

Abstract
Background
Short QT syndrome (SQTS) is a rare inheritable disease associated with sudden cardiac death. Data on long‐term outcomes of families with SQTS are limited.
Methods and Results
Seventeen patients with SQTS in 7 independent families (48% men; median age, 42.4 years; corrected QT interval, 324.9±40.8 ms) were followed up for 13.5±2.5 years. A history of sudden cardiac death was documented in 71% of families. A large number of them showed sudden cardiac deaths at a younger age, with a predominance of men (67%). Five patients had syncope (29%) and 9 (53%) had atrial fibrillation or atrial flutter. An SQTS‐related gene was found in 76% of the patients as follows: KCNH2 (SQTS 1) in 4, CACNA1C (SQTS 4) in 3, and CACNb2 (SQTS 5) in 6. Five patients (29%) received an implantable cardioverter‐defibrillator and 5 patients received long‐term prophylaxis with hydroquinidine. During follow‐up, 1 patient received an appropriate implantable cardioverter‐defibrillator shock attributable to ventricular fibrillation. The patient received no further implantable cardioverter‐defibrillator shocks after treatment with hydroquinidine.
Conclusions
The risk of sudden cardiac death in SQTS families is high. However, after appropriate risk assessment and individualized treatment options (hydroquinidine and/or implantable cardioverter‐defibrillator), the long‐term outcome is relatively benign when patients are seen at a reference center.
Keywords: arrhythmia (heart rhythm disorders), arrhythmia (mechanisms), sudden cardiac arrest
Subject Categories: Arrhythmias, Sudden Cardiac Death
Clinical Perspective
What Is New?
The risk of sudden cardiac death in families with short QT syndrome is high.
Implantable cardioverter‐defibrillator implantation and cascade family screening is recommended in patients at high risk.
What Are the Clinical Implications?
The long‐term outcome of short QT syndrome is rather benign when patients undergo appropriate risk assessment and individualized treatment.
Introduction
An alteration of corrected QT (QTc) interval, either shortening or prolongation, is associated with sudden cardiac death (SCD).1 Short QT syndrome (SQTS) is a rare, inheritable cardiac channelopathy associated with abbreviated QTc interval and risk of SCD. The prevalence of SQTS is 0.02% to 0.1% with a male predominance.2 The first SQTS cases were reported in 2000.3 Despite progress in the past decade, the diagnostic and treatment approach may challenge physicians because of its low prevalence and few published cases. There is agreement that in channelopathies, survivors of SCD are at high risk for recurrent ventricular tachyarrhythmia events, and therefore implantable cardioverter‐defibrillator (ICD) therapy is recommended.4
The clinical presentation of patients with SQTS ranges from asymptomatic, palpitations, syncope, dizziness, atrial fibrillation, and SCD.5, 6 Family screening is mandatory when a patient receives a diagnosis of SQTS.7, 8
Six subtypes of SQTS have been described. A gain of potassium ion channels was found in SQTS 1 to 3 affecting genes KCNH2, KCNQ1, and KCNJ2. In SQTS 4, SQT5, and SQTS6, a loss of calcium channels was found, affecting subunits of calcium channel encoding genes including CACNA1C, CACNB2, and CACNA2D1.9, 10 Overlap syndromes, such as SQTS accompanied by Brugada syndrome (BrS), have been reported.11, 12 Hyperkalemia or hypercalcemia, myocardial ischemia, acidosis, and carnitine deficiency are associated with SQTS. Hyperthermia can also induce a shortened QT interval. Furthermore, drugs such as digitalis, acetylcholine, catecholamines or ATP‐sensitive K+ channel activators, and antiepileptic drugs may induce QT shortening.13 Short QT intervals have also been associated with epilepsy, particularly during ictal and postictal states.14, 15, 16
Data on long‐term outcomes of families with SQTS and risk stratification are limited. Therefore, we sought to present the clinical profiles, diagnostic assessments, and outcomes of 7 families with SQTS that were consecutively admitted to our hospital. They have been followed up for 13.5±2.5 years.
Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure and will be provided on request to the corresponding author. SQTS was defined according to Gollob criteria published in 2011 and European Society of Cardiology criteria published in 2015.10, 17 In addition, family screening of first‐ and second‐degree relatives was evaluated. Baseline characteristics of patients with SQTS excluding those with SCD but without ECG documentation are presented. This study was conducted in compliance with the Declaration of Helsinki regarding investigations in human subjects. Informed consent was not required by the ethics committee of the Medical Faculty Mannheim.
Syncope was defined as loss of transient consciousness in the absence of other causes. Arrhythmic events were documented as ventricular fibrillation (VF)/ventricular tachycardia requiring resuscitation and/or defibrillation.
Of 10 families admitted to our hospital with suspected SQTS, 7 families fulfilled the SQTS criteria. From these 7 families, cascade family screening led to the diagnosis of SQTS in 10 affected relatives. QT interval was measured using the tangent method in the precordial lead presenting the highest T‐wave amplitude in V2 or V3.
Genetic Screening
For genetic screening of SQTS 1 to 6, DNA was analyzed using next‐generation sequencing of the affected genes KCNH2, KCNQ1, KCNJ2, CACNA1C, CACNB2, and CACNA2D1. Blood samples were analyzed consistent with the local ethics committee of the University Hospital Mannheim. The diagnosis of SQTS was independently reviewed by 2 experienced cardiologists.
Statistical Analysis
Data are presented as mean±SD for continuous variables with a normal distribution, median (interquartile range) for continuous variables with a nonnormal distribution, and frequency (percentage) for categorical variables. The Kolmogorov–Smirnov test was used to assess normal distribution. A Kaplan‐Meier curve was created indicating the risk of SCD and age. Statistical analysis was performed with SPSS statistical software version 23.0 (IBM) for all analyses.
Results
Demographics of 17 Patients With SQTS
Baseline characteristics of patients with SQTS are illustrated in Table 1. We found 7 independent families with SQTS including 2 families with overlap syndrome with BrS. The mean age was 42 years (interquartile range, 0–67 years). The QTc interval was 324.9±40.8. Most patients and their relatives presented with symptoms including syncope (29%), palpitations (47%), atrial fibrillation (41%), and atrial flutter (12%). Cardiac arrest at admission was found in 3 patients. 12 Twelve patients with SCD underwent cascade family screening. In 6 of the 12 patients, SCD was documented during resting situations. Five patients were discharged on the antiarrhythmic drug hydroquinidine by expert consensus, 2 patients refused starting hydroquinidine, and 2 patients stopped using hydroquinidine because of poor compliance. Three patients started and continued hydroquinidine use and showed a QTc prolongation with no shocks or life‐threatening arrhythmia during follow‐up. One patient who experienced VF and was treated successfully by ICD shock also started hydroquinidine. No further VF events were documented during follow‐up.
Table 1.
Baseline Characteristics of Patients With SQTS at Admission
| Variables | N=17 |
|---|---|
| Demographics | |
| Age, median (IQR) | 42.45 (0–67) |
| Men, No. (%) | 8 (48) |
| Symptoms, No. (%) | |
| Syncope | 5 (29) |
| Palpitation | 8 (47) |
| SCD | 3 (17.6) |
| Atrial flutter | 2 (12) |
| Atrial fibrillation | 7 (41) |
| ECG data, mean±SD | |
| QTc interval, ms | 324.9±40.8 |
| Quinidine, No. (%) | |
| Yes | 6 (35) |
| ECG after quinidine, mean±SD | |
| QTc interval, ms | 409±41.63 |
| ICD implantation, No. (%) | |
| Yes | 5 (29) |
| No | 11 (65) |
| Event recorder | 1 (6) |
| Genetic screening, No. (%) | |
| Unknown mutation | 4 (24) |
| Known mutation | 13 (76) |
| CaCN2b | 6 (35) |
| CaCNA1C | 3 (17) |
| KCNH2 | 4 (23) |
| SCN5a | 1 (6) |
| Positive ajmalin test, No. (%) | 7 (41) |
ICD indicates implantable cardioverter‐defibrillator; IQR, interquartile range; QTc, corrected QT; SCD, sudden cardiac death; SQTS, short QT syndrome.
Genetic Screening
Genetic screening confirmed affected genes in 3 families: SQTS 1 consistent with a KCNH2 mutation (N588K) in 4 patients, SQTS 4 consistent with a CACNA1C mutation (C1442T) in 3 patients, and SQTS 5 consistent with a CACNB2b (G490R) mutation in 6 patients. Table 2 illustrates patient characteristics according to SQTS genotype.
Table 2.
Characteristics of Different Types of SQTS
| Sex | Gene | Mutation | Current | Clinical Symptoms | Supraventricular Arrhythmias | SQTS Type |
|---|---|---|---|---|---|---|
| Female | KCNH2 | N588K | IKr | Palpitation | Atrial fibrillation | 1 |
| Female | KCNH2 | N588K | IKr | None | 1 | |
| Male | KCNH2 | N588K | IKr | Syncope | Atrial fibrillation | 1 |
| Female | KCNH2 | N588K | IKr | SCD | Atrial fibrillation | 1 |
| Female | CaCNA1C | G490R | ICa,L | None | 4 | |
| Female | CaCNA1C | G490R | ICa,L | Palpitation | 4 | |
| Male | CaCNA1C | G490R | ICa,L | Palpitation | Atrial fibrillation | 4 |
| Male | CaCN2b | C1422T | ICa,L |
Syncope SCD |
Atrial fibrillation Atrial flutter |
5 |
| Male | CaCN2b | C1422T | ICa,L | Syncope | 5 | |
| Male | CaCN2b | C1422T | ICa,L | None | 5 | |
| Female | CaCN2b | C1422T | ICa,L | None | 5 | |
| Female | CaCN2b | C1422T | ICa,L | None | 5 | |
| Female | CaCN2b | C1422T | ICa,L | None | 5 |
ICa,L indicates L‐type calcium; IKr, delayed rectifier potassium; SCD, sudden cardiac death; SQTS, short QT syndrome.
Clinical Profiles and Follow‐Up Data of Families With SQTS
The following symptoms were presented in all families: palpitations (6 families), SCD (5 families), atrial fibrillation (5 families), syncope (4 families), and atrial flutter (2 families). Affected genes were documented in 3 families (Table 3). The mean follow‐up time was 13.5±2.5 years. All families except for 1 that were lost to follow‐up were followed for at least 8 years.
Table 3.
Baseline Characteristics of SQTS Families
| Variables | Families (N=7) |
|---|---|
| Demographics, No. (%) | |
| Syncope | 4 (57.1) |
| Palpitation | 6 (85.7) |
| SCD | 5 (71.4) |
| Atrial flutter | 2 (28.6) |
| Atrial fibrillation | 5 (71.4) |
| Affected gene | 3 (42.8) |
SCD indicates sudden cardiac death; SQTS, short QT syndrome.
Family 1
Patients from family 1 were referred for syncope, palpitations, and presence of SCD in their family (Figure 1A). Four of these family members showed an abbreviated QTc interval. One of these patients had SCD at the age of 62 years (QTc interval, 250 ms). Echocardiogram and cardiac magnetic resonance imaging excluded structural heart disease. An ICD was implanted in 3 patients with confirmed genotype‐phenotype SQTS (Figure 1B). Two patients were started on hydroquinidine. One patient had VF 19 months after ICD implantation that was terminated by an appropriate ICD shock. After this event, the patient was started on hydroquinidine. Follow‐up of this family over 16 years was without further events. Studying the phenotype of cardiomyoctyes from induced pluripotent stem cells of a patient from this family confirmed the malignant phenotype and the abbreviated action potential duration.18 Next‐generation sequencing using blood cells and fibroblasts from skin biopsy and reprogrammed pluripotent stem cell samples confirmed a pathogenic mutation in the HERG gene (Figure 1C).
Figure 1.
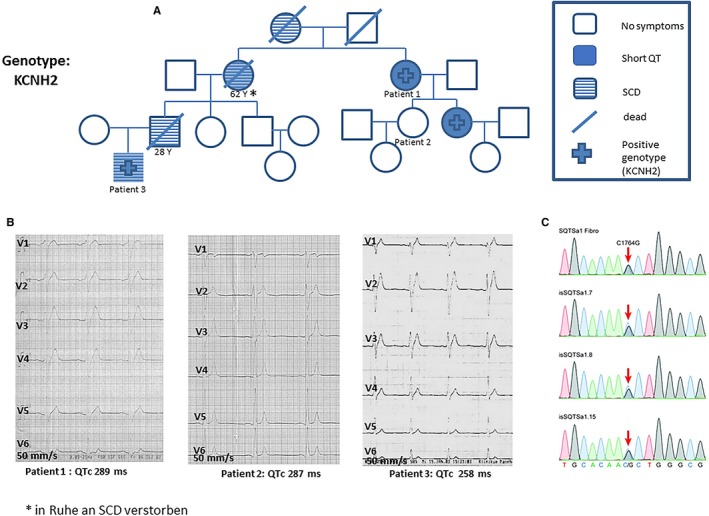
A, Pedigree of a family with short QT syndrome (SQTS) with 4 cases of sudden cardiac death (SCD). B, Representative ECGs of 3 affected patients with SQTS showed an abbreviated corrected QT (QTc) interval. C, Identified missense mutation (C to G substitution at nucleotide 1764) in the KCNH2 gene using next‐generation sequencing from induced pluripotent stem cells reprogrammed from a skin biopsy of 1 patient.
Family 2
The pedigree of family 2 is presented in Figure 2A. A 41‐year‐old man presented with atrial fibrillation and an abbreviated QT interval (QTc, 347 ms). His baseline ECG results showed an elevation of V1 to V2 consistent with suspected BrS. His family history confirmed SCD of his brother at age 44. An autopsy was not performed (Figure 2B). Following intravenous ajmaline, increased ST‐segment elevation in leads V1 to V3 consistent with BrS was confirmed. During electrophysiology study, sustained ventricular tachycardia had been inducible by 2 extrastimuli. An ICD was implanted and hydroquinidine treatment was initiated. Cascade family screening showed affected daughters. Their genetic screening confirmed a pathogenic mutation in CACNB2b (G490R) mutation consistent with SQTS 4. No events occurred during a follow‐up of over 13 years.
Figure 2.
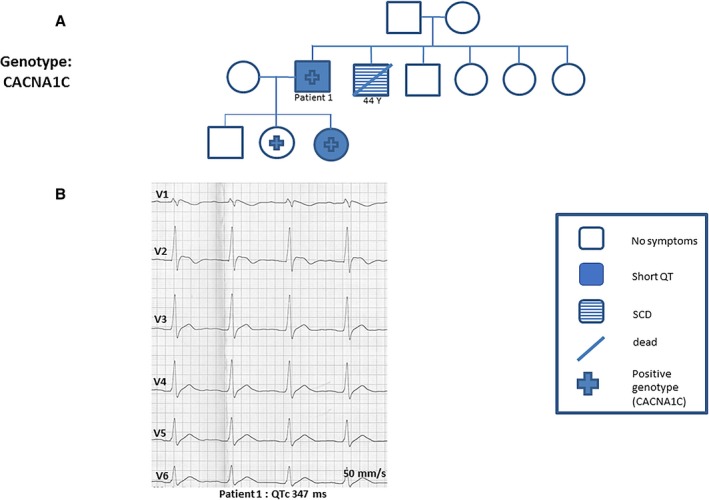
A, A 41‐year‐old man was admitted with atrial fibrillation and abbreviated QT interval (corrected QT [QTc], 347 ms). His family history confirmed a case of sudden cardiac death (SCD) of his brother at age 45 years. Cascade family screening and genetic screening confirmed short QT syndrome type 4. B, Representative ECGs of this patient showed a QTc interval of 347 ms. His baseline ECG results showed an elevation of V1 to V2 consistent with suspected Brugada syndrome.
Family 3
A 25‐year‐old patient was admitted to the hospital with aborted SCD. Results from his ECG showed an abbreviated QTc interval and ST‐segment elevation in V1 (Figure 3B). Ajmaline test confirmed BrS 1 ECG with coved ST‐segment elevation. An ICD was implanted and hydroquinidine prophylaxis was applied. Cascade family screening confirmed SQTS in 5 other first‐ or second‐degree relatives (Figure 3A). Genetic screening of this family confirmed a mutation of CACNB2b consistent with SQTS 5. No events have been documented during 15 years of follow‐up.
Figure 3.
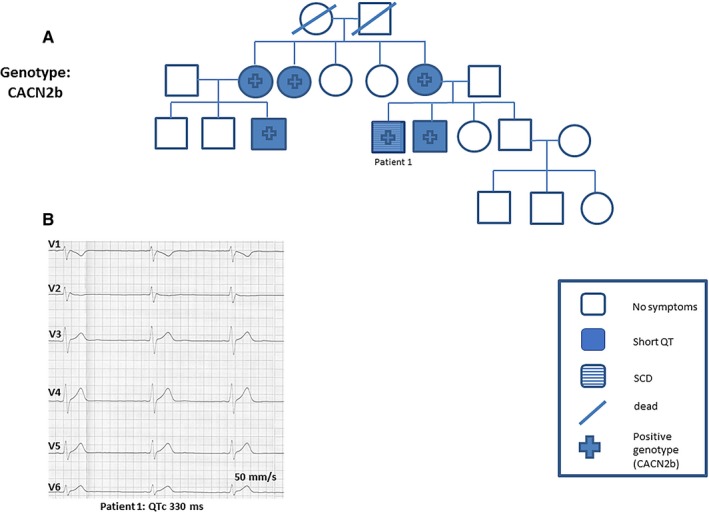
A and B, Patient 1 was admitted to our hospital after cardiopulmonary resuscitation. His baseline ECG findings were suggestive of short QT syndrome (SQTS). Cascade family screening and genetic screening confirmed a mutation of CACNB2, a subunit of voltage calcium channel, consistent with SQTS type 5. QTc indicates corrected QT; SCD, sudden cardiac death.
Family 4
A 67‐year‐old woman was admitted to our hospital with palpitations. ECG results on admission showed an abbreviated QT interval (QTc, 320 ms). Her 40‐year‐old son had died suddenly. Further medical history confirmed 3 cases of SCD at the age of 18, 40, and 55 years, 2 of them at rest (Figure 4. Follow‐up of this family over 9 years did not document any additional events.
Figure 4.
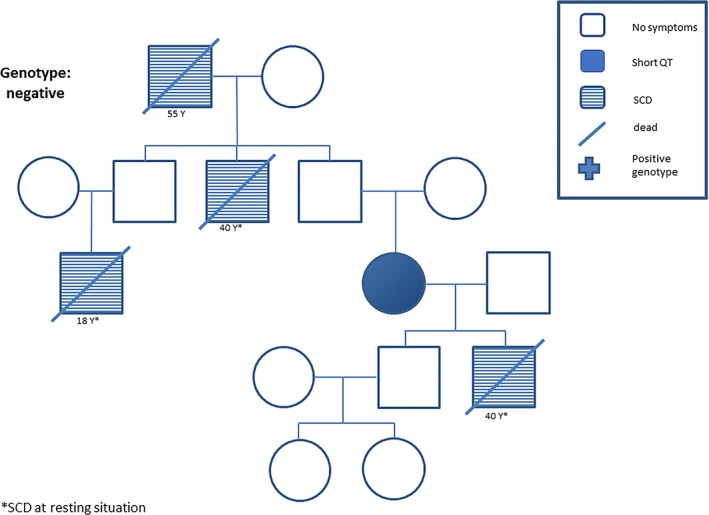
Pedigree of a family with short QT syndrome (SQTS) with 4 cases of sudden cardiac death (SCD) at rest. No SQTS genes were affected. *SCD at rest.
Family 5
A 62‐year‐old patient was admitted with recurrent syncope and palpitations. His baseline ECG results showed a QTc of 272 ms (Figure 5B). Family history confirmed 2 cases of SCD (Figure 5A). During electrophysiological study, VF was shown to be inducible. ICD implantation and starting hydroquinidine was recommended but not accepted by the patient. Follow‐up over 20 years showed no further cases of SCD. The patient continues to experience recurrent palpitation and atrial fibrillation.
Figure 5.
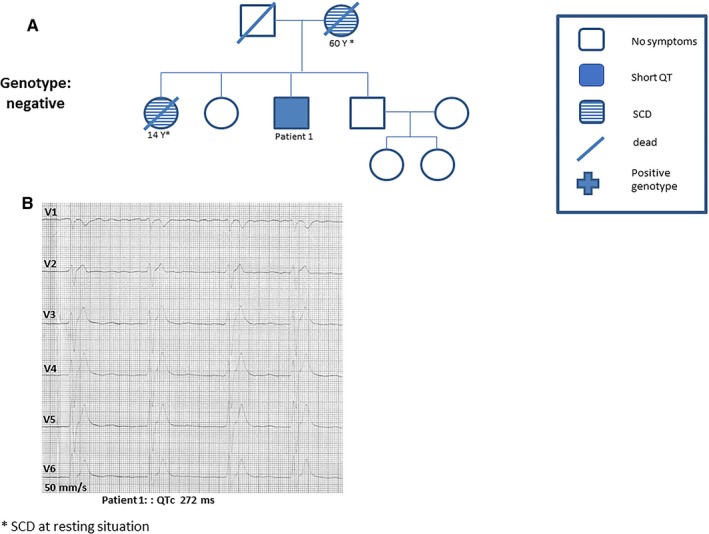
A, A 62‐year‐old patient was admitted with recurrent syncope and palpitation. B, The baseline ECG findings presented shortened corrected QT (QTc) interval <300 ms. *Sudden cardiac death (SCD) at rest.
Family 6
A 34‐year‐old woman was admitted to the hospital with recurrent syncope. Her ECG results showed a shortened QT interval (QTc, 320 ms) (Figure 6A). Other causes of syncope were excluded using Holter monitoring, magnetic resonance imaging, and echocardiogram. A Reveal recorder was implanted. This patient was lost to follow‐up.
Figure 6.
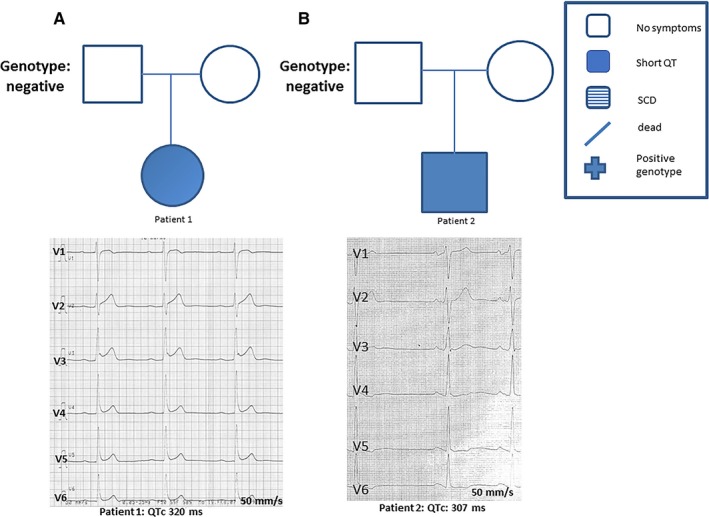
A and B, Two patients with short QT syndrome from independent families presented with a corrected QT interval (QTc) <330 ms. One patient was admitted with recurrent syncope and the other with atrial fibrillation. SCD indicates sudden cardiac death.
Family 7
A 28‐year‐old man was admitted to the hospital with recurrent palpitations. Atrial flutter and atrial fibrillation were diagnosed. His ECG results showed a QTc of 307 ms. Family screening of his parents did not reveal any cases of SCD (Figure 6B). The patient has had no further symptoms after more than 15 years of treatment with hydroquinidine.
Discussion
We have described the clinical profile and short‐ and long‐term risk of SCD in 7 families with SQTS and found the following: (1) the risk of SCD in families with SQTS is high; (2) up to 86% of patients have symptoms, with a higher prevalence of palpitations followed by atrial fibrillation, syncope, and SCD; (3) ICD implantation is recommended in patients at high risk with a family history of SCD; (4) family screening of patients with SQTS is mandatory to avoid SCD in affected individuals; and (5) appropriate risk assessment and individualized treatment options (hydroquinidine/ICD) make the long‐term outcome relatively benign when patients are seen at a reference center.
SQTS is an inheritable disease and is associated with the risk of SCD. Gollob et al10 has recommended a QTc <370 ms as a cutoff for diagnosis of SQTS. However, expert consensus suggests QTc <330 ms as a stand‐alone criterion for SQTS diagnosis or a possible diagnosis if QTc is <360 ms in combination with other risk factors including family history or presence of pathogenic mutation.17
The presence of a genetic basis for SQTS has been described in up to 39% of published cases of families with SQTS.19 Here, we describe a genetic basis in up to 42% of our families, which is consistent with published data. None of our patients presented mutations in KCNQ1, KCNJ2, or the rarely described CACNA2D1. This low prevalence might be attributable to the low number of described families with SQTS and the incomplete understanding of the pathophysiology of SQTS caused by the absence of suitable SQTS models. However, recently published data have described a new genetic association of SQTS within the anion exchanger solute carrier family 4 member 3 (SLC4A3) gene.20 Its functionality has been characterized in Zebrafish embryos.20
Atrial fibrillation has been reported in up to 11% of families with SQTS.19 Our data, however, show a prevalence of 41% among the families. At least 7 of 17 patients with SQTS experienced atrial fibrillation.
At admission, 2 patients with a genetic and phenotype SQTS presented with SCD (12%). Five patients with SQTS received an ICD. Among them, 1 patient, who received an ICD as primary prophylaxis because of short QT time <280 ms and a family history of SCD, had VF 19 months after ICD implantation. Nevertheless, SCD did not occur in any of these patients during follow‐up. While Mazzanti et al11 presented a prevalence of 42% of SCD events in patients with SQTS, among them 9 patients with recurrent SCD, our data do not confirm this observation.
In all 7 families in our study, at least 1 symptom was the cause of admission and led to the diagnosis of SQTS. SCD was the leading cause of admission in our hospital in 4 families, atrial fibrillation was the leading cause for 2 families, and recurrent syncope was the leading cause in 1 family.
Of the patients with SQTS treated in our center during a median follow‐up of 164.5 months (interquartile range, 12–189 months), none died. In all families, 9 relatives had SCD without ECG documentation, a large part of them at younger ages (Figure 7. The present study shows a higher incidence of SCD in patients with SQTS in 12 cases of SCD in 7 independent families with SQTS, compared with previously published data.11, 21 Most patients with SCD were men (67%). This raises the predominance of SQTS in men and higher penetrance of SCD in this cohort as consistent with BrS. The present study on the long‐term follow‐up of families with SQTS had a median follow‐up of 164.5 months, a longer period compared with previously published data, with a follow‐up of 56 months.11, 22
Figure 7.
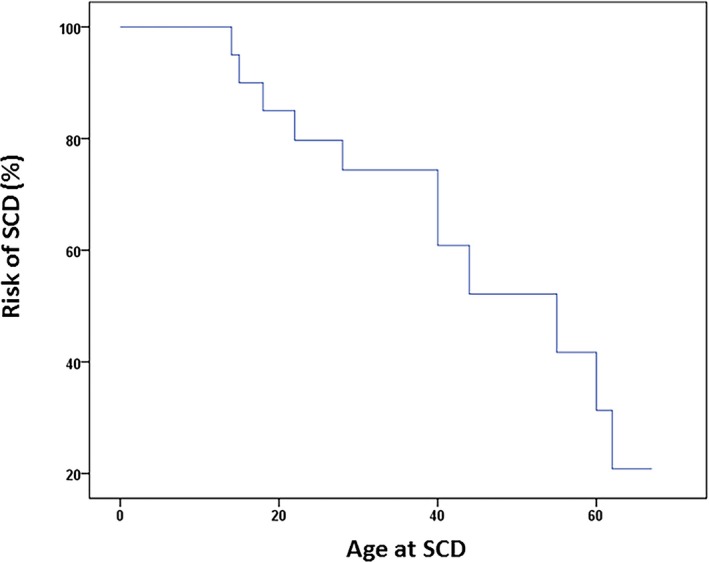
Survival curve presenting the risk of sudden cardiac death (SCD).
Conclusions
Our data present an update of SQTS in 17 patients among 7 families with SQTS. The presence of SCD was documented in 12 cases among the family members. Most patients with SQTS presented with symptoms such as palpitations, atrial fibrillation, syncope, and SCD. Family screening is mandatory in affected patients. However, undergoing risk assessment and individualized treatment (hydroquinidine/ICD) can reduce the long‐term outcome when seen at a reference center.
Disclosures
None.
(J Am Heart Assoc. 2018;7:e010073 DOI: 10.1161/JAHA.118.010073.)
References
- 1. Algra A, Tijssen JG, Roelandt JR, Pool J, Lubsen J. QT interval variables from 24 hour electrocardiography and the two year risk of sudden death. Br Heart J. 1993;70:43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guerrier K, Kwiatkowski D, Czosek RJ, Spar DS, Anderson JB, Knilans TK. Short QT interval prevalence and clinical outcomes in a pediatric population. Circ Arrhythm Electrophysiol. 2015;8:1460–1464. [DOI] [PubMed] [Google Scholar]
- 3. Gussak I, Brugada P, Brugada J, Wright RS, Kopecky SL, Chaitman BR, Bjerregaard P. Idiopathic short QT interval: a new clinical syndrome? Cardiology. 2000;94:99–102. [DOI] [PubMed] [Google Scholar]
- 4. Priori SG, Blomstrom‐Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, Kirchhof P, Kjeldsen K, Kuck KH, Hernandez‐Madrid A, Nikolaou N, Norekval TM, Spaulding C, Van Veldhuisen DJ. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36:2793–2867. [DOI] [PubMed] [Google Scholar]
- 5. Giustetto C, Di Monte F, Wolpert C, Borggrefe M, Schimpf R, Sbragia P, Leone G, Maury P, Anttonen O, Haissaguerre M, Gaita F. Short QT syndrome: clinical findings and diagnostic‐therapeutic implications. Eur Heart J. 2006;27:2440–2447. [DOI] [PubMed] [Google Scholar]
- 6. Gaita F, Giustetto C, Bianchi F, Wolpert C, Schimpf R, Riccardi R, Grossi S, Richiardi E, Borggrefe M. Short QT Syndrome: a familial cause of sudden death. Circulation. 2003;108:965–970. [DOI] [PubMed] [Google Scholar]
- 7. Brugada J, Gussak I, Brugada P. Short QT syndrome: a predictable story. Cardiology. 2014;128:231–233. [DOI] [PubMed] [Google Scholar]
- 8. Suzuki H, Hoshina S, Ozawa J, Sato A, Minamino T, Aizawa Y, Saitoh A. Short QT syndrome in a boy diagnosed on screening for heart disease. Pediatr Int. 2014;56:774–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Perez Riera AR, Paixao‐Almeida A, Barbosa‐Barros R, Yanowitz FG, Baranchuk A, Dubner S, Palandri Chagas AC. Congenital short QT syndrome: landmarks of the newest arrhythmogenic cardiac channelopathy. Cardiol J. 2013;20:464–471. [DOI] [PubMed] [Google Scholar]
- 10. Gollob MH, Redpath CJ, Roberts JD. The short QT syndrome: proposed diagnostic criteria. J Am Coll Cardiol. 2011;57:802–812. [DOI] [PubMed] [Google Scholar]
- 11. Mazzanti A, Kanthan A, Monteforte N, Memmi M, Bloise R, Novelli V, Miceli C, O'Rourke S, Borio G, Zienciuk‐Krajka A, Curcio A, Surducan AE, Colombo M, Napolitano C, Priori SG. Novel insight into the natural history of short QT syndrome. J Am Coll Cardiol. 2014;63:1300–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, Gelber P, Bonaros EP Jr, Burashnikov E, Wu Y, Sargent JD, Schickel S, Oberheiden R, Bhatia A, Hsu LF, Haissaguerre M, Schimpf R, Borggrefe M, Wolpert C. Loss‐of‐function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST‐segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schimpf R, Veltmann C, Papavassiliu T, Rudic B, Goksu T, Kuschyk J, Wolpert C, Antzelevitch C, Ebner A, Borggrefe M, Brandt C. Drug‐induced QT‐interval shortening following antiepileptic treatment with oral rufinamide. Heart Rhythm. 2012;9:776–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Workman AJ, MacKenzie I, Northover BJ. A K(ATP) channel opener inhibited myocardial reperfusion action potential shortening and arrhythmias. Eur J Pharmacol. 2001;419:73–83. [DOI] [PubMed] [Google Scholar]
- 15. Roussel J, Labarthe F, Thireau J, Ferro F, Farah C, Roy J, Horiuchi M, Tardieu M, Lefort B, Francois Benoist J, Lacampagne A, Richard S, Fauconnier J, Babuty D, Le Guennec JY. Carnitine deficiency induces a short QT syndrome. Heart Rhythm. 2016;13:165–174. [DOI] [PubMed] [Google Scholar]
- 16. Surges R, Taggart P, Sander JW, Walker MC. Too long or too short? New insights into abnormal cardiac repolarization in people with chronic epilepsy and its potential role in sudden unexpected death. Epilepsia. 2010;51:738–744. [DOI] [PubMed] [Google Scholar]
- 17. Priori SG, Blomstrom‐Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, Kirchhof P, Kjeldsen K, Kuck KH, Hernandez‐Madrid A, Nikolaou N, Norekval TM, Spaulding C, Van Veldhuisen DJ. 2015 Task Force for the Management of Patients with Ventricular A and the Prevention of Sudden Cardiac Death of the European Society of C. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Europace. 2015;17:1601–1687. [DOI] [PubMed] [Google Scholar]
- 18. El‐Battrawy I, Lan H, Cyganek L, Zhao Z, Li X, Buljubasic F, Lang S, Yucel G, Sattler K, Zimmermann WH, Utikal J, Wieland T, Ravens U, Borggrefe M, Zhou XB, Akin I. Modeling short QT syndrome using human‐induced pluripotent stem cell‐derived cardiomyocytes. J Am Heart Assoc. 2018;7:e007394 DOI: 10.1161/JAHA.117.007394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bjerregaard P. Proposed diagnostic criteria for short QT syndrome are badly founded. J Am Coll Cardiol. 2011;58:549–550; author reply 550–551. [DOI] [PubMed] [Google Scholar]
- 20. Thorsen K, Dam VS, Kjaer‐Sorensen K, Pedersen LN, Skeberdis VA, Jurevicius J, Treinys R, Petersen I, Nielsen MS, Oxvig C, Morth JP, Matchkov VV, Aalkjaer C, Bundgaard H, Jensen HK. Loss‐of‐activity‐mutation in the cardiac chloride‐bicarbonate exchanger AE3 causes short QT syndrome. Nat Commun. 2017;8:1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Villafane J, Atallah J, Gollob MH, Maury P, Wolpert C, Gebauer R, Watanabe H, Horie M, Anttonen O, Kannankeril P, Faulknier B, Bleiz J, Makiyama T, Shimizu W, Hamilton RM, Young ML. Long‐term follow‐up of a pediatric cohort with short QT syndrome. J Am Coll Cardiol. 2013;61:1183–1191. [DOI] [PubMed] [Google Scholar]
- 22. Giustetto C, Schimpf R, Mazzanti A, Scrocco C, Maury P, Anttonen O, Probst V, Blanc JJ, Sbragia P, Dalmasso P, Borggrefe M, Gaita F. Long‐term follow‐up of patients with short QT syndrome. J Am Coll Cardiol. 2011;58:587–595. [DOI] [PubMed] [Google Scholar]