

Abstract
Methyl isocyanate (MIC) is a highly toxic industrial chemical causing acute lethality after inhalation. The objective of this study was to determine whether alterations in hemostasis also occur in the immediate hours after exposure. Male rats were exposed to MIC (125-500 ppm) by nose-only vapor inhalation for 30 min. Arterial O2 saturation was monitored prior to exposure, and hourly thereafter. Rats were euthanized at 1, 2, 4 and 8 hr and plasma analyzed for recalcification clotting time, Tissue Factor (TF) activity and protein levels. Hypoxemia, as assessed by pulse oximetry, was an early feature of MIC inhalation. In contrast to sham or low (125 ppm) concentrations, 250 and 500 ppm MIC caused significant declines in blood oxygen saturation (% SpO2) at 1 hr, which remained at deficit during the post exposure period. Commensurate with hypoxemia, plasma clotting time was significantly accelerated 1 hr after MIC inhalation (sham treatment: 955 ± 62.8 seconds; 125 ppm MIC: 790 ± 62 seconds; 250 ppm: 676 ± 28.0 seconds; 500 ppm: 581 ± 175 seconds). This procoagulant effect was transient, with no difference observed between sham and all MIC groups by 8 hr. Similarly, elevated TF activity and protein were detected in plasma 1 hr after MIC inhalation, each of which showed a progressive decline back to control levels at later timepoints. This study demonstrates that MIC inhalation resulted in hypoxemia and transient hypercoagulability of blood. Accelerated clotting occurred rapidly and was likely due to intravascular TF, which initiates the extrinsic coagulation pathway.
Keywords: Methyl isocyanate, Bhopal agent, Tissue Factor, coagulation, Tissue Factor pathway inhibitor (TFPI), inhalation, respiratory
Introduction
The mechanisms of airway injury by toxic inhaled chemicals (TICs) are incompletely understood, and therapies for such injuries are often lacking, resulting in high morbidity and mortality in exposed individuals. Accidental discharge of TICs can be extremely hazardous as evidenced by the release of methyl isocyanate (MIC) at Bhopal, India, in 1984. During that incident, more than 500 people perished in the immediate vicinity before emergency response arrival. At least 2,000 additional individuals died in the immediate days after exposure [1]. Reports from the media indicated that many deaths occurred within minutes to a few hours after MIC had drifted into Bhopal [1]. Due to the emergent disaster, however, no systematic autopsy data has been published.
In the aftermath of the catastrophe, studies have attempted to determine the cause of early mortality. Studies have estimated the MIC concentration to be on the order of 1,000 ppm at Bhopal ground-level locations, which is consistent with the reported large numbers of deaths and injuries of humans and animals [2]. In a series of published papers, Union Carbide investigators performed MIC exposure studies on female rats and guinea pigs to assess whether certain physiological mechanisms were responsible for acute mortality [3]. Unlike reports of the human condition, they did not observe any loss of consciousness or convulsions in either rodent model after 15 min exposure to 100- 1,000 ppm MIC. The possibility that MIC exposure could elicit disseminated intravascular coagulation (DIC) was also evaluated in these studies [4]. Histological examination of the lung did reveal fibrin deposition in the lumina of small vessels in both species as an early (30 min) post exposure event. However, in animal subjects analyzed at later time points (1, 2 and 4 hr), these lesions were absent. Due to their absence and the lack of any significant decrease in platelet counts, a link between MIC and DIC was not substantiated. Limitations of this study are that functional coagulation parameters beyond platelet counts were not reported, and DIC was not evaluated in any peripheral tissue besides the lung. Studies by Kolb et al have suggested that acute toxicity of MIC could be mediated by its effects on the plasma membrane of endothelial cells, resulting in fluid loss from the vascular compartment, with subsequent hypovolemia and hypotension. [5, 6] MIC toxicity due to inadequate circulating volume and perfusion is an attractive hypothesis since these events can lead to rapid endothelial damage and organ failure. However, since these latter studies were performed by subcutaneous injection of MIC, it is uncertain whether hypovolemia of the same magnitude would also occur in response to inhalation exposure. Also inconclusive was whether the observed fluid loss was due to endothelial injury and vascular leak or inflammation-mediated increase in permeability and third spacing. It was also hypothesized that MIC could form glutathione conjugates, which could spontaneously revert to MIC after leaving the lung, facilitating multisystem organ toxicity.[7] Because our understanding of MIC toxicity remains incomplete, a more detailed understanding of the mechanism(s) of cardiopulmonary injuries following MIC inhalation is needed.
Previously we have identified several adverse effects resulting from inhalation of agents similar to MIC, including the alkylating agents half-mustard (2-choroethyl ethyl sulfide, CEES) and sulfur mustard. Among these findings were in vitro and in vivo evidence of activation of Tissue Factor (TF), activation of the clotting cascade, and fibrin deposition in the airways [8]. In addition, we found activation of intravascular coagulation resulting in thrombotic events in the lung vasculature [9]. Leakage of plasma contents, including blood-clotting factors, into airways from the bronchial circulation further contributed to extravascular coagulation there. Finally, the persistence of such fibrinous lesions in airways was assured by the inhibition of multiple fibrinolytic pathways following inhalation of these TICs [10]. Here we report an early procoagulant effect, including a rapid, yet transient, expression of TF in the circulating plasma from MIC-exposed rats.
Material and methods
Reagents
Tissue Factor Pathway Inhibitor; TFPI; Tifacogin, was acquired from Novartis; Basel, Switzerland, was produced in E coli, and was a recombinant form of endogenous human TFPI protein. Hirudin was purchased commercially from Prospec; East Brunswick, NJ.
Laboratory animals and handling
Male Sprague-Dawley rats (250-300 g) were obtained from Charles River (Kingston, NY). Upon arrival, each animal was assigned a unique identifier number and acclimated to laboratory conditions for 5-7 days prior to study, including acclimation to nose-only exposure chambers at intervals for 72 hours before study. Food and water were provided ad libitum. To evaluate MIC toxicity, 40 rats were randomized by body weight into 5 exposure regimens: sham exposure, 125, 250, 500 ppm MIC. All procedures were performed according to approved Institutional Animal Care and Use Committee protocols of MRIGlobal (Kansas City, MO).
MIC synthesis and rat inhalation exposure model.
Methyl isocyanate (MIC) was prepared at MRIGlobal (Kansas City, MO) by use of the Curtius Rearrangement method, which involved refluxing acetyl chloride with sodium azide in toluene until all starting materials were consumed. The product was shown to be 99.2% pure MIC by gas chromatography-flame ionization detection (GC-FID). During the exposure of rats, the entire inhalation system was placed in a fume hood. A nose-only (CH Technologies; Westwood, NJ) inhalation system was used for the exposure of rats. A proprietary custom vapor diffusion system (MRIGlobal) was used to generate MIC vapor. Rats were acclimated to the exposure system for three sequential days prior to MIC exposure. The vapor was then delivered to the plenum of the exposure system in a mixture of dry nitrogen (50 mL/min UHP nitrogen) that was then blended with and HEPA-carbon-filtered dry air (10-15 L/min, depending on the desired concentration). To assure consistent MIC concentrations, gas constituents were monitored continuously in real time during exposures by Fourier transform infrared spectroscopy (FTIR). This was performed by accessing the vapor via a 3-way valve downstream of the central exposure plenum. Adjustments in the amount of MIC delivery were made as necessary to limit deviation in the height of the absorption peak for MIC in the atmosphere to less than 1% during the course of exposure. Exhaust from the system was then scrubbed in 10% NaOH and next exhausted through the fume hood via a pump. Conscious rats were exposed for 30 min, followed by in cage recovery for 1, 2, 4, or 8 hr. MIC concentrations used in the study were selected on the basis of pilot studies demonstrating that the lethal concentration (LC50) at 8 hr for MIC was 1,000 ppm. Sham animals were challenged by exposure to an atmosphere devoid of MIC by use of the identical nose-only system, in the same fume hood where MIC inhalation was performed.
Pulse oximetry
Oxygen saturation of conscious rats was monitored using the MouseOx pulse oximeter (Starr Life Science, Pittsburgh, PA). The large rat (300 g) sensor collar was used. Three measurements per time point were taken.
Plasma isolation and clotting time measurements
Blood samples were obtained after each subject was administered a terminal dose of anesthesia (ketamine/xylazine) and unresponsive to toe pinch. An abdominal incision was performed, followed by direct visualization of the descending aorta and placement of a butterfly catheter. Blood was then collected into a pre-citrated (0.5 mL of 3.2% sodium citrate) 5 mL syringe, and filled to 5 ml of volume, followed by exsanguination of the rat. Citrated blood was not refrigerated, centrifuged within 15 minutes of collection at 3,000 g for 15 minutes at 20°C, and the upper ¾ of plasma was immediately removed, aliquoted and frozen at −80°C. To measure clotting time, 100 μL of plasma was mixed with an equal volume of 0.9% saline, of which 50 μL was transferred in triplicate to wells on a 96-well plate already containing 100 μL of saline per well. The clotting reaction was then initiated by adding 50 μL of 30 mM CaCl2. Absorbance values at 405 nm were measured at 20-s intervals in a SpectraMax 340 plate reader (37°C). A well was considered clotted at the first time point when absorbance reached within 97% or greater of its maximal value during the 20-min run.
Tissue factor activity assay
TF activity was assayed as FVII-dependent activation of FX as illustrated in the diagram provided in Figure 1A. Platelet poor plasma was thawed and 10 μL added to a 96-well plate containing 2 μL of hirudin (25 μg/ml in 0.9% saline; Prospec; East Brunswick, NJ) and 13 μL of saline. The well contents were gently mixed, followed by the addition of 100 μL of reaction buffer (125 mM NaCl, 50 mM tris, 1 mM CaCl2 (pH 8.3)) and 25 μL of reaction buffer containing 1.25 mg/ml of chromogenic substrate (S-2765; Chromogenix; Bedford, MA). For reactions requiring inhibition of TF/FVIIa pathway, TFPI was added to the reaction buffer to achieve a 0.5 μg/ml final concentration. After all additions, plates were mixed by the plate reader (Molecular Devices, Sunnyvale CA) and kinetic absorbance readings (405 nm) recorded every 30 s for 1 hr. Representative kinetic curves of TF activity assay in rat plasma obtained after sham or MIC exposure are depicted in Figure 1B. The reaction rate for plasma incubated with hirudin+TFPI was subtracted from mean reaction rate of triplicate wells containing plasma + hirudin only, and this value (rate of absorbance increase) was expressed relative to the activity of Innovin recombinant human thromboplastin reagent (Innovin; Baxter, Unterschleissheim, Germany). A standard curve of serial dilutions of the Innovin was assayed during each plated experiment. The activity of 1x Innovin (manufacturer’s specifications) was designated as 1,000 Units of relative TF activity. To generate the standard curve, variable amounts of Innovin were reacted with fixed amounts of FVII (25 ng; Enzyme Research Labs; South Bend, IN) and FX (50 ng; Enzyme Research Labs) in 125 μl reaction buffer, followed by 25 μl of reaction buffer containing 1.25 mg/ml of chromogenic substrate.
Figure 1.

(A) Tissue Factor (TF) activity assay diagram. TF is the physiologic initiator of blood coagulation. An assay was developed (Figure 1A) to measure the amidolytic activity of the TF/FVIIa complex in plasma by quantifying the amount of FXa generated. To detect FXa, plasma is incubated with a substrate that undergoes FXa-dependent cleavage, releasing a yellow para-nitroaniline (pNA) chromophore. However, thrombin also increases as a function of FX activation, and is also capable of cleaving the FXa substrate (red hatched line) or clotting the plasma in the reaction. Only after inhibition of thrombin by hirudin does absorbance change become directly proportional to TF activity. A second inhibitor, Tissue factor pathway inhibitor (TFPI) protein, is used to inhibit TF-dependent FX activation. Determining rates of substrate cleavage in the presence and absence of these two inhibitors allows TF activity to be measured. (B) Representative TF activity assay measurements from rat plasma obtained after sham or MIC exposure. As shown, TF activity was measured in plasma by performing 6 separate reactions for each subject. Reaction 1 and 2 contained plasma incubated with chromogenic substrate (CS) only, reaction 3, 4, and 5 contained plasma + CS + hirudin, and reaction 6 was plasma incubated with CS + hirudin and TFPI. The difference in slope between red and blue curves represents plasma TF activity.
Western blot analysis
Platelet-poor plasma was thawed, transferred to a new tube containing an equal volume of EBC lysis buffer {20 mM NaCl; 0.5% (v/v) Nonidet P-40; 5 μg/ml leupeptin; 10 μg/ml aprotinin; 50 μg/ml PMSF; 50 mM Tris-Cl (pH 8.0)} and rocked at 4°C for 20 minutes. Samples were then diluted to 1:50 dilution in 0.9% saline containing 5× Laemmli buffer and loaded onto a 4–15% gradient SDS-polyacrylamide gel. After transfer, membranes were initially stained with Ponceau S, blocked for 1 hr in Tris-buffered saline with 0.5% Tween 20 (TBST) containing 5% powdered milk, followed by overnight incubation with a rabbit monoclonal anti-TF Ab (ab151748; Abcam, Cambridge, MA). Blots were washed in TBST 20 for 1 hr, followed by incubation with a goat anti-rabbit secondary (1:7,000; Southern Biotechnology, Birmingham, AL).
Statistics
Statistical analyses utilized Prism 4 software. Means were compared by Kruskal-Wallis test followed by Dunn’s post-test. P values < 0.05 were considered significant.
Results
Acute hypoxemia in response to MIC inhalation.
These studies were designed to assess the impact of MIC exposure on arterial oxygen saturation of blood. Conscious rats were given nose-only inhalation of either air (sham), or neat MIC (resulting in final inhaled concentrations of 125, 250 or 500 ppm) and allowed to recover for 8 hr. As shown in Figure 2, oxygen delivery to tissues, as measured by pulse oximetry, was decreased in an inverse inhalation dose-related manner. All rats had peripheral arterial oxygen saturation (SpO2) levels above 90% prior to MIC inhalation. At 1 hr after exposure, little change in SpO2 was evident in sham or 125 ppm MIC-exposed rats. Mean SpO2 of the 125 ppm group did decrease relative to sham group levels by 3 hour, but this decline remained modest (at or above 90% SpO2) over the course of the study. In contrast, a pronounced decline in SpO2 was evident at 1 hr in groups exposed to 250 or 500 ppm MIC. Following this 1 hr nadir, there was a slight recovery in SpO2 by 2 hr that was evident in both groups. After 2-3 hr, no additional rebound in oxygenation was observed in the 250 or 500 ppm groups, with all subjects demonstrating significantly lower SpO2 throughout the remainder of the 8 hr study.
Figure 2.

Effect of acute MIC nose-only inhalation exposure on oxygen delivery to tissues in rat as assessed by pulse oximetry. Rats were exposed to 0, 125, 250, or 500 ppm MIC for 30 minutes, followed by an 8 hr observation period. Pulse oximetry was performed immediately prior to exposure, and hourly thereafter to assess arterial oxygen saturation. The dotted horizontal line indicates the usual threshold for oxygen supplementation therapy in human patients. *P < 0.05 and denotes significant difference from sham-exposed (0 ppm) group at 1 hr.
Protracted time course studies beyond 8 hours’ observation after MIC inhalation were not conducted. Bronchoalveolar lavage and histopathology studies were performed and that reporting is beyond the scope of the current presentation. At 8 hours after MIC inhalation (125 and 250 ppm), BALF fluids revealed scant inflammation, modest but variable sloughed epithelium, and variable microhemorrhage. These findings of sloughed epithelium, inflammation and microhemorrhage were somewhat more pronounced at 500 ppm. Histopathology and airway microdissection revealed significant upper airway fibrin deposition, leading to partial airway obstruction of central trachea and mainstem bronchi, most severe at 500 ppm and 250 ppm, but relatively minor at 125 ppm MIC (data not shown).
Accelerated rate of plasma clotting time in response to MIC inhalation.
The clotting times of platelet-poor plasma from rats were determined to assess whether MIC inhalation had a procoagulant effect on systemic circulating blood. Shown in Figure 3 are representative clotting curves obtained 1 hr after sham- or MIC-exposure (500 ppm). These hemodynamic measurements were performed by monitoring the sigmoidal absorbance increase of plasma, which was proportional to sample viscosity and translucence. Absorbance values eventually reached a plateau phase, at which time the samples had attained a semisolid clotted state. At 1 hr after exposure (Figure 4), plasma from rats receiving sham treatment exhibited a mean clotting time of 955 ± 62.8 seconds. In rats exposed to 125 ppm MIC, mean clotting time was 790 ± 62 seconds. In contrast, 250 ppm and 500 ppm MIC inhalation significantly reduced mean clotting time to 676 ± 28.0 and 581 ± 175 seconds respectively. At 2 hr after exposure, clotting times of all MIC-exposed groups were significantly more rapid than sham exposure, (sham: 900 ± 46 sec; 125 ppm 720 ± 60.5 sec; 250 ppm 733.3 ± 52.4 sec; 500 ppm 675.0 ± 61.4 sec). The procoagulant effect of MIC exposure began to wane by 4 hr, (sham: 970.0 ± 25.69 sec; 125 ppm 950 ± 142.1 sec; 250 ppm 770 ± 74.8 sec; 500 ppm 665.0 ± 56.8 sec). By 8 hr, there was no significant difference between sham treatment (974 ± 44 sec) and MIC exposed groups (125 ppm 932.5 ± 136.5 sec; 250 ppm 822.2 ± 105.4 sec; 500 ppm 850 ± 213.5 sec).
Figure 3.
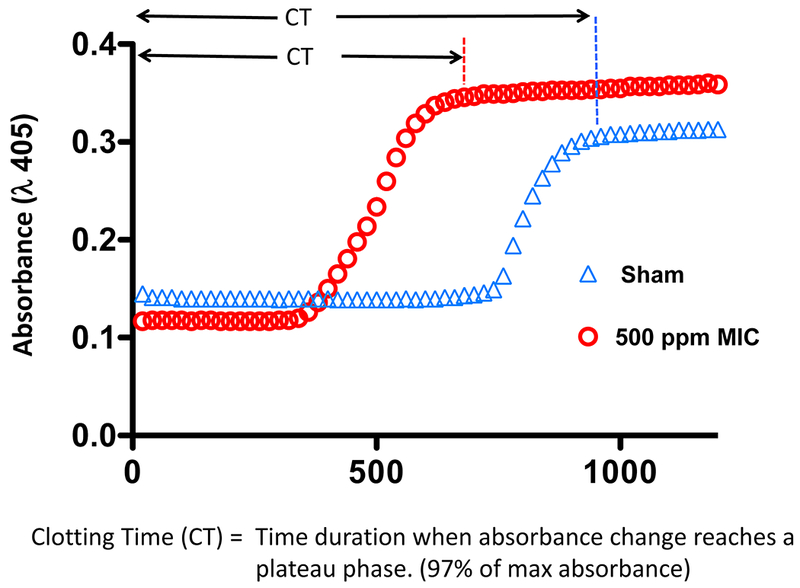
Effect of MIC inhalation on clotting time of rat plasma. Clotting time was assessed in 96-well format by recalcification of platelet-poor plasma and spectrophotometric measurement of the time-dependent absorbance increase that occurs due to clot formation. Two representative plasma clotting curves are presented: One was obtained from a plasma sample obtained after sham exposure (blue triangles; 0 ppm) and a second curve was from plasma obtained after MIC exposure (red circles; 500 ppm). Absorbance values eventually reached a plateau phase, at which time samples have attained a semisolid clotted state that is visible without magnification. The clotting time (CT) of the reaction was defined as the time to reach this plateau in absorbance (97% of the absorbance maximum).
Figure 4.

Effect of MIC inhalation exposure concentration (0–500 ppm, 30 min) on clotting time. Plasma clotting time was measured at 1, 2, 4 and 8 hr after sham exposure (0 ppm) or MIC inhalation (125, 250, 500 ppm). Reactions were initiated by addition of CaCl2, with spectrophotometric measurement of time-dependent absorbance increases. Each plotted symbol represents the mean clotting time in seconds of three replicates ± SEM for an individual rat subject. *P < 0.05 and denotes a significant difference for MIC-exposed versus sham-exposed (0 ppm) plasma samples. NS indicates that there was no difference between sham- and MIC-exposed groups at 8 hr (n = 4–8 per group).
TF activity and protein levels in plasma were rapidly increased after MIC inhalation.
TF activity in sham and 500 ppm MIC-exposed rat plasma samples was measured using a modified chromogenic assay (described in Figure 1). As shown in Figure 5, at 1 hr, the activity of MIC samples (1.81 ± 0.26 Activity Units-AU) was nearly 4-fold higher than the sham group (0.49 ± 0.08 AU). By 4 hours after exposure, however, differences in TF activity between sham and MIC were no longer significant (sham: 0.65 ± 0.1; MIC: 1.47 ± 0.29 AU), and by 8 hr this had nearly returned to baseline levels (sham: 0.56 ± 0.12; MIC: 0.996 ± 0.09). Consistent with TF activity change, Western blot analysis (Figure 6) revealed elevated TF protein in plasma at 1 hr after MIC exposure. By 8 hr post MIC exposure, however, no differences in TF levels were observed between sham and MIC samples.
Figure 5.

MIC inhalation caused a transient elevation of TF activity in plasma. Rats were exposed to sham (0 ppm) or MIC (500 ppm) and TF activity analyzed at 1, 4 and 8 hr post-exposure. Each symbol represents mean ± SE of 3 determinations for each individual plasma sample (n = 4 rats in sham and 6 rats in MIC group). *P < 0.05 compared with corresponding sham-exposed control fraction. TF activity was expressed relative to the activity of recombinant human TF (Innovin; 1 × strength designated as 1,000 units arbitrary).
Figure 6.

Effect of sham or MIC exposure on TF protein expression in rat plasma. (A and B) Western blotting was performed to detect TF 1 and 8 hours after exposure. Gel loading for each blot (from left to right) consisted of 4 plasma samples from sham- and 4 from MIC-exposed rats. (C) Ponceau S-staining of the nitrocellulose membrane imaged in 6B is shown to demonstrate equal loading of loading of plasma samples.
Discussion
The present study demonstrates the acute effects of MIC inhalation on blood coagulation in rats. Early time points (1- 8 hr) were analyzed in an effort to reflect the rapid onset of cardiorespiratory illness and early mortality of the Bhopal disaster. Indeed, the prompt onset of decreased tissue oxygen delivery in our study indicated the presence of a severe cardiopulmonary and/or circulatory disease process(es). The principal findings of this study were that MIC inhalation elicited a rapid and transient rise in TF activity, which may have been responsible for the coinciding procoagulant activity of blood. Acute MIC toxicity studies have been performed previously in rat and guinea pig. Studies by Dodd et al demonstrated lowered PaO2, an effect of compromised gas exchange caused by sloughing of epithelial cells from bronchial, bronchiolar, and alveolar regions epithelium [3, 11]. Our results were consistent with these earlier studies in that we observed marked hypoxemia at early time points. At higher concentration MIC exposures, a decline in SpO2 was evident as early as 1 hr post exposure, and pulse oximetery values remained at this low level for the remainder of the 8 hr study. Although early onset hypoxemia was demonstrated at the higher concentration exposures in the present study, monitoring was terminated prior to the onset of mortality. Herein we provide new data demonstrating that MIC inhalation can elicit a transient rise in systemic TF activity and activation of the coagulation pathway (accelerated plasma clotting). A growing body of evidence suggests that active circulating TF, probably borne on microparticles, can directly cause intravascular coagulation and thrombosis [12, 13].
To determine TF coagulant activity in our MIC plasma samples, we employed a Factor Xa-dependent chromogenic assay. Factor X (FX) is a vitamin K-dependent plasma glycoprotein that plays a pivotal role in hemostasis. In its zymogen form it can be activated by either the TF-FVIIa complex or the factor IXa-VIIIa pairing, resulting in the proteolytic cleavage of FX on its heavy chain. Such cleavage splits off the carboxyl terminus, releasing the activation peptide (FXa), which can subsequently convert prothrombin to thrombin in the presence of phospholipids. Our current assay measures the ability of TF/FVIIa complex to activate FX by use of a purportedly specific FXa substrate that releases a yellow chromophore upon cleavage. However, thrombin also increases as a function of FX activation, and thrombin can also cause low-level cleavage of this substrate (data not shown). Hirudin is therefore introduced into the assay to inhibit thrombin-specific cleavage and to prevent clotting of the plasma during the reaction. The endogenous anticoagulant TFPI was also used to verify that the assay was measuring activity of the extrinsic pathway blood factors. Structurally, TFPI is comprised of several Kunitz domains, each of which bind and inhibit TF, FVIIa, and FX [14, 15]. Assay specificity toward measurement of TF-dependent FX activity was demonstrated by the observation that all plasma samples from MIC subjects possessed negligible activity when TFPI was introduced into the reaction (depicted in Figure 4B). An additional advantage of the current assay was that it measured functional TF activity present in plasma, and not merely levels of TF antigen as detected by ELISA, which may not correlate with activity due to TF encryption or delipidation [16].
The transient effect of MIC on clotting time and TF activity may relate to the efficiency of blood hemostasis in mammalian systems. For instance, the early TF-induced coagulation abnormalities experienced by rats in this study may be compensated for by endogenous inhibitor systems. The first of these is circulating TFPI. After clotting is initiated by the coupling of TF with FVII and FX at cell membrane surfaces, TFPI binds and inhibits these entities by formation of a quaternary complex, neutralizing the ability to activate thrombin. Our in vitro results demonstrate that TF activity in plasma samples can be suppressed by addition of supplemental TFPI. Secondly, nascent microthrombi containing TF can be eliminated from vasculature by the fibrinolytic action of plasmin. When the circulating burden of TF does not exceed the neutralizing capacity of TFPI and fibrinolytic pathways, DIC is considered to be “compensated”. During early periods of compensated DIC, clotting factors and platelet levels may continue to be normal. In contrast, sepsis- or trauma-associated coagulopathies can develop “decompensated” DIC because continuous TF availability can exhaust TFPI levels in blood, leading to rampant thrombin activity and ungovernable fibrin formation.
On the basis of our recent findings, several distinctions between MIC and SM inhalation toxicity can be made. MIC causes extremely rapid onset distress, hypoxemia and injury, whereas SM has a latency period of 4 or more hours before the onset of hypoxemia and morbidity. Changes in coagulation after sulfur mustard inhalation can be delayed relative to MIC, requiring up to 8 hr before they are detected. Additionally, the procoagulant effect of SM may persist for 24 hr or longer after initial exposure. Possible explanations for these differences are that SM must undergo transition to a sulfonium ion intermediate before it is able to alkylate biomolecules, and is more lipophilic than MIC. Differences in these properties, as well as chemical reactivities, may influence distribution of inhaled vapors, site of deposition, and half-life in the airway. In conclusion, MIC inhalation gave rise to increased circulating TF activity and blood coagulation. A limitation of the current study was that it did not delineate from what cell and tissue type(s) the TF was derived. Because of its rapid elevation, TF may not have been a manifestation of apoptosis, or caused by a change in gene expression [17]. Although speculative, one possibility is that MIC inhalation caused rapid destruction of airway epithelium and subsequent release of membrane-derived particles containing TF. Consequently, these microparticles may have gained entrance into the systemic vasculature and initiated coagulation. It has previously been demonstrated that the bronchial vasculature is a site of leakage and ingress of clotting factors and procoagulant matter after chemical inhalation [18]. The procoagulant effects of MIC were transient, however, and suggested that healthy subjects might be able to compensate by neutralizing the early TF release.
Acknowledgement:
This research is supported by the CounterACT Program, National Institutes of Health (NIH), Office of the Director, and the National Institute of Environmental Health Sciences (NIEHS), Grant Numbers 5U54 ES015678 (CWW) and 1 U54 ES027698-01 (CWW) and NIH grant AR055073. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Environmental Health Sciences or the National Institutes of Health.
Footnotes
Declaration of Interest: The authors report no declarations of conflict of interest.
References:
- [1].Dhara VR, Dhara R, The Union Carbide disaster in Bhopal: a review of health effects, Archives of environmental health 57(5) (2002) 391–404. [DOI] [PubMed] [Google Scholar]
- [2].Havens J, Walker H, Spicer T, Bhopal atmospheric dispersion revisited, J Hazard Mater 233-234 (2012) 33–40. [DOI] [PubMed] [Google Scholar]
- [3].Dodd DE, Frank FR, Fowler EH, Troup CM, Milton RM, Biological effects of short-term, high-concentration exposure to methyl isocyanate. I. Study objectives and inhalation exposure design, Environ Health Perspect 72 (1987) 13–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Troup CM, Dodd DE, Fowler EH, Frank FR, Biological effects of short-term, high-concentration exposure to methyl isocyanate. II. Blood chemistry and hematologic evaluations, Environ Health Perspect 72 (1987) 21–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kolb WP, Savary JR, Troup CM, Dodd DE, Tamerius JD, Biological effects of short-term, high-concentration exposure to methyl isocyanate. VI. In vitro and in vivo complement activation studies, Environ Health Perspect 72 (1987) 189–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jeevarathinam K, Selvamurthy W, Ray US, Mukhopadhyay S, Thakur L, Acute toxicity of methyl isocyanate, administered subcutaneously in rabbits: changes in physiological, clinico-chemical and histological parameters, Toxicology 51(2-3) (1988) 223–40. [DOI] [PubMed] [Google Scholar]
- [7].Slatter JG, Rashed MS, Pearson PG, Han DH, Baillie TA, Biotransformation of methyl isocyanate in the rat. Evidence for glutathione conjugation as a major pathway of metabolism and implications for isocyanate-mediated toxicities, Chem Res Toxicol 4(2) (1991) 157–61. [DOI] [PubMed] [Google Scholar]
- [8].Rancourt RC, Veress LA, Guo X, Jones TN, Hendry-Hofer TB, White CW, Airway tissue factor-dependent coagulation activity in response to sulfur mustard analog 2-chloroethyl ethyl sulfide, Am J Physiol Lung Cell Mol Physiol 302(1) (2012) L82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].McGraw MD, Rioux JS, Garlick RB, Rancourt RC, White CW, Veress LA, From the Cover: ImpairedProliferation and Differentiation of the Conducting Airway Epithelium Associated With Bronchiolitis Obliterans After Sulfur Mustard Inhalation Injury in Rats, Toxicol Sci 157(2) (2017) 399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rancourt RC, Ahmad A, Veress LA, Rioux JS, Garlick RB, White CW, Antifibrinolytic mechanisms in acute airway injury after sulfur mustard analog inhalation, Am J Respir Cell Mol Biol 51(4) (2014) 559–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fowler EH, Dodd DE, Troup CM, Biological effects of short-term, high-concentration exposure to methyl isocyanate. V. Morphologic evaluation of rat and guinea pig lungs, Environ Health Perspect 72 (1987) 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mann KG, Krudysz-Amblo J, Butenas S, Tissue factor controversies, Thromb Res 129 Suppl 2 (2012) S5–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hisada Y, Thalin C, Lundstrom S, Wallen H, Mackman N, Comparison of microvesicle tissue factor activity in non-cancer severely ill patients and cancer patients, Thromb Res 165 (2018) 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hjort PF, Intermediate reactions in the coagulation of blood with tissue thromboplastin; convertin, accelerin, prothrombinase, Scand J Clin Lab Invest 9 Suppl 27 (1957) 1–183. [PubMed] [Google Scholar]
- [15].Lwaleed BA, Bass PS, Tissue factor pathway inhibitor: structure, biology and involvement in disease, J Pathol 208(3) (2006) 327–39. [DOI] [PubMed] [Google Scholar]
- [16].Hansen CB, van Deurs B, Petersen LC, Rao LV, Discordant expression of tissue factor and its activity in polarized epithelial cells. Asymmetry in anionic phospholipid availability as a possible explanation, Blood 94(5) (1999) 1657–64. [PubMed] [Google Scholar]
- [17].Greeno EW, Bach RR, Moldow CF, Apoptosis is associated with increased cell surface tissue factor procoagulant activity, Lab Invest 75(2) (1996) 281–9. [PubMed] [Google Scholar]
- [18].Veress LA, O’Neill HC, Hendry-Hofer TB, Loader JE, Rancourt RC, White CW, Airway obstruction due to bronchial vascular injury after sulfur mustard analog inhalation, Am J Respir Crit Care Med 182(11) (2010) 1352–61. [DOI] [PMC free article] [PubMed] [Google Scholar]