

Abstract
Background
Cardiac autonomic neuropathy is thought to cause adverse cardiovascular effects in diabetes mellitus. Pulmonary vein ganglia (PVG), which have been implicated in normal and abnormal heart rhythm regulation, have not been fully investigated in type 1 diabetes mellitus (T1D). We examined the functional and anatomical effects of T1D on PVG and studied the details of T1D‐induced remodeling on the PVG structure and function.
Methods and Results
We used a mouse model of T1D (Akita mouse), immunofluorescence, isolated Langendorff‐perfused hearts, and mathematical simulations to explore the effects of T1D on PVG. Whole‐mount atrial immunofluorescence of choline acetyltransferase and tyrosine hydroxylase labeling showed that sympathetic and parasympathetic somas of the PVG neurons were significantly hypotrophied in T1D hearts versus wild type. Stimulation of PVG in isolated Langendorff‐perfused hearts caused more pronounced P‐P interval prolongation in wild type compared with Akita hearts. Propranolol resulted in a comparable P‐P prolongation in both phenotypes, and atropine led to more pronounced P‐P interval shortening in wild type compared with Akita hearts. Numerical modeling using network simulations revealed that a decrease in the sympathetic and parasympathetic activities of PVG in T1D could explain the experimental results.
Conclusions
T1D leads to PVG remodeling with hypotrophy of sympathetic and parasympathetic cell bodies and a concomitant decrease in the PVG sympathetic and parasympathetic activities.
Keywords: autonomic nervous system, diabetes mellitus, pulmonary vein
Subject Categories: Electrophysiology
Clinical Perspective
What Is New?
Type 1 diabetes mellitus leads to hypotrophy of pulmonary vein ganglia somas and to decreased pulmonary vein ganglia sympathetic and parasympathetic activities.
What are the Clinical Implications?
Pulmonary vein ganglia autonomic disbalance can contribute to heart rate dysregulation in diabetic cardiac autonomic neuropathy.
Restoring pulmonary vein ganglia sympathovagal balance could have therapeutic effects on diabetic heart rhythm control.
Introduction
Type 1 diabetes mellitus (T1D) is increasing at an alarming rate of about 4% per year.1 Consequently, T1D is becoming a significant public health burden.1, 2 Cardiac autonomic neuropathy (CAN) is a serious complication in patients with diabetes mellitus.3 It is estimated that in T1D, the prevalence of CAN is ≈40%.3 CAN is characterized by both cholinergic/parasympathetic4, 5 and noreadrenergic/sympathetic dysfunction.5, 6 CAN has been proposed to increase the risk of silent myocardial infarction and sudden cardiac death2, 5 and serious cardiac arrhythmias including atrial fibrillation (AF).7, 8
Multiple studies have suggested that there is a significant association between T1D and AF.9, 10, 11, 12, 13 CAN has been proposed to contribute to AF in the diabetic heart; however, the exact pathways are elusive.7, 8 AF initiation and perpetuation mechanisms include triggers that can originate from the pulmonary veins14, 15 with involvement of the pulmonary vein ganglia (PVG).16, 17, 18 Nevertheless, it remains unclear if PVG are remodeled in diabetes mellitus and if such remodeling could affect heart rate regulation. We used a murine model of T1D (Akita)19 to study the functional and anatomical effects of T1D on the PVG. The Akita mouse is characterized by a mutation in the ins2 gene, resulting in Ins2 cystein 96 to tyrosine mutation. This leads to an insulin transport defect causing destruction of pancreatic islet cells and subsequent development of the T1D phenotype.19
In this study, we investigated, in detail—using confocal microscopy, whole‐heart electrophysiology, and numerical simulations—whether T1D remodeled the PVG structure and function.
Methods
Numerical simulation material relevant to this study is available on reasonable request to the corresponding author.
Animals
Our study conformed to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication no. 85‐23, revised 1996) and was approved by the University Committee on Use and Care of Animals of the University of South Florida. Adult wild‐type (WT; n=16) and Akita (n=16) mice, aged 3 to 5 months, were purchased from the Jackson Laboratory and used for experiments. Animals were heparinized, and on induction with CO2 and following loss of reflexes, cervical dislocation was performed and the heart was quickly excised via thoracotomy.
Whole‐Mount Atria Preparations
The samples were prepared as described previously.20 The atria were gently dissected open, flattened, and fixed for 30 minutes in 4% paraformaldehyde at room temperature, and then rinsed 3×10 minutes in cold PBS.
Immunofluorescence
As detailed earlier,20, 21 the specimens were permeabilized for 1 hour with 0.5% Triton‐X in PBS and blocked in 5% normal donkey serum in PBS for 2 hours at room temperature. The preparations were incubated in a mixture of primary antibodies (polyclonal goat anti–choline acetyltransferase [anti‐ChAT; 1:100] and polyclonal rabbit anti–tyrosine hydroxylase [anti‐TH; 1:500]) overnight at 4°C. After washing, they were incubated in a mixture of secondary antibodies (Cy3‐labeled donkey antigoat [1:300] and FITC‐labeled donkey antirabbit [1:100]) for 4 hours at room temperature. Subsequently, the preparations were mounted on glass slides using Vectashield Mounting Medium (Vector Laboratories), covered with coverslips, and sealed with clear nail polish. For all antibodies used in this study, negative controls were processed as outlined and did not show any fluorescent staining.
Confocal microscopy and image analysis
Fluorescent images were acquired using a Nikon A1R confocal microscope (Nikon Instruments) and the built‐in NIS‐Elements AR v4.20.01 software (Nikon Instruments). Images of intracardiac PVG were acquired. Fluorescence images in the Cy3 and FITC channels consisted of a series of z‐stacks, 1‐μm thickness. Image analysis was performed using Fiji software.22 The number of cells and the phenotype (ChAT positive or TH positive) was determined by manually counting the cell bodies throughout the optical projection in 3 dimensions. Cells were either ChAT positive only or biphenotypic (positive for both TH and ChAT). Purely TH‐positive cells were uncommon, as shown earlier.20, 21 Ganglionic area was determined in maximum‐intensity Z‐projection, whereas neuronal size was determined by randomly selecting 10 to 20 neurons per ganglia and determining their area. Cell body areas of ChAT‐positive and of biphenotypic (ChAT and TH positive) ganglionic neurons were calculated, and cumulative frequency distributions of ChAT‐positive and of biphenotypic soma areas were plotted in Prism software (GraphPad Software).
Karnovsky and Roots method for acetylcholinesterase staining
Isolated Akita and WT hearts, cleaned from blood, were perfused for 10 minutes with 4% paraformaldehype in 0.1M PBS (pH=7.4). The hearts were dissected in ice‐cold PBS to expose the posterior left atrium and the sinoatrial node (SAN) region. Subsequently, the hearts were further incubated in paraformaldehyde at 4°C overnight. Afterward, the samples were washed by incubation with PBS at 4°C on a rocker. We used the Karnovsky and Roots method21, 23 to stain for acetylcholinesterase, allowing the visualization of both sympathetic and parasympathetic nerve elements. The staining medium contained 6 mmol/L Na acetate, 2 mmol/L acetylthiocholine iodide, 15 mmol/L sodium citrate, 3 mmol/L CuSO4, 0.5 mmol/L K3Fe(CN)6, and 1% Triton‐ X (pH=5.6). The hearts were incubated in the Karnovsky and Roots medium for up to 12 hours at 4°C, and the incubation medium was renewed every 3 to 6 hours. The stained hearts were visualized using a dissection microscope coupled to a 5‐megapixel CMOS camera (Moticam).
ECG Recordings and PVG Stimulation in Isolated Langedroff‐Perfused Hearts
The hearts of 13 WT and 13 Akita mice were isolated and Langendorff‐perfused, as described earlier.21 In short, the excised heart was quickly cannulated and retrogradely perfused with HEPES‐buffered tyrode solution bubbled with 100% O2, and the preparation was maintained at 36±1°C. Volume‐conducted ECG in lead II configuration was recorded using the PowerLab digitizer and the animal BioAmp from AD Instruments. As we have done previously,21 PVG was stimulated via a bipolar electrode using high‐frequency monophasic pulses (200 Hz, 300 μs, and 400 μA; Stimulus Isolator, AD Instruments) applied in trains of durations 300, 500, 700, and 1200 ms.21 Intervals between trains were at least 2 minutes. Stimuli were subthreshold for atrial myocardial excitation. The P‐P intervals were measured before and after stimulation in the absence and presence of 100 μmol/L propranolol and/ or 10 μmol/L atropine in the perfusate.
Numerical Network Simulations
Network simulations were performed using a previously described “integrate‐and‐fire” modeling approach24 based on the SYSTM11 program by MacGregor,25 implemented using the C language in the UNIX environment. The excitability of each cell population was regulated by synaptic currents, an injected current, and noise added to provide variability. Slow activity‐dependent dynamics of the threshold for spike generation were incorporated in some populations. Electrical stimulation of the system was simulated as synaptic inputs from “fiber populations” turned on at a specified time in the simulation run. Parameters for the cell populations in the simulated circuit are in Table 1. The variables used for each population were as described by Rybak.24 Briefly, the resting thresholds for each population were normally distributed around the value of the resting threshold with a standard deviation equal to the variability parameter value (Table 1). Noise amplitude is the conductance added to synaptic conductances produced by each cell's internal noise generator, which acts like 2 synapses, one with an equilibrium potential of 70 mV above and the other 70 mV below resting threshold. These inputs have a time constant of 1.5 ms and firing probabilities of 0.05 per time step. The DC injected current, specified in millivolts, raises the membrane potential by an amount that is inversely proportional to the membrane conductance and is interpreted as the current that is required to raise the membrane potential by the specified number of millivolts when the membrane conductance is at its resting value. At other membrane conductances, the effect on the membrane potential is inversely proportional to the conductance. Parameter values for the fiber population that generated the stimulus and for connections among the cell populations are in Table 2. Excitatory synapses had an equilibrium potential of 115.0 mV; inhibitory synapses had an equilibrium potential of −25.0 mV.
Table 1.
Population Parameters for Simulated Circuit
| Cell Parameters | Population | |||
|---|---|---|---|---|
| SA Node | ChAT | ChAT+TH | Stimulus | |
| Cells in population, n | 1 | 15 | 8 | 10 |
| Resting threshold, mV | 10 | 10 | 10 | 10 |
| Resting threshold variability, ±mV | 0 | 2 | 2 | 2 |
| Membrane time constant, ms | 5 | 9 | 9 | 9 |
| Postspike increase in potassium conductance | 20 | 20 | 20 | 20 |
| Postspike potassium conductance time constant | 100 | 7 | 7 | 10 |
| Adaptation threshold increase | 0.1 | 0.0 | 0.0 | 0.3 |
| Adaptation time constant, ms | 25 | 500 | 500 | 500 |
| Noise amplitude | 0.0 | 0.005 | 0.01 | 0.2 |
| DC injected current, mV | 11.7 | 10.5 | 10.1 | 0.0 |
Table 2.
Connectivity parameters for the simulated circuit
| Connectivity Parameters | Simulation Parameter Value | ||||
|---|---|---|---|---|---|
| Fiber connections to stimulus population | Excitatory fibers | ||||
| Fibers, n | 100 | ||||
| Terminals, n | 9 | ||||
| Firing probability at each simulation time step (1.0 ms) | 0.05 | ||||
| Synaptic weight | 0.002 | ||||
| Synaptic type and time constant (ms) | Excitatory | ||||
| Time constant (ms) | 100 | ||||
| Cell populations | Stim→ChAT | Stim→ChAT+TH | Ach→ChAT+TH | ChAT→SAN | ChAT+TH→SAN |
| Synaptic type | Excitatory | Excitatory | Inhibitory | Inhibitory | Excitatory |
| Time constant (ms) | 5 | 5 | 2 | 500 | 5 |
| Min. conduction time (ms) | 2 | 2 | 2 | 2 | 2 |
| Max. conduction time (ms) | 2 | 2 | 6 | 6 | 6 |
| Terminals |
WT: 13 AK: 15 |
WT: 12 AK: 12 |
WT: 14 AK: 8 |
WT: 5 AK: 7 |
3 |
| Synaptic weight |
WT: 0.027 Akt: 0.035 |
WT: 0.0044 Akt: 0.01 |
WT: 0.001 Akt: 0.000005 Atropine: 0 |
WT: 0.000005 Akt: 0.000002 Atropine: 0 |
WT: 0.0005 Akt: 0.0002 PR: 0 |
Ach indicates acetylcholinesterase; Akt, Akita; ChAT, choline acetyltransferase; PR, propranolol; Stim, stimulation; TH, tyrosine hydroxylase; WT, wild type.
Statistical Analysis
Data were plotted and presented as average±SE. A 2‐sample t test, or Wilcoxon rank sum test, as well as paired a t test or Wilcoxon signed rank test were used, as appropriate. When Wilcoxon tests were applied, the median and interquartile range (IQR) were also reported. Significance was taken at P<0.05. Analysis was performed in SAS/STAT v14.3 (SAS Institute). Gaussian cumulative distributions were fitted to cell body areas using GraphPad Prism v7.0b.
Results
Somas of PVG Neurons Are Hypotrophied in the Diabetic Heart
To quantify possible remodeling of the PVG in the Akita diabetic heart, we performed immunofluorescence confocal microscopy of the mouse atria in whole‐mount preparations stained with the sympathetic marker TH and the parasympathetic marker ChAT. A WT heart is shown in Figure 1. We then quantified the areas of somas in PVG of WT and Akita hearts. Somas were either ChAT positive (parasympathetic) or biphenotypic ChAT and TH positive (sympathetic), as we and others have shown previously.20, 21 Figure 2 is a high‐magnification image of the PVG boxed in white in Figure 1. Green is TH (Figure 2A, left panel), red is ChAT (middle panel), and the right panel is the merged image. Cumulative frequency distributions of ganglionic cell areas in WT and Akita PVG are shown in Figure 2B, in which ChAT and biphenotypic somas of PVG neurons in Akita hearts were atrophied and had a significantly smaller mean area compared with their WT counterparts. In Figure 2B (left), the cumulative frequency distribution of ChAT‐positive cell body area showed that the mean area was 130.2±0.22 μm2, R 2=0.99 (N=3 hearts, n=160 cells), for WT and 118.4±0.25 μm2, R 2=0.99 (N=3, n=161 cells), for Akita (P<0.001, WT versus Akita). The cumulative frequency distribution of biphenotypic cell body areas in Figure 2B (right) showed that the mean soma area was 141.7±0.161 μm2, R 2=0.99 (N=3, n=152 cells), for WT and 127±0.36 μm2, R 2=0.99 (N=3, n=150 cells), for Akita (P<0.001, WT versus Akita). These results indicated that the parasympathetic and biphenotypic somas of PVG are hypotrophied in the Akita heart.
Figure 1.
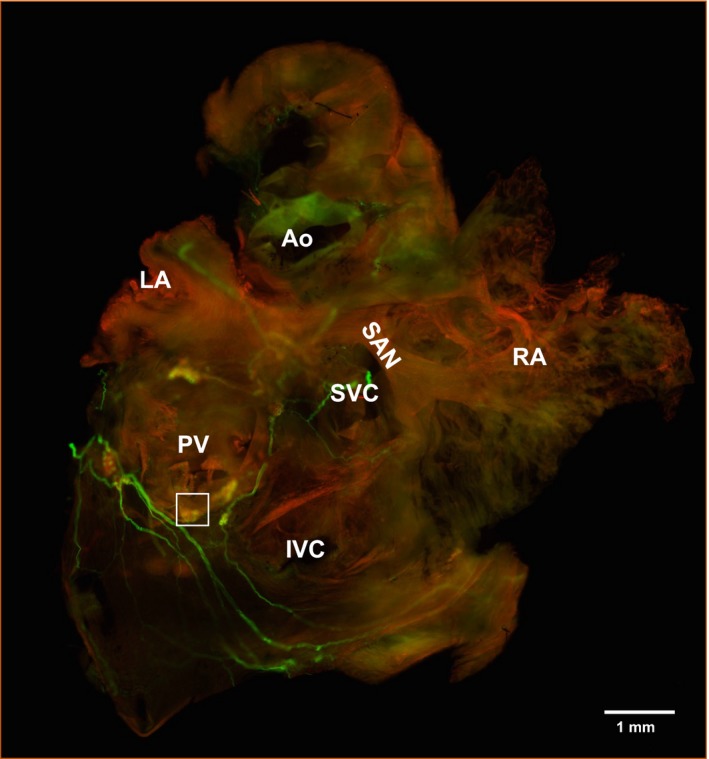
Immunofluorescence of murine atrial intrinsic ganglia. Whole‐mount atrial preparation from a wild‐type heart stained with tyrosine hydroxylase (green) and choline acetyltransferase (red). The pulmonary vein ganglion boxed in white is shown at higher magnification in Figure 2. Ao indicates aorta; IVC, inferior vena cava; LA, left atrium; PV, pulmonary veins; RA, right atrium; SAN, sinoatrial node; SVC, superior vena cava.
Figure 2.
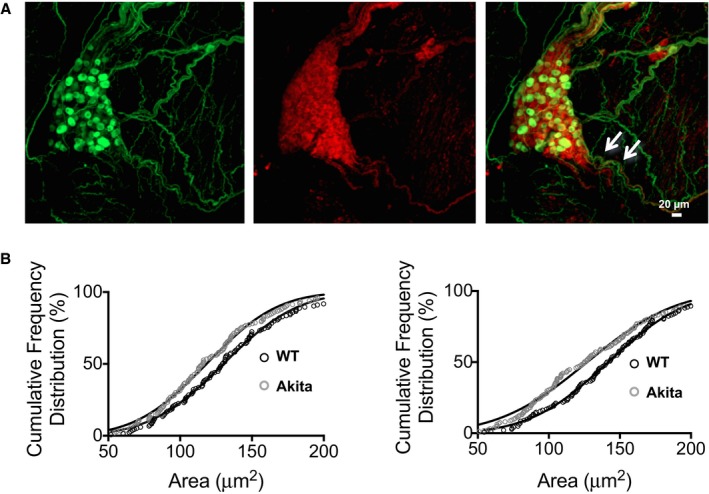
Quantification of pulmonary vein ganglia (PVG) soma size. A, Immunofluorescence of mouse PVG. Tyrosine hydroxylase (TH, green) and choline acetyltransferase (ChAT, red). The PVG boxed in white in Figure 1 is shown at higher magnification. B, The white arrows point to nerve containing ChAT‐ and TH‐positive fibers. Cumulative frequency distribution of ganglionic neuronal cell body areas of ChAT‐positive and of biphenotypic (ChAT‐ and TH‐positive) cells. Cells that are only TH positive were not common. The left panel shows cumulative frequency distribution of ChAT‐positive cell body area, where the mean soma size was 130.2±0.22 μm2, R 2=0.99 (N=3 hearts, n=160 cells) for wild‐type (WT) and 118.4±0.25 μm2, R 2=0.99 (N=3, n=161 cells) for Akita hearts (P<0.001). The right panel shows cumulative frequency distribution of biphenotypic cell body areas, where the mean soma size was 141.7±0.161 μm2, R 2=0.99 (N=3, n=152 cells) for WT and 127±0.36 μm2, R 2=0.99 (N=3, n=150 cells) for Akita hearts (P<0.001).
Innervation of the SAN by the PVG Is Intact in the Diabetic Heart
PVG innervates the SAN in humans and in rodents, and these nerve connections are similar between the 2 species.21, 26 We used qualitative acetylcholinesterase staining, as we have done previously,21, 26 to investigate whether diabetes mellitus disrupts the connections between PVG and the SAN. Figure 3A and 3B are images of stained WT and Akita hearts. PVG in the posterior left atrium are indicated by white double arrows. Nerves emerge from these ganglia, advance into the right atrium, and proceed to innervate the SA node (triple arrow heads). Figure 3C and 3E are enlarged views of the SAN area (boxed in white), showing intact innervation originating from the PVG in both WT and Akita hearts. Figure 3D and 3F show ×20 magnification of the PVG (white double arrows) in Figure 3A and 3B. These experiments suggested that diabetes mellitus did not cause anatomical disruption of the nerves that emerge from the PVG and advance to innervate the SAN.
Figure 3.
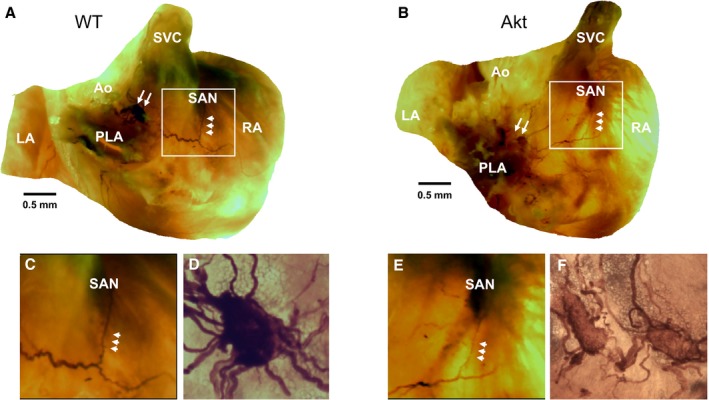
Acetylcholinesterase staining of wild‐type (WT) and Akita (Akt) atria. A and B, Stained WT and Akita hearts respectively. Pulmonary vein ganglia (PVG) are indicated by white double arrows. Triple arrowheads indicate nerves projecting from PVG to the sinoatrial node (SAN). C and E, Enlarged views of the SAN area boxed in white in WT and Akita hearts. D and F, ×20 magnification of the PVG (white double arrows) in panels A and B. Ao indicates aorta; LA, left atrium; PLA, posterior left atrium; RA, right atrium; SVC, superior vena cava.
Diabetes Mellitus Alters the Regulation of Heart Rate by PVG
We demonstrated in an earlier study that PVG stimulation modulated the heart rate. In this study, we investigated whether diabetes mellitus affected the regulation of the heart rate by PVG. In isolated Langendorff‐perfused hearts, PVG was stimulated with a bipolar electrode using high‐frequency monophasic pulses applied in trains of durations 300, 500, 700, and 1200 ms.21 This stimulation regimen stimulates neurons but is subthreshold for cardiomyocytes.21 Figure 4A shows ECG traces of WT and Akita hearts. When a 700‐ms stimulus was applied to the PVG region, the P‐P interval after stimulation was longer in the WT compared with the Akita heart. Figure 4B shows the P‐P intervals of the 20 beats immediately following PVG stimulation in WT and Akita hearts. In control conditions, the P‐P intervals after PVG stimulation were significantly longer in the WT hearts compared with Akita (P<0.001). In the presence of propranolol, which blocks sympathetic signaling, the P‐P intervals were prolonged in the WT and Akita hearts (P=0.3). We then blocked parasympathetic signaling with atropine to investigate the unopposed effects of the sympathetic PVG tone. Stimulation caused a more pronounced decrease in the P‐P interval of the WT hearts compared with Akita (P<0.001). In Figure 4C (left), the averaged P‐P intervals of the 3 beats before PVG stimulation and those of 3 beats right after PVG stimulation are plotted. In control, stimulation prolonged the WT P‐P interval from 205.6±7.5 ms (median: 211.3 ms; IQR: 173.8‐248.8 ms) before stimulation to 317.8±11.5 ms (median: 315 ms; IQR: 276.9‐353.1 ms) after stimulation (P<0.001). In Akita hearts, the P‐P interval was prolonged, from 167.8±5.7 ms (median: 182.4 ms; IQR: 155.6‐209.2 ms) to 218.9±5.1 ms (median: 209.6 ms; IQR: 190‐229.2 ms; P<0.001). In the presence of 1 μmol/L propranolol (Figure 4C, middle), the WT P‐P interval was prolonged significantly, from 235.2±13.4 ms (median: 208.4 ms; IQR: 163.8‐253 ms) before stimulation to 407.1±22.6 ms (median: 432.4 ms; IQR: 351.3‐514.6 ms) after stimulation (P<0.001). In Akita, the prolongation was also significant, from 211.9±7.5 ms (median: 212.5 ms; IQR: 174.6‐250.4 ms) to 348.9±16.1 ms (median: 312.8 ms; IQR: 232.9‐392.8 ms; P<0.001). Finally, with 10 μmol/L atropine (Figure 4C, right), stimulation significantly shortened the P‐P interval in WT hearts from 199.5±2.5 ms (median: 201.5 ms; IQR: 196.8‐206.3 ms) before to 176.4±6.4 ms (median: 176 ms; IQR: 160.8‐191.3 ms) after stimulation (P=0.003) and in Akita hearts from 202.3±6.76 ms (median: 210 ms; IQR: 184.5‐235.5 ms) before to 195.9±7.1 ms (median: 182 ms, IQR: 156‐208 ms) after stimulation (P=0.031).
Figure 4.
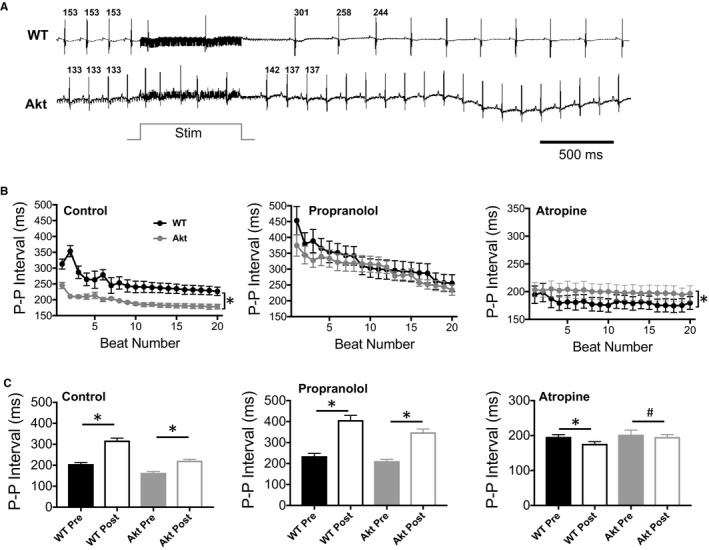
Modulation of the P‐P interval in the isolated Langendorff‐perfused wild‐type (WT) and Akita (Akt) mouse hearts by pulmonary vein ganglia (PVG) stimulation. A, ECG traces from WT and Akita hearts. The P‐P intervals of the 3 beats before and after stimulation were measured. Stim=700‐ms stimulation. B, P‐P intervals of the 20 beats following 700‐ms stimulation of the PVG in control (WT, n=7; Akt, n=7), propranolol (WT, n=8; Akt, n=8), or atropine (WT, n=5; Akt, n=5). *P<0.01, WT vs Akita hearts, Wilcoxon rank sum test. C, Average of 3 P‐P intervals preceding PVG stimulation (pre) and the average of the 3 P‐P intervals immediately following PVG stimulation of the experiments shown in panel B during control and treatment with propranolol or atropine. # P<0.05, *P<0.01, pre vs post, Wilcoxon signed rank test.
We then computed the change in P‐P interval (difference between the average of 3 beats before and 3 beats after PVG stimulation) as a function of stimulus train duration in the control (Figure 5A) and in the presence of propranolol (Figure 5B), atropine (Figure 5C), or propranolol plus atropine (Figure 5D). Figure 5A shows that at stimulus of 300 ms (P=0.046), 700 ms (P=0.0022), and 1200 ms (P=0.0018), the increase in P‐P interval was significantly larger in WT than Akita hearts. After sympathetic blockade with propranolol (Figure 5B), although not significant (P=0.095 at 300‐ms stimulus duration, P=0.791 at 500 ms, P=0.109 at 700 ms, and P=0.244 at 1200 ms), the increase in P‐P interval tended to be larger in WT animals at 700‐ and 1200‐ms trains. After parasympathetic blockade with atropine (Figure 5C), the decrease in P‐P interval was significantly more pronounced in WT than Akita hearts at 300‐ms (P=0.022), 500‐ms (P=0.034), and 1200‐ms (P=0.012) trains. Blocking both sympathetic and parasympathetic signaling (Figure 5D) blunted the effects of PVG stimulation on the P‐P interval, and thus there was no difference in WT versus Akita hearts (P=0.122 at 300 ms, P=0.576 at 500 ms, P=0.096 at 700 ms, and P=0.467 at 1200 ms).
Figure 5.
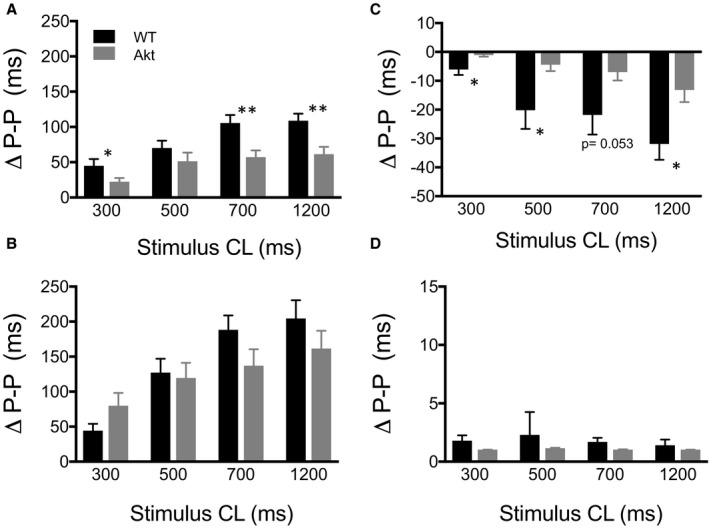
Average change in P‐P interval of the first 3 beats after stimulation. Change in P‐P interval after stimulation with 300‐, 500‐, 700‐, and 1200‐ms stimulus trains in (A) control (wild type [WT], n=7; Akita [Akt], n=7), (B) propranolol (WT, n=8; Akt, n=8), (C) atropine (WT, n=5; Akt, n=5), and (D) propranolol plus atropine (WT, n=4; Akt, n=4). *P<0.05, **P<0.01, WT vs Akita hearts, t test. CL indicates cycle length.
The results of Figures 4 and 5 indicate that T1D altered the sympathovagal balance of the PVG tone. PVG contain parasympathetic (ChAT‐positive) somas (Figures 1 and 2) and sympathetic (TH‐ and ChAT‐positive) somas (Figures 1 and 2).20, 21 When PVG was stimulated in control conditions, P‐P interval prolongation was more pronounced in WT compared with Akita hearts. Initially, this indicated to us that the net output of the Akita PVG is shifted more toward a sympathetic tone and that this could be achieved by a simple increase in the sympathetic activity of the Akita PVG neurons. However, when parasympathetic signaling was blocked with atropine, leaving sympathetic activity unopposed, stimulation caused a significant decrease of the P‐P interval of the WT but not the Akita hearts. This implied that the sympathetic activity of PVG neurons is in fact reduced in Akita compared with WT hearts. Consequently, we hypothesized that the stronger net sympathetic tone of the Akita PVG (Figures 4A–4C and 5A) could be due to a decrease in both the sympathetic and parasympathetic activities of the PVG neurons. To test this hypothesis, we performed mathematical modeling using network simulations.
Modeling the Effects of Diabetes Mellitus on the Regulation of the P‐P Interval by PVG
Network simulations were performed using a previously described modeling approach.24, 25 The circuit in Figure 6A was formed by an excitatory population of 10 cells (Stim) that simulated the excitation provided by the bipolar electrode in the experiment. The ganglion was modeled as a population of 15 ChAT‐positive parasympathetic cells and 8 biphenotypic ChAT‐ and TH‐positive sympathetic cells. Ganglionic parasympathetic neurons are hypothesized to have inhibitory connections with both the SAN cell and the ganglionic sympathetic neurons. Ganglionic sympathetic neurons are hypothesized to excitatory connections with the SA node. Thus, the fiber projecting from the ganglion to the SAN cell has sympathetic and parasympathetic fibers, as also seen in the immunofluorescence experiment of Figure 2A and as we have previously shown.21 Each cell population was regulated by synaptic currents, an injected current, and noise added to provide variability. Slow activity‐dependent dynamics of the threshold for spike generation were incorporated in some populations. Electrical stimulation of the system was simulated as synaptic inputs from a cell population (Stim) turned on at a specified time in the simulation run. The population parameters are in Table 1, and the connectivity parameters are in Table 2. In WT settings, stimulation initiation (black arrow in Figure 6B) resulted in acceleration of firing the ChAT and biphenotypic ganglionic cell population and in the slowing down the SAN cell firing rate. Diabetes mellitus was modeled as smaller synaptic weights of the sympathetic and parasympathetic synaptic parameters compared with WT to simulate a reduction in the sympathetic and parasympathetic activities of the PVG neurons (table 2). Figure 6C shows the simulations results, and Figure 6D shows the experimental results at 700‐ms stimulus duration (from Figure 5). In simulations, under control conditions, the change in P‐P interval (Δ P‐P) is larger in WT compared with Akita hearts. The effects of propranolol in WT and Akita hearts were simulated as reduction to 0 in the synaptic weight of the excitatory synapse between sympathetic cells and the SAN cell (Table 2). This resulted in more pronounced prolongation of the P‐P interval in WT compared with Akita hearts. Atropine in WT and Akita hearts was simulated as a decrease to 0 in the synaptic weight of the inhibitory synapse between parasympathetic cells and the SAN cell and in the inhibitory synapse interconnecting the sympathetic and parasympathetic neurons in the PVG (Table 2). This resulted in more pronounced reduction the P‐P interval in WT compared with Akita hearts. The simulation results (Figure 6C) agree with the experimental results reproduced in Figure 6D.
Figure 6.
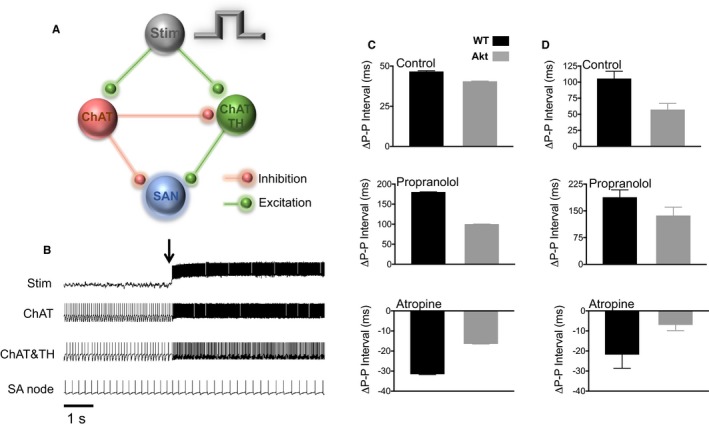
Numerical model. A, A ganglion was modeled as having 15 choline acetyltranferase positive cells, and 8 biphenotypic (tyrosine hydroxylase and choline acetyltranferase positive cells). The nerve projecting from the ganglion to the sinoatrial node (SAN) cell has sympathetic and parasympathetic fibers. B, Action potentials from the different components of the circuit. Stim shows the time when stimulation is on. ChAT and ChAT plus TH populations increased their firing rates in response to stimulation, and SAN cell action potential firing slowed down. C, Simulated SAN action potential cycle length changes (Δ P‐P) in wild‐type (WT) and Akita (Akt) cases without and with propranolol or atropine. D, Experimental data from Figure 5 (700‐ms stimulus duration) showing good agreement between numerical and experimental data. ChAT indicates choline acetyltransferase; TH, tyrosine hydroxylase.
Discussion
We show that T1D induces dystrophic changes in the PVG where parasympathetic and sympathetic neuronal cell bodies atrophy in the diabetic heart. In addition, stimulation of the PVG in the isolated Langendorff‐perfused heart resulted in blunted P‐P interval prolongation in the diabetic heart compared with WT. This suggested that the net output of the Akita PVG tone could be sympathetically shifted. PVG stimulation in the presence of propranolol led to P‐P interval prolongation in both the diabetic and WT hearts, and atropine caused a larger decrease in the WT P‐P interval. Treatment with atropine and propranolol together abrogated the effects of PVG stimulation on the P‐P interval. Mathematical modeling using network simulations suggested that in diabetes mellitus, a decrease in the PVG sympathetic and parasympathetic activities could explain the experimental results.
Our whole‐mount atrial immunofluorescence of ChAT and TH labeling showed a significant decrease in the surface area of PVG somas in diabetic compared with WT hearts. This correlates with what has been shown in previous studies27, 28, 29 of streptozocin‐induced T1D mice and rats and in human hearts30 where cell bodies of intrinsic cardiac neurons displayed dystrophic features. The mechanism behind this remodeling is poorly understood, although it was hypothesized that the extracellular hyperosmolarity caused by hyperglycemia could cause neuronal osmotic shrinkage, which might affect neuronal activity.27, 31 It was recently shown by Mabe et al29 that heart rate dysregulation in chemically induced T1D via streptozocin injection in mice was upstream of atrial/nodal tissue, and neurodystrophic changes of atrial innervation at the level of nerve endings were not observed.
Remodeling of the intrinsic cardiac and extrinsic nervous systems could occur in diabetic CAN. Moreover, the intrinsic cardiac nervous system, which includes PVG, plays a role in heart rate regulation,32 and its malfunction may facilitate arrhythmogenesis.15, 33, 34 PVG has significant sympathetic and parasympathetic connections to the SAN in humans and in rodents, and these connections are very similar between the 2 species.21, 26 It has been shown in patients that T1D could result in a more dominant sympathetic regulation of heart rate.35, 36, 37, 38, 39 Our results under control conditions suggested a stronger net sympathetic tone at the level of the PVG output in the diabetic mice.
PVG is thought to play a role in the initiation and perpetuation of AF. Triggers of AF can originate from the pulmonary veins,14 and studies suggested that ablation of PVG can terminate AF.16, 17, 18 Consequently, the alteration of sympathetic and parasympathetic balance within the PVG can conceivably participate in the mechanism of heart rate dysregulation and diabetic AF. However, it should be noted that the pathophysiological changes in diabetes mellitus leading to increased susceptibility to AF do not only revolve around autonomics. Electrical, electromechanical, and structural remodeling take place.40 For instance, diabetic cardiomyopathy is characterized by myocardial hypertrophy, myocardial lipotoxicity, oxidative stress, cellular apoptosis, interstitial fibrosis, contraction–relaxation dysfunction, and mitochondrial dysfunction.40 These processes can also contribute to the initiation and maintenance of AF in the diabetic heart.
T1D is increasing at an alarming rate.1 It is estimated that 5% to 15% of adults diagnosed with type 2 diabetes mellitus are misdiagnosed and actually have T1D.2 In diabetic CAN, both branches of the autonomic nervous system were shown to be affected.5 Perturbations of the cardiac sympathovagal balance by diabetes mellitus could contribute to arrhythmogenesis.2, 5 It was shown that in patients with T1D, β‐blockade modified the sympathovagal balance; it reduced the sympathetic tone, reduced inflammation, and normalized heart rate variability.41 In addition, parasympathetic variables of heart rate variability improved in T1D patients with CAN treated with quinapril, an angiotensin‐converting enzyme inhibitor.42 It thus seems plausible that modulation of cardiac autonomics in diabetes mellitus could have therapeutic effects.43 It remains to be determined whether long‐term cardiac autonomic modulation,44 specifically, restoring the PVG sympathovagal balance, reduces the occurrence of adverse effects in cardiac rhythm and function in T1D.
Limitations
Our experiments were performed in the isolated heart. Although likely, it is still unclear whether PVG remodeling by diabetes mellitus could affect regulation of the heart rate in vivo. It has been shown using ECG telemetry that parameters of heart rate variability are affected by diabetes mellitus in the Akita mouse45 and that those changes do suggest sympathetic and parasympathetic dysfunction similar to our experiments in this study.45 At present, however, it is difficult to isolate, in vivo, the effects of diabetes mellitus on the PVG regulation of the heart rate in the mouse. In addition, the intrinsic cardiac nervous system is extensive and is composed of ganglionated plexi located in several regions of the atria.32 These ganglionated plexi have been shown to participate in modulating SAN activity.32 In this study, we focused on the PVG given their demonstrated role in normal and abnormal heart rhythm regulation, namely, AF.16, 17, 18
Sources of Funding
This work was funded in part by National Heart, Lung, and Blood Institute grants R21HL138064 and R01HL129136 to Noujaim.
Disclosures
None.
(J Am Heart Assoc. 2018;7:e008919 DOI: 10.1161/JAHA.118.008919)
References
- 1. Minges KE, Whittemore R, Grey M. Overweight and obesity in youth with type 1 diabetes. Annu Rev Nurs Res. 2013;31:47–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Skyler JS, Bakris GL, Bonifacio E, Darsow T, Eckel RH, Groop L, Groop PH, Handelsman Y, Insel RA, Mathieu C, McElvaine AT, Palmer JP, Pugliese A, Schatz DA, Sosenko JM, Wilding JP, Ratner RE. Differentiation of diabetes by pathophysiology, natural history, and prognosis. Diabetes. 2017;66:241–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kempler P, Tesfaye S, Chaturvedi N, Stevens LK, Webb DJ, Eaton S, Kerenyi Z, Tamas G, Ward JD, Fuller JH; Group EICS . Autonomic neuropathy is associated with increased cardiovascular risk factors: the EURODIAB IDDM Complications Study. Diabet Med. 2002; 19:900–909. [DOI] [PubMed] [Google Scholar]
- 4. Chessa M, Butera G, Lanza GA, Bossone E, Delogu A, De Rosa G, Marietti G, Rosti L, Carminati M. Role of heart rate variability in the early diagnosis of diabetic autonomic neuropathy in children. Herz. 2002;27:785–790. [DOI] [PubMed] [Google Scholar]
- 5. Vinik AI, Ziegler D. Diabetic cardiovascular autonomic neuropathy. Circulation. 2007;115:387–397. [DOI] [PubMed] [Google Scholar]
- 6. Scognamiglio R, Casara D, Avogaro A. Myocardial dysfunction and adrenergic innervation in patients with Type 1 diabetes mellitus. Diabetes Nutr Metab. 2000;13:346–349. [PubMed] [Google Scholar]
- 7. Bissinger A. Cardiac autonomic neuropathy: why should cardiologists care about that? J Diabetes Res. 2017;2017:5374176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bissinger A, Grycewicz T, Grabowicz W, Lubinski A. The effect of diabetic autonomic neuropathy on P‐wave duration, dispersion and atrial fibrillation. Arch Med Sci. 2011;7:806–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dahlqvist S, Rosengren A, Gudbjornsdottir S, Pivodic A, Wedel H, Kosiborod M, Svensson AM, Lind M. Risk of atrial fibrillation in people with type 1 diabetes compared with matched controls from the general population: a prospective case‐control study. Lancet Diabetes Endocrinol. 2017;5:799–807. [DOI] [PubMed] [Google Scholar]
- 10. Movahed MR, Hashemzadeh M, Jamal MM. Diabetes mellitus is a strong, independent risk for atrial fibrillation and flutter in addition to other cardiovascular disease. Int J Cardiol. 2005;105:315–318. [DOI] [PubMed] [Google Scholar]
- 11. Huxley RR, Alonso A, Lopez FL, Filion KB, Agarwal SK, Loehr LR, Soliman EZ, Pankow JS, Selvin E. Type 2 diabetes, glucose homeostasis and incident atrial fibrillation: the Atherosclerosis Risk in Communities study. Heart. 2012;98:133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huxley RR, Filion KB, Konety S, Alonso A. Meta‐analysis of cohort and case‐control studies of type 2 diabetes mellitus and risk of atrial fibrillation. Am J Cardiol. 2011;108:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aksnes TA, Schmieder RE, Kjeldsen SE, Ghani S, Hua TA, Julius S. Impact of new‐onset diabetes mellitus on development of atrial fibrillation and heart failure in high‐risk hypertension (from the VALUE Trial). Am J Cardiol. 2008;101:634–638. [DOI] [PubMed] [Google Scholar]
- 14. Haissaguerre M, Jais P, Shah DC, Takahashi A, Hocini M, Quiniou G, Garrigue S, Le Mouroux A, Le Metayer P, Clementy J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med. 1998;339:659–666. [DOI] [PubMed] [Google Scholar]
- 15. Choi EK, Zhao Y, Everett TH, Chen PS. Ganglionated plexi as neuromodulation targets for atrial fibrillation. J Cardiovasc Electrophysiol. 2017;28:1485–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lemola K, Chartier D, Yeh YH, Dubuc M, Cartier R, Armour A, Ting M, Sakabe M, Shiroshita‐Takeshita A, Comtois P, Nattel S. Pulmonary vein region ablation in experimental vagal atrial fibrillation: role of pulmonary veins versus autonomic ganglia. Circulation. 2008;117:470–477. [DOI] [PubMed] [Google Scholar]
- 17. Lu Z, Scherlag BJ, Lin J, Yu L, Guo JH, Niu G, Jackman WM, Lazzara R, Jiang H, Po SS. Autonomic mechanism for initiation of rapid firing from atria and pulmonary veins: evidence by ablation of ganglionated plexi. Cardiovasc Res. 2009;84:245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stavrakis S, Nakagawa H, Po SS, Scherlag BJ, Lazzara R, Jackman WM. The role of the autonomic ganglia in atrial fibrillation. JACC Clin Electrophysiol. 2015;1:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kayo T, Koizumi A. Mapping of murine diabetogenic gene mody on chromosome 7 at D7Mit258 and its involvement in pancreatic islet and beta cell development during the perinatal period. J Clin Invest. 1998;101:2112–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rysevaite K, Saburkina I, Pauziene N, Vaitkevicius R, Noujaim SF, Jalife J, Pauza DH. Immunohistochemical characterization of the intrinsic cardiac neural plexus in whole‐mount mouse heart preparations. Heart Rhythm. 2011;8:731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zarzoso M, Rysevaite K, Milstein ML, Calvo CJ, Kean AC, Atienza F, Pauza DH, Jalife J, Noujaim SF. Nerves projecting from the intrinsic cardiac ganglia of the pulmonary veins modulate sinoatrial node pacemaker function. Cardiovasc Res. 2013;99:566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open‐source platform for biological‐image analysis. Nat Methods. 2012;9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Karnovsky MJ, Roots L. A “Direct‐Coloring” thiocholine method for cholinesterases. J Histochem Cytochem. 1964;12:219–221. [DOI] [PubMed] [Google Scholar]
- 24. Rybak IA, O'Connor R, Ross A, Shevtsova NA, Nuding SC, Segers LS, Shannon R, Dick TE, Dunin‐Barkowski WL, Orem JM, Solomon IC, Morris KF, Lindsey BG. Reconfiguration of the pontomedullary respiratory network: a computational modeling study with coordinated in vivo experiments. J Neurophysiol. 2008;100:1770–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. MacGregor RJ. Neural and Brain Modeling. San Diego: Academic Press; 1987. [Google Scholar]
- 26. Singh S, Johnson PI, Lee RE, Orfei E, Lonchyna VA, Sullivan HJ, Montoya A, Tran H, Wehrmacher WH, Wurster RD. Topography of cardiac ganglia in the adult human heart. J Thoracic Cardiovasc Surg. 1996;112:943–953. [DOI] [PubMed] [Google Scholar]
- 27. Lin M, Ai J, Harden SW, Huang C, Li L, Wurster RD, Cheng ZJ. Impairment of baroreflex control of heart rate and structural changes of cardiac ganglia in conscious streptozotocin (STZ)‐induced diabetic mice. Auton Neurosci. 2010;155:39–48. [DOI] [PubMed] [Google Scholar]
- 28. Menard CE, Durston M, Zherebitskaya E, Smith DR, Freed D, Glazner GW, Tian G, Fernyhough P, Arora RC. Temporal dystrophic remodeling within the intrinsic cardiac nervous system of the streptozotocin‐induced diabetic rat model. Acta Neuropathol Commun. 2014;2:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mabe AM, Hoover DB. Remodeling of cardiac cholinergic innervation and control of heart rate in mice with streptozotocin‐induced diabetes. Auton Neurosci. 2011;162:24–31. [DOI] [PubMed] [Google Scholar]
- 30. Tsujimura T, Nunotani H, Fushimi H, Inoue T. Morphological changes in autonomic ganglionic cells of the heart in diabetic patients. Diabetes Res Clin Pract. 1986;2:133–137. [DOI] [PubMed] [Google Scholar]
- 31. Henneman E, Somjen G, Carpenter DO. Excitability and inhibitability of motoneurons of different sizes. J Neurophysiol. 1965;28:599–620. [DOI] [PubMed] [Google Scholar]
- 32. Armour JA. Potential clinical relevance of the ‘little brain’ on the mammalian heart. Exp Physiol. 2008;93:165–176. [DOI] [PubMed] [Google Scholar]
- 33. Tan AY, Chen PS, Chen LS, Fishbein MC. Autonomic nerves in pulmonary veins. Heart Rhythm. 2007;4:S57–S60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tan AY, Li H, Wachsmann‐Hogiu S, Chen LS, Chen PS, Fishbein MC. Autonomic innervation and segmental muscular disconnections at the human pulmonary vein‐atrial junction: implications for catheter ablation of atrial‐pulmonary vein junction. J Am Coll Cardiol. 2006;48:132–143. [DOI] [PubMed] [Google Scholar]
- 35. Rosengard‐Barlund M, Bernardi L, Fagerudd J, Mantysaari M, Af Bjorkesten CG, Lindholm H, Forsblom C, Waden J, Groop PH; FinnDiane Study Group . Early autonomic dysfunction in type 1 diabetes: a reversible disorder? Diabetologia. 2009; 52:1164–1172. [DOI] [PubMed] [Google Scholar]
- 36. Pop‐Busui R, Kirkwood I, Schmid H, Marinescu V, Schroeder J, Larkin D, Yamada E, Raffel DM, Stevens MJ. Sympathetic dysfunction in type 1 diabetes: association with impaired myocardial blood flow reserve and diastolic dysfunction. J Am Coll Cardiol. 2004;44:2368–2374. [DOI] [PubMed] [Google Scholar]
- 37. Javorka M, Javorkova J, Tonhajzerova I, Javorka K. Parasympathetic versus sympathetic control of the cardiovascular system in young patients with type 1 diabetes mellitus. Clin Physiol Funct Imaging. 2005;25:270–274. [DOI] [PubMed] [Google Scholar]
- 38. Jaiswal M, Urbina EM, Wadwa RP, Talton JW, D'Agostino RB Jr, Hamman RF, Fingerlin TE, Daniels S, Marcovina SM, Dolan LM, Dabelea D. Reduced heart rate variability among youth with type 1 diabetes: the SEARCH CVD study. Diabetes Care. 2013;36:157–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Koivikko ML, Tulppo MP, Kiviniemi AM, Kallio MA, Perkiomaki JS, Salmela PI, Airaksinen KE, Huikuri HV. Autonomic cardiac regulation during spontaneous nocturnal hypoglycemia in patients with type 1 diabetes. Diabetes Care. 2012;35:1585–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Goudis CA, Korantzopoulos P, Ntalas IV, Kallergis EM, Liu T, Ketikoglou DG. Diabetes mellitus and atrial fibrillation: pathophysiological mechanisms and potential upstream therapies. Int J Cardiol. 2015;184:617–622. [DOI] [PubMed] [Google Scholar]
- 41. Lanza GA, Pitocco D, Navarese EP, Sestito A, Sgueglia GA, Manto A, Infusino F, Musella T, Ghirlanda G, Crea F. Association between cardiac autonomic dysfunction and inflammation in type 1 diabetic patients: effect of beta‐blockade. Eur Heart J. 2007;28:814–820. [DOI] [PubMed] [Google Scholar]
- 42. Kontopoulos AG, Athyros VG, Didangelos TP, Papageorgiou AA, Avramidis MJ, Mayroudi MC, Karamitsos DT. Effect of chronic quinapril administration on heart rate variability in patients with diabetic autonomic neuropathy. Diabetes Care. 1997;20:355–361. [DOI] [PubMed] [Google Scholar]
- 43. Aronson D. Pharmacologic modulation of autonomic tone: implications for the diabetic patient. Diabetologia. 1997;40:476–481. [DOI] [PubMed] [Google Scholar]
- 44. Pitocco D, Zaccardi F, Infusino F, Nerla R, Ghirlanda G, Lanza GA. Comment on: Koivikko et al. Autonomic cardiac regulation during spontaneous nocturnal hypoglycemia in patients with type 1 diabetes. Diabetes Care 2012; 35:1585–1590. Diabetes Care. 2013;36:e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rajab M, Jin H, Welzig CM, Albano A, Aronovitz M, Zhang Y, Park HJ, Link MS, Noujaim SF, Galper JB. Increased inducibility of ventricular tachycardia and decreased heart rate variability in a mouse model for type 1 diabetes: effect of pravastatin. Am J Physiol Heart Circ Physiol. 2013;305:H1807–H1816. [DOI] [PMC free article] [PubMed] [Google Scholar]