSEAUSerGuideFrame.html). We used standard procedures as described by the GSEA user guide to compute overlaps between our gene set and gene sets in the Molecular Signature Database.
To compare the level of similarity among the samples and their replicates, we used two methods: classical multidimensional scaling or principal-component analysis and Euclidean distance-based sample clustering. The downstream statistical analyses and generating plots were performed in R environment (v3.1.1) (https://www.r-project.org/).
Identification of transcripts expressed by human prostate cancer and shKlf4 prostate cancer stem cells
To identify transcripts that are differentially expressed between human primary and metastatic prostate cancers (Chandran et al., 2007; GEO: GSE6919) and indolent and aggressive prostate cancers (Mortensen et al., 2015; GEO: GSE46602), datasets have been re-analyzed with GEO2R. Transcripts that were significantly up- or downregulated (p < 0.01) in these two datasets have been extracted and intersected with transcripts that are differentially expressed between shCon and shKlf4 murine prostate cancer cells. Mouse (GRCm38.p5) and human (GRCh38.p10) gene names have been converted with ENSEMBL BioMart before intersecting the gene sets for the generation of area proportional Venn diagrams with Venneuler software in R. Statistical significance in the gene overlap was determined by Montecarlo simulations with 100,000 iterations and 21,000 expressed transcripts.
ChIP-seq data and analysis
For ChIP-seq analyses, 1×107 cells of the Akt transformed prostate stem cell line with endogenous Klf4 were fixed in PBS containing 1% of formaldehyde for 8 min at room temperature on a rotating platform. The reaction was quenched with 0.125M glycine for 5 min. After washing twice with cold PBS, cells were collected and the chromatin prepared according to the ChIP-IT High Sensitivity protocol (Active Motif, CA). Cell pellets were re-suspended in 10ml of chromatin prep buffer supplemented with a proteinase inhibitor cocktail (PIC) and PMSF (100μM) and nuclei were extracted using a chilled Dounce homogenizer. Nuclei were re-suspended in ChIP buffer supplemented with PIC and PMSF (1mM) and sonicated using a Diagenode Bioruptor 300 at maximum intensity and pulsed for 30sec on and 30sec off for 30 cycles. Prior to ChIP, chromatin size was confirmed by reverse-crosslinking of 2.5% of the input by incubation with 10μg of RNase for 30min at 37°C followed by addition of 20μg proteinase K and incubation for 30min at 55°C and 2 hours at 80°C, making sure we had achieved an average length between 200-1200. Immunoprecipitation was performed overnight at 4°C on 30 μg of sheared chromatin using 4 μg of KLF4 antibody (R&D systems, MN). Chromatin samples were washed 5 times before reversing crosslinks. DNA was then extracted using a DNA purification column (Active Motif, CA) and eluted in 37μl of DNA Purification Elution Buffer (Active Motif, CA) for ChIP-seq analysis. Sample quality and DNA concentration was assessed by Qubit (Invitrogen, CA). ChIP-seq libraries were made using the ThruPLEX DNA-seq kit (Rubicon Genomics, MI) using 200pg of ChIP DNA or KAPA Hyper Prep kit (Kapa Biosystems, MA) using 1ng of ChIP DNA. Libraries were size selected prior to PCR amplification using AMPure XP beads (Beckman Coulter, CA). Multiplexed libraries were run on an Illumina HiSeq 2500 instrument, Genome Analyzer using the 50-base pair single read method.
All of the reads from the ChIP Sequencing for each sample were mapped to the mouse reference genome (mm10/GRCm38.74) using Bowtie2 (v2.2.4) and duplicate reads were removed using Picard tools (v.1.126) (http://broadinstitute.github.io/picard/). Low quality mapped reads (MQ < 20) were removed from the analysis. The read per million (RPM) normalized BigWig files were generated using BEDTools (v.2.17.0) and the bedGraphToBigWig tool (v.4). Peak calling was performed using MACS (v1.4.2) and peak count tables were created using BEDTools. Differential Binding (DB) analysis was performed using DESeq1 (v3.0). ChIPseeker (v1.8.0) R package and HOMER (v4.8) were used for peak annotations and motif discovery was performed using HOMER. ngs.plot (v2.47) and ChIPseeker was used for TSS binding site visualizations and quality controls.
Differentially expressed genes that were bound by Klf4 (from our ChIP-Seq data) were used for pathway analysis. KEGG pathway analysis and Gene Ontology (GO) analysis was performed using the ClusterProfiler R package (v3.0.0). To compare the level of similarity among the samples and their replicates, we used two methods: classical multidimensional scaling or principal-component analysis and Euclidean distance-based sample clustering. The downstream statistical analyses and generated plots were performed in R environment (v3.1.1) (https://www.r-project.org/).
Survival analysis and selection of prognostic biomarker gene signature
To ascertain that expression of KLF4 is correlated with survival, raw CEL files were downloaded from GSE16560 (Sboneret al., 2010) and GSE21032 (Taylor et al., 2010). Background correction and quantile normalization was performed using the rma function from affy (v1.54) R package. Probe IDs were then annotated with their gene symbols and KLF4 gene expression of all the samples was used for survival analysis using Kaplan-Meier curves.
In order to identify prognostically significant genes we examined the expression of genes that were bound and upregulated by KLF4 (168 genes) (Figure 7G) and were represented in the 26,446 genes used in the MSKCC cBioPortal mRNA analysis. This was done using a cohort of 94 patients with a low Gleason score (Gleason 6 and 7(3+4)) as well as 131 patients with primary prostate cancer and 29 matched normal prostate tissues obtained from the MSKCC cBioPortal (Taylor et al., 2010). Gene expression Z-scores were calculated for each gene using the 29 normal expression profiles as base distribution.
Good-prognosis genes
Out of 168 genes (see above) we found that 21 genes were upregulated (Z-Score > 2) in the MSKCC cohort. From these 21 genes, we enumerated and evaluated all 5-gene combinations. For each patient, we examined gene expression Z-Scores of each 5-gene combination. If, for any gene out of the 5 gene combinations, the patient had a Z-Score larger than 2, the patient was assigned to the group indicating an altered gene expression; the remaining individuals were assigned to the unaltered group. Based on this binary grouping, we used the log-rank test to obtain the separation power of each 5-gene combination. Using the separation power (p value from log-rank test), we selected the best gene list based on its ability to minimize the disease-free recurrence in the MSKCC patients with low Gleason scores (Gleason 6 and 7(3+4)). In this cohort 9 out of 94 patients with low Gleason scores had a biochemical relapse over the 10-year follow-up period. Kaplan-Meier plots were performed using the ‘Survival’ R package (Terry and Therneau, 2000; Therneau, 2015) in R version 3.3.2. The Chi-square independence test was used to determine if KLF4 was one of the best individual predictor genes. This gene signature was then examined in the cohort of 131 patients (all Gleason scores) with primary prostate cancer, of whom 27 had a biochemical relapse over the 10-year follow-up period.
We validated our gene signatures on Sboner’s dataset (Sboner et al., 2010) that contains 162 patients with low Gleason score (GL 6 and 7(3+4)) and 281 patients (all Gleason scores) with survival information using Z-scores as described above for the Taylor dataset. Kaplan-Meier plots were performed based on the Z-scores of the gene signatures.
Generation and comparison of gene signatures
All gene signature combinations of up to 7 genes (adding more genes becomes computationally unfeasible) were constructed starting from the predefined gene list of 168 KLF4 target genes, that were upregulated by re-expression of KLF4 and bound by KLF4 (Figure 6G, Table S5). More specifically, we compared the performance of gene signatures constructed based only on the 168 KLF4 target genes to the performance of gene signatures constructed from randomly selected gene lists of the same size (a total of 10,000 such lists were randomly selected). We tested genes (KLF4 was included as we found that indolence or aggressiveness of prostate tumors is dependent on its levels) that were upregulated by KLF4 re-expression in the RNA-seq experiments (168 genes), without knowing a priori the survival status of patients. In Figures S6, S7, S8A, S8B, we report and statistically compare the survival analysis p value distributions of the gene signatures generated from the KLF4 target genes versus those generated by the randomly selected gene lists. For the statistical comparison, we used the Wilcoxon Rank Sum test. This comparison was done independently in the MSKCC and Sboner patient cohorts and demonstrates that signatures constructed from genes identified in our mouse experiments as KLF4 targets, have significantly lower p values in both the MSKCC and Sboner cohorts (Gleason scores 6 and 7(3+4)), compared to the control gene lists. Finally, we also evaluated sensitivity (the ability to identify as many of the non-relapsed patients as possible) of the signature combinations derived from the 168 mouse genes as a function of signature size (Figure S8C, 8D). Patient probability of survival was also modeled using the Cox proportional hazard model fitted on: (a) Gleason score alone, (b) Gleason score plus our 5-gene signature, and, (c) Gleason score plus our 4-gene signature (after excluding KLF4). A similar analysis was done using the D-Amico classification (takes into account the Gleason score, clinical T stage and PSA levels). Different models were compared against each other using the ANOVA chi-square test.
TCGA data analysis
The read count tables for the PRAD RNA-Seq samples (n = 506) in The Cancer Genome Atlas (TCGA) were downloaded from the National Cancer Institute’s (NCI) Genomic Data Commons (GDC) (https://gdc.cancer.gov/). Of these samples, primary prostate tumor samples (n = 455) were used. Among the 455 patients, 44 are Gleason 6, 231 are Gleason 7 (136 Gleason 3+4 and 95 Gleason 4+3), 59 are Gleason 8, 118 are Gleason 9 and 3 are Gleason 10. All the read count tables were then corrected for their library size differences based on their geometric library size factors using the DESeq2 package (v3.0). Normalized KLF4 gene expression of the samples was separated into different groups by the sample’s Gleason score and their boxplots were generated. The normalized gene expression levels were then log transformed (log2), scaled (z-scores) and used for generating boxplots.
QUANTIFICATION AND STATISTICAL ANALYSIS
Each experiment was repeated 3-4 times. Results are presented as means ± SD of a representative experiment. Statistical differences between grouped quantitative datasets were measured using the 2-tailed unpaired Student’s t test. A p value of < 0.05 is considered statistically significant.
To compare the level of similarity among the samples and their replicates in RNA-seq and ChIP-seq data, we used two methods: classical multidimensional scaling or principal-component analysis and Euclidean distance-based sample clustering. The downstream statistical analyses and generated plots were performed in R environment (v3.1.1) (https://www.r-project.org/).
Patient survival data was examined using Kaplan-Meier survival analysis using the Log-rank test. Comparison of gene expression in patients with tumors having different Gleason scores was performed using the Student’s t test.
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-sequencing data and ChIP-sequencing data reported in this paper is GEO: GSE114772.
Supplementary Material
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Ki67 Antibody | Abcam | Cat# ab16667; RRID:AB_302459 |
| Anti-KLF4 Antibody | Santa Cruz Biotechnology | Cat# sc-20691; RRID:AB_669567 |
| Anti-KLF4 Antibody | R&D Systems | Cat# AF3158; RRID:AB_2130245 |
| Anti-Slug Antibody | Cell Signaling Technology | Cat# 9585P; RRID:AB_10828257 |
| Anti-Snail Antibody | Cell Signaling Technology | Cat# 3879S; RRID:AB_2255011 |
| Anti-Zeb1 Antibody | Santa Cruz Biotechnology | Cat# sc-25388; RRID:AB_2217979 |
| Anti-E-cadherin Antibody | Cell Signaling Technology | Cat# 3195; RRID:AB_2291471 |
| Anti-pAKT Antibody | Cell Signaling Technology | Cat# 4060; RRID:AB_2315049 |
| Anti-CK5 Antibody | Covance Antibody Products | Cat# PRB-160P; RRID:AB_10063444 |
| Anti-CK8 Antibody | Covance Antibody Products | Cat# MMS-162P; RRID:AB_291334 |
| PerCP/Cy5.5 Anti-CD49 Antibody | BioLegend | Cat# 313617; RRID:AB_1575054 |
| Pacific Blue Anti-Sca-1 Antibody | BioLegend | Cat# 108120; RRID:AB_493273 |
| Anti-mouse IgG, HRP Linked Antibody | R&D Systems | Cat# HAF007; RRID:AB_357234 |
| Anti-rabbit IgG, HRP Linked Antibody | R&D Systems | Cat# HAF008; RRID:AB_357235 |
| Goat-anti-mouse IgG (H+L) Secondary Antibody, Alexa Fluor 488 | Molecular probes | Cat# A11017; RRID:AB_143160 |
| Goat-anti-mouse IgG (H+L) Secondary Antibody, Alexa Fluor 594 | Molecular probes | Cat# A11020; RRID:AB_141974 |
| Goat-anti-rabbit IgG (H+L) Secondary Antibody, Alexa Fluor 488 | Molecular probes | Cat# AA11070; RRID:AB_142134 |
| Bacteria and Virus Strains | ||
| Stabl3 competent cells | Thermo Fisher Scientific | Cat# C737303 |
| pMD2.G packaging vector | Addgene | Plasmid# 12259 |
| psPAX2 packageing vector | Addgene | Plasmid# 12260 |
| Experimental Models: Cell Lines | ||
| Akt-transformed prostate stem cell line | This paper | N/A |
| WPE-stem cell line | ATCC | Cat# ATCC CRL-2887 |
| Biological Samples | ||
| Xenografts of cells in athymic nude mice | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Puromycin | Sigma-Aldrich | Cat# p9620 |
| Dox | Sigma-Aldrich | Cat# D9891 |
| Critical Commercial Assays | ||
| SuperScript™ III Reverse Transcriptase | Thermo Fisher Scientific | Cat# 18080044 |
| SYBR™ Green Master Mix | Thermo Fisher Scientific | cat# A25780 |
| Deposited Data | ||
| RNA-sequencing Data | This paper | GEO: GSE114772 |
| ChIP-sequencing Data | This paper | GEO: GSE114772 |
| Experimental Models | ||
| Prostate proximal primary cells | This paper | N/A |
| Akt transformed prostate stem cell line | This paper | N/A |
| Primer: mouse Klf4 FW: 5′-ATCACGAAGTGGTGAAGTTC-3′ | This paper | N/A |
| Primer: mouse Klf4 RV: 5′-TGCTGTAGGAAGCTCATCTC-3′ | This paper | N/A |
| Primer: Actin FW: 5′-GTCGACACAGTGGCCATCAGCAGTT-3′ | This paper | N/A |
| Primer: Actin RV: 5′-TTCCACAGAGCCCTTCTGGTT-3′ | This paper | N/A |
| Recombinant DNA | ||
| FUW-tetO-BFP-hKLF4 vector | This paper | N/A |
| FUW-tetO-BFP vector | This paper | N/A |
| GIPZ mouse Klf4 shRNA lentiviral vector | Open Biosystem | Cat# RMM4532-EG16600 |
| GIPZ human KLF4 shRNA lentiviral vector | Open Biosystem | Cat# RHS4430-101025579 |
| Scramble shRNA lentiviral vector | Open Biosystem | Cat# RHS4349 |
| pMXs-Klf4-IP retroviral vector | Addgene | Plasmid ID: 15920 |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ii// |
| Adobe Illustrator CC 2014 | Adobe software | https://www.adobe.com/products/illustrator.html |
| Adobe Photoshop CC 2014 | Adobe software | https://www.adobe.com/products/photoshop.html |
| STAR aligner (v2.5.0c) | Dobin et al., 2013 | https://www.ncbi.nlm.nih.gov/pubmed/23104886 |
| BEDTools(v2.17.0) | Quinlan and Hall, 2010 | https://www.ncbi.nlm.nih.gov/pubmed/20110278 |
| HTSeq (v0.60) | Anders et al., 2015 | https://www.ncbi.nlm.nih.gov/pubmed/25260700 |
| DESeq2 (v3.0) | Love et al., 2014 | https://www.ncbi.nlm.nih.gov/pubmed/?term=Moderated+estimation+of+fold+change+and+dispersion+for+RNA-seq+data+with+DESeq2 |
| Clusterprofilter R package (v3.0.0) | Yu et al., 2012 | https://www.ncbi.nlm.nih.gov/pubmed/22455463 |
| Bowtie2 (v2.2.4) | Langmead and Salzberg, 2012 | https://www.ncbi.nlm.nih.gov/pubmed/22388286 |
| DESeq1 (v3.0) | Anders and Huber, 2010 | https://www.ncbi.nlm.nih.gov/pubmed/20979621 |
| ChIPseeker (v1.8.0) | Yu et al., 2015 | https://www.ncbi.nlm.nih.gov/pubmed/25765347 |
| HOMER (v4.8) | Heinz et al., 2010 | https://www.ncbi.nlm.nih.gov/pubmed/?term=Simple+Combinations+of+Lineage-Determining+Transcription+Factors+Prime+cis-Regulatiory+Elements+Required+for+Macrophage+and+B+Cell+Identities |
| ngs.plot (v2.47) | Shen et al., 2014 | https://www.ncbi.nlm.nih.gov/pubmed/24735413 |
| MACS (v1.4.2) | Zhang et al., 2008 | https://www.ncbi.nlm.nih.gov/pubmed/18798982 |
| Affy (v1.54) R package | Gautier et al., 2004 | https://www.ncbi.nlm.nih.gov/pubmed/14960456 |
Highlights.
KLF4 regulates adult prostate stem cell homeostasis and is relevant in cancer biology
KLF4 regulates indolence, malignant transformation, and tumor progression
KLF4 inhibits expression of molecules linked to aggressive human prostate cancer
Downstream target genes of KLF4 make up a significant molecular prognostic signature
ACKNOWLEDGMENTS
We thank Dr. O. Witte (UCLA) for the Akt constructs, Dr. D. Fenyo (NYU School of Medicine) for help with the Monte Carlo simulations, and Susan Logan (NYU School of Medicine) and Michael Garabedian (NYU School of Medicine) for helpful discussions. This research was supported by the NIH, United States (R01CA132641 to E.L.W.; R01CA181111 to M.S.; T32CA16002 to M.C.; and T32CA009161 and T32AR064184 to A.S.-P.), and the American Cancer Society, United States (RSG-16-033-01-DDC to M.S.). This research was also supported by the Department of Urology and the Kimmel Center for Stem Cell Biology. We thank the Perlmutter Cancer Center (P30CA016087) for partial support of core funding (Applied Bioinformatics Laboratories, Genome Technology Center, Experimental Pathology Research Laboratory, and Cytometry and Cell Sorting Laboratory).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes nine figures and seven tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.11.065.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala G, Thompson T, Yang G, Frolov A, Li R, Scardino P, Ohori M, Wheeler T, and Harper W (2004). High levels of phosphorylated form of Akt-1 in prostate cancer and non-neoplastic prostate tissues are strong predictors of biochemical recurrence. Clin. Cancer Res 10, 6572–6578. [DOI] [PubMed] [Google Scholar]
- Bhagirath D, Zhao X, Mirza S, West WW, Band H, and Band V (2016). Mutant PIK3CA Induces EMT in a Cell Type Specific Manner. PLoS One 11, e0167064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum R, Gupta R, Burger PE, Ontiveros CS, Salm SN, Xiong X, Kamb A, Wesche H, Marshall L, Cutler G, et al. (2009). Molecular signatures of prostate stem cells reveal novel signaling pathways and provide insights into prostate cancer. PLoS One 4, e5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger PE, Xiong X, Coetzee S, Salm SN, Moscatelli D, Goto K, and Wilson EL (2005). Sca-1 expression identifies stem cells in the proximal region of prostatic ducts with high capacity to reconstitute prostatic tissue. Proc. Natl. Acad. Sci. USA 102, 7180–7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger PE, Gupta R, Xiong X, Ontiveros CS, Salm SN, Moscatelli D, and Wilson EL (2009). High aldehyde dehydrogenase activity: a novel functional marker of murine prostate stem/progenitor cells. Stem Cells 27, 2220–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran UR, Ma C, Dhir R, Bisceglia M, Lyons-Weiler M, Liang W, Michalopoulos G, Becich M, and Monzon FA (2007). Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer 7, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Sun Y, Wei Y, Zhang P, Rezaeian AH, Teruya-Feldstein J, Gupta S, Liang H, Lin HK, Hung MC, and Ma L (2012). LIFR isa breast cancer metastasis suppressor upstream of the Hippo-YAP pathway and a prognostic marker. Nat. Med 18, 1511–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetty R, Serra S, and Asa SL (2008). Loss of membrane localization and aberrant nuclear E-cadherin expression correlates with invasion in pancreatic endocrine tumors. Am. J. Surg. Pathol 32, 413–419. [DOI] [PubMed] [Google Scholar]
- Cui H, Darmanin S, Natsuisaka M, Kondo T, Asaka M, Shindoh M, Higashino F, Hamuro J, Okada F, Kobayashi M, et al. (2007). Enhanced expression of asparagine synthetase under glucose-deprived conditions protects pancreatic cancer cells from apoptosis induced by glucose deprivation and cisplatin. Cancer Res 67, 3345–3355. [DOI] [PubMed] [Google Scholar]
- D’Amico AV, Whittington R, Malkowicz SB, Schultz D, Blank K, Broderick GA, Tomaszewski JE, Renshaw AA, Kaplan I, Beard CJ, and Wein A (1998). Biochemical outcome after radical prostatectomy, external beam radiation therapy, or interstitial radiation therapy for clinically localized prostate cancer. JAMA 280, 969–974. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forootan FS, Forootan SS, Gou X, Yang J, Liu B, Chen D, Al Fayi MS, Al-Jameel W, Rudland PS, Hussain SA, and Ke Y (2016). Fatty acid activated PPARγ promotes tumorigenicity of prostate cancer cells by up regulating VEGF via PPAR responsive elements of the promoter. Oncotarget 7, 9322–9339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier L, Cope L, Bolstad BM, and Irizarry RA (2004). affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315. [DOI] [PubMed] [Google Scholar]
- Goldstein AS, Lawson DA, Cheng D, Sun W, Garraway IP, and Witte ON (2008). Trop2 identifies a subpopulation of murine and human prostate basal cells with stem cell characteristics. Proc. Natl. Acad. Sci. USA 105, 20882–20887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonnissen A, Isebaert S, and Haustermans K (2013). Hedgehog signaling in prostate cancer and its therapeutic implication. Int. J. Mol. Sci 14, 13979–14007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto K, Salm SN, Coetzee S, Xiong X, Burger PE, Shapiro E, Lepor H, Moscatelli D, and Wilson EL (2006). Proximal prostatic stem cells are programmed to regenerate a proximal-distal ductal axis. Stem Cells 24, 1859–1868. [DOI] [PubMed] [Google Scholar]
- Grambsch PM, and Therneau TM (1994). Proportional Hazards Tests and Diagnostics Based on Weighted Residuals. Biometrika 81, 515–526. [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irshad S, Bansal M, Castillo-Martin M, Zheng T, Aytes A, Wenske S, Le Magnen C, Guarnieri P, Sumazin P, Benson MC, et al. (2013). A molecular signature predictive of indolent prostate cancer. Sci. Transl. Med 5, 202ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isayeva T, Moore LD, Chanda D, Chen D, and Ponnazhagan S (2009). Tumoristatic effects of endostatin in prostate cancer is dependent on androgen receptor status. Prostate 69, 1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen F, Kraft J, Schroeder C, Hube-Magg C, Kluth M, Lang DS, Simon R, Sauter G, Izbicki JR, Clauditz TS, et al. (2017). Up-regulation of Biglycan is Associated with Poor Prognosis and PTEN Deletion in Patients with Prostate Cancer. Neoplasia 19, 707–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson EA, Are A, Greicius G, Kuo IC, Kelly D, Arulampalam V, and Pettersson S (2005). The Wnt/beta-catenin signaling pathway targets PPARgamma activity in colon cancer cells. Proc. Natl. Acad. Sci. USA 102, 1460–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S, Xu J, and Derynck R (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol 15, 178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson DA,Xin L, Lukacs RU, Cheng D, and Witte ON (2007). Isolation and functional characterization of murine prostate stem cells. Proc. Natl. Acad. Sci. USA 704, 181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson DA, Zong Y, Memarzadeh S, Xin L, Huang J, and Witte ON (2010). Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proc. Natl. Acad. Sci. USA 707, 2610–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YN, Abou-Kheir W, Yin JJ, Fang L, Hynes P, Casey O, Hu D, Wan Y, Seng V, Sheppard-Tillman H, et al. (2012). Critical and reciprocal regulation of KLF4 and SLUG in transforming growth factor β-initiated prostate cancer epithelial-mesenchymal transition. Mol. Cell. Biol 32, 941–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 75, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs RU, Goldstein AS, Lawson DA, Cheng D, and Witte ON (2010a). Isolation, cultivation and characterization of adult murine prostate stem cells. Nat. Protoc 5, 702–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs RU, Memarzadeh S, Wu H, and Witte ON (2010b). Bmi-1 is a crucial regulator of prostate stem cell self-renewal and malignant transformation. Cell Stem Cell 7, 682–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddipati R, and Katz JP (2016). KLF4 Initiates Acinar Cell Reprogramming and Is Essential for the Early Stages of Pancreatic Carcinogenesis. Cancer Cell 29, 247–248. [DOI] [PubMed] [Google Scholar]
- Merlos-Suárez A, Barriga FM, Jung P, Iglesias M, Céspedes MV, Rossell D, Sevillano M, Hernando-Momblona X, da Silva-Diz V, Muñoz P, et al. (2011). The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell 8, 511–524. [DOI] [PubMed] [Google Scholar]
- Mortensen MM, Høyer S, Lynnerup AS, Ørntoft TF, Søensen KD, Borre M, and Dyrskjøt L (2015). Expression profiling of prostate cancer tissue delineates genes associated with recurrence after prostatectomy. Sci. Rep 5, 16018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentyala S, Whyard T, Pentyala S, Muller J, Pfail J, Parmar S, Helguero CG, and Khan S (2016). Prostate cancer markers: an update. Biomed. Rep 4, 263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfarr CM, Mechta F, Spyrou G, Lallemand D, Carillo S, and Yaniv M (1994). Mouse JunD negatively regulates fibroblast growth and antagonizes transformation by ras. Cell 76, 747–760. [DOI] [PubMed] [Google Scholar]
- Qi Y, Yu J, Han W, Fan X, Qian H, Wei H, Tsai YH, Zhao J, Zhang W, Liu Q, et al. (2016). A splicing isoform of TEAD4 attenuates the Hippo-YAP signalling to inhibit tumour proliferation. Nat. Commun 7, ncomms11840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland BD, and Peeper DS (2006). KLF4, p21 and context-dependent opposing forces in cancer. Nat. Rev. Cancer 6, 11–23. [DOI] [PubMed] [Google Scholar]
- Rowland BD, Bernards R, and Peeper DS (2005). The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat. Cell Biol 7, 1074–1082. [DOI] [PubMed] [Google Scholar]
- Salm SN, Koikawa Y, Ogilvie V, Tsujimura A, Coetzee S, Moscatelli D, Moore E, Lepor H, Shapiro E, Sun TT, and Wilson EL (2000a). Generation of active TGF-beta by prostatic cell cocultures using novel basal and luminal prostatic epithelial cell lines. J. Cell. Physiol 784, 70–79. [DOI] [PubMed] [Google Scholar]
- Salm SN, Koikawa Y, Ogilvie V, Tsujimura A, Coetzee S, Moscatelli D, Moore E, Lepor H, Shapiro E, Sun TT, and Wilson EL (2000b). Transforming growth factor-beta is an autocrine mitogen for a novel androgen-responsive murine prostatic smooth muscle cell line, PSMC1. J. Cell. Physiol 785,416–424. [DOI] [PubMed] [Google Scholar]
- Salm SN, Burger PE, Coetzee S, Goto K, Moscatelli D, and Wilson EL (2005). TGF-beta maintains dormancy of prostatic stem cells in the proximal region of ducts. J. Cell Biol 770, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sboner A, Demichelis F, Calza S, Pawitan Y, Setlur SR, Hoshida Y, Perner S, Adami HO, Fall K, Mucci LA, et al. (2010). Molecular sampling of prostate cancer: a dilemma for predicting disease progression. BMC Med. Genomics 3, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Shao N, Liu X, and Nestler E (2014). ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 75, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M, Cui J, Du J, Wei D, Jia Z, Zhang J, Zhu Z, Gao Y, and Xie K (2014). A novel KLF4/LDHA signaling pathway regulates aerobic glycolysis in and progression of pancreatic cancer. Clin. Cancer Res 20, 4370–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BA, Sokolov A, Uzunangelov V, Baertsch R, Newton Y, Graim K, Mathis C, Cheng D, Stuart JM, and Witte ON (2015). A basal stem cell signature identifies aggressive prostate cancer phenotypes. Proc. Natl. Acad. Sci. USA 772, E6544–E6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. (2010). Integrative genomic profiling of human prostate cancer. Cancer Cell 78, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry M, and Therneau PMG (2000). Modeling Survival Data: Extending the Cox Model (Springer; ). [Google Scholar]
- Tetreault MP, Yang Y, and Katz JP (2013). Krüppel-like factors in cancer. Nat. Rev. Cancer 73, 701–713. [DOI] [PubMed] [Google Scholar]
- Therneau TM (2015). A Package for Survival Analysis in S Version 2.38, https://cran.r-project.org/package=survival. [Google Scholar]
- Tokar EJ, Qu W, Liu J, Liu W, Webber MM, Phang JM, and Waalkes MP (2010). Arsenic-specific stem cell selection during malignant transformation. J. Natl. Cancer Inst 702, 638–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomii C, Inokuchi M, Takagi Y, Ishikawa T, Otsuki S, Uetake H, Kojima K, and Kawano T (2017). TPX2 expression is associated with poor survival in gastric cancer. World J. Surg. Oncol 75, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimura A, Koikawa Y, Salm S, Takao T, Coetzee S, Moscatelli D, Shapiro E, Lepor H, Sun TT, and Wilson EL (2002). Proximal location of mouse prostate epithelial stem cells: a model of prostatic homeostasis. J. Cell Biol 757, 1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez JM, Zhang L, Su Q, Dakhova O, Zhang Y, Shahi P, Spencer DM, Creighton CJ, Ittmann MM, and Xin L (2012). Notch and TGFβ form a reciprocal positive regulatory loop that suppresses murine prostate basal stem/progenitorcell activity. Cell Stem Cell 77, 676–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Hoogen C, van der Horst G, Cheung H, Buijs JT, Pelger RC, and van der Pluijm G (2011). The aldehyde dehydrogenase enzyme 7A1 is functionally involved in prostate cancer bone metastasis. Clin. Exp. Metastasis 28, 615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Garcia AJ, Wu M, Lawson DA, Witte ON, and Wu H (2006). Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation. Proc. Natl. Acad. Sci. USA 703, 1480–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Tang H, Thayanithy V, Subramanian S, Oberg AL, Cunningham JM, Cerhan JR, Steer CJ, and Thibodeau SN (2009). Gene networks and microRNAs implicated in aggressive prostate cancer. Cancer Res 69, 9490–9497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Place RF, Huang V, Wang X, Noonan EJ, Magyar CE, Huang J, and Li LC (2010). Prognostic value and function of KLF4 in prostate cancer: RNAa and vector-mediated overexpression identify KLF4 as an inhibitor of tumor cell growth and migration. Cancer Res. 70, 10182–10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, He Y, Deng W, Lang L, Yang H, Jin B, Kolhe R, Ding HF, Zhang J, Hai T, and Yan C (2018). Atf3 deficiency promotes genome instability and spontaneous tumorigenesis in mice. Oncogene 37, 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei P, Zhang N, Xu Y, Li X, Shi D, Wang Y, Li D, and Cai S (2013). TPX2 is a novel prognostic marker for the growth and metastasis of colon cancer. J. Transl. Med 77, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei D, Wang L, Yan Y, Jia Z, Gagea M, Li Z, Zuo X, Kong X, Huang S, and Xie K (2016). KLF4 Is Essential for Induction of Cellular Identity Change and Acinar-to-Ductal Reprogramming during Early Pancreatic Carcinogenesis. Cancer Cell 29, 324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin L, Lawson DA, and Witte ON (2005). The Sca-1 cell surface marker enriches for a prostate-regenerating cell subpopulation that can initiate prostate tumorigenesis. Proc. Natl. Acad. Sci. USA 102, 6942–6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin L, Teitell MA, Lawson DA, Kwon A, Mellinghoff IK, and Witte ON (2006). Progression of prostate cancer by synergy of AKT with genotropic and nongenotropic actions of the androgen receptor. Proc. Natl. Acad. Sci. USA 103, 7789–7794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Li Z, Kong X, Jia Z, Zuo X, Gagea M, Huang S, Wei D, and Xie K (2016). KLF4-Mediated Suppression of CD44 Signaling Negatively Impacts Pancreatic Cancer Stemness and Metastasis. Cancer Res. 76, 2419–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, and He QY (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biology 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong Y, Goldstein AS, and Witte ON (2015). Preparation of Urogenital Sinus Mesenchymal Cells for Prostate Tissue Recombination Models. Cold Spring Harb. Protoc 2015, 988–990. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-sequencing data and ChIP-sequencing data reported in this paper is GEO: GSE114772.