

Abstract
Structure−activity relationship (SAR) studies of numerous opioid ligands have shown that introduction of a methyl or ethyl group on the tertiary amino group at position 17 of the epoxymorphinan skeleton generally results in a mu opioid receptor (MOR) agonist while introduction of a cyclopropylmethyl group typically leads to an antagonist. Furthermore, it has been shown that introduction of heterocyclic ring systems at position 6 can favor antagonism. However, it was reported that 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(2′-indolyl)acetamido]morphinan (INTA), which bears a cyclopropylmethyl group at position 17 and an indole ring at position 6, acted as a MOR agonist. We herein report a SAR study on INTA with a series of its complementary derivatives to understand how introduction of an indole moiety with α or β linkage at position 6 of the epoxymorphinan skeleton may influence ligand function. Interestingly, one of INTA derivatives, compound 15 (NAN) was identified as a MOR antagonist both in vitro and in vivo. Molecular modeling studies revealed that INTA and NAN may interact with different domains of the MOR allosteric binding site. In addition, INTA may interact with W293 and N150 residues found in the orthosteric site to stabilize MOR activation conformation while NAN does not. These results suggest that INTA and NAN may be bitopic ligands and the type of allosteric interactions with the MOR influence their functional activity. These insights along with our enriched comprehension of the “message-address” concept will to benefit future ligand design.
Keywords: Opioids, SAR, bitopic, allosteric, functional conversion, message-address concept
Graphical Abstract

INTRODUCTION
Since opioid binding sites were first proposed in the early 1950s and 1960s, and discovered in mammalian brain tissue in 1973, extensive pharmacological studies have identified several types of opioid receptors.1−5 To date, four opioid receptors have been cloned, the mu opioid receptor (MOR), kappa opioid receptor (KOR), delta opioid receptor (DOR), and the opioid receptor-like orphan receptor (ORL) or nociceptin/orphanin FQ receptor (NOP).6−9 Opioid receptors share high sequence homology and belong to the superfamily of seven transmembrane-spanning G protein-coupled receptors (GPCRs).10 GPCRs play important roles in mediating the actions of many known neurotransmitters and hormones.11,12 Effects of opioids are mainly inhibitory resulting in significant inhibition of nerve firing and reduction in neurotransmitter release. The rewarding, dependence-producing, and analgesic effects of opioids result from activation of MORs in several brain regions.13−15 KOR agonists have been shown to produce dysphoric and psychotomimetic effects,16 while DOR activation results in inhibition of anxiety and stress.17,18 As a result, some opioid agonists and antagonists have been applied in the treatment of a number of diseases.19 In general, a compound is characterized as an opioid agonist only if its effect is competitively inhibited by an opioid antagonist.20−22
Structure−activity relationship (SAR) studies of opioids have revealed that introduction of a methyl or ethyl group on the 17-amino group of the epoxymorphinan skeleton generally results in a MOR agonist while introduction of an allyl or cyclopropylmethyl group at the same position typically leads to an opioid antagonist. Also, a hydroxyl group at position 14 may strengthen the binding to the opioid receptors, like naltrexone.23−25 In addition, some compounds with hetero-cyclic rings at position 6 of the epoxymorphinan group such as 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α (isoquinoline-3-carboxamido)morphinan (NAQ) and 17-cyclopropylm e t h y l - 3, 1 4 β - dihydroxy - 4, 5 α - epoxy - 6 β - (4 ′ -pyridylcarboxamido)morphinan (NAP) have been shown to be MOR selective antagonists (Figure 1).26−33 However, introduction of an allyl or cyclopropylmethyl group on the 17-amino group of the epoxymorphinan skeleton may also lead to an opioid agonist.34−36 For example, Le Naour et al. reported that 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(2′-indolyl)acetamido]morphinan (INTA), which is similar to NAQ and NAP structure-wise, actually acted as an opioid agonist and activated KOR-DOR and KOR-MOR heterodimers in HEK293 cells (Figure 1).34 It was shown that INTA produced potent antinociception and was devoid of tolerance and dependence while it showed no aversive effects in the conditioned place preference assay. Similar to NAQ and NAP, INTA carries a heterocyclic ring at position 6 and a cyclopropylmethyl group at position 17, thus it was intriguing how INTA acted as an agonist while NAQ and NAP acted as antagonists. Therefore, a series of compounds were prepared as INTA complementary analogues in order to understand how introduction of an indole moiety with an α or β linkage at position 6 of the epoxymorphinan skeleton would influence ligand function.
Figure 1.

Structural similarities between INTA and some known MOR ligands.
Interestingly, we identified 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(indole-7-carboxamido)morphinan (NAN) as a potent MOR antagonist. Per the “message-address concept”, ligands that bind opioid receptors have distinct pharmacophores responsible for efficacy (message) or selectivity (address).37−39 Both NAN and INTA bear an identical epoxymorphinan message portion with subtle differences in the linker and address regions of the molecule. Thus, it would be expected that they carry similar efficacies at the opioid receptors; however, the opposite was observed.
Studies have shown that allosteric ligands of the MOR show varying abilities to modulate the orthosteric ligand’s affinity, efficacy, or both.40,41 Allosteric ligands bind to a site on the receptor, termed as allosteric binding site, that is distinct from the site that endogenous orthosteric ligands bind (orthosteric binding site).42−44 An allosteric ligand can act as a positive allosteric modulator (PAM) which enhances the affinity or efficacy of an orthosteric ligand, a negative allosteric modulator (NAM) which inhibits the intrinsic affinity or efficacy, or a silent allosteric modulator (SAM) which occupies the allosteric binding site and has no effect on the affinity or efficacy. In addition, bitopic (dual steric) ligands which bind to both the orthosteric and allosteric binding sites of a single receptor monomer have been shown to have higher affinity, increase or decrease intrinsic efficacy, improve off-rates and induce functional selectivity.42,43,53,45−52 Thus, bitopic compounds may combine the advantages of orthosteric and allosteric ligands and carry unique pharmacological properties, e.g., higher selectivity.
We herein describe our effort to understand the SAR of INTA analogues as well as to investigate the molecular mechanisms of how structurally similar MOR ligands, NAN and INTA, result in the opposite functional effects on the MOR by application of classic “message-address” concept in addition to allosteric modulation of GPCRs.
RESULTS AND DISCUSSION
Chemistry.
The INTA analogues were synthesized in a similar way as described previously.27,31 Briefly, stereoselective reductive amination of naltrexone with benzyl amine followed by catalytic hydrogenation under acidic conditions produced 6α-naltrexamine (6α-NTA) dihydrochloride salt in a total yield of 67%, while reductive amination of naltrexone with dibenzyl amine followed by catalytic hydrogenation under acidic conditions led to 6β-naltrexamine (6β-NTA) dihydrochloride salt in a total yield of 59%.54 A variety of commercially available indole carboxylic acids were coupled with 6α-NTA and 6β-NTA using the EDCI/HOBt coupling method. The reaction mixture was then treated with K2CO3 to obtain the 6- monosubstituted NTA derivatives with yields ranging from 23 to 81% which were then converted to HCl salts (Scheme 1).
Scheme 1.

Synthetic Route for INTA Analogues
Biological Results.
The binding affinity and selectivity of these naltrexamine derivatives in three opioid receptors (mouse MOR, KOR, and DOR) were determined using monocloned opioid receptor-expressed Chinese hamster ovary (CHO) cells as described previously27,28,31 through radio-ligand binding assay. The [35S]-GTPγS functional assay using mouse MOR was applied to determine whether the synthesized compounds acted as agonist, partial agonist, or antagonist of the MOR by determining their efficacy for G-protein activation relative to a full agonist, DAMGO. An in vivo study was further conducted in mice using the tail immersion assay to determine whether the compounds had antinociceptive effects or antagonized the antinociceptive effects of morphine.
In Vitro Radioligand Binding and [35S]-GTPγS Functional Assays.
The binding affinities of the synthesized compounds for the MOR, KOR, and DOR together with their efficacy at the mouse MOR (mMOR) were compared with those of INTA. [3H]Naloxone was used to label the mMOR, while [3H]diprenorphine was used to label both the mKOR and mDOR. The [35S]-GTPγS functional assay was used to determine the potency and efficacy of these derivatives at the mMOR, and the results were interpreted as potency (EC50) and efficacy (%Emax relative to DAMGO). The binding, selectivity, potency, and efficacy data for the 6β-INTA analogues are summarized in Table 1, and the results for the 6α-INTA analogues are summarized in Table 2.
Table 1.
Opioid Receptor Binding Affinity and MOR [35S]-GTPγS Functional Assay Results for 6β-INTA Analogues
| Compd. | R | Ki (nM) | Selectivity | MOR [35S]-GTPγS assay | ||||
|---|---|---|---|---|---|---|---|---|
| μ | κ | δ | κ/μ | δ/μ | EC50 (nM) | % Emax of DAMGO | ||
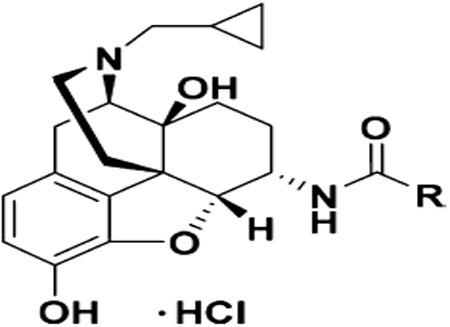
| 1 (INTA) |  |
0.29 ± 0.04 | 0.18 ±0.00 | 1.54 ±0.47 | 0.6 | 5.3 | 0.21 ±0.01 | 92.42 ±2.81 |
| 2 |  |
0.19 ±0.02 | 0.16 ±0.01 | 7.17 ±1.23 | 0.8 | 37.7 | 0.22 ± 0.07 | 79.48 ±7.41 |
| 3 | 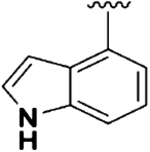 |
0.24 ± 0.03 | 1.94 ±0.30 | 49.84 ± 12.69 | 8.1 | 207.6 | 8.16 ±4.72 | 16.22 ± 1.40 |
| 4 | 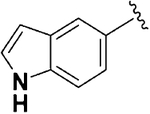 |
0.26 ± 0.04 | 0.51 ±0.04 | 17.67 ±4.78 | 2.0 | 68.0 | 2.98 ± 1.25 | 22.32 ±3.50 |
| 5 |  |
0.25 ± 0.04 | 0.17 ±0.01 | 6.23 ±0.83 | 0.7 | 24.9 | 0.19 ±0.02 | 71.79 ±4.37 |
| 6 |  |
0.19 ±0.01 | 0.52 ± 0.09 | 30.45 ±9.49 | 2.7 | 160.3 | 1.96 ± 1.01 | 37.94 ±5.85 |
| 7 |  |
0.24 ± 0.03 | 0.95 ±0.11 | 37.02 ± 1.21 | 4.0 | 154.3 | 1.44 ±0.75 | 48.27 ±4.45 |
| 8 |  |
0.17 ±0.01 | 0.39 ±0.05 | 77.10 ±28.29 | 2.3 | 453.5 | 1.73 ± 1.10 | 45.08 ±5.18 |
| 9 |  |
0.20 ± 0.03 | 0.18 ±0.04 | 25.30 ±4.34 | 0.9 | 126.5 | 1.04 ± 0.17 | 36.29 ±4.11 |
Table 2.
Opioid Receptor Binding Affinity and MOR [35S]-GTPγS Functional Assay Results for 6α-INTA Analogues
| Compd. | R | Ki (nM) | Selectivity | MOR [35S]-GTPγS assay | ||||
|---|---|---|---|---|---|---|---|---|
| μ | κ | δ | κ/μ | δ/μ | EC50 (nM) | % Emax of DAMGO | ||
| 10 |  |
0.36 ±0.03 | 0.93 ±0.13 | 14.24 ±2.78 | 2.6 | 39.5 | 6.75 ±3.20 | 36.91 ±2.40 |
| 11 |  |
0.29 ± 0.04 | 0.98 ±0.13 | 10.54 ±2.89 | 3.4 | 36.3 | 1.00 ±0.13 | 33.34 ±3.51 |
| 12 |  |
0.26 ± 0.04 | 1.49 ±0.35 | 9.26 ±2.83 | 5.7 | 35.6 | 4.99 ±2.19 | 28.90 ±0.95 |
| 13 |  |
0.76 ±0.11 | 3.45 ±0.99 | 26.72 ± 7.72 | 4.5 | 35.2 | 1.55 ±0.08 | 32.24 ± 1.26 |
| 14 | 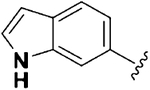 |
0.43 ± 0.05 | 1.63 ±0.28 | 12.78 ±3.36 | 3.8 | 29.7 | 6.06 ± 1.25 | 26.62 ± 0.66 |
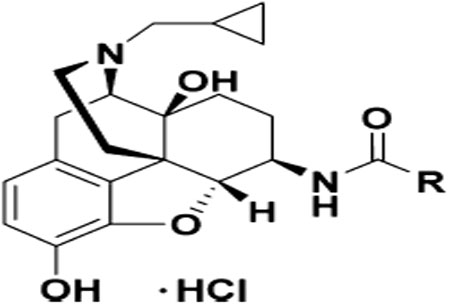
| 15 (NAN) |  |
0.23 ± 0.02 | 1.69 ±0.35 | 13.69 ± 1.39 | 7.3 | 59.5 | 3.85 ±2.32 | 19.11 ±3.31 |
| 16 | 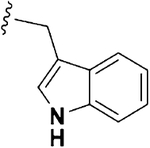 |
0.84 ±0.12 | 3.14 ±0.45 | 9.17 ±2.67 | 3.7 | 10.9 | 4.75 ±0.31 | 34.14 ±0.97 |
| 17 |  |
0.44 ± 0.04 | 1.36 ±0.18 | 25.07 ±6.83 | 3.1 | 57.0 | 2.09 ±0.19 | 41.55 ±6.14 |
| 18 | 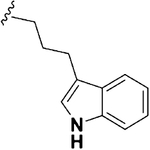 |
0.29 ± 0.04 | 0.15 ±0.03 | 6.73 ± 0.40 | 0.5 | 23.2 | 1.89 ± 1.04 | 54.89 ±6.35 |
As shown in Table 1, compounds 1−9 showed subnano-molar affinity for the MOR and KOR while having a lower affinity at the DOR except for compound 3 that bound to KOR with nanomolar affinity. Particularly, INTA showed similar affinity for the MOR and KOR and was about 5-fold selective for MOR over DOR and 9-fold selective for KOR over DOR. These results corroborated the findings from Le Naour et. al, where they reported that INTA produced similar stimulation in the MOR and KOR and the least stimulation in the DOR.34 INTA showed the lowest selectivity for the MOR over the DOR compared to other compounds.
Among the 6α-INTA analogues (compounds 10−18), it was observed that all the compounds bound to the MOR with subnanomolar affinity while having relatively lower affinity at the KOR, and even lower at the DOR. The reason for the greater binding affinity of these ligands for the MOR and KOR compared to other opioid antagonist with different hetero-cyclic rings attached to position 6 (examples include NAP and NAQ) could be due to additional hydrogen bonding interactions between the hydrogen atom at position 1 of the indole ring and an acceptor group in the binding pocket.31 It has been shown that methylation of the indole 1-nitrogen atom led to reduction in activation of singly or coexpressed opioid receptors.34 Even though MOR and DOR have a relatively higher percent sequence identity (62%) than that of MOR and KOR (57%), the percent sequence identities for TM3 and TM7 where the address region is located is relatively greater for the MOR and KOR (TM3 = 100%, TM7 = 86%) than those of the MOR and DOR (TM3 = 90%, TM7 = 78%).26 This could account for the overall similar binding affinities at the MOR and KOR over the DOR.
It was also noticed that the position of substitution on the indole ring (compounds 1−6 and 10−15) did not result in a significant change in the MOR binding affinity except for compound 13. Also, increasing the alkyl chain length of the indole analogues (compounds 7−9 and 16−18) at position 3 did not result in significant change in their MOR binding affinity, except a slightly lower affinity of compound 16. This suggests that the binding pocket of the address site of the MOR may be spacious enough to allow the rotation and extension of the indole group.
Not surprisingly, INTA was identified as a MOR full agonist with subnanomolar potency (Table 1) in the [35S]-GTPγS functional assay, while compound 2 and 5 also showed high MOR efficacies relative to DAMGO. In addition to INTA, compounds 2 and 4 have previously been synthesized by Le Naour et al. and the results we obtained were similar to theirs.34 For other compounds, even though they carried the same substitution (indole moiety) as the side chain, most of them showed moderate to low efficacy at the MOR. Particularly, compound 3 and compound 15 (NAN) acted as MOR antagonists since both had efficacies lower than 20%. Based on our previous studies, we have observed that compounds with less than 20% relative efficacies for G-protein activation in MOR-expressing CHO cells tend to be antagonists in vivo.26,28,31
Furthermore, INTA and NAN together with their respective diastereomers (compound 10 and compound 6) were compared to understand how the stereochemistry at position 6 of the epoxymorphinan skeleton would affect binding affinity and efficacy. It was observed that the configuration of the linker at position 6 did not lead to a significant difference in their MOR binding affinity but influenced their potency and efficacy on the MOR more evidently. For instance, changing the linker of INTA from β-configuration to α-configuration (compound 10) did not result in a significant change in MOR binding affinity (INTA Ki = 0.29 ± 0.04 nM, compound 10 Ki = 0.36 ± 0.03 nM) but produced a 32-fold and 2.5-fold reduction in potency and efficacy, respectively (INTA EC50 = 0.21 ± 0.01 nM, INTA Emax = 92.42 ± 2.81%, compound 10 EC50 = 6.75 ± 3.20 nM, compound 10 Emax = 36.91 ± 2.40%). Meanwhile, changing the configuration of the linker in NAN from α to β (compound 6) did not result in a significant change in MOR binding affinity either (NAN Ki = 0.23 ± 0.02 nM, compound 6 Ki = 0.19 ± 0.01 nM), but resulted in an increase in efficacy (NAN Emax = 19.11 ± 3.31%, compound 6 Emax = 37.94 ± 5.85%). In all, it seemed that changing the linker from the α-configuration to the β-configuration at position 6 resulted in an increase in efficacy on the MOR without affecting binding affinity on the receptor significantly.
Warm-Water Immersion Assay.
To corroborate the findings of the [35S]-GTPγS functional assay, an in vivo assay was conducted using the warm-water immersion assay to evaluate whether the compounds synthesized produced acute antinociceptive effects (opioid agonists) or antagonized morphine’s antinociceptive effect (opioid antagonists). The warm-water immersion assay was performed as described previously.27 The test was conducted 20 min after injection of the compounds because morphine’s effect starts to peak 20 min after s.c. administration. The results were interpreted as the percentage of maximum possible effect (% MPE). A higher % MPE indicates a stronger antinociception effect by the ligand. Figure 2A and B shows the antinociceptive effects of these INTA analogues at a single dose of 10 mg/kg. Similar to the report from Le Naour et. al, among all the ligands tested, INTA, identified as the most potent and efficacious MOR agonists in the [35S]-GTPγS functional assay, also acted as an opioid agonist with the highest antinociception in the warm-water immersion assay. The % MPE values of the remaining INTA analogues, except for compounds 6 and 7, were less than 20%, which corresponded to their lower efficacy observed in the [35S]-GTPγS functional assay. The low % MPE values for compounds 2 and 5, which showed high potency and efficacy in the [35S]-GTPγS binding assay, could be due to their poor pharmacokinetic properties such as low CNS permeability, since the tail immersion response is mainly mediated at the level of the spinal cord in the CNS.55
Figure 2.

Warm-water tail immersion assay in mice (n = 5) at 56 ± 0.1 °C. All tested compounds were administered subcutaneously (s.c.). Antinociceptive effects of (A) INTA analogues based on 6β-naltrexamine and (B) INTA analogues based on 6α-naltrexamine. Compounds (10 mg/kg) were injected at time 0. Twenty minutes after injection, tail withdrawal response was assessed with warm water. Antagonism of the antinociceptive effect of morphine by (C) INTA analogues based on 6β-naltrexamine and (D) INTA analogues based on 6α-naltrexamine. Tested compounds (10 mg/kg) were injected at time 0. Five minutes later, morphine (10 mg/kg) was administered. Twenty minutes after morphine injection, tail flick was tested using warm water. *P < 0.05 compared to vehicle; **P < 0.01 compared to vehicle; ***P < 0.001. ns indicates P > 0.05. In (A) and (B), all compounds except 1 had significantly less antinociception than morphine. While in (B) and (C), only compounds 6, 7, 15, and naltrexone significantly antagonized morphine’s antinociception compared to distilled water treated mice.
Figure 2C and D shows the inhibition of morphine’s antinociceptive effect at 10 mg/kg in the presence of each INTA indole analogue at a single dose of 10 mg/kg. As expected, NAN, which showed a high potency and low efficacy in the [35S]-GTPγS functional assay, was identified as the most potent opioid antagonist and its AD50 value was determined as 2.07 (0.40−10.74) mg/kg (95% CL) from a following dose− response study (Figure 3). We also observed that compound 3 which had a low efficacy in the [35S]-GTPγS functional assay did not significantly inhibit morphine’s antinociceptive effect. This could be due to its poor pharmacokinetic properties preventing it from getting into the CNS.
Figure 3.

Dose dependent studies on NAN for opioid antagonist effect. NAN had an AD50 value of 2.07 (0.40−10.74) mg/kg (95% CL).
Molecular Modeling Study.
SAR studies of numerous opioids have revealed that a cyclopropylmethyl group on the tertiary amino group at position 17 typically leads to opioid antagonism as observed with naltrexone56 as well as NAN. On the contrary, INTA, which also carries a cyclopropylmethyl group at position 17, acted as an MOR agonist. Hence, docking studies were conducted to understand how these two structurally similar compounds, INTA and NAN (Figure 4), with the identical “message” structure but slightly different “address” portions produced almost opposite functional effects. The ligands INTA and NAN were both sketched using SybylX2.1, and then docked into the agonist-bound (PDB ID: 5C1M)57 and antagonist-bound (PDB ID: 4DKL)58 MORs using GOLD 5.4, respectively.59 The highest scored (CHEMPLP) docking solutions were chosen as the optimal binding poses, named INTA_MORactive and NAN_MORinactive complexes.
Figure 4.

Chemical structures of INTA (A) and NAN (B) together with their atom notation derived from the complexes obtained from docking studies. The “message” and “address” portions of the molecules are marked out in different colors.
From the docking results, it was observed that the epoxymorphinan moiety (“message” portion) of both INTA and NAN bound to the MOR orthosteric binding site in a similar fashion as reported for other known MOR ligands.26,60 The “message” portions formed ionic interactions with D147, hydrogen bonding interactions with Y148, and hydrophobic interactions with residues M151, W293, and H297. Meanwhile, the indole ring (“address” portion) of INTA seemed to be able to form hydrophobic interactions with H54 (Figure 5A), while this interaction did not exist in the NAN_-MORinactive complex. Moreover, the “address” portions of both INTA and NAN could form π−π stacking interactions with W318 located at the allosteric binding site, but the interaction in the INTA_MORactive complex was stronger than that in the NAN_MORinactive complex based on the distances between the ligand and the residue (Table 3). Another observation from the docking study of INTA was that in order to accommodate the β-configuration of the indole side chain in the protein, the cyclohexyl ring that the side chain attached to had to adopt a twisted boat conformation. In the case of NAN, the cyclohexyl ring actually was in a twisted chair conformation to retain the α-configuration of the indole side chain.
Figure 5.

Binding poses of (A) INTA in the agonist-bound (5C1M) and (B) NAN in the antagonist-bound (4DKL) MOR crystal structures from docking results. Protein shown as cartoon model in light-green (A) and light-pink (B); INTA, NAN, and key amino acid residues shown as stick model. Carbon atoms: INTA (cyan), NAN (magenta), amino acid residues in (A) (green) and in (B) (orange); oxygen atoms (red); nitrogen atoms (blue). Two-dimensional representations of binding modes of (A) INTA and (B) NAN in 5C1M and 4DKL, respectively, are also shown.
Table 3.
Measured Shortest Distances between Atoms on Critical Amino Acid Residues and Atoms on the Ligands from INTA-MORactive and NAN-MORinactive Complexes before and after MD Simulations
| complex | atom of ligand | atom of residue | distance from docking study (Å) | distance from MD (Å) |
|---|---|---|---|---|
| INTA_MORactive | N37@INTA | ND1@H54 | 3.0 | 4.3 |
| N21@INTA | OD2@D147 | 4.0 | 3.1 | |
| O15@INTA | OH@Y148 | 3.4 | 4.0 | |
| C37@INTA | CB@N150 | 5.5 | 4.5 | |
| C5@INTA | CE@M151 | 3.8 | 4.7 | |
| C4@INTA | CZ3@W293 | 3.8 | 4.1 | |
| C6@INTA | CE1@H297 | 3.3 | 4.0 | |
| C63@INTA | NZ@K303 | 3.1 | 4.3 | |
| C58@INTA | CH2@W318 | 3.2 | 3.9 | |
| N21@NAN | OD2@D147 | 3.0 | 3.1 | |
| O15@NAN | OH@Y148 | 3.0 | 3.5 | |
| C37@NAN | CG@N150 | 6.5 | 6.8 | |
| NAN_MORinactive | C26@NAN | CE@M151 | 3.6 | 4.2 |
| C34@NAN | CD2@L232 | 8.6 | 4.8 | |
| O54@NAN | NZ@K233 | 5.2 | 4.5 | |
| C52@NAN | CE@K233 | 5.3 | 3.9 | |
| C4@NAN | CZ3@W293 | 5.4 | 5.7 | |
| C6@NAN | CE1@H297 | 4.5 | 5.4 | |
| C63@NAN | NZ@K303 | 3.6 | 6.9 | |
| C63@NAN | CZ2@W318 | 3.7 | 5.4 |
Many studies have proved that molecular dynamics (MD) simulation is an effective method to explore the binding modes between proteins and their ligands.61−64 Therefore, an MD simulation study was further conducted on the INTA_-MORactive and NAN_MORinactive complexes using NAMD 2.8 to investigate the potential allosteric modulation mechanisms of the “address” portions of INTA and NAN to the binding of their “message” portions with the orthosteric binding site of the MOR.65 Prior to conducting the MD simulations, the two ligand−receptor complexes were first embedded in a lipid (POPC) bilayer membrane using VMD 1.9.3.66 Next, each complex was solvated by TIP3 water layers. After that, any water molecules and POPC lipid molecules at a distance of 0.75 Å or less from the ligand−receptor complex were removed. Finally, sodium and chloride ions were added to the solvated system to make the ion concentration approximately 0.1 nM. A representative ligand−receptor complex embedded in a POPC bilayer membrane and solvated by a water box is displayed in the Supporting Information (Figure S1A).
Here, 10 ns MD simulations were then conducted for the INTA_MORactive and NAN_MORinactive complexes. After the simulation, the root-mean-square deviation (rmsd) values of all the protein backbone atoms relative to the respective starting structures were calculated to evaluate the stability of the two systems (Figure S1B). The average rmsd values of the INTA_MORactive and NAN_MORinactive complexes were determined as 1.45 and 1.31 Å, respectively.
Previous studies have shown that when a system’s rmsd value is less than 3 Å, it indicates that the system has achieved dynamic equilibrium.67−69 In addition, it seemed that the rmsd values of both systems remained stable after 5 ns MD simulations (Figure S1B). Therefore, it can be concluded that both systems achieved stability after 10 ns MD simulations and snapshots from the last 4 ns of MD simulations were collected to analyze the interactions between the ligand and the receptor.
As shown in Figure 6, owing to the hydrophobic interactions with W318 and H54, the “address” portion of INTA still interacted with the same domain (K303 and W318) of the allosteric binding site in the active MOR after 10 ns MD simulations (Table 3, Figure S2). On the other hand, the “address” portion of NAN formed new hydrophobic interactions with L232 and K233 and its binding domain seemed to shift from K303 and W318 to L232 and K233 (a residue that formed a covalent bond with β-FNA in the MOR inactive crystal structure)58 after 10 ns MD simulations (Table 3). As a consequence, the cyclopropylmethyl group of INTA was buried deeply into the orthosteric binding site, and much closer to N150 than that of NAN (Table 3) to induce a hydrophobic interaction. This hydrophobic interaction could stabilize the conformation of the side chain of N150, which may further help secure the hydrogen bonding interaction between the side chain of N150 and the backbone carbonyl of I146 in the INTA_MORactive complex. Such a postulation was based on the fact that the distances between the nitrogen atom at the side chain of N150 and the backbone carbonyl of I146 in the INTA_MORactive complex was 3.2 Å, (Figure 6A) very close to the same measurement in the BU72-bound active MOR crystal structure (2.8 Å) respectively.57
Figure 6.

Binding modes of INTA_MORactive (A) and NAN_MORinactive (B) complexes after MD simulations. Protein shown as cartoon model in light-green (INTA_MORactive complex) and light-pink (NAN_MORinactive complex); INTA, NAN, and key amino acid residues shown as stick model. Carbon atoms: INTA (cyan); NAN (magenta); key amino acid residues in INTA_MORactive complex (green) and in NAN_MORinactive complex (orange). Oxygen atoms (red); nitrogen atoms (blue). The red dashed lines represent the critical distances discussed. Two-dimensional representations of binding modes of (A) INTA and (B) NAN in INTA_MORactive and in NAN_MORinactive complexes, respectively, are also shown.
Another observation from the binding of the “address” portion of both INTA and NAN with the allosteric binding site was that the distance between the aromatic moiety of epoxymorphinan skeleton in INTA and W293 (4.1 Å) was 1.6 Å shorter than that of NAN to the same residue (5.7 Å) (Table 3, Figure 6). As previous studies reported, the hydrophobic interaction between an agonist and W293 could stabilize the conformation of the side chain of W293 in its active state.57 Therefore, this may also provide another possible explanation on INTA acting as an agonist.
Bitopic ligands typically carry two distinct parts that interact with the orthosteric binding site and allosteric binding site and are connected by a linker. Since the indole side chain (the “address” portion of the two molecules) attached to the “message” portion of the epoxymorphinan skeleton through opposite configurations, we further looked into how different linkers would influence their function on the MOR. As INTA has a β-configuration linker while NAN has an α one, this led to opposite conformations of the cyclohexyl ring in the skeletons in order to accommodate the “address” portion of the molecules when the “message” portions of NAN and INTA bound to the orthosteric binding site of the MOR. This may further make the “message” portion and cyclopropylmethyl group of INTA reach deeply into the orthosteric binding site and form interactions with W293 and N150, respectively, to stabilize their active conformations.
CONCLUSION
In summary, a series of 6α- and 6β-indolylacetamidonaltrexamine derivatives were prepared to understand the SAR of INTA. NAN, as one of INTA analogues, was identified as an MOR antagonist from in vitro radioligand competition binding and functional assays. Further in vivo studies showed that NAN antagonized morphine’s antinociceptive effects while INTA produced potent antinociception. Molecular modeling studies were conducted to further understand how two structurally similar compounds, NAN and INTA, showed almost opposite functional activities. The hydrophobic interactions between the “address” portion of INTA and residues W318 and H54 at the allosteric binding site may cause the “message” portion and cyclopropylmethyl group of INTA to reach deeply into the orthosteric binding site and form favorable interactions with W293 and N150 to stabilize the MOR in its active state conformation. On the other hand, the “address” portion of NAN formed hydrophobic interactions with residues L232 and K233 at another allosteric binding site, which prevented the message portion of NAN to reach deep enough into the orthosteric binding site. In other words, the indole moiety of INTA may be acting as a PAM while that of NAN may be acting as a NAM at the MOR. Since NAN and INTA have the same “message” portion that bind to the orthosteric binding site and distinct “address” portions that may interact with the allosteric binding site, NAN and INTA may be called bitopic ligands. These results further suggested that even though two ligands may carry identical “message” portion, they could “deliver” totally opposite function if the “address” portion (even with some minor structural differences) of the compounds do not interact with the same domain of the allosteric binding site of MOR. It is believed that such an elaboration of the “message-address” concept may enhance our comprehension of structure−activity relationship of opioid ligands and provide insight for future rational molecule design.
METHODS
Chemical Synthesis.
General Methods.
Chemical reagents were purchased from either Sigma-Aldrich or Alfa Aesar. Reactions were monitored using Analtech Uniplate F254 TLC plates. Compounds were purified using silica gel columns (230−400 mesh, Merck). The IR spectra were obtained using a Thermo Scientific smart iTR instrument. Proton (400 MHz) and carbon-13 (100 MHz) nuclear magnetic resonance (NMR) spectra were acquired at ambient temperature with tetramethylsilane as the internal standard on a Bruker Ultrashield 400 Plus spectrometer. MS analysis was performed using the PerkinElmer AxION TOF mass spectrometer. HPLC analysis was done with a Varian ProStar 210 system on Microsorb-MV 100–5 C8/C18 column (250 mm × 4.6 mm) at 254 nm, eluting with acetonitrile (0.1% TFA)/water (40/60) at 1 mL/min over 30 min. All above analytical methods were used to determine purity of the newly synthesized compounds, and their purity is confirmed as ≥95%.
General Procedure for Amide Coupling Reaction.
The carboxylic acid derivatives (3 equiv), hydrobenzotriazole (HOBt) (3 equiv), 4 Å molecular sieves, triethylamine (9 equiv), N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride (EDCI) (3 equiv), and dimethylformamide (DMF) (2 mL) were added into a three-necked flask on an ice−water bath and stirred for 15 min. Naltrexamine (1 equiv) suspended in 2 mL of DMF was added dropwise and then stirred on the water bath for 24 h. When the reaction was completed, the reaction mixture was filtered with Celite and the filtrate was concentrated to remove DMF. MeOH (7 mL) and K2CO3 (3 equiv) were added to the concentrate and stirred at ambient temperature. When hydrolysis of the ester at position 3 was complete, the mixture was filtered through Celite again and concentrated to remove MeOH. The mixture was then purified using column chromatography with dichloromethane/MeOH (20:1) and 1% NH4OH to give the free base.27 Upon confirmation by 1HNMR and 13C NMR, the free base was then transformed into hydrochloride salt by dissolving in MeOH (0.1 mL) and dichloro-methane (2 mL), adding HCl methanol solution (1.25 M, 4 equiv) on an ice−water bath, and stirring for 5 min. Diethyl ether (10 mL) was then added. Two hours later, the precipitate was collected by filtration and dried in vacuum to give the target compound as a hydrochloride salt, which was characterized by 1H NMR, 13C NMR, IR, MS and the purity determined using HPLC (for spectra, see the Supporting Information).
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(indole-2-carboxamido)morphinan (1).
The title compound was prepared by following the general procedure in 32% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.60 (s, 1H, exchangeable), 9.33 (s, 1H, exchangeable) 8.88 (s, 1H, exchangeable), 8.73 (d, J = 7.92 Hz, 1H, exchangeable), 7.62 (d, J = 7.96 Hz, 1H), 7.42 (d, J = 8.24 Hz, 1H), 7.18 (m, 2H), 7.04 (t, J = 7.52 Hz, 1H), 6.73 (d, J = 8.08 Hz, 1H), 6.66 (d, J = 7.96 Hz, 1H), 6.24 (s, 1H, exchangeable), 4.84 (d, J = 7.72 Hz, 1H), 3.89 (s, 1H), 3.71 (m, 1H), 3.33 (m, 2H), 3.11−3.06 (m, 2H), 2.88 (m, 1H), 2.50 (m, 2H), 1.91 (q, J = 11.84 Hz, 1H), 1.80 (d, J = 13.56 Hz, 1H) 1.63 (m, 1H), 1.49−1.40 (m, 2H), 1.09 (m, 1H), 0.68 (m, 1H), 0.60 (m, 1H), 0.52 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 160.73, 142.02, 141.09, 136.23, 131.30, 129.62, 126.97, 125.26, 123.46, 121.52, 120.64, 119.83, 119.39, 117.87, 112.23, 102.63, 89.85, 69.62, 61.67, 56.69, 50.66, 46.37, 45.61, 29.37, 23.78, 22.91, 5.62, 5.07, 2.55. Mp 235−237 °C. HRMS (m/z) [M]+ calculated for C29H31N3O4 485.2315, found [MH]+ 486.2411. IR (ATR, cm−1) νmax 3212, 2167, 1975, 1617, 1551, 1506, 1315, 1239,1125, 1034, 815, 747, 668.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(indole-3-carboxamido)morphinan (2).
The title compound was prepared by following the general procedure in 27% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.64 (s, 1H, exchangeable), 9.34 (s, 1H, exchangeable), 8.89 (s, 1H, exchangeable), 8.13−8.10 (m, 3H, 1 proton exchangeable), 7.42 (d, J = 7.88 Hz, 1H), 7.16−7.06 (m, 2H), 6.73 (d, J = 8.08 Hz, 1H), 6.65 (d, J = 8.12 Hz, 1H), 6.24 (s, 1H, exchangeable), 4.82 (d, J = 7.76 Hz, 1H), 3.90 (d, J = 4.28 Hz, 1H), 3.70 (m, 1H), 3.36 (m, 1H), 3.12−3.05 (m, 3H), 2.88 (m, 2H), 1.89 (q, J = 12.88 Hz, 1H), 1.78 (d, J = 13.2 Hz, 1H), 1.63 (m, 1H), 1.48−1.41 (m, 2H), 1.09 (m, 1H), 0.68 (m, 1H), 0.50 (m, 1H), 0.52 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 164.46, 142.18, 141.03, 135.90, 129.70, 127.60, 125.86, 121.98, 120.89, 120.63, 120.43, 119.29, 117.89, 111.79, 110.30, 90.17, 69.69, 61.70, 56.70, 50.21, 46.38, 45.60, 29.40, 27.30, 23.99, 22.93, 5.61, 5.12, 2.53. Mp 272−274 °C. HRMS (m/z) [M]+ calculated for C29H31N3O4 485.2315, found [MH]+ 486.2411. IR (ATR, cm−1) νmax 3267, 1671, 1621, 1539, 1505, 1455, 1313, 1176, 1119.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(indole-4-carboxamido)morphinan (3).
The title compound was prepared by following the general procedure in 31% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.31 (s, 1H, exchangeable), 9.34 (s, 1H, exchangeable), 8.87 (s, 1H, exchangeable), 8.39 (d, J = 8.12 Hz, 1H, exchangeable), 7.55 (d, J = 8.04 Hz, 1H), 7.48 (d, J = 7.16 Hz, 1H), 7.43 (m, 1H), 7.15 (t, J = 7.72 Hz, 1H), 6.88 (s, 1H), 6.73 (d, J = 8.12 Hz, 1H), 6.66 (d, J = 8.16 Hz, 1H), 6.19 (s, 1H, exchangeable), 4.89 (d, J = 7.76 Hz, 1H), 3.88 (d, J = 4.92 Hz, 1H), 3.77−3.72 (m, 1H), 3.32 (m, 1H), 3.13−3.04 (m, 2H), 2.87 (m, 1H), 1.93 (q, J = 12.08 Hz, 1H), 1.78 (d, J = 13.68 Hz, 1H), 1.65 (m, 1H), 1.46 (m, 2H), 1.09 (m, 2H), 0.70−0.68 (m, 1H), 0.60 (m, 1H), 0.53−0.50 (m, 1H), 0.43−0.41 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 167.77, 142.15, 141.02, 136.35, 129.67, 126.30, 126.09, 125.75, 120.64, 120.19, 119.34, 118.50, 117.90, 114.31, 101.78, 89.92, 69.68, 61.68, 56.68, 50.89, 46.37, 45.61, 29.43, 27.29, 23.66, 22.90, 5.58, 5.11, 2.50. Mp 259−261 °C. HRMS (m/z) [M]+ calculated for C29H31N3O4 485.2315, found [MH]+ 486.2434. IR (ATR, cm−1) νmax 3207, 2166, 1639, 1503, 1455, 1319, 1237, 1195, 1123, 1037, 917.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(indole-5-carboxamido)morphinan (4).
The title compound was prepared by following the general procedure in 61% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.36 (s, 1H, exchangeable), 9.32 (s, 1H, exchangeable) 8.88 (s, 1H, exchangeable), 8.48 (d, J = 8.08 Hz, 1H, exchangeable), 8.18 (s, 1H), 7.66 (dd, J1 = 1.56 Hz, J2 = 8.56 Hz, 1H), 7.42 (m, 2H), 6.73 (d, J = 8.12 Hz, 1H), 6.65 (d, J = 8.16 Hz, 1H), 6.53 (t, J1 = 2.04 Hz, J2 = 2.00 Hz, 1H), 6.20 (s, 1H, exchangeable), 4.89 (d, J = 7.80 Hz, 1H), 3.88 (d, J = 4.96 Hz, 1H), 3.71 (m, 1H), 3.35 (m, 2H), 3.11−3.04 (m, 2H), 2.87 (m, 1H), 1.90 (q, J = 12.6 Hz, 1H), 1.78 (d, J = 13.76 Hz, 1H), 1.62 (m, 1H), 1.49−1.39 (m, 2H), 1.09 (m, 1H), 0.68 (m, 1H), 0.59 (m, 1H), 0.52 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.77, 142.30, 141.30, 137.41, 129.77, 126.99, 126.60, 125.29, 120.56, 120.49, 119.91, 119.16, 117.92, 110.80, 102.05, 90.06, 69.80, 61.81, 56.74, 51.11, 46.52, 45.62, 29.44, 27.41, 23.93, 23.06, 5.72, 5.09, 2.65. Mp 290−292 °C. HRMS (m/z) [M]+ calculated for C29H31N3O4 485.2315, found [MH]+ 486.2434. IR (ATR, cm−1) νmax 3125, 2162, 1980, 1622, 1595, 1547, 1360, 1311, 1249, 1124, 1037, 919, 760, 672.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(indole-6-carboxamido)morphinan (5).
The title compound was prepared by following the general procedure in 29% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.43 (s, 1H, exchangeable), 9.33 (s, 1H, exchangeable), 8.87 (s, 1H, exchangeable), 8.54 (d, J = 7.4 Hz, 1H, exchangeable), 7.99 (s, 1H), 7.58 (s, 2H), 7.50 (s, 1H), 6.73 (d, J = 6.36 Hz, 1H), 6.66 (d, J = 7.68 Hz, 1H), 6.49 (s, 1H), 6.20 (s, 1H, exchangeable), 4.90 (d, J = 5.92 Hz, 1H), 3.88 (s, 1H), 3.71 (m, 1H), 3.17−3.06 (m, 2H), 2.89 (m, 2H), 2.5 (s, 1H), 1.90 (m, 1H), 1.78 (m, 1H), 1.62 (m, 1H), 1.49−1.39 (m, 2H), 1.09 (m, 2H), 0.68 (m, 1H), 0.60 (m, 1H), 0.52 (m, 1H), 0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.85, 142.16, 140.23, 135.05, 129.76, 127.74, 127.20, 119.34, 118.39, 117.95, 116.98, 111.16, 101.20, 90.74, 69.62, 61.76, 58.35, 51.50, 49.99, 39.96, 39.76, 30.60, 29.99, 24.65, 22.25, 3.72, 3.46. Mp 254−256 °C. HRMS (m/z) [M]+ calculated for C29H31N3O4 485.2315, found [MH]+ 486.2396. IR (ATR, cm−1) νmax 3223, 2152, 1970, 1608, 1551, 1312, 1035, 920, 820, 740.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(indole-7-carboxamido)morphinan (6).
The title compound was prepared by following the general procedure in 56% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.07 (s, 1H, exchangeable), 9.37 (s, 1H, exchangeable), 8.90 (s, 1H, exchangeable), 8.75 (d, J = 8.04 Hz, 1H, exchangeable), 7.78−7.75 (m, 2H), 7.35 (m, 1H), 7.10 (t, J = 7.60 Hz, 1H), 6.74 (d, J = 8.12 Hz, 1H), 6.67 (d, J = 8.16 Hz, 1H), 6.49 (m, 1H), 6.26 (s, 1H, exchangeable), 4.93 (d, J = 7.8 Hz, 1H), 3.91 (d, J = 4.84 Hz, 1H), 3.82−3.78 (m, 2H), 3.18−3.06 (m, 3H), 2.91−2.86 (m, 1H), 2.02−1.93 (q, J = 11.56 Hz, 1H), 1.82 (d, J = 13.64 Hz, 1H), 1.65 (m, 1H), 1.56−1.41 (m, 2H), 1.10 (t, J = 7.00 Hz, 2H), 0.71−0.68 (m, 1H), 0.62−0.60 (m, 1H), 0.55−0.52 (m, 1H), 0.44−0.42 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 164.50, 139.51, 138.46, 131.48, 127.15, 126.67, 123.95, 121.72, 118.20, 117.28, 117.02, 115.79, 115.40, 113.77, 98.68, 87.35, 67.21, 62.45, 59.20, 54.25, 48.23, 43.86, 43.16, 26.96, 24.75, 21.14, 20.39, 12.43, 3.07, 2.65. Mp 246−248 °C. HRMS (m/z) [M]+ calculated for C29H31N3O4 485.2315, found [MH]+ 486.2411. IR (ATR, cm−1) νmax 3090, 2162, 1979, 1633, 1540, 1327, 1248, 1040.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[2-(indol-3-yl)acetamido)morphinan (7).
The title compound was prepared by following the general procedure in 49% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.89 (s, 1H, exchangeable), 9.32 (s, 1H, exchangeable), 8.82 (s, 1H, exchangeable), 8.20 (d, J = 7.92 Hz, 1H, exchangeable), 7.54 (d, J = 7.76 Hz, 1H), 7.34 (d, J = 8.04 Hz, 1H), 7.18 (d, J = 2.08 Hz, 1H), 7.06 (t, J = 7.08 Hz, 1H), 6.98 (t, J = 7.24 Hz, 1H), 6.70 (d, J = 8.12 Hz, 1H), 6.62 (d, J = 8.16 Hz, 1H), 6.14 (s, 1H, exchangeable), 4.60 (d, J = 7.84 Hz, 1H), 3.82 (d, J = 5.2 Hz, 1H), 3.50 (q, J = 14.96 Hz, 2H), 3.40 (m, 1H), 3.32 (m, 1H), 3.28 (m, 1H), 3.07−3.01 (dd, J1 = 5.96 Hz, J2 = 19.24 Hz, 2H), 2.84 (m, 1H), 2.43−2.39 (m, 2H), 1.68 (m, 2H), 1.49−1.28 (m, 3H), 1.07 (m, 1H), 0.67−0.66 (m, 1H), 0.60−0.56 (m, 1H), 0.51−0.48 (m, 1H), 0.41− 0.39 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 170.94, 170.86, 141.99, 140.99, 135.88, 129.55, 127.03, 123.57, 121.02, 120.58, 119.36, 118.53, 118.42, 117.83, 111.28, 108.49, 99.49, 69.56, 61.54, 56.62, 46.32, 45.49, 32.78, 29.17, 27.21, 23.45, 22.82, 5.55, 5.08, 2.46. Mp 292−294 °C. HRMS (m/z) [M]+ calculated for C30H33N3O4 499.2471, found [MH]+ 500.2539. IR (ATR, cm−1) νmax 3270, 1666, 1549, 1463, 1303,1274, 1232, 1174, 1124, 1030, 898.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[3-(indol-3-yl) propanamido)morphinan (8).
The title compound was prepared by following the general procedure in 70% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.77 (s, 1H, exchangeable), 9.34 (s, 1H, exchangeable), 8.83 (s, 1H, exchangeable), 8.14 (d, J = 7.8 Hz, 1H, exchangeable), 7.52 (d, J = 7.8 Hz, 1H), 7.32 (d, J = 8.04 Hz, 1H), 7.10 (s, 1H), 7.06 (t, J = 7.96 Hz, 1H), 6.97 (t, J = 7.88 Hz, 1H), 6.72 (d, J = 8.12 Hz, 1H), 6.63 (d, J = 8.16 Hz, 1H), 6.18 (s, 1H, exchangeable), 4.55 (d, J = 7.8 Hz, 1H), 3.85 (s, 1H), 3.45 (m, 1H), 3.34−3.29 (m, 2H), 3.09−3.00 (m, 2H), 2.92−2.83 (m, 3H), 2.45−2.42 (m, 4H), 1.71−1.68 (m, 2H), 1.53−1.50 (m, 1H), 1.42 (d, J =9.76 Hz, 1H), 1.33 (t, J = 12.32 Hz, 1H), 1.08 (m, 1H), 0.68−0.63 (m, 1H), 0.60−0.57 (m, 1H), 0.52−0.48 (m, 1H), 0.42−0.38 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 141.98, 140.93, 135.97, 129.53, 126.86, 121.96, 120.98, 120.64, 119.41, 118,26, 117.89, 113.57, 111.27, 89.91, 69.58, 61.58, 56.67, 50.47, 46.31, 45.49, 39.62, 29.21, 27.21, 23.43, 22.83, 20.85, 5.55, 5.11, 2.47, 0.012. Mp 279−281 °C. HRMS (m/z) [M]+ calculated for C31H35N3O4 513.2628, found [MH]+ 514.2714. IR (ATR, cm−1) νmax 3187, 2160, 1640, 1541, 1505, 1450, 1325, 1271, 1234, 1124, 1032, 854.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[4-(indol-3-yl) butanamido)morphinan (9).
The title compound was prepared by following the general procedure in 39% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.79 (s, 1H, exchangeable), 9.35 (s, 1H, exchangeable), 8.84 (s, 1H, exchangeable), 8.09 (d, J = 7.88 Hz, 1H, exchangeable) 7.51 (d, J = 7.84 Hz, 1H), 7.33 (d, J = 8.04 Hz, 1H), 7.12 (d, J = 2.08 Hz, 1H), 7.08 (t, J = 7.46 Hz, 1H), 6.97 (t, J = 7.42 Hz, 1H), 6.72 (d, J = 8.12 Hz, 1H), 6.64 (d, J = 8.16 Hz, 1H), 6.18 (s, 1H, exchangeable), 4.56 (d, J = 7.84 Hz, 1H), 3.85 (d, J = 4.84 Hz, 1H), 3.09−3.03 (dd, J1 = 5.88 Hz, J2 = 19.48 Hz, 2H), 2.88−2.84 (m, 1H), 2.68 (t, J = 7.48 Hz, 2H), 2.44−2.41 (m, 2H), 2.16 (t, J = 7.32 Hz, 2H), 1.89−1.85 (m, 2H), 1.75−1.70 (m, 2H), 1.56−1.53 (m, 1H), 1.43 (d, J = 9.68 Hz, 1H), 1.38−1.31 (m, 1H), 1.12−1.05 (m, 2H), 0.69−0.67 (m, 1H), 0.61−0.58 (m, 1H), 0.53−0.50 (m, 1H), 0.43−0.40 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 142.02, 140.99, 136.07, 129.56, 127.05, 122.03, 120.88, 120.60, 119.35, 118.28, 118.18, 117.87, 114.11, 111.27, 89.88, 69.59, 67.88, 61.59, 56.65, 50.44, 46.33, 45.49, 29.24, 27.22, 26.15, 24.05, 23.53, 22.85, 15.00, 5.56, 5.10, 2.48. Mp 210−212 °C. HRMS (m/z) [M]+ calculated for C32H37N3O4 527.2784, found [MH]+ 528.2874. IR (ATR, cm−1) νmax 3268, 1671, 1633, 1505, 1455, 1423, 1316, 1178, 1127, 1033.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(indole-2-carboxamido)morphinan (10).
The title compound was prepared by following the general procedure in 81% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.72 (s, 1H, exchangeable), 9.22 (s, 1H, exchangeable), 8.92 (s, 1H, exchangeable), 8.10 (d, J = 7.72 Hz, 1H, exchangeable), 7.62 (d, J = 8.00 Hz, 1H), 7.45 (d, J = 8.24 Hz, 1H), 7.25 (s, 1H), 7.20 (t, J = 7.24 Hz, 1H), 7.05 (t, J = 7.44 Hz), 6.74 (d, J = 8.04 Hz, 1H), 6.58 (d, J = 8.12 Hz, 1H), 6.43 (s, 1H, exchangeable), 4.78 (d, J = 3.68 Hz, 1H), 4.65 (m, 1H), 3.98 (d, J = 6.48 Hz, 1H), 3.39−3.37 (m, 2H), 3.09−2.99 (m, 3H), 2.73 (m, 1H), 1.99 (m, 1H), 1.65 (d, J = 11.52 Hz, 1H), 1.56−1.43 (m, 2H), 1.19−1.07 (m, 3H), 0.71−0.61 (m, 2H), 0.52−0.49 (m, 1H), 0.51−0.40 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 160.45, 146.16, 138.79, 136.50, 131.46, 128.74, 126.95, 123.40, 122.16, 121.44, 119.73, 119.15, 118.55, 112.26, 103.77, 87.41, 69.41, 61.07, 59.70, 57.08, 45.57, 45.28, 30.23, 29.27, 23.57, 19.56, 5.70, 5.16, 2.60. Mp 269−271 °C. HRMS (m/z) [M]+ calculated for C29H31N3O4 485.2315, found [MH]+ 486.2414. IR (ATR, cm−1) νmax 3146, 1633, 1544.97, 1505, 1457, 1341, 1312, 1235, 1173, 1116, 989, 810, 746.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(indole-3-carboxamido)morphinan (11).
The title compound was prepared by following the general procedure in 29% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.65 (s, 1H, exchangeable), 9.21 (s, 1H, exchangeable), 8.85 (s, 1H, exchangeable), 8.14 (d, J = 2.88 Hz, 1H, exchangeable), 8.11 (d, J = 7.56 Hz, 1H), 7.45 (t, J = 6.68 Hz, 2H), 7.18−7.09 (m, 2H), 6.71 (d, J = 8.08 Hz, 1 H), 6.58 (d, J = 8.12 Hz, 1H), 6.27 (s, 1H, exchangeable), 4.80 (d, J = 3.8 Hz, 1H), 4.66−4.60 (m, 1H), 3.91 (d, J = 6.68 Hz, 1H), 3.12−3.05 (m, 2H), 2.95 (m, 1H), 2.74 (m, 1H), 1.90 (m, 1H), 1.66 (d, J = 10.64 Hz, 1H), 1.56−1.44 (m, 2H), 1.11−1.06 (m, 2H), 0.71−0.61 (m, 2H), 0.51−0.40 (m, 2H). 13CNMR (100 MHz, DMSO-d6) δ 164.32, 145.79, 138.41, 135.91, 128.82, 128.09, 125.73, 122.18, 122.07, 120.62, 119.31, 118.06, 111.90, 110.04, 87.85, 69.23, 60.97, 56.99, 45.13, 44.82, 39.02, 30.12, 29.16, 23.36, 19.59, 5.55, 5.19, 2.41. Mp 250−252 °C. HRMS (m/z) [M]+ calculated for C29H31N3O4 485.2315, found [MH]+ 486.2357. IR (ATR, cm−1) νmax 3269, 1667, 1540, 1504, 1459, 1318, 1174, 1118.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(indole-4-carboxamido)morphinan (12).
The title compound was prepared by following the general procedure in 81% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.37 (s, 1H, exchangeable), 9.18 (s, 1H, exchangeable), 8.92 (s, 1H, exchangeable), 7.65 (d, J = 8.00 Hz, 1H, exchangeable), 7.57 (d, J = 8.00 Hz, 1H), 7.46−7.44 (m, 2H), 7.16 (t, J = 7.72 Hz, 1H), 6.85 (s, 1H), 6.71 (d, J = 8.04 Hz, 1H), 6.58 (d, J = 8 Hz, 1H), 6.33 (s, 1H, exchangeable), 4.83 (d, J = 3.24 Hz, 1H), 4.66 (m, 1H), 3.94 (d, J = 6.08 Hz, 1H), 3.32 (m, 1H), 3.10 (m, 2H), 3.05 (m, 2H), 2.74 (m, 2H), 1.94 (m, 1H), 1.67 (d, J = 12.92 Hz, 1H), 1.58 (m, 1H), 1.47 (m, 1H), 1.09 (m, 2H), 0.69 (m, 1H), 0.62 (m, 1H), 0.49 (m, 1H), 0.41 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 167, 145.62, 137.71, 136.30, 130.81, 126.53, 126.19, 125.36, 125.20, 120.34, 118.93, 118.77, 117.22, 114.36, 101.16, 88.85, 69.20, 61.27, 58.73, 54.59, 46.52, 45.99, 42.74, 29.27, 22.31, 20.32, 9.03, 3.80, 3.34. Mp 220−222 °C. HRMS (m/z) [M]+ calculated for C29H31N3O4 485.2315, found [MH]+ 486.2391. IR (ATR, cm−1) νmax 3212, 2152, 1980, 1606, 1505, 1462, 1318, 1118, 771.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(indole-5-carboxamido)morphinan (13).
The title compound was prepared by following the general procedure in 85% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.39 (s, 1H, exchangeable), 9.21 (s, 1H, exchangeable), 8.89 (s, 1H, exchangeable), 8.16 (s, 1H), 7.80 (d, J = 7.76 Hz, 1H), 7.65 (d, J = 8.44 Hz, 1H), 7.44 (d, J = 8.84 Hz, 1H, exchangeable), 7.43 (s, 1H), 6.72 (d, J = 8.04 Hz, 1H), 6.58 (d, J = 8.08 Hz, 1H), 6.55 (s, 1H), 6.37 (s, 1H, exchangeable), 4.79 (d, J = 3.52 Hz, 1H), 4.66−4.61 (m, 1H), 3.95 (d, J = 6.24 Hz, 1H), 3.35 (s, 2H), 3.09−2.98 (m, 4H), 2.73 (m, 2H), 1.97−1.93 (m, 1H), 1.65 (d, J = 12.04 Hz, 1H), 1.56−1.43 (m, 2H), 1.18−1.09 (m, 2H), 0.69−0.62 (m, 2H), 0.50 (m, 1H), 0.41 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 167.01, 146.10, 138.81, 137.43, 128.80, 126.92, 126.64, 125.33, 122.09, 120.66, 120.08, 119.06, 118.34, 110.85, 102.03, 87.55, 69.41, 61.02, 57.02, 45.79, 45.21, 39.54, 30.23, 29.29, 23.54, 19.61, 5.71, 5.17, 2.58. Mp 240−242 °C. HRMS (m/z) [M]+ calculated for C29H31N3O4 485.2315, found [MH]+ 486.2421. IR (ATR, cm−1) νmax 3147, 1627, 1603, 1522, 1502, 1464, 1351, 1323, 1118, 1036, 771, 755, 726.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(indole-6-carboxamido)morphinan (14).
The title compound was prepared by following the general procedure in 75% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.43 (s, 1H, exchangeable), 9.21 (s, 1H, exchangeable), 8.88 (s, 1H, exchangeable), 7.98 (s, 1H), 7.85 (d, J = 7.72 Hz, 1H, exchangeable), 7.61 (d, J = 8.28 Hz, 1H), 7.54 (dd, J1 = 1.32 Hz, J2 = 8.28 Hz, 1H), 7.50 (t, J = 2.72 Hz, 1H), 6.73 (d, J = 8.04 Hz, 1H), 6.58 (d, J = 8.12 Hz, 1H), 6.49 (d, J = 1.88 Hz, 1H), 6.35 (s, 1H, exchangeable), 4.81 (d, J = 3.76 Hz, 1H), 4.63 (m, 1H), 3.94 (d, J = 6.76 Hz, 1H), 3.3 (m, 2H), 3.11−3.05 (m, 2H), 2.98 (m, 1H), 2.72 (m, 1H), 1.94 (m, 1H), 1.65 (d, J = 10.96 Hz, 1H), 1.57−1.43 (m, 2H), 1.23−1.09 (m, 3H), 0.71−0.59 (m, 2H), 0.50 (m, 1H), 0.40 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 167.16, 145.92, 138.53, 134.98, 129.89, 128.76, 127.88, 127.01, 122.15, 119.46, 119.24, 118.21, 118.10, 111.31, 101.26, 87.53, 69.29, 61.04, 57.03, 45.73, 45.17, 39.28, 30.17, 29.16, 23.44, 19.44, 5.60, 5.18, 2.49. Mp 241−243 °C. HRMS (m/z) [M]+ calculated for C29H31N3O4 485.2315, found [MH]+ 486.2371. IR (ATR, cm−1) νmax 3225, 2162, 1975, 1605, 1540, 1503, 1318, 1119, 1066, 780, 735.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(indole-7-carboxamido)morphinan (15).
The title compound was prepared by following the general procedure in 22% yield. 1H NMR (400 MHz, DMSO-d6) δ 11.13 (s, 1H, exchangeable), 9.25 (s, 1H, exchangeable), 8.91 (s, 1H, exchangeable), 8.14 (d, J = 7.52 Hz, 1H, exchangeable), 7.74 (m, 2H), 7.36 (t, J = 2.72 Hz, 1H), 7.09 (t, J = 7.60 Hz, 1H), 6.71 (d, J = 8.04 Hz, 1H), 6.58 (d, J = 8.12 Hz, 1H), 6.50 (m, 1H), 6.39 (s, 1H, exchangeable), 4.88 (d, J = 3.68 Hz, 1H), 4.70 (m, 1H), 3.95 (d, J = 6.64 Hz, 1H), 3.12−3.04 (m, 3H), 2.94 (m, 1H), 2.73 (m, 2H), 2.00−1.92 (m, 1H), 1.68 (d, J = 11.20 Hz, 1H), 1.56−1.45 (m, 3H), 1.24−1.18 (m, 1H), 1.08 (m, 1H), 0.70 (m, 1H), 0.62 (m, 1H), 0.50 (m, 1H), 0.41 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.63, 145.93, 138.61, 133.95, 129.03, 128.69, 126.40, 123.86, 122.10, 120.24, 119.20, 118.17, 118.06, 116.80, 101.06, 87.30, 69.31, 69.01, 57.00, 45.61, 45.18, 30.22, 29.15, 23.45, 19.27, 5.62, 5.16, 2.53. Mp 227−229 °C. HRMS (m/z) [M]+ calculated for C29H31N3O4 485.2315, found [MH]+ 486.2408. IR (ATR, cm−1) νmax 3060, 2166, 1634, 1585, 1504, 1456, 1312, 1280, 1123, 1030, 984.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-[2-(indol-3-yl)acetamido)morphinan (16).
The title compound was prepared by following the general procedure in 80% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.85 (s, 1H, exchangeable), 9.20 (s, 1H, exchangeable), 8.78 (s, 1H, exchangeable), 7.80 (d, J = 7.96 Hz, 1H, exchangeable), 7.58 (d, J = 7.92 Hz, 1H), 7.33 (d, J = 8.08 Hz, 1H), 7.20 (d, J = 2.28 Hz, 1H), 7.06 (t, J = 7.50 Hz, 1H), 6.96 (t, J = 7.88 Hz, 1H), 6.71 (d, J = 8.08 Hz, 1H), 6.56 (d, J = 8.16 Hz, 1H), 6.15 (s, 1H, exchangeable), 4.60 (d, J = 3.88 Hz, 1H), 4.40 (m, 1H), 3.84 (d, J = 6.84 Hz, 1H), 3.58 (s, 2H), 3.37 (s, 2H), 3.07−3.00 (m, 2H), 2.91 (m, 1H), 2.69 (m, 1H), 1.18 (m, 1H), 1.61 (d, J = 10.76 Hz, 1H), 1.40 (m, 2H), 1.10 (m, 2H), 0.68 (m, 1H), 0.59 (m, 1H), 0.46 (m, 1H), 0.39 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 174.63, 185.37, 146.81, 139.41, 138.03, 131.58, 128.34, 126.14, 125.26, 122.75, 120.82, 120.16, 119.49, 118.91, 112.80, 109.25, 90.01, 71.39, 63.20, 60.22, 45.06, 37.47, 34.11, 33.81, 32.11, 29.98, 23.93, 21.85, 9.43, 5.13, 4.01. Mp 242−244. HRMS (m/z) [M]+ calculated for C30H33N3O4 499.2471, found [MH]+ 500.2539. IR (ATR, cm−1) νmax 3218, 1640, 1506, 1317, 1234, 117, 1032, 746.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-[3-(indol-3-yl) propanamido)morphinan (17).
The title compound was prepared by following the general procedure in 55% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.78 (s, 1H, exchangeable), 9.20 (s, 1H, exchangeable), 8.85 (s, 1H, exchangeable), 7.71 (d, J = 8.08 Hz, 1H exchangeable), 7.53 (d, J = 7.88 Hz, 1H), 7.32 (d, J = 8.04 Hz, 1H), 7.12 (d, J = 2.16 Hz, 1H), 7.05 (t, J = 7.08 Hz, 1H), 6.97 (t, J = 7.88 Hz, 1H), 6.71 (d, J = 8.08 Hz, 1H), 6.55 (d, J = 8.16 Hz, 1H), 6.28 (s, 1H, exchangeable), 4.57 (d, J = 3.84 Hz, 1H), 4.42 (m, 1H), 3.90 (d, J = 6.80 Hz, 1H), 3.25 (m, 1H), 3.17 (s, 2H), 3.07−3.01 (m, 2H), 2.93 (t, J = 7.72 Hz, 3H), 2.74−2.66 (m, 1H), 2.53 (m, 1H), 2.44 (dd, J1 =4.76 Hz, J2 = 13.36 Hz, 1H), 1.90−1.82 (m, 1H), 1.60 (d, J = 10.92 Hz, 1H), 1.42−1.36 (m, 2H), 1.11−1.06 (m, 1H), 0.96−0.90 (m, 1H), 0.70−0.59 (m, 2H), 0.50−0.46 (m, 1H), 0.42−0.37 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 169.37, 169.29, 143.31, 136.02, 133.54, 126.26, 124.50, 119.45, 118.46, 116.73, 115.85, 115.57, 111.26, 108.78, 85.09, 66.74, 58.46, 54.49, 45.97, 42.61, 42.29, 33.51, 27.59, 26.55, 20.90, 18.48, 17.08, 3.12, 2.70. Mp 202−204 °C. HRMS (m/z) [M]+ calculated for C31H35N3O4 513.2628, found [MH]+ 514.2691. IR (ATR, cm−1) νmax 3267, 3146, 2157, 1670, 1616, 1543, 1504, 1463, 1427, 1343, 1316, 1172, 1118, 1031.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-[4-(indol-3-yl) butanamido)morphinan (18).
The title compound was prepared by following the general procedure in 34% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.79 (s, 1H, exchangeable), 9.17 (s, 1H, exchangeable), 8.86 (s, 1H, exchangeable), 7.64 (d, J = 8.04 Hz, 1H, exchangeable), 7.51 (d, J = 7.84 Hz, 1H), 7.32 (d, J = 8.04 Hz, 1H), 7.11 (d, J = 2.16 Hz, 1H), 7.05 (t, J = 7.04 Hz, 1H), 7.96 (t, J = 7.88 Hz, 1H), 6.71 (d, J = 8.08 Hz, 1H), 6.54 (d, J = 8.12 Hz, 1H), 6.29 (s, 1H, exchangeable), 4.59 (d, J = 3.88 Hz, 1H), 4.45−4.38 (m, 1H), 3.91 (d, J = 6.80 Hz, 1H), 3.32 (d, J = 19.72 Hz, 1H), 3.25 (m, 1H), 3.02 (m, 2H), 2,96 (m, 1H), 2.69 (t, J = 7.4 Hz, 3H), 2.43 (dd, J1 =4.92 Hz, J2 = 13.32 Hz, 1H), 2.22 (t, J = 7.32 Hz, 2H), 1.92−1.83 (m, 1H), 1.60 (d, J = 13.16 Hz, 1H), 1.38 (m, 2H), 1.07 (m, 1H), 0.99−0.87 (m, 1H), 0.69−0.59 (m, 2H), 0.50−0.45 (m, 1H), 0.41−0.36 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 173.08, 146.69, 139.36, 137.00, 129.64, 127.98, 123.04, 122.95, 121.78, 120.15, 119.20, 119.07, 118.97, 115.05, 112.19, 88.45, 70.11, 61.85, 57.90, 45.98, 45.66, 35.98, 30.97, 29.95, 27.11, 25.11, 24.28, 20.41, 15.92, 6.46, 6.08, 3.34. Mp 191−193 °C. HRMS (m/z) [M]+ calculated for C32H37N3O4 527.2784, found [MH]+ 528.2876. IR (ATR, cm−1) νmax 3267, 3124, 2162, 1671, 1623, 1506, 1456, 1373, 1119.
Biological Evaluation. Drugs.
Morphine sulfate was purchased from Mallinckrodt (St. Louis, MO) or provided by NIDA. Naloxone and naltrexone were purchased from Sigma-Aldrich (St. Louis, MO). All drugs and test compounds were dissolved in pyrogen-free isotonic saline (Baxter Healthcare, Deerfield, IL) or sterile-filtered distilled/deionized water. All radioligands were purchased from PerkinElmer, Boston, MA.
Animals.
Male Swiss Webster mice were used for the warm-water immersion assay. The mice weighed 23−35 g and were housed five to a cage in animal care quarters maintained at 22 °C on a 12 h light− dark cycle with food and water available ad libitum. The mice were transferred to the test room and the tests were then conducted 18 h later. Protocols and procedures were approved by the Institutional Animal Care and Use Committee at Virginia Commonwealth University Medical Center and complied with the recommendations of the International Association for the Study of Pain.
In Vitro Competitive Radioligand Binding Assay.
The competition binding assay was conducted to determine the affinity of the synthesized compounds to the MOR, KOR and DOR. [3H]NLX was used to label MOR while [3H]DPN was used to label both DOR and KOR. The K 3 d and Bmax values for [H]NLX at MOR and [3H]DPN at the KOR and DOR had been determined previously in Dr. Selley’s laboratory. Both the radioligand binding assay and the [35S]-GTPγS binding assay were conducted using monoclonal mice opioid receptor expressed in Chinese hamster ovary (CHO) cell lines. In the radioligand binding assay, 30 μg of membrane protein was incubated with the corresponding radioligand in the presence of different concentrations of test compounds in TME buffer (50 mM Tris, 3 mM MgCl2, and 0.2 mM EGTA, pH 7.7) for 1.5 h at 30 °C. The bound radioligand was separated by filtration using the Brandel harvester. Specific (i.e., opioid receptor-related) binding at the MOR, KOR, and DOR was determined as the difference in binding obtained in the absence and presence of 5 μM naltrexone, U50,488 and SNC80, respectively. The IC50 values were determined and converted to Ki values using the Cheng−Prusoff equation.70
In Vitro [35S]-GTPγS Functional Assay.
The [35S]-GTPγS functional assay was conducted to determine the efficacy of the compounds at the MOR.71 In this assay, 10 μg of MOR-CHO membrane protein was incubated with 10 μM GDP, 0.1 nM [35S]-GTPγS and varying concentrations of the compound under investigation for 1.5 h in a 30 °C water bath. GTPγS was used instead of GTP because GTPγS cannot be hydrolyzed by GTPase. Therefore, when GTPγS binds to the Gα subunit it remains bound and the Gα subunit remains in the active form. Since GTPγS is radiolabeled, the amount of GTPγS that is bound to the Gα subunit can be quantified to determine the relative efficacy of the compounds. The Bradford protein assay was utilized to determine and adjust the concentration of protein required for the assay. Nonspecific binding was determined with 20 μM unlabeled GTPγS. TME buffer (50 mM Tris-HCl, 3 mM MgCl2, 0.2 mM EGTA, pH 7.4) with 100 mM NaCl was used to increase agonist stimulated binding and the final volume in each assay tube was 500 μL. Furthermore, 3 μM of DAMGO was included in the assay as maximally effective concentration of a full agonist for MOR. After the incubation, the bound radioactive ligand was separated from the free radioligand by filtration through a GF/B glass fiber filter paper and rinsed three times with ice-cold wash buffer (50 mM Tris-HCl, pH 7.2) using the Brandel harvester. The results were determined by utilizing a scintillation counter. All assays were determined in triplicate and repeated at least three times. Percent DAMGO-stimulated [35S]-GTPγS binding was defined as (net-stimulated binding by ligand/net-stimulated binding by 3 μM DAMGO) × 100%.
Warm Water Immersion Assay.
Antinociception for the 6α- and 6β-naltrexamine derivatives were determined using the warm-water tail immersion assay described by Coderre and Rollman.72 The assay was conducted using male Swiss Webster mice. The water bath temperature was maintained at 56 ± 0.1 °C. The baseline latency (control) was determined before injecting the compounds into the mice. The average baseline latency obtained for this experiment was 3.0 ± 0.1 s and only mice with a baseline latency of 2−4 s were used. To test for agonism, the tail immersion was done 20 min (time that morphine effect starts to peak) after injecting the indole analogues of 6α and 6β-naltrexamine. To prevent tissue damage, a 10 s maximum cut off time was imposed. Antinociceptive response was calculated as the percentage maximum possible effect (%MPE), where %MPE = [(test − control)/(10 − control)] × 100. In the antagonism study, the 6α and 6β-naltrexamine derivatives were given 5 min before morphine. The tail immersion test was then conducted 20 min after giving morphine. %MPE was calculated for each mouse using at least five mice per drug. AD50 values were calculated using the least-squares linear regression analysis followed by calculation of 95% confidence interval by Bliss method.
Statistical Analysis.
One-way ANOVA followed by the posthoc Dunnett test were performed to assess significance using Prism 6.0 software (GraphPad Software, San Diego, CA).
Molecular Modeling Study.
Molecular modeling studies were conducted to observe the binding modes of NAN and INTA with the MOR which helped in understanding their molecular mechanism of action. Chemical structures of the compounds were sketched in SybylX2.1, and Gasteiger−Hückel charges were assigned before energy minimization (10,000 iterations) with the Tripos Force Fields. The X-ray crystal structure of the antagonist-bound (4DKL)58 and agonist-bound (5C1M)57 MOR were retrieved from the Protein Data Bank (PDB) and prepared for docking by adding hydrogen atoms and deleting water molecules and bound ligands inside the binding pocket. Automated docking was done utilizing the genetic algorithm docking program GOLD 5.4. The binding site was defined to include all atoms within 10 Å of the γ-carbon atom of D147 along with a distance constraint between the 17-N of the ligand’s epoxymorphinan skeleton and the carboxylate group of D147. The best CHEM-PLP scored solutions were chosen for further analyses. Pictures of the binding modes were generated using PyMOL Molecular Graphics System, version 1.7.4.5 Schrödinger, LLC. The distances in the active and inactive MOR crystal structures were measured using PyMOL Molecular Graphics System, version 1.7.4.5 Schrödinger, LLC. The best scored solutions of NAN and INTA in MOR were further analyzed to obtain a 2D image using LigPlot+.73 In the molecular dynamics (MD) study, the best CHEM-PLP-scored solutions were selected and the force field parameter and topology files for NAN and INTA were generated utilizing CGenFF.74−76 Coordinates for the spatial arrangement of the receptors within the lipid bilayer were retrieved from the Orientations of Proteins in Membranes (OPM) database. The simulation system, consisting of the receptor−ligand complex embedded in a lipid (POPC) bilayer surrounded with saline solution (0.1 nM NaCl) was created in VMD 1.9.366 using the CHARMM force field topology file. All simulations were performed under hybrid CHARMM force field parameters that included protein, lipids and ligand with a time-step of 2 fs (fs). Periodic boundary conditions were employed, and Particle Mesh Ewald (PME) summation was used to calculate long-range electrostatic interactions. Nonbonded interactions were calculated with a smooth cutoff at 14 Å with a frequency of 2 fs. The temperature was maintained at 310 K via Langevin dynamics. All molecular modeling simulations were performed using NAMD 2.8.65 MD simulations were carried out in four stages. In the first stage, equilibration of the fluidlike lipid bilayer was performed via minimization (1000 iterations) followed by NPT equilibration (pressure equilibration, 0.5 ns) of the lipid tails only. In the second stage, an NPT equilibration of the system was run for a period of 1 ns with harmonic constraints placed on protein, INTA, and NAN atoms (5 kcal/(mol-Å)). The harmonic restraint was released in stage 3 and the entire system was equilibrated using the NVT canonical ensemble for a further 1 ns. The final production run was conducted for 10 ns using an NVT ensemble. The snapshots from the last 4 ns MD simulations were selected for further analysis and the distances in the receptor−ligand complexes were calculated using VMD 1.9.3.66
Supplementary Material
Acknowledgments
Funding
The work was funded by a PHS grant from NIH/NIDA, DA024022 (Y.Z.). This work was also supported by the VCU Center for High Performance Computing (CHiPC).
ABBREVIATIONS USED
- CHO
chinese hamster ovary
- DAMGO
[D-Ala2-MePhe4-Gly(ol)5]enkephalin
- DOR
delta opioid receptor
- DPN
diprenorphine
- β-FNA
β-funaltrexamine
- INTA
17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(2′-indolyl)-acetamido]morphinan
- KOR
kappa opioid receptor
- MOR
mu opioid receptor
- NAN
17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(indole-7-carboxamido)morphinan
- NAP
17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(4′-pyridyl)carboxamido]morphinan
- NAQ
17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(isoquinoline-3-carboxamido)morphinan
- norBNI
nor-binaltorphimine
- NLX
naloxone
- NTI
naltrindole
- NTX
naltrexone
- TFF
TRIPOS force field
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.8b00349.
Spectra data for the compounds (1H NMR, 13C NMR, MS, IR, and HPLC graphs) (PDF)
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Drug Abuse or the National Institutes of Health.
The authors declare no competing financial interest.
REFERENCES
- (1).Beckett AH, and Casy AF (1954) Synthetic analgesics: stereochemical considerations. J. Pharm. Pharmacol 6, 986–1001. [DOI] [PubMed] [Google Scholar]
- (2).Portoghese PS (1965) A new concept on the mode of interaction of narcotic analgesics with receptors. J. Med. Chem 8, 609–616. [DOI] [PubMed] [Google Scholar]
- (3).Terenius L (1973) Stereospecific interaction between narcotic analgesics and a synaptic plasm membrane fraction of rat cerebral cortex. Acta Pharmacol. Toxicol 32, 317–320. [DOI] [PubMed] [Google Scholar]
- (4).Simon EJ, Hiller JM, and Edelman I (1973) Stereospecific binding of the potent narcotic analgesic (3H) etorphine to rat-brain homogenate. Proc. Natl. Acad. Sci. U. S. A 70, 1947–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Pert BC, and Snyder HS (1973) Opiate receptor: demonstration in nervous tissue. Science 179, 1011–1014. [DOI] [PubMed] [Google Scholar]
- (6).Lord JA, Waterfield AA, Hughes J, and Kosterlitz HW (1977) Endogenous opioid peptides: multiple agonists and receptors. Nature 267, 495–499. [DOI] [PubMed] [Google Scholar]
- (7).Bunzow JR, Saez C, Mortrud M, Bouvier C, Williams JT, Low M, and Grandy DK (1994) Molecular cloning and tissue distribution of a putative member of the rat opioid receptor gene family that is not a μ, δ or κ opioid receptor type. FEBS Lett 347, 284–288. [DOI] [PubMed] [Google Scholar]
- (8).Mollereau C, Parmentier M, Mailleux P, Butour JL, Moisand C, Chalon P, Caput D, Vassart G, and Meunier JC (1994) ORL1, a novel member of the opioid receptor family: cloning, functional expression and localization. FEBS Lett 341, 33–38. [DOI] [PubMed] [Google Scholar]
- (9).Meunier J, Mollereau C, Toll L, Suaudeau C, Moisand D, Alvinerie P, Butour JL, Guillemot JC, Ferrara P, Monsarrat B, et al. (1995) Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature 377, 532–535. [DOI] [PubMed] [Google Scholar]
- (10).Katritch V, Cherezov V, and Stevens RC (2013) Structure-function of the G protein−coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol 53, 531–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Law PY, Wong YH, and Loh HH (2000) Molecular mechanisms and regulation of opioid receptor signaling. Annu. Rev. Pharmacol. Toxicol 40, 389–430. [DOI] [PubMed] [Google Scholar]
- (12).Jin W, Lee NM, Loh HH, and Thayer SA (1994) Opioids mobilize calcium from inositol 1, 4, 5-trisphosphate-sensitive stores in NG108−15 cells. J. Neurosci 14, 1920–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Li AH, and Wang H-L (2001) G protein-coupled receptor kinase 2 mediates μ-opioid receptor desensitization in GABAergic neurons of the nucleus raphe magnus. J. Neurochem 77, 435–444. [DOI] [PubMed] [Google Scholar]
- (14).von Zastrow M, Svingos A, Haberstock-Debic H, and Evans C (2003) Regulated endocytosis of opioid receptors: cellular mechanisms and proposed roles in physiological adaptation to opiate drugs. Curr. Opin. Neurobiol 13, 348–353. [DOI] [PubMed] [Google Scholar]
- (15).Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, Le Meur M, Dolle P, Tzavara E, Hanoune J, Roques BP, and Kieffer BL (1996) Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the μ-opioid-receptor gene. Nature 383, 819–823. [DOI] [PubMed] [Google Scholar]
- (16).Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, and Rothman RB (2002) Salvinorin A: a potent naturally occurring nonnitrogenous κ opioid selective agonist. Proc. Natl. Acad. Sci. U. S. A 99, 11934–11939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Saitoh A, Yoshikawa Y, Onodera K, and Kamei J (2005) Role of δ-opioid receptor subtypes in anxiety-related behaviors in the elevated plus-maze in rats. Psychopharmacology 182, 327–334. [DOI] [PubMed] [Google Scholar]
- (18).Baamonde A, Dauge V, Ruiz-Gayo M, Fulga IG, Turcaud S, Fournie-Zaluski M-C, and Roques BP (1992) Antidepressant-type effects of endogenous enkephalins protected by systemic RB 101 are mediated by opioid δ and dopamine D1 receptor stimulation. Eur. J. Pharmacol 216, 157–166. [DOI] [PubMed] [Google Scholar]
- (19).Foley KM (1993) Opioids. Neurol. Clin 11, 503–522. [PubMed] [Google Scholar]
- (20).Zimmerman DM, and Leander JD (1990) Selective opioid receptor agonists and antagonists: research tools and potential therapeutic agents. J. Med. Chem 33, 895–902. [DOI] [PubMed] [Google Scholar]
- (21).Schmidhammer H (1998) Opioid receptor antagonists In Progress in Medicinal Chemistry (Ellis GP, Luscombe DK, and Oxford AW, Eds.), 35th ed., pp 84–125, Elsevier Science, Amsterdam. [PubMed] [Google Scholar]
- (22).Eguchi M (2004) Recent advances in selective opioid receptor agonists and antagonists. Med. Res. Rev 24, 182–212. [DOI] [PubMed] [Google Scholar]
- (23).Portoghese PS (1992) The role of concepts in structure-activity relationship studies of opioid ligands. J. Med. Chem 35, 1927–1937. [DOI] [PubMed] [Google Scholar]
- (24).Feinberg AP, Creese I, and Snyder SH (1976) The opiate receptor: a model explaining structure-activity relationships of opiate agonists and antagonists. Proc. Natl. Acad. Sci. U. S. A 73, 4215–4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Cheney BV (1988) Structure-activity relationships for drugs binding to the agonist and antagonist states of the primary morphine receptor. J. Med. Chem 31, 521–531. [DOI] [PubMed] [Google Scholar]
- (26).Zaidi SA, Arnatt CK, He H, Selley DE, Mosier PD, Kellogg GE, and Zhang Y (2013) Binding mode characterization of 6α-and 6β-N-heterocyclic substituted naltrexamine derivatives via docking in opioid receptor crystal structures and site-directed mutagenesis studies: Application of the ‘message−address’ concept in development of mu opioid receptor selective antagonists. Bioorg. Med. Chem 21, 6405–6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yuan Y, Zaidi SA, Stevens DL, Scoggins KL, Mosier PD, Kellogg GE, Dewey WL, Selley DE, and Zhang Y (2015) Design, syntheses, and pharmacological characterization of 17-cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(isoquinoline-3′-carboxamido)morphinan analogues as opioid receptor ligands. Bioorg. Med. Chem 23, 1701–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yuan Y, Li G, He H, Stevens DL, Kozak P, Scoggins KL, Mitra P, Gerk PM, Selley DE, Dewey WL, and Zhang Y (2011) Characterization of 6α-and 6β-N-heterocyclic substituted naltrexamine derivatives as novel leads to development of mu opioid receptor selective antagonists. ACS Chem. Neurosci 2, 346–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yuan Y, Elbegdorj O, Beletskaya IO, Selley DE, and Zhang Y (2013) Structure activity relationship studies of 17-cyclopropylmethyl-3, 14β-dihydroxy-4, 5α-epoxy-6α-(isoquinoline-3′-carboxamido) morphinan (NAQ) analogues as potent opioid receptor ligands: preliminary results on the role of electronic characteristics for affinity and function. Bioorg. Med. Chem. Lett 23, 5045–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Siemian JN, Obeng S, Zhang Y, Zhang Y, and Li J-X (2016) Antinociceptive interactions between the imidazoline I2 receptor agonist 2-BFI and opioids in rats: role of efficacy at the μ-opioid receptor. J. Pharmacol. Exp. Ther 357, 509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Li G, Aschenbach LC, Chen J, Cassidy MP, Stevens DL, Gabra BH, Selley DE, Dewey WL, Westkaemper RB, and Zhang Y (2009) Design, synthesis, and biological evaluation of 6α-and 6β-N-heterocyclic substituted naltrexamine derivatives as μ opioid receptor selective antagonists. J. Med. Chem 52, 1416–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Cornelissen JC, Obeng S, Rice KC, Zhang Y, Negus SS, and Banks ML (2018) Application of receptor theory to the design and use of fixed-proportion mu-opioid agonist and antagonist mixtures in rhesus monkeys. J. Pharmacol. Exp. Ther 365, 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Obeng S, Yuan Y, Jali A, Selley DE, and Zhang Y (2018) In vitro and in vivo functional profile characterization of 17-cyclopropylmethyl-3, 14β-dihydroxy-4, 5α-epoxy-6α-(isoquinoline-3-carboxamido) morphinan (NAQ) as a low efficacy mu opioid receptor modulator. Eur. J. Pharmacol 827, 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Le Naour M, Lunzer MM, Powers MD, Kalyuzhny AE, Benneyworth MA, Thomas MJ, and Portoghese PS (2014) Putative kappa opioid heteromers as targets for developing analgesics free of adverse effects. J. Med. Chem 57, 6383–6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ben Haddou T, Beni S, Hosztafi S, Malfacini D, Calo G, Schmidhammer H, and Spetea M (2014) Pharmacological investigations of N-substituent variation in morphine and oxymorphone: opioid receptor binding, signaling and antinociceptive activity. PLoS One 9, e99231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Huang P, Kehner GB, Cowan A, and Liu-Chen L-Y (2001) Comparison of pharmacological activities of buprenorphine and norbuprenorphine: norbuprenorphine is a potent opioid agonist. J. Pharmacol. Exp. Ther 297, 688–695. [PubMed] [Google Scholar]
- (37).Chavkin C, and Goldstein A (1981) Specific receptor for the opioid peptide dynorphin: structure-activity relationships. Proc. Natl. Acad. Sci. U. S. A 78, 6543–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Portoghese P (1989) Bivalent ligands and the message-address concept in the design of selective opioid receptor antagonists. Trends Pharmacol. Sci 10, 230–235. [DOI] [PubMed] [Google Scholar]
- (39).Portoghese PS, Sultana M, Nagase H, and Takemori AE (1988) Application of the message-address concept in the design of highly potent and selective non-peptide. delta. opioid receptor antagonists. J. Med. Chem 31, 281–282. [DOI] [PubMed] [Google Scholar]
- (40).Burford NT, Clark MJ, Wehrman TS, Gerritz SW, Banks M, O’Connell J, Traynor JR, and Alt A (2013) Discovery of positive allosteric modulators and silent allosteric modulators of the μ-opioid receptor. Proc. Natl. Acad. Sci. U. S. A 110, 10830–10835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Kathmann M, Flau K, Redmer A, Tränkle C, and Schlicker E (2006) Cannabidiol is an allosteric modulator at mu- and delta-opioid receptors. Naunyn-Schmiedeberg’s Arch. Pharmacol 372, 354–361. [DOI] [PubMed] [Google Scholar]
- (42).Remesic M, Hruby VJ, Porreca F, and Lee YS (2017) Recent advances in the realm of allosteric modulators for opioid receptors for future therapeutics. ACS Chem. Neurosci 8, 1147–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Christopoulos A (2014) Advances in G protein-coupled receptor allostery: from function to structure. Mol. Pharmacol 86, 463–478. [DOI] [PubMed] [Google Scholar]
- (44).Burford NT, Watson J, Bertekap R, and Alt A (2011) Strategies for the identification of allosteric modulators of G-protein-coupled receptors. Biochem. Pharmacol 81, 691–702. [DOI] [PubMed] [Google Scholar]
- (45).Fronik P, Gaiser BI, and Sejer Pedersen D (2017) Bitopic ligands and metastable binding sites: opportunities for G protein-coupled receptor (GPCR) medicinal chemistry. J. Med. Chem 60, 4126–4134. [DOI] [PubMed] [Google Scholar]
- (46).Keov P, Valant C, Devine SM, Lane JR, Scammells PJ, Sexton PM, and Christopoulos A (2013) Reverse engineering of the selective agonist TBPB unveils both orthosteric and allosteric modes of action at the M1 muscarinic acetylcholine receptor. Mol. Pharmacol 84, 425–437. [DOI] [PubMed] [Google Scholar]
- (47).Baltos J-A, Gregory KJ, White PJ, Sexton PM, Christopoulos A, and May LT (2016) Quantification of adenosine A1 receptor biased agonism: implications for drug discovery. Biochem. Pharmacol 99, 101–112. [DOI] [PubMed] [Google Scholar]
- (48).Soriano-Ursúa MA, Trujillo-Ferrara JG, Arias-Montaño JA, and Villalobos-Molina R (2015) Insights into a defined secondary binding region on β-adrenoceptors and putative roles in ligand binding and drug design. MedChemComm 6, 991–1002. [Google Scholar]
- (49).Mohr K, Schmitz J, Schrage R, Tränkle C, and Holzgrabe U (2013) Molecular alliance-from orthosteric and allosteric ligands to dualsteric/bitopic agonists at G Protein coupled receptors. Angew. Chem., Int. Ed 52, 508–516. [DOI] [PubMed] [Google Scholar]
- (50).Gentry PR, Sexton PM, and Christopoulos A (2015) Novel allosteric modulators of G protein-coupled receptors. J. Biol. Chem 290, 19478–19488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Feng Z, Hu G, Ma S, and Xie X-Q (2015) Computational advances for the development of allosteric modulators and bitopic ligands in G protein-coupled receptors. AAPS J 17, 1080–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Christopoulos A (2014) Advances in G protein-coupled receptor allostery: from function to structure. Mol. Pharmacol 86, 463–478. [DOI] [PubMed] [Google Scholar]
- (53).Bradley SJ, and Tobin AB (2016) Design of next-generation G protein−coupled receptor drugs: linking novel pharmacology and in vivo animal models. Annu. Rev. Pharmacol. Toxicol 56, 535–559. [DOI] [PubMed] [Google Scholar]
- (54).Sayre LM, and Portoghese PS (1980) Stereospecific synthesis of the 6α- and 6β-amino derivatives of naltrexone and oxymorphone. J. Org. Chem 45, 3366–3368. [Google Scholar]
- (55).Caggiula AR, Epstein LH, Perkins KA, and Saylor S (1995) Different methods of assessing nicotine-induced antinociception may engage different neural mechanisms. Psychopharmacology 122, 301–306. [DOI] [PubMed] [Google Scholar]
- (56).Pasternak GW, and Pan Y-X (2013) Mu opioids and their receptors: evolution of a concept. Pharmacol. Rev 65, 1257–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, Kato HE, Livingston KE, Thorsen TS, Kling RC, Granier S, Gmeiner P, Husbands SM, Traynor JR, Weis WI, Steyaert J, Dror RO, and Kobilka BK (2015) Structural insights into μ-opioid receptor activation. Nature 524, 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, and Granier S (2012) Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 485, 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Jones G, Willett P, Glen RC, Leach AR, and Taylor R (1997) Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol 267, 727–748. [DOI] [PubMed] [Google Scholar]
- (60).Zhang Y, Sham YY, Rajamani R, Gao J, and Portoghese PS (2005) Homology modeling and molecular dynamics simulations of the mu opioid receptor in a membrane−aqueous system. ChemBioChem 6, 853–859. [DOI] [PubMed] [Google Scholar]
- (61).Wang H, Zaidi SA, and Zhang Y (2017) Binding mode analyses of NAP derivatives as mu opioid receptor selective ligands through docking studies and molecular dynamics simulation. Bioorg. Med. Chem 25, 2463–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Izrailev S, Stepaniants S, Balsera M, Oono Y, and Schulten K (1997) Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys. J 72, 1568–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Woo H-J, and Roux B (2005) Calculation of absolute protein−ligand binding free energy from computer simulations. Proc. Natl. Acad. Sci. U. S. A 102, 6825–6830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Wang W, Donini O, Reyes CM, and Kollman PA (2001) Biomolecular simulations: recent developments in force fields, simulations of enzyme catalysis, protein-ligand, protein-protein, and protein-nucleic acid noncovalent interactions. Annu. Rev. Biophys. Biomol. Struct 30, 211–243. [DOI] [PubMed] [Google Scholar]
- (65).Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, and Schulten K (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Humphrey W, Dalke A, and Schulten K (1996) VMD: visual molecular dynamics. J. Mol. Graphics 14, 33–38. [DOI] [PubMed] [Google Scholar]
- (67).Hou T, McLaughlin W, Lu B, Chen K, and Wang W (2006) Prediction of binding affinities between the human amphiphysin-1 SH3 domain and its peptide ligands using homology modeling, molecular dynamics and molecular field analysis. J. Proteome 5, 32–43. [DOI] [PubMed] [Google Scholar]
- (68).Gohlke H, Kiel C, and Case DA (2003) Insights into protein−protein binding by binding free energy calculation and free energy decomposition for the Ras−Raf and Ras−RalGDS complexes. J. Mol. Biol 330, 891–913. [DOI] [PubMed] [Google Scholar]
- (69).Wang J, Hou T, and Xu X (2006) Recent advances in free energy calculations with a combination of molecular mechanics and continuum models. Curr. Comput.-Aided Drug Des 2, 287–306. [Google Scholar]
- (70).Yung-Chi C, and Prusoff WH (1973) Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- (71).Selley DE, Liu Q, and Childers SR (1998) Signal transduction correlates of mu opioid agonist intrinsic efficacy: receptor-stimulated [35S] GTPγS binding in mMOR-CHO cells and rat thalamus. J. Pharmacol. Exp. Ther 285, 496–505. [PubMed] [Google Scholar]
- (72).Coderre TJ, and Rollman GB (1983) Naloxone hyperalgesia and stress-induced analgesia in rats. Life Sci 32, 2139–2146. [DOI] [PubMed] [Google Scholar]
- (73).Wallace AC, Laskowski RA, and Thornton JM (1995) LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng., Des. Sel 8, 127–134. [DOI] [PubMed] [Google Scholar]
- (74).Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, and Mackerell AD (2010) CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Vanommeslaeghe K, and MacKerell AD (2012) Automation of the CHARMM general force field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model 52, 3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Vanommeslaeghe K, Raman EP, and MacKerell AD (2012) Automation of the CHARMM general force field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model 52, 3155–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.