

Abstract
Reactive astrogliosis and early synaptic degeneration are two characteristic hallmarks in AD brains, but a direct link between the two events has not been established. Here we show that CIP2A, a cancerous protein with high expression level in astrocytes, is upregulated in AD patients and 3xTg-AD transgenic mice. Overexpression of CIP2A in astrocytes through AAV infection both in cultured cells and in mice brains results in activation of astrocytes, increased production of cytokines and Aβ, and synaptic degeneration indicated by decreased levels of synaptic proteins, spine loss and impairment in LTP. As a result of synaptic degeneration, CIP2A overexpression in astrocytes in vivo induces significant deficits in visual episodic memory detected by novel objective recognition test and spatial memory detected by Morris water maze. We conclude that CIP2A-promoted astrogliosis induces synaptic degeneration and cognitive deficits in Alzheimer’s disease.
Keywords: CIP2A, reactive astrogliosis, synaptic degeneration, cognitive deficit
1. Introduction
Alzheimer’s disease (AD) is the most common type of dementia. With the rapid increase of ageing population in the world, AD has become a major public health concern. The two characteristic pathological hallmarks in AD brains including neurofibrillary tangles (NFTs) and senile plaques (SPs) have been well recognized and studied (Braak and Braak, 1992). Besides NFTs and SPs, a loss of synapse and impairment in synaptic function were observed in patients with early stage AD or mild cognitive impairment (MCI) -- the preclinical stage of AD (Scheff et al., 2006). It has been shown that the severity of synaptic degeneration correlates with the degree of cognitive deficits (Crews et al., 2010; Scheff et al., 2006). However, the upstream factors that lead to the early synaptic degeneration in AD remain less clear.
Reactive astrogliosis, which refers to the activation of astrocytes in response to injury or disease in the central nervous system (CNS), is also an early event in AD brain. Astrogliosis is characterized by increased gene expression of a number of astrocyte structural proteins such as glial fibrillary acid protein (GFAP); morphological changes such as hypertrophy of the cell soma and processing; and proliferation. Typical changes of astrogliosis were observed in the brains of AD patients, as well as in APPswe transgenic mouse brains before Aβ plaque deposition (Carter et al., 2012; Rodriguez-Vieitez et al., 2015), with the GFAP expression level correlating with the Braak staging (Simpson et al., 2010). Reactive astrocytes were observed clustering around amyloid plaques (Duyckaerts et al., 2009; Wisniewski and Wegiel 1991). Recently, Liddelow et al. reported the toxicity of an A1 subtype reactive astrocytes on synapses. They showed that A1-astrocyte-conditioned medium (A1-ACM) reduced the synapse number and even induced neuronal death when used in high concentrations. Further exploration indicated that A1 astrocytes were abundant in AD brains (Liddelow et al., 2017). Thus, reactive astrocytes, especially the “harmful” A1 type astrocytes may play a key role in early synaptic degeneration in AD. However, the molecular mechanism inducing reactive astrogliosis in AD brains has not been elucidated.
In the present study, we show that upregulation of Cancerous Inhibitor of PP2A (CIP2A) participates in the harmful reactive astrogliosis in AD. CIP2A expression levels are increased in AD patients and transgenic mice. When CIP2A overexpression is induced in astrocytes in culture system or in mouse brains, activation of astrocytes, release of cytokines and Aβ, and synaptic toxicity are observed. Cognitive impairment of the mice is prominent. We conclude that CIP2A-promoted astrogliosis induces synaptic degeneration and cognitive deficits in Alzheimer’s disease.
2. Materials and methods
2.1. Antibodies and reagents
Rabbit polyclonal antibody (pAb) against CIP2A (1:1000 for Western blotting) was from ABclonal (Boston, USA); another pAb of CIP2A (1:1000 for Western blotting and 1:200 for immunofluorescence) was from Cell Signaling (Danvers, MA, USA). Mouse monoclonal antibody (mAh) DM1A and GFAP, and pAb against PSD95 were all purchased from Cell Signaling (Danvers, MA, USA). PAb against synapsin I and synaptophysin were from Abcam (Cambridge, UK). MAb GAPDH was from YEASEN (Pudong, Shanghai, China). Neurobasal, B27, DMEM-high glucose and protein marker were from Invitrogen (Grand Island, NY, USA). DMEM/F12 and fetal bovine serum (FBS) were from GIBCO (Grand Island, New York, USA). Rat or mouse IL-1α, IL-6, TNF-α ELISA kits and Aβ40/Aβ42 ELISA kits were from Elabscience Biotechnology (Wuhan, China). LDH cytotoxicity assay kit was from Promega (Madison, FL, USA). Nissl staining solution was from Goodbio technology (Wuhan, China). AAV-GFAP-EGFP-CIP2A-3FLAG and AAV-Vector were constructed and prepared by Obio Technology (Shanghai, China).
2.2. Human brain tissues, animals and AAV delivering
The human brain tissues were obtained from the National Institutes of Health (NIH) NeuroBioBank. For animal experiments, Sprague-Dawley rats and C57BL/6J mice were from the Experimental Animal Center of Tongji Medical College, Huazhong University of Science and Technology. 3xTg-AD mice carrying human mutated APP, PS1 and tau genes were from Jackson Lab (Stock No: 008880). Rats and mice were kept under standard laboratory conditions: 12 h light and 12 h dark with water and food ad libitum. All animal experiments were approved by the Animal Care and Use Committee of Huazhong University of Science and Technology, and performed in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
For AAV injection, C57 mice were deeply anesthetized with isoflurane, AAV virus particles (1.0 μl at 0.1 μl/min) was injected into the bilateral CA3 region (1.8 mm behind bregma, 2.2 mm beside bregma, 2 mm deep). After the injection, the mice were placed on a heating plate for recovery, and then were kept under standard laboratory conditions.
2.3. Primary astrocyte and neuron culture
Primary cortical astrocytes were isolated from newborn Sprague-Dawley rats. Briefly, tissues were dissected, dissociated, and incubated with 5 ml of D-Hanks containing 0.25% trypsin for 15 min, centrifuged at 1000 g for 5 min after addition of 4 ml of the medium containing high glucose DMEM with 10% fetal bovine serum. Then the cells were resuspended, and plated onto bottles which were previously coated with poly-D-lysine. Cells were cultured in a humidified incubator with 5% CO2 at 37°C. During the culture, the medium was changed every three days. After 7-9 days, the astrocytes were sorted at shake cultivation which was rotating at 200-220 r/min overnight. Then the medium was discarded and the cells were incubated with 2 ml DMEM containing 0.25% trypsin for 5 min, centrifuged at 1000 g for 5 min after addition of 4 ml of the medium containing high glucose DMEM with 10% fetal bovine serum. Then the cells were resuspended, about 5×105 cells were plated onto each well of 12-well plates for Western blotting, and 1×105 cells were plated onto each glass cover slip for cell imaging. Both the plates and the glass cover slips were previously coated with poly-D-lysine. Primary neuron culture was performed following the method described previously (Sun et al., 2016). At the end of treatments, cells were collected and lyzed in RIPA buffer for further biological detections or fixed with 4% paraformaldehyde for immunofluorescence imaging. Purity of the cultured astrocytes was confirmed through using qPCR to detect the expression levels of cell type specific genes (Astrocyte: GFAP; Neuron: Eno2; Microglia: Cd11b).
2.4. Western blotting
For Western blotting, brain tissue homogenates or cell lysates were boiled at 100°C for 5 min in the loading buffer (50 mM Tris-HCl, pH 7.6, 2% SDS, 10% glycerol, 10 mM DTT, and 0.2% bromphenol blue). The proteins were electrophoresed in 10% SDS-polyacrylamide gel and the separated proteins were transferred to nitrocellulose membranes (Amersham Biosciences). The membranes were then blocked with 5% nonfat milk dissolved in TBS-Tween-20 (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.2% Tween-20) for 1 h and probed with primary antibody at 4°C overnight. Then the blots were detected using secondary antibodies at room temperature for 1 h and visualized using the Odyssey Infrared Imaging System (LicorBiosciences, Lincoln, NE, USA). The protein bands were quantitatively analyzed by Image J software (Rawak Software, Inc. Germany).
2.5. Fluorescence imaging and confocal microscopy
For cultured astrocytes, cells were fixed with 4% paraformaldehyde for 15 min, permeabilized in 0.5% Triton X-100 for 10 min, followed by incubation with 3% bovine serum albumin (BSA) to block nonspecific sites. For mouse brain slices, sectioned slices were incubated with 3% bovine serum albumin (BSA) to block nonspecific sites. After blocking, primary antibody incubation was performed overnight at 4°C. Alexa 488 and 543-conjugated secondary antibody (1:200, A-21206, A-10036, Invitrogen) were used for double fluorescence labeling. All the images were observed under the LSM710 confocal microscope (Zeiss, Germany).
2.6. ELISA assay of IL-1α, IL-6, TNF-α and Aβ40/42
The culture medium or brain tissue homogenates which were lyzed in RIPA buffer were collected. The levels of IL-1α, IL-6, TNF-α, Aβ40 and Aβ42 were detected by ELISA following the construction offered by the assay kit manufacturer (Elabscience Biotechnology, Wuhan, China).
2.7. LDH assay and CCK-8 cell number counting
The LDH assay was performed using a LDH cytotoxicity assay kit (Promega, Madison, FL, USA) according to the manufacturer’s instructions. Briefly, culture media from primary cortical neuronal cultures (50 μl per sample) was mixed with assay buffers (50 μl in wells of 96-well plate and incubated for 30 min at room temperature in darkness, then 50 μl stopping buffer were added to every well, the OD values were detected by microplate reader at 490 nm and 630 nm, the final OD values = OD490 nm − OD630 nm. The cell number of astrocytes was counted by CCK8 cell counting kit (Beyotime Biotechnology, Shanghai, China) according to the manufacturer’s instructions. CCK-8 assay buffers (10 μl were added in 100 μl culturing cells which were in wells of 96-well plate, and then incubated in a humidified incubator with 5% CO2 at 37°C for two hours, finally the OD values were detected by microplate reader at 450 nm.
2.8. Behavior Tests
2.8.1. Novel objective recognition test (NORT)
The mice were habituated to the arenas (50 cm × 50 cm ×50 cm plastic container) for 5 min without objects 24 h prior to the test. Arenas were cleaned with 70% ethanol between each habituation period. The day after the mice re-entered the arenas from the same starting point and were granted 5 min to familiarize themselves with the A object and B object. After each period the arena and objects were cleaned with 70% ethanol. One hour after the familiarization period, B object was replaced with C object, and the mice were granted 5 min to explore both objects. The exploring time on each object was recorded. After 24 h, C object was replaced with D object, and the mice were granted 5 min to explore both objects. The exploring time on each object was recorded. The recognition index was calculated by TA/(TA+TB), TB/(TA+TB), TC/(TA+TC),TD/(TA+TD). The discrimination index was calculated by (TC-TA)/(TA+TC), (TD-TA)/(TA+TD). TA, TB, TC, TD were respectively the time mice exploring the object A, B, C and D.
2.8.2. Morris water maze test (MWM)
Spatial learning and memory were detected by Morris Water Maze (MWM). The MWM task was performed as previously described (Wang et al., 2014). Briefly, a circular arena (120 cm×50 cm) made of white polyvinyl chloride (PVC) was filled with water (23 ± 2 °C). An escape platform (10×10×15 cm) was placed into the pool, 1.5 cm below the water surface and 10 cm from the wall. The water was made opaque by the addition of a white titanium dioxide. The test room contained several permanent extra-maze cues such as posters, a flag or other objects on the walls. A video-tracking camera above the center of the pool surface monitored the trajectory of the mice. The video signal was transmitted to a computer in an adjacent room. Learning consisted of six consecutive daily acquisition sessions, each of them consisting of four trials, with a maximum trial duration of 60 s. Latency time (s) to find the hidden platform were recorded during each trial of each learning session. If the mice found the platform within the maximum trial time allowed, it was left on the platform for 20 s. If the mice did not find the platform within the time limit, it was gently placed on the platform for a 20 s period. Probe tests were carried out 1 hour or 48 hours after acquisition.
2.9. Nissl staining
The mice were deeply anesthetized and perfused intracardially with 400 ml normal saline, followed with fixing in 400 ml 4% formaldehyde for 3 days. The fixed brains were transferred into 20%, 25%, 30% sucrose for each one day successively to be dehydrated. Then the brains were embedded in OCT (optimum cutting temperature compound, Sakura, USA), frozen, and sectioned at 25 ×m using freezing microtome (Leica 1950, Wetzlar, Germany).Then brain slices were immersed in Nissl staining solution for 30 s, followed with 100% ethanol for 1 min, and xylene for 15 min successively. Finally, the brain slices were mounted with neutral balsam. Images were observed under the microscope (Nikon, Tokyo, Japan).
2.10. Golgi staining
The mice were anesthetized and perfused intracardially with 400 ml normal saline containing 0.5% sodium nitrite, followed with 400 ml 4% formaldehyde and the Golgi dye solution containing 5% chloral hydrate, 4% formaldehyde, and 5% potassium dichromate. After perfusion, the brains were dissected into 5 mm × 5 mm sections and transferred to a vial containing Golgi dye solution for 3 days in dark, then immersed in solution containing 1% silver nitrate for another 3 days. The brains were serially sectioned into 100 μm thick slices using a vibrating microtome (Leica, VT1000S, Germany). Images were observed under the microscope (Nikon, Tokyo, Japan).
2.11. Acute slice preparation and LTP recording
For recording hippocampal LTP, mice were decapitated under deep anesthesia, the brain was quickly removed and placed in ice-cold oxygenated artificial cerebral spinal fluid (aCSF) containing 119 mM NaCl, 2.5 mM KCl, 26.2 mM NaHCO3, 1 mM NaH2PO4, 11 mM glucose, 1.3 mM MgSO4, and 2.5 mM CaCl2 (pH 7.4). Horizontal 350 μm thick brain slices were cut in ice-cold aCSF using a vibrating microtome (Leica, VT1000S, Germany). Slices were then transferred to a recovery chamber at least 1.5 h with oxygenated aCSF at room temperature until recordings were performed.
For LTP recording, acute brain slices were transferred to a recording chamber and submerged in aCSF. Slices were laid down in a chamber with an 8 × 8 microelectrode array (Parker Technology, Beijing, China) in the bottom planar (each 50 × 50 μm in size, with an interpolar distance of 150 μm) and kept submerged in aCSF. Signals were acquired using the MED64 System (Alpha MED Sciences, Panasonic). The fEPSPs in CA1 neurons were recorded by stimulating CA3 neurons. LTP was induced by applying three trains of high-frequency stimulation (HFS; 100 Hz, 1s duration). The LTP magnitude was quantified as the percentage change in the fEPSP slope (10%–90%) taken during the 60 min interval after LTP induction.
2.12. Statistical analysis
Data are expressed as mean ± SEM and analyzed using GraphPad Prism 5 statistical software (USA, GraphPad Software). The one-way analysis of variance (ANOVA) procedure followed by Tukey was used to determine the differences among groups. For the comparison between two groups, student-t test was used. The significance was set at p < 0.05. All results shown correspond to individual representative experiments.
3. Results
3.1. CIP2A is expressed in astrocytes and upregulated in AD patients and transgenic mouse brains
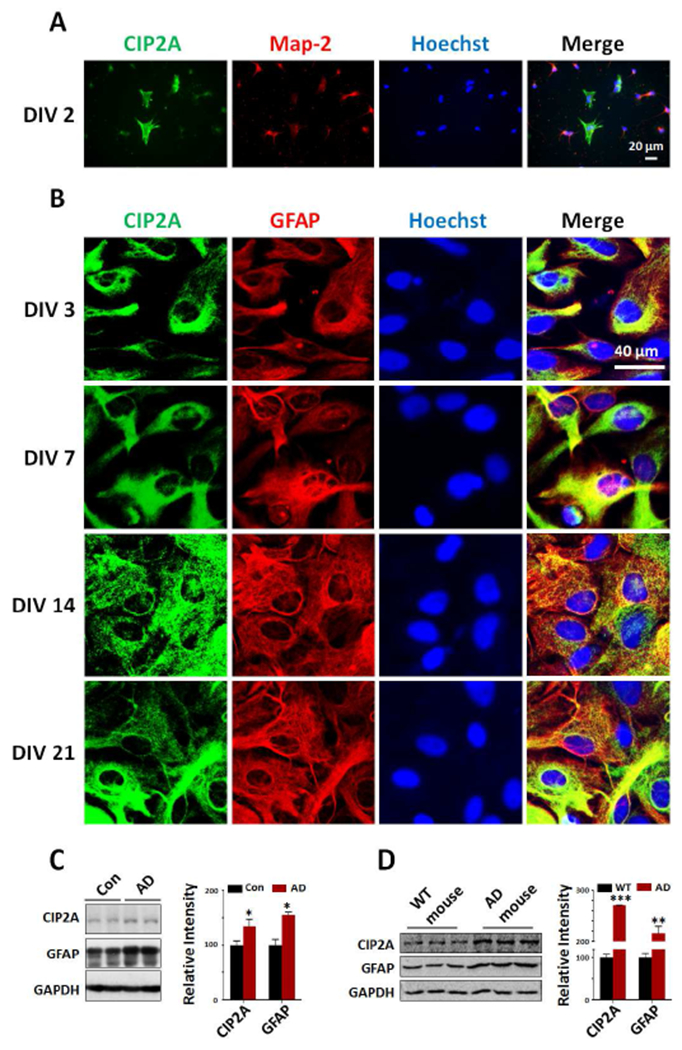
To explore the role of CIP2A in astrocytes and astrogliosis in AD, we first examined the expression of CIP2A in cultured astrocytes in vitro. In co-cultures of primary neurons and astrocytes (DIV 2), we found that CIP2A was expressed at considerable level in astrocytes (Map-2 negative) when comparing to that in neurons (Map-2 positive) (Fig. 1A). For a detailed observation of CIP2A in astrocytes, we examined the CIP2A immunostaining in cultured primary astrocytes on different days in vitro. Before experiments, the cell purity was confirmed through detecting the expression levels of cell type specific genes (Supplementary Fig. 1). In cultured primary astrocytes, CIP2A expression was observed in cytoplasm and maintained at relative high levels (Fig. 1B). Western blotting and PCR further confirmed the expression of CIP2A in astrocytes (Supplementary Fig.2). These data indicated that CIP2A may play a physiological or pathophysiological role in astrocytes.
Fig.1.
CIP2A is expressed in astrocytes and upregulated in AD brains. (A) Primary neurons and astrocytes from rat hippocampus were cocultured for 2 days (DIV 2) and immunostained with antibodies against CIP2A (green), Map-2 (red) and Hoechst (blue). Scale bar = 20 μm. (B) Rat primary astrocytes (DIV 3, 7, 14 and 21) were immunostained with antibodies against CIP2A (green), GFAP (red) and Hoechst (blue). Scale bar = 40 μm. (C) Left: CIP2A and GFAP expression levels in the temporal cortex from AD patients were detected by Western blotting. GAPDH was used as a loading control. Right: Quantitative analysis of the CIP2A and GFAP levels. *p < 0.05 versus Control, n = 5 control group, n = 4 AD group. (D) Left: CIP2A and GFAP expression levels in the hippocampus from 3xTg-AD and control mice (male, 9 months old) were detected by Western blotting. GAPDH was used as a loading control. Right: Quantitative analysis of the CIP2A and GFAP levels. **p < 0.01, ***p < 0.001 versus WT mice, n = 3. Data are mean ± SEM.
In AD brains, reactive astrogliosis is observed and suggested to participate in AD pathogenesis (Birch, 2014). To evaluate the potential effect of CIP2A in astrogliosis in AD, we detected the expression of GFAP as an astrogliosis marker, as well as CIP2A levels in AD patients and 3xTg-AD mice brains. The results showed that both CIP2A and GFAP levels were increased in AD patients and mouse brains (Fig. 1C, D). These findings suggest that CIP2A may play a role in reactive astrogliosis in AD brains.
3.2. Overexpression of CIP2A induces reactive astrogliosis
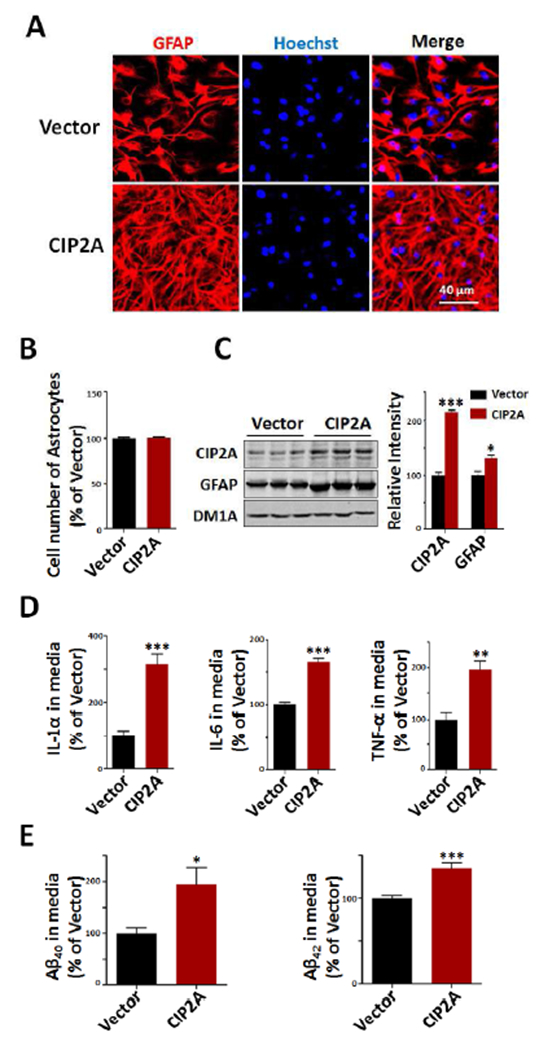
To further validate the hypothesis that CIP2A promotes astrogliosis, we overexpressed CIP2A through AAV-virus infection in cultured rat primary astrocytes. Staining of astrocytes with anti-GFAP showed that CIP2A overexpression induced a significant morphological change of reactive astrogliosis: cell soma hypertrophy and more processes (Fig.2A). CIP2A did not increase the cell number of astrocytes, indicating that CIP2A does not promote cell proliferation in our experimental system (Fig.2B). Consistent with the findings in AD mouse and patients brains, CIP2A overexpression resulted in increased expression of GFAP protein in astrocytes (Fig.2C), further supporting a role of CIP2A in astrogliosis.
Fig.2.
Overexpression of CIP2A induces reactive astrogliosis. Primary rat astrocytes (DIV15) were infected with AAV-CIP2A for 5 days. (A) Immunofluorescence staining of GFAP (red) and Hoechst (blue). Scale bar = 40 μm. (B) The numbers of astrocytes were counted in control and CIP2A overexpression group, n = 9 well per group. (C) Left: CIP2A and GFAP expression levels were detected by Western blotting. DM1A was used as a loading control. Right: Quantitative analysis of the CIP2A and GFAP levels, n = 3 per group. (D) IL-1α, IL-6 and TNF-α in culture media were detected by ELISA, n = 9 well per group. (E) Aβ40 and Aβ42 levels in culture media were detected by ELISA, n = 9 well per group. *p < 0.05, **p < 0.01, ***p < 0.001 versus Vector. Data are mean ± SEM.
Previous studies have identified that reactive astrocytes may be neurotoxic through producing inflammatory cytokines (Sofroniew, 2009; Gruol, 2016); we thus measured the release of cytokines from CIP2A-overexpressed astrocytes. The results showed that cytokines like IL-1α, IL-6 and TNF-α in the culture media were significantly increased in CIP2A group compared to control group (Fig.2D). Several research groups have reported that reactive astrocytes can produce significant amounts of Aβ (Frost, 2017; Zhao et al. 2011), so we measured the Aβ levels in the medium. We found that both Aβ40 and Aβ42 levels were dramatically increased in CIP2A group (Fig.2E). Taking together, these results indicate that overexpression of CIP2A induces reactive astrogliosis, leading to release of inflammatory cytokines and overproduction of Aβ.
3.3. CIP2A -induced astrogliosis promotes synaptic degeneration
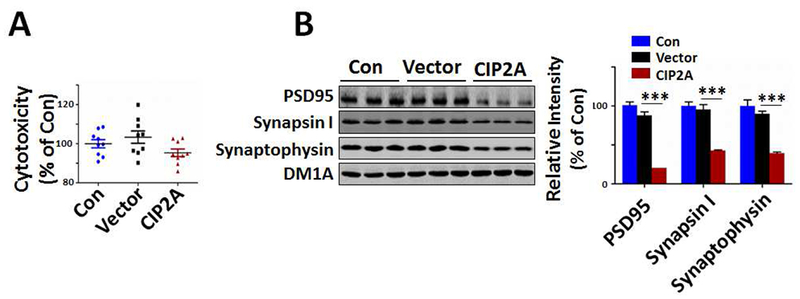
Activated astrocytes-derived toxic factors could promote neuronal death in retinal ganglion cells (RGCs), differentiated oligodendrocytes, and dopaminergic neurons when these cells were treated with reactive astrocyte-conditioned medium (Liddelow et al., 2017). To verify whether neurotoxic factors released in CIP2A-induced astrogliosis also promote neurodegeneration, we incubated rat primary cortical neurons (DIV 10) with CIP2A-overexpressed astrocytes-conditioned medium (CIP2A-ACM) for 5 days. LDH assay showed that CIP2A-ACM did not induce marked cell death comparing to control astrocytes-conditioned medium (Vector-ACM) and maintenance medium (Con) (Fig.3A). However, Western blotting analysis revealed a significant reduction in synaptic proteins including PSD95, synapsin I and synaptophysin (Fig.3B), indicating that CIP2A-induced astrogliosis promotes synaptic degeneration.
Fig.3.

CIP2A-induced astrogliosis promotes synaptic degeneration in primary neurons. Rat primary cortical neurons (DIV 10) were cultured in the conditional media from CIP2A-overexpressed asrocytes (DIV 10, infected with AAV-CIP2A for 5 days) for 4 days. (A) Cell viability was detected by LDH cytotoxicity assay kit. n = 9 well per group. (B) Left: PSD95, synapsin I and synaptophysin expression levels were detected by Western blotting using the antibodies indicated. DM1A was used as a loading control. Right: Quantitative analysis of the PSD95, synapsin I and synaptophysin levels. *** p < 0.001 versus Vector, n = 3 per group. Data are mean ± SEM.
3.4. Overexpression of CIP2A in astrocytes induces AD-like cognitive deficits
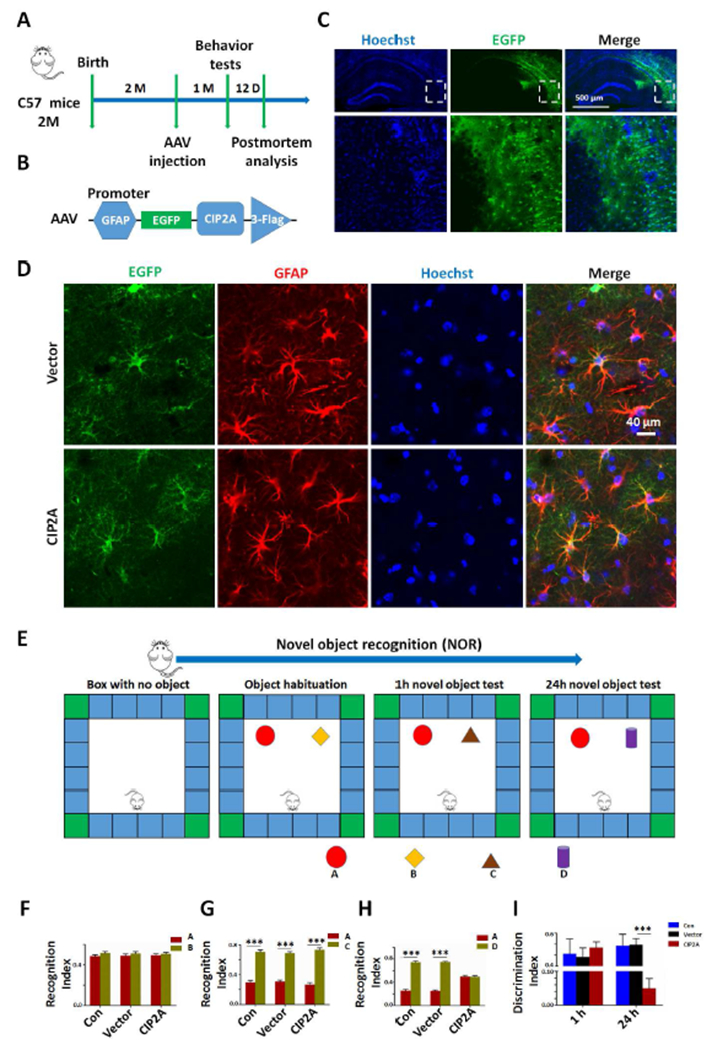
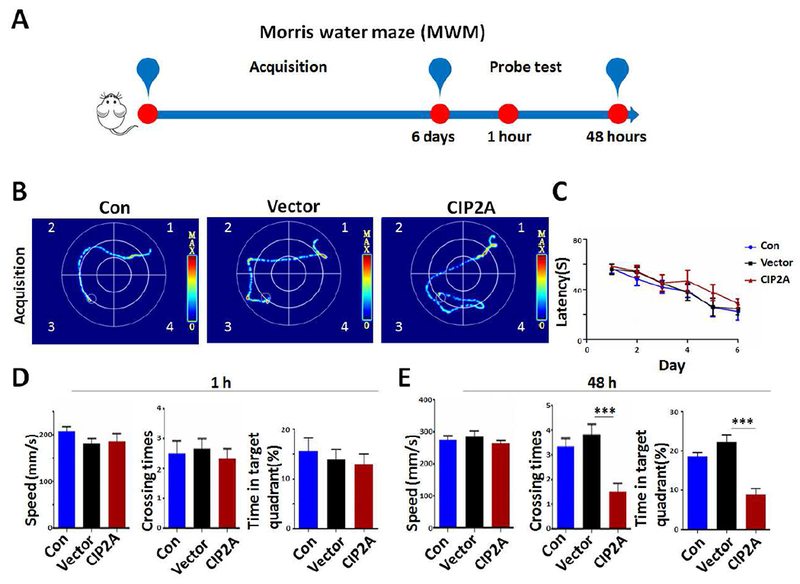
Synaptic degeneration underlies cognitive deficits in AD, to explore the effect of CIP2A overexpression in astrocytes on cognitive function, we performed bilateral intrahippocampal injection of PBS (Con), AAV-GFAP-Vector (Vector) or AAV-GFAP-CIP2A (CIP2A) virus to C57/BL6 mice aged 8 weeks (Fig.4A). A GFAP promoter in the AAV vector was designed to induce specific expression of target protein in astrocytes (Fig.4B). After 4 weeks, overexpression of CIP2A in hippocampus was confirmed by fluorescence microscopy (Fig.4C). It was observed that almost all GFP-expressing (green) cells were co-stained with GFAP imunofluorescence (red). The virus infect efficiency in astrocytes was 53% in control group and 47% in CIP2A-overexpressed group, respectively (Fig.4D). Behavioral tests were performed to evaluate the learning and memory ability of the mice. In novel object recognition test (NOR, Fig.4E), the recognition index was comparable among the three groups (Fig.4F) in the acquisition trial. In the test trial 1 h after the acquisition trial, mice in the three groups showed increased interest to the new objective with comparable recognition index (Fig. 4G). However, in the test trials 24 h after the acquisition trial, recognition index to new object was significantly decreased in the CIP2A-overexpressed mice (Fig. 4H), indicating impaired long-term memory. The analysis of the discrimination index at 1 h and 24 h test trials showed the same effects (Fig. 4I). These results suggest that CIP2A overexpression in astrocytes induces deficits in visual episodic memory. Next, we examined the effect of astrocytic CIP2A on the reference spatial memory using the Morris water maze (MWM, Fig.5A). In the acquisition training phase, the three group mice had the same latency, showing that learning ability was not impaired in AAV-GFAP-CIP2A virus-infected mice (Fig. 5B,C). In the test trials at 1 h, the three group mice showed the same speed, crossing times and swimming time in the target quadrant, indicating that astrocytic CIP2A overexpression has no effect on motion ability and short spatial memory (Fig.5D). In the test trials at 48 h, CIP2A virus-infected mice showed decreased crossing times and swimming time in the target quadrant compared to the PBS-injected or Vector virus-infected mice, with no changes in swimming speed, suggesting that CIP2A overexpression in astrocytes causes disruption in long-term spatial memory (Fig.5E). Taken together, overexpression of CIP2A in astrocytes promotes AD-like cognitive deficits.
Fig.4.
Overexpression of CIP2A in astrocytes induces visual episodic memory deficit in mice. (A) Timeline of the experiment. (B) The structure diagram of AAV-CIP2A. (C) AAV-CIP2A was injected into the hippocampal CA3 region of the C57/BL6 mice, and CIP2A expression was observed (green) 4 weeks after injection. Cell nuclei were stained with Hoechst (Blue) to show the hippocampus. Scale bar = 500 μm. (D) Brain slices in C were immunostained with anti-GFAP to show astrocytes; virus infection efficiency was obtained through calculating the ratio of co-stained (red and green) cells/ GFAP-positive (red) cells. (E) The experimental design of novel object recognition test (NOR). One day before the memory acquisition, the mice were habituated to the arenas for 5 min, as indicated in the first box. The second box showed the acquisition trial. The third and forth box showed the test trial conducted 1 h and 24 h after the acquisition trial. (F) The recognition index of object A and B in the acquisition trial. (G) The recognition index of object A and C in the test trial 1 h post the acquisition trial. *** p < 0.001 unfamiliar object C versus familiar object A. (H) The recognition index of object A and D in the test trial 24 h post the acquisition trial. *** p < 0.001 unfamiliar object D versus familiar object A. (I) The discrimination index of 1 h test trial and 24 h test trial. *** p < 0.001 versus Vector group, n = 6 mice per group. Data are mean ± SEM.
Fig.5.
Overexpression of CIP2A in astrocytes induces spatial memory deficit in mice. (A) The experimental design of Morris water maze test (MWM). (B) The representative searching trace on day 6 of the training. (C) The latency to reach the hidden platform. (D) Left: Swimming speed after removing the platform (1 h) during the probe trial. Middle: The number of times of crossing the position of the platform. Right: The percentage of time spent in target quadrant. Quadrant 3 is the target quadrant. (E) Left: Swimming speed after removing the platform (48 h) during the probe trial. Middle: The number of times to cross the position of the platform. Right: The percentage of time spent in target quadrant. *** p < 0.001 versus Vector, n = 6 mice per group. Data are mean ± SEM.
3.5. Overexpression of CIP2A in astrocytes causes neuronal loss, synaptic degeneration and impairments in synaptic plasticity in mouse brains
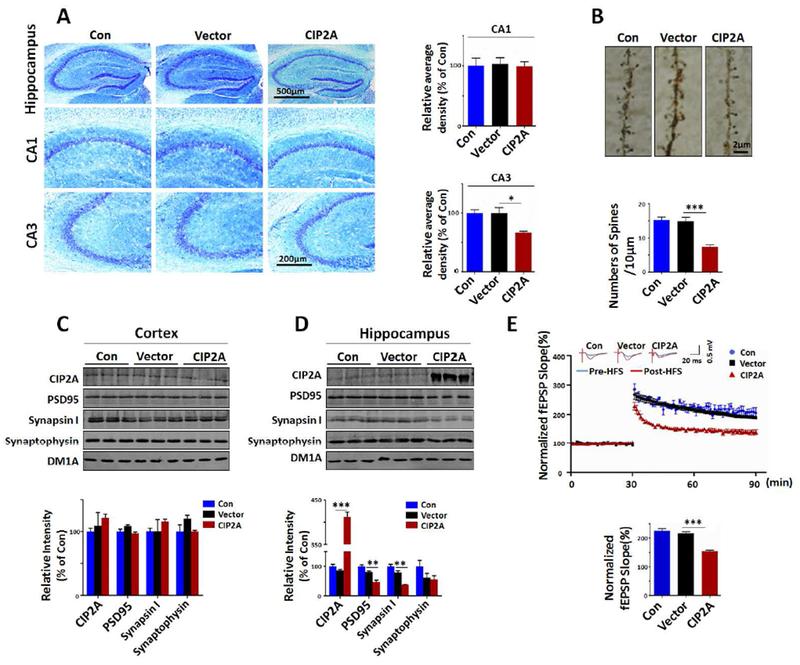
To investigate whether CIP2A overexpression in astrocytes induce neurodegeneration in mouse brains, we used Nissl staining to show neuron survival and found that the number of neurons was decreased in hippocampal CA3 region of CIP2A-overexpressed mice compared to the Con or Vector virus-infected mice (Fig.6A). Furthermore, the dendritic spine number of CIP2A virus-infected mice was significantly decreased comparing with the other groups (Fig.6B). Consistent with the experiment results in cultured cells, the protein levels of PSD95, synapsin I and synaptophysin were decreased markedly in the hipppcampus of CIP2A-overexpressing group but not in other two groups (Fig. 6C, D). Next, we recorded the long-term potentiation (LTP), a classic form of synaptic plasticity in brain hippocampal slices prepared from the three groups of mice. We found that the magnitude of LTP was significantly decreased in CIP2A-overexpressing group (Fig.6E). These in vivo data reinforced the idea that overexpression of CIP2A in astrocytes causes neuronal loss, synaptic degeneration and impairments in synaptic plasticity.
Fig.6.
Overexpression of CIP2A in astrocytes induces neuronal loss, synaptic degeneration and impairment in hippocampal LTP in mouse brains. (A) Left: Nissl staining of the brain slices from Con, AAV-Vector and AAV-CIP2A infected mouse brains. Right: Quantitative analysis of the neuron numbers in CA1 and CA3 regions of hippocampus, n = 3 brain slices per mouse, three mice per group. (B) Top: Representative dendritic spines of neurons in hippocampal CA3 region from Golgi impregnated hippocampus. Bottom: Quantitative analysis of averaged spine density (mean spine number per 10 μm dendrite segment), n = 10, from three mice per group. (C) Top: CIP2A, PSD95, synapsin I and synaptophysin expression levels were detected by Western blotting in mouse cortex. DM1A was used as a loading control. Bottom: Quantitative analysis of the protein levels, n = 3 per group. (D) Top: CIP2A, PSD95, synapsin I and synaptophysin expression levels were detected by Western blotting in mouse hippocampus. DM1A was used as a loading control. Bottom: Quantitative analysis of the protein levels, n = 3 per group. (E) Top: Normalized CA3-CA1 fEPSP mean slope recorded from the CA1 dendritic region in acute hippocampal slices. Bottom: Quantitative analysis of the normalized fEPSP slope, n = 15, 3 mice per group, 5 brain slices per mouse were recorded. * p < 0.05, ** p < 0.01, ***p < 0.001 versus Vector. Data are mean ± SEM.
3.6. Overexpression of CIP2A in astrocytes results in increased production of inflammatory cytokines and Aβ in mouse brains
To determine the active molecules responsible for neuronal and synaptic degeneration in CIP2A-overexpressed mice, we examined the inflammatory cytokines and Aβ production in mouse brain homogenates. IL-1α, TNF-α and Aβ40 levels were increased in hippocampus of CIP2A virus-infected mice comparing to the other groups, hippocampal IL-6 and Aβ42 levels showed no difference among the three groups. Comparable levels of the cytokines and Aβ were detected in brain cortex without CIP2A overexpression (Fig.7A, B). These results indicate that expression of CIP2A in astrocytes induces increase in inflammatory cytokines and Aβ production in vivo.
Fig.7.
Overexpression of CIP2A in astrocytes induces increased production of inflammatory cytokines and Aβ in mouse brains. (A) IL-1α, IL-6 and TNF-α in brain homogenates from the cortex and hippocampus of the mice were detected by ELISA, n = 6 per group. (B) Aβ40 and Aβ42 levels in brain homogenates were detected by ELISA, n = 6 per group. *** p < 0.001 versus Vector. Data are mean ± SEM. (C) Cultured primary astrocytes were treated with Aβ oligomers (2 μM) for 24 h, CIP2A levels were detected by Western blotting. Left, representative blots. Right, Quantitative analysis of the protein levels, n = 3 per group. *p < 0.05 versus Con. Data are mean ± SEM. (D) Cell culture media in (C) were collected, IL-1α, IL-6 and TNF-α in culture media were detected by ELISA, n = 9 well per group. * p < 0.05, *** p < 0.001, **** p < 0.0001 versus Con. Data are mean ± SEM. (E) A working model: In AD brains, CIP2A is up-regulated in astrocytes and induces reactive astrogliosis, the latter, results in overproduction of toxic cytokines and Aβ (identified in the present study), and probably reduced release of neurotrophins, thus promotes neuronal degeneration. Aβ further stimulates CIP2A overexpression and thus forms a vicious cycle.
3.7. Aβ induces CIP2A upregulation and release of inflammatory cytokines in primary astrocytes
The upstream factor inducing CIP2A upregulation in astrocytes in AD patients and transgenic mouse brains needs further exploration. Since Aβ itself can active astrocytes, we then explored the possibility that Aβ activated astrocytes through up-regulating CIP2A. Incubation of cultured primary astrocytes with Aβ oligomers resulted in increased CIP2A expression (Fig.7C) and cytokines release (Fig.7D), indicating that Aβ is a causative factor inducing CIP2A upregulation in astrocytes.
4. Discussion
Reactive astrogliosis has long been suspected to participate in AD pathogenesis, with the exact role being in debate (Birch, 2014). Active astrocyte has been shown to be beneficial through clearing Aβ by both phagocytosis and secretion of Aβ-degrading proteases (Dorfman et al., 2010; Koistinaho et al., 2004; Pihlaja et al., 2011; Yin et al., 2006). Consistently, attenuating astrocyte activation accelerates plaque deposition in APP/PS1 mice (Kraft et al., 2013). On the other side, there is a great deal of research showing harmful effects of active astrocytes in AD. Activated astrocyte is a significant source of Aβ (Forst and Li, 2017; Zhao et al., 2011). Aβ itself can reversely activate astrocytes and induce release of neurotoxic inflammatory factors such as cytokines and nitric oxide (Akama and Van Eldik, 2000; Hu et al., 1998; White et al., 2005). An in vivo study shows that reducing astrocyte activation ameliorates neurologic pathologies, and improves synaptic function and cognition in a mouse model of AD (Furman et al., 2012).
By analyzing transcriptome databases obtained from reactive astrocytes from different mouse injury models, Zamanian et al. found two subtypes of reactive astrocytes dependent on the type of inducing injury: Reactive astrocytes in ischemia exhibited a molecular phenotype that may be beneficial or protective, whereas reactive astrocytes induced by LPS exhibited a phenotype that may be detrimental (Zamanian et al., 2012). Thus, the conflicting findings on the role of astrogliosis in AD may derive from the distinct strategies for activating astrocytes in different studies. Further exploration by the same group showed that LPS-induced A1 phenotype reactive astrocytes lose the ability to promote neuronal survival, outgrowth, synaptogenesis and phagocytosis, and induce neuronal death. More importantly, A1 astrocytes make up a large proportion of astrocytes in AD brains (Liddelow et al., 2017). Thus, understanding the molecular steps leading to “harmful” A1 type astrocytes will help us find effective therapeutic target in preventing the detrimental effect of astrogliosis.
In the present study, we explored the role of CIP2A in inducing reactive astrogliosis in AD. CIP2A is first discovered as an oncoprotein overexpressed in a variety of peripheral tumors (Junttila et al., 2007; Khanna et al., 2013; Soo Hoo et al., 2002).Through specific inhibition of Protein Phosphatase 2A (PP2A), CIP2A selectively regulates the phosphorylation and stabilization of well-established cancer drivers such as MYC and E2F1, thus promotes the development of cancer (Junttila et al., 2007; Laine et al., 2013). Despite considerable levels of CIP2A expression in normal adult human and mouse brains (Junttila et al., 2007; Kerosuo et al. 2010), its function in the brain remains largely unknown. Our interest in CIP2A in astrocytes was originated from the observation that this protein was expressed at a comparable level in primary astrocytes when comparing to that in neurons. Also, in AD patients and transgenic mice, we observed that an increased CIP2A expression was correlated with GFAP levels. These data imply a potential role of CIP2A in reactive astrogliosis in AD.
We thus overexpressed CIP2A in cultured astrocytes, and found that upregulation of CIP2A induced typical morphological and molecular changes of astrogliosis. More importantly, CIP2A overexpression in culture system resulted in significant release of inflammatory cytokines such as IL-1, IL-6 and TNF-α, a phenotype of “harmful” A1 astrocytes. Further detection identified that CIP2A promoted Aβ production, which is also an identified event in activated astrocytes (Zhao et al., 2011). These findings indicate that CIP2A can promote the inducement of detrimental reactive astrocytes.
Both excessive Aβ and cytokines are destructive to synapses (Koffie et al., 2011; Liu et al., 2017; Rossi et al., 2012).To test the neurotoxicity of the CIP2A-promoted reactive astrocytes, we incubated the primary neurons with CIP2A-overexpressed astrocyte-conditioned medium (CIP2A-ACM). The CIP2A-ACM only induced modest but not significant cytotoxicity. However, functional synaptic proteins, such as PSD95, synapsin I and synaptophysin were all reduced markedly. These findings indicate that CIP2A-promoted astrogliosis may induce synaptic degeneration.
To further confirm this hypothesis in vivo, we injected AAV with GFAP promoter to the mouse hippocampus to induce specific CIP2A overexpression in astrocytes, and then test the cognition and the biochemical changes in the brain of the mice. Consistent with the findings in cells, upregulation of CIP2A in hippocampal astrocytes resulted in deficits in long term visual episodic and spatial memory, loss in neurons and dendritic spines, reduced expression of synaptic proteins, and impairment in hippocampal LTP. Detection of cytokine and Aβ in the brain homogenates also revealed an elevation in IL-1α, TNF-α and Aβ40 in the hippocampus of CIP2A-overexpressed mice.
The upstream factor that induces CIP2A upregulation in astrocytes in AD patients and transgenic mouse brains remains unknown. With the knowledge that Aβ can active astrocytes, we suspected that Aβ might be one causative factor. To identify this hypothesis, we treated primary astrocytes with Aβ oligomers and found that Aβ induced CIP2A overexpression and cytokines release, indicating that Aβ is an upstream factor inducing CIP2A upregulation in astrocytes in AD.
In summary, under normal physiologic conditions, astrocytes exert supportive function to neurons; However in AD brains, upregulated CIP2A induces neurotoxic A1 phenotype astrocytes which promote synaptic degeneration through releasing toxic cytokines and Aβ, the latter, further stimulates CIP2A overexpression and thus forms a vicious cycle to promote disease development (Fig. 7E). As a peripheral tumor promoter, CIP2A has already been druggable in cancer therapy, thus it deserves further investigation as potential therapeutic target in Alzheimer’s disease. Further studies such as CIP2A depletion in astrocytes will help us identify the role of CIP2A in astrocytes and its potential as a drug target.
Supplementary Material
Highlights.
CIP2A is upregulated in AD
CIP2A upregulation in astrocytes induces astrogliosis, release of cytokines and Aβ
CIP2A upregulation in astrocytes induces synaptic degeneration and memory deficits.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant numbers 81471304, 31771189) (RL), Natural Science Foundation of Hubei Province, China (grant number 2017CFA065) (RL), Integrated Innovative Team for Major Human Diseases Program of Tongji Medical College, HUST (JZW) and NIH (grant number R01MH079407) (HYM).
Funding information
National Natural Science Foundation of China, Grant Number: 31771189 and 81471304 (RL); Natural Science Foundation of Hubei Province, China, Grant Number: 2017CFA065 (RL); Integrated Innovative Team for Major Human Diseases Program of Tongji Medical College, HUST (JZW); NIH grant, Grant Number: RO1MH079407 (HYM)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement
The authors have declared that no conflict of interest exists.
References
- Akama KT, Van Eldik LJ, 2000. Beta-amyloid stimulation of inducible nitric-oxide synthase in astrocytes is interleukin-1beta- and tumor necrosis factor-alpha (TNF alpha)-dependent, and involves a TNF alpha receptor-associated factor- and NF kappa B-inducing kinase-dependent signaling mechanism. J. Biol. Chem 275, 7918–7924. [DOI] [PubMed] [Google Scholar]
- Birch AM, 2014. The contribution of astrocytes to Alzheimer’s disease. Biochem. Soc. Trans 42, 1316–1320. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E, 1992. The human entorhinal cortex: normal morphology and lamina-specific pathology in various diseases. Neurosci. Res 15, 6–31. [DOI] [PubMed] [Google Scholar]
- Carter SF, Schöll M, Almkvist O, Wall A, Engler H, Långström B, Nordberg A, 2012. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J. Nucl. Med 53, 37–46. [DOI] [PubMed] [Google Scholar]
- Crews L, Masliah E, 2010. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum. Mol. Genet 19, R12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorfman VB, Pasquini L, Riudavets M, López-Costa JJ, Villegas A, Troncoso JC, Lope F, Castaño EM, Morelli L, 2010. Differential cerebral deposition of IDE and NEP in sporadic and familial Alzheimer’s disease. Neurobiol. Aging 31, 1743–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C, Delatour B, Potier MC, 2009. Classification and basic pathology of Alzheimer disease. Acta Neuropathol 118, 5–36. [DOI] [PubMed] [Google Scholar]
- Frost GR, Li YM, 2017. The role of astrocytes in amyloid production and Alzheimer’s disease. Open bio. 7, 170228–170242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman JL, Sama DM, Gant JC, Beckett TL, Murphy MP, Bachstetter AD, Van Eldik LJ, Norris CM, 2012. Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer’s disease. J. Neurosci 32, 16129–16140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruol DL, 2016. Impact of Increased Astrocyte Expression of IL-6, CCL2 or CXCL10 in Transgenic Mice on Hippocampal Synaptic Function. Brain Sci. 6, pii: E19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Akama KT, Krafft GA, Chromy BA, Van Eldik LJ, 1998. Amyloid-beta peptide activates cultured astrocytes: morphological alterations, cytokine induction and nitric oxide release. Brain Res. 785, 195–206. [DOI] [PubMed] [Google Scholar]
- Junttila MR, Puustinen P, Niemela M, Ahola R, Arnold H, Bottzauw T, Ala-aho R, Nielsen C, Ivaska J, Taya Y, Lu SL, Lin S, Chan EK, Wang XJ, Grènman R, Kast J, Kallunki T, Sears R, Kähäri VM, Westermarck J, 2007. CIP2A inhibits PP2A in human malignancies. Cell 130, 51–62. [DOI] [PubMed] [Google Scholar]
- Kerosuo L, Fox H, Perala N, Ahlqvist K, Suomalainen A, Westermarck J,, Sariola H, Wartiovaara K, 2010. CIP2A increases self-renewal and is linked to Myc in neural progenitor cells. Differentiation 80, 68–77. [DOI] [PubMed] [Google Scholar]
- Khanna A, Kauko O, Böckelman C, Laine A, Schreck I, Partanen JI, Szwajda A, Bormann S, Bilgen T, Helenius M, Pokharel YR, Pimanda J, Russel MR, Haglund C, Cole KA, Klefström J, Aittokallio T, Weiss C, Ristimäki A, Visakorpi T, Westermarck J, 2013. Chk1 targeting reactivates PP2A tumor suppressor activity in cancer cells. Cancer Res. 73, 6757–6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Hyman BT, Spires-Jones TL, 2011. Alzheimer’s disease: synapses gone cold. Mol. Neurodegener 6, 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM, 2004. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat. Med 10, 719–726. [DOI] [PubMed] [Google Scholar]
- Kraft AW, Hu X, Yoon H, Yan P, Xiao Q, Wang Y, Lee JM, 2013. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 27, 187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine A, Sihto H, Come C, Rosenfeldt MT, Zwolinska A, Niemelä M, Khanna A, Chan EK, Kähäri VM, Kellokumpu-Lehtinen PL, Sansom OJ, Evan GI, Junttila MR, Ryan KM, Marine JC, Joensuu H, Westermarck J, 2013. Senescence sensitivity of breast cancer cells is defined by positive feedback loop between CIP2A and E2F1. Cancer Discov. 3, 182–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres A, 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhou LJ, Wang J, Li D, Ren WJ, Peng J, Wei XI, Xu T, Xin WJ, Pang RP, Li YY, Qin ZH, Murugan M, Mattson MP, Wu LJ, Liu XG 2017. TNF-α Differentially Regulates Synaptic Plasticity in the Hippocampus and Spinal Cord by Microglia-Dependent Mechanisms after Peripheral Nerve Injury. J. Neurosci 37, 871–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihlaja R, Koistinaho J, Kauppinen R, Sandholm J, Tanila H, Koistinaho M, 2011. Multiple cellular and molecular mechanisms are involved in human Aβ clearance by transplanted adult astrocytes. Glia 59, 1643–1657. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Vieitez E, Ni R, Gulyás B, Tóth M, Häggkvist J, Halldin A, Marutle A, Nordberg A, 2015. Astrocytosis precedes amyloid plaque deposition in Alzheimer APPswe transgenic mouse brain: a correlative positron emission tomography and in vitro imaging study. Eur. J. Nucl. Med. Mol. Imaging 42, 1119–11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi S, Furlan R, De Chiara V, Motta C, Studer V, Mori F, Musella A, Bergami A, Muzio L, Bemardi G, Battistini L, Martino G, Centonze D, 2012. Interleukin-1β causes synaptic hyperexcitability in multiple sclerosis. Ann. Neurol 71, 76–83. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, Mufson EJ, 2006. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobio. Aging 27, 1372–1384. [DOI] [PubMed] [Google Scholar]
- Simpson JE, Ince PG, Lace G, Forster G, Shaw PJ, Matthews F, Savva G, Brayne C, Wharton SB, 2010. Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobio. Aging 31, 578–590. [DOI] [PubMed] [Google Scholar]
- Sun XY, Tuo QZ, Liuyang ZY, Xie AJ, Feng XL, Yan X, Qiu M, Li S, Wang XL, Cao FY, Wang XC, Wang JZ, Liu R, 2016. Extrasynaptic NMDA receptor-induced tau overexpression mediates neuronal death through suppressing survival signaling ERK phosphorylation. Cell Death Dis. 7, e2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, 2009. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 32, 638–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoo L. Soo, Zhang JY, Chan EK, 2002. Cloning and characterization of a novel 90 kDa ‘companion’ auto-antigen of p62 overexpressed in cancer. Oncogene 21, 5006–5015. [DOI] [PubMed] [Google Scholar]
- Wang XL, Zeng J, Feng J, Tian YT, Liu YJ, Qiu M, Yan X, Yang Y, Xiong Y, Zhang ZH, Wang Q, Wang JZ, Liu R, 2014. Helicobacter pylori filtrate impairs spatial learning and memory in rats and increases β-amyloid by enhancing expression of presenilin-2. Front. Aging Neurosci 6, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JA, Manelli AM, Holmberg KH, Van Eldik LJ, Ladu MJ, 2005. Differential effects of oligomeric and fibrillar amyloid-beta 1–42 on astrocyte-mediated inflammation. Neurobio. Dis 18, 459–465. [DOI] [PubMed] [Google Scholar]
- Wisniewski HM, Wegiel J, 1991. Spatial relationships between astrocytes and classical plaque components. Neurobio. Aging 12, 593–600. [DOI] [PubMed] [Google Scholar]
- Yin KJ, Cirrito JR, Yan P, Hu X, Xiao Q, Pan X, Bateman R, Song H, Hsu FF, Turk J, Xu J, Hsu CY, Mills JC, Holtzm DM, Lee JM, 2006. Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J. Neurosci 26, 10939–10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA, 2012. Genomic analysis of reactive astrogliosis. J. Neurosci 32, 6391–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, O’Connor T, Vassar R, 2011. The contribution of activated astrocytes to Aβ production: implications for Alzheimer’s disease pathogenesis. J. Neuroinflammation 8, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.