

Acute myeloid leukemia (AML) is the most common form of acute leukemia in adults, but hereditary predisposition to AML has been insufficiently studied. Previous investigations of AML heritability have focused on families with a clear clustering of AML, and have identified highly penetrant germline mutations that segregate with disease1–2. The importance of these findings has been highlighted by the addition of “myeloid neoplasm with germline predisposition” to the World Health Organization classification of myeloid neoplasms, and the recommendation for “germline testing for predisposing variants in appropriate cases of AML” in the 2017 European LeukemiaNet guidelines. However, these AML risk variants are exceedingly rare in the general population, and in some cases have only been observed in the families in which they were first discovered. The role of more common polymorphisms in AML susceptibility is not clear. Despite evidence for an elevated risk of AML in first-degree relatives of AML patients3,4, low-penetrance common predisposition to AML remains understudied. Genome-wide association studies (GWASs) for AML risk variants have been limited to studies with small sample sizes (fewer than 200 cases), or investigations testing only small numbers of candidate single nucleotide polymorphisms (SNPs)5,6. In contrast, large-scale GWASs have identified risk alleles associated with other leukemias and uncovered numerous associations between SNPs and various benign hematologic traits7–9. We therefore conducted a GWAS for AML risk using three separate patient cohorts comprising 1533 adults of European ancestry with de novo AML compared to 3969 controls.
The discovery phase of the GWAS was performed using two independent American case-control sets. After filtering samples for quality control and ancestry, 1183 AML cases and 2369 controls of European ancestry (USA1: 894 cases, 1714 controls; USA2: 289 cases, 655 controls) were tested for 16,058,266 autosomal variants. Standard quality control measurements showed there was no systematic bias in the association testing, as evidenced by a genomic inflation factor λ<1 and a quantile-quantile plot in which the observed P-values only deviated from the expected P-values at the tail end (Supplementary Figure S1). The association test results from the combined USA1 and USA2 sample sets meta-analysis showed five independent loci with 11 polymorphisms that were suggestively associated with AML (P<10−6) (Supplementary Table S2).
We validated the SNPs with the lowest P-values using an independent set of German AML cases and controls by MassARRAY multiplexed genotyping. We validated SNPs from 19q13, 19p13 and 13q22 since these three loci contained SNPs with the lowest overall P-values and also contained two or more SNPs with P<10-6. Six SNPs were genotyped in 350 German AML patient samples and compared with 1600 previously genotyped population-matched controls from the German LIFE-Adult study10. For the 19q13 locus rs75797233 was genotyped, for the 13q22 locus four SNPs were genotyped (rs6562807, rs139878336, rs2039647, and rs4356363), and for the 19p13 locus rs57706619 was genotyped because primers could not be designed for genotyping rs11670628 and rs2240811, and the genotype of rs57706619 is concordant with the genotypes of rs11670628 and rs2240811 in 99% of genomes from the study population. The 19p13 SNP rs57706619 and the 19q13 SNP rs75797233 showed support for association with AML (P<0.05) when tested individually in the German sample set (Supplementary Table S3). We performed a fixed-effects meta-analysis to combine the German sample set with the two American sample sets and calculate overall P-values for rs57706619, rs75797233 and the 13q22 SNP rs2039647 (Table 1). The combined meta-analysis P-value for rs75797233 was 4.15×10−8 (odds ratio=2.28), which meets the commonly accepted threshold of P<5×10−8 to be considered significant in a GWAS, making this the first reported SNP associated with AML risk in a GWAS. Figure 1a shows overall P-values from all tested polymorphisms in the genome.
Table 1.
Association test results combining USA1, USA2 and German sample sets, for the most significant SNPs associated with AML on 19q13, 19p13 and 13q22
| SNP (cytoband) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Study population (cases / controls) |
Nearest gene | Positiona | RA | OA | RAFcase | RAFcon | OR (95% CI) | P-valueb |
| rs75797233 (19q13) | BICRA | 19:48099347 | T | A | ||||
|
combined analysis (I2=0) |
0.031 | 0.018 | 2.28 (1.70–3.06) | 4.15×10−8 | ||||
| USA1 (832/1653) | 0.025 | 0.013 | 2.28 (1.54–3.36) | 3.78×10−5 | ||||
| USA2 (277/611) | 0.040 | 0.020 | 2.58 (1.40–4.74) | 0.0039 | ||||
| Germany (346/1565) | 0.036 | 0.023 | 1.97 (1.01–3.83) | 0.046 | ||||
| rs57706619 (19p13) | B3GNT3 | 19:17915881 | T | C | ||||
|
combined analysis
(I2=0.70) |
0.42 | 0.35 | 1.31 (1.18–1.45) | 2.47×10−7 | ||||
| USA1 (718/1394) | 0.40 | 0.36 | 1.21 (1.07–1.37) | 0.003 | ||||
| USA2 (270/615) | 0.45 | 0.35 | 1.51 (1.24–1.87) | 5.26×10−5 | ||||
| Germany (336/991) | 0.43 | 0.35 | 1.44 (1.02–1.70) | 0.038 | ||||
| rs2039647 (13q22) | KLF12 | 13:74763378 | G | A | ||||
| combined analysis (I2=0) | 0.25 | 0.21 | 1.34 (1.21–1.51) | 2.08×10−7 | ||||
| USA1 (882/1714) | 0.26 | 0.21 | 1.41 (1.23–1.62) | 9.92×10−7 | ||||
| USA2 (279/615) | 0.24 | 0.20 | 1.23 (0.98–1.65) | 0.077 | ||||
| Germany (350/1600) | 0.24 | 0.22 | 1.18 (0.90–1.56) | 0.23 |
Abbreviations: SNP, single nucleotide polymorphism; RA, risk allele; OA, other allele; RAFcase, risk allele frequency in the cases; RAFcon, risk allele frequency in the controls; OR, odds ratio; CI, confidence interval; T, thymine; A, adenine; C, cytosine; G, guanine; I2, heterogeneity statistic representing the fraction of variability due to heterogeneity between study groups.
Position is given according to GRCh37 human genome build.
Association testing between variants and disease was performed using logistic regression assuming an additive genetic model. Combined analyses were performed using fixed-effects.
Figure 1.
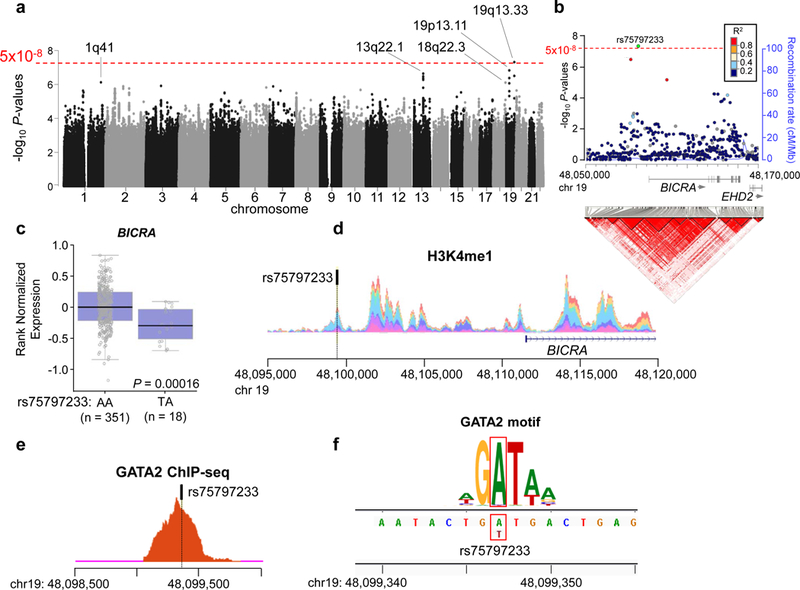
Genome-wide association test from combined analysis of USA1, USA2 and German sample sets reveals the rs75797233 polymorphism near BICRA is associated with acute myeloid leukemia (AML) risk. a Manhattan plot shows -log10 P-values (Y-axis) for all analyzed polymorphisms in the genome. The threshold for significance in a genome-wide association study (5×10−8) is represented by the red dotted line. b Regional association and linkage disequilibrium plot of the AML risk locus at 19q13. Top, significance of association with AML (-log10 P-values) for all tested polymorphisms is shown on the Y-axis, and the threshold for genome-wide significance (5×10−8) is represented by the red dotted line. Polymorphisms are colored according to their linkage disequilibrium (R2) with rs75797233 (colored in green). The unbroken blue line behind the circles representing the polymorphisms is the recombination rate. The arrows under gene names show direction of transcription. Bottom, linkage disequilibrium plot shows the pairwise linkage disequilibrium between all polymorphisms in the region. The red triangles indicate the locations of polymorphisms that are likely to be co-inherited. Linkage disequilibrium is plotted according to 1000 Genomes European population (November 2014 release), and genome is plotted according to human genome build GRCh37. c Blood expression levels of BICRA stratified by rs75797233 genotype (AA is homozygous for the non-risk allele and AT is heterozygous). d Layered plot for monomethylation of histone H3 lysine 4 (H3K4me1) shows a peak encompassing rs75797233, which implies an open chromatin state. e Transcription factor chromatin immunoprecipitation sequencing (ChIP-seq) for GATA2 shows a clear peak that encompasses rs75797233, which indicates GATA2 binds to this region. f Position weight matrix shows that the location of rs75797233 (indicated by the red boxes) is within a consensus GATA2 binding motif. The height of different letters at the same position is proportional to their importance for transcription factor binding in the motif.
To better characterize the rs75797233 AML risk SNP we examined its genomic context. rs75797233 is located 13kb 5′ of BRD4 Interacting Chromatin Remodeling Complex Associated Protein (BICRA also called GLTSCR1) in a 9kb haplotype block of variants often inherited together (Figure 1b). BICRA was first identified nearly two decades ago as a candidate tumor suppressor gene for glioma11. More recent studies have shown that BICRA directly interacts with the scaffold protein BRD4 to regulate transcriptional profiles12, and notably, BRD4 plays a critical role in AML disease maintenance13. BICRA was also recently identified as a key component in a novel SWI/SNF chromatin remodeling complex, and was shown to impact on cancer cell proliferation14.
We first used the GTEx project database to determine if rs75797233 genotype correlates with BICRA expression. Indeed, individuals who have the rs75797233 AML risk allele (thymine, [T]) have significantly lower expression of BICRA in blood compared to individuals who are homozygous for the non-risk rs75797233 allele (adenine, [A]) (Figure 1c). We hypothesized that this correlation is because rs75797233 influences transcription factor binding in a BICRA promoter or enhancer region. We examined the ENCODE database and found that rs75797233 is marked by histone H3 lysine 4 mono-methylation (H3K4me1), indicating open chromatin and accessible DNA (Figure 1d). Chromatin immunoprecipitation (ChIP) sequencing data confirmed that the GATA2 transcription factor binds to DNA in this region (Figure 1e). Using motifbreakR we determined that the non-risk allele of rs75797233 (A) is a critical residue in a GATA2 binding site, and that GATA2 is not predicted to bind this site when the rs75797233 risk allele (T) is present (Figure 1f).
To validate these data, lymphoblastoid cell lines generated from eight non-leukemic donors (four heterozygous for rs75797233 and four homozygous for the non-risk allele rs75797233 [A]) were assessed for BICRA expression using quantitative reverse transcription PCR, and assessed for GATA2 binding to rs75797233 via ChIP quantitative PCR and Sanger sequencing. Cell lines heterozygous for rs75797233 had significantly reduced BICRA expression compared to homozygous cell lines (Supplementary Figure S2a), and showed reduced binding of GATA2 to the rs75797233 locus (Supplementary Figure S2b). Preferential binding of GATA2 to the (A) allele of rs75797233 was confirmed by Sanger sequencing of amplified immunoprecipitated chromatin from heterozygous cell lines. The sequenced PCR product had a higher peak height of rs75797233 (A) compared to (T), and individually sequenced PCR amplicons from DNA topoisomeriase I (TOPO) cloning contained more (A) alleles than (T) alleles (Supplementary Figure S2c, d). Together, these data suggest that rs75797233 might associate with AML risk through mechanisms involving regulation of BICRA by GATA2.
We also examined the genomic context of the suggestive association loci at 13q22 and 19p13. The suggestive association polymorphisms at 13q22 are located in a 28kb haplotype block that overlaps with the long non-coding RNA LINC00402, and is immediately 5′ of Krüppel Like Factor 12 (KLF12) (Supplementary Figure S3a). The locus at 19p13 comprises a 14kb haplotype block that includes polymorphisms both immediately 5′ and within the first intron of UDP-GlcNAc:βGal β−1,3-N-Acetylglucosaminyltransferase 3 (B3GNT3), which encodes a type II transmembrane protein involved in poly-N-acetyllactosamine synthesis (Supplementary Figure S3b). Interestingly, one of the suggestive AML risk SNPs in the 19p13 locus, rs2240811, is within a region of open chromatin marked by H3K4me1 near the transcription start site of B3GNT3 (Supplementary Figure S4a), and ENCODE ChIP sequencing data indicate that multiple transcription factors likely occupy this region (Supplementary Figure S4b).
In this work we sought to expand the investigation of AML heritability to common (i.e. minor allele frequencies [MAF] > 1%) polymorphisms in the general population. We identified rs75797233 as the first common risk allele for AML, with a MAF of 1.8% in the controls and 3.1% in the cases. The overall per-allele odds ratio was >2, and notably, the individual odds ratios and MAFs were similar between the USA1, USA2 and German sample sets, indicating that the AML risk locus is not specific to a single population (Table 1). Because our study was performed using pretreatment bone marrow and blood samples from AML patients it is important to note the loci significantly or suggestively associated with AML risk are not within regions that are often somatically deleted or amplified in AML patients. Although our sample sets comprised a substantial collection of AML cases, additional association testing on more patients will be necessary to validate the significance of the 13q22 and 19p13 loci, and could also be used to explore the significance of the suggestive association loci at 1q41 and 18q22.3.
The expression of BICRA is significantly lower in blood and lymphoblastoid cell lines from individuals with an rs75797233 risk allele, and the presence of the risk allele results in loss of a GATA2 binding site. However, the mechanistic link between BICRA expression and development of AML is not yet clear. BICRA is not somatically mutated in AML, and the relationship between rs75797233 and BICRA expression within the bone marrow cells that contribute to leukemic transformation needs to be assessed. Future characterization of rs75797233 and the 19q13 locus might yield novel insights into the role of BICRA in AML biology, similar to how other factors important for leukemia biology were discovered through their associations with expression quantitative trait locus SNPs15. In summary, our study sheds light on the role of common polymorphisms in low-penetrance heritability of AML and identifies rs75797233 as the first common low-penetrance AML risk allele.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to acknowledge: the patients who consented to provide material for this study; Jan Lockman and Barbara Fersch for administrative help; Donna Bucci and Wacharaphon Vongchucherd of The Alliance NCTN Biorepository and Biospecimen Resource for sample processing and storage services; the Ohio Supercomputer Center for computational resources; and Lisa J. Sterling of The Ohio State University Comprehensive Cancer Center for data management. This work was supported in part by National Institutes of Health National Cancer Institute grants CA180821 and CA180882 (to the Alliance for Clinical Trials in Oncology), CA196171, CA077658, CA180850, CA180861, CA140158, CA016058, a Pelotonia idea grant, and the Leukemia Clinical Research Foundation. The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest
REFERENCES
- 1.Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, Li XC, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet 2011;43: 1012–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith ML, Cavenagh JD, Lister TA, Fitzgibbon J. Mutation of CEBPA in familial acute myeloid leukemia. N Engl J Med 2014; 351: 2403–2407. [DOI] [PubMed] [Google Scholar]
- 3.Hemminki K, Jiang Y. Familial myeloid leukemias from the Swedish Family-Cancer Database. Leuk Res 2002; 26: 611–613. [DOI] [PubMed] [Google Scholar]
- 4.Goldin LR, Kristinsson SY, Liang XS, Derolf AR, Landgren O, Björkholm M. Familial aggregation of acute myeloid leukemia and myelodysplastic syndromes. J Clin Oncol 2012; 30: 179–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lv H, Zhang M, Shang Z, Li J, Zhang S, Lian D, Zhang R, et al. Genome-wide haplotype association study identify the FGFR2 gene as a risk gene for acute myeloid leukemia. Oncotarget 2017; 8: 7891–7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Knight JA, Skol AD, Shinde A, Hastings D, Walgren RA, Shao J, et al. Genome-wide association study to identify novel loci associated with therapy-related myeloid leukemia susceptibility. Blood 2009; 113: 5575–5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Bernardo MC, Crowther-Swanepoel D, Broderick P, Webb E, Sellick G, Wild R, et al. A genome-wide association study identifies six susceptibility loci for chronic lymphocytic leukemia. Nat Genet 2008; 40: 1204–1210. [DOI] [PubMed] [Google Scholar]
- 8.Kim DH, Lee ST, Won HH, Kim S, Kim MJ, Kim HJ, et al. A genome-wide association study identifies novel loci associated with susceptibility to chronic myeloid leukemia. Blood 2011; 117: 6906–6911 [DOI] [PubMed] [Google Scholar]
- 9.Papaemmanuil E, Hosking FJ, Vijayakrishnan J, Price A, Olver B, Sheridan E, et al. Loci on 7p122, 10q212 and 14q112 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet 2009; 41: 1006–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loeffler M, Engel C, Ahnert P, Alfermann D, Arelin K, Baber R, et al. The LIFE-Adult-Study: objectives and design of a population-based cohort study with 10,000 deeply phenotyped adults in Germany. BMC Public Health 2015; 15: 691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith JS, Tachibana I, Pohl U, Lee HK, Thanarajasingam U, Portier BP, et al. Genomics 2000; 64: 44–50. [DOI] [PubMed] [Google Scholar]
- 12.Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW, et al. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol Cell Biol 2011; 31: 2641–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roe JS, Vakoc CR. The essential transcriptional function of BRD4 in acute myeloid leukemia. Cold Spring Harb Symp Quant Biol 2016; 81: 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alpsoy A, Dykhuizen EC. Glioma tumor suppressor candidate region gene 1 (GLTSCR1) and its paralog GLTSCR1-like form SWI/SNF chromatin remodeling subcomplexes. J Biol Chem 2018; 11: 3892–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maharry SE, Walker CJ, Liyanarachchi S, Mehta S, Patel M, Bainazar MA, et al. Dissection of the major hematopoietic quantitative trait locus in chromosome 6q23.3 identifies miR-3662 as a player in hematopoiesis and acute myeloid leukemia. Cancer Discov 2016; 6: 1036–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.