

Abstract
Background
Whether chronic obstructive sleep apnea (OSA) could promote epicardial adipose tissue (EAT) secretion of profibrotic adipokines, and thereby contribute to atrial fibrosis, and the potential therapeutic effects of metoprolol remain unknown.
Methods and Results
A chronic OSA canine model was established by repeatedly clamping the endotracheal tube for and then reopening it for 4 hours every other day for 12 weeks. In a metoprolol treatment group, metoprolol succinate was administered daily for 12 weeks. The EAT infiltration and left atrial fibrosis were examined. The expressions of adipokines secreted by EAT and hypoxic 3T3‐L1 adipocytes were detected. The changes in collagen synthesis, transforming growth factor‐β1 expression, and cell differentiation and proliferation in cardiac fibroblasts induced by hypoxic 3T3‐L1 adipocyte‐derived conditioned medium were further analyzed. Chronic OSA induced infiltration of EAT into the left atrium. OSA enhanced the profibrotic effect of EAT on the adjacent atrial myocardium. Moreover, OSA induced profibrotic cytokine secretion from EAT. We also found that hypoxia induced adipokine secretion in cultured adipocytes, and the medium conditioned by the hypoxic adipocytes increased collagen and transforming growth factor‐β1 protein expression and cell proliferation of cardiac fibroblasts. More importantly, metoprolol attenuated infiltration of EAT and alleviated the profibrotic effect of EAT by inhibiting adipokine secretion. Metoprolol also inhibited hypoxia‐induced adipokine secretion in adipocytes and thereby blocked the hypoxic adipocyte–derived conditioned medium–induced fibrotic response of cardiac fibroblasts.
Conclusions
Chronic OSA enhanced the profibrotic effect of EAT on the neighboring atrial myocardium by stimulating the secretion of profibrotic adipokines from EAT, which was significantly attenuated by metoprolol. This study gives insights into mechanisms underlying OSA‐induced atrial fibrillation and also provides experimental evidence for the protective effects of metoprolol.
Keywords: atrial fibrillation, atrial fibrosis, epicardial adipose tissue, metoprolol, obstructive sleep apnea
Subject Categories: Atrial Fibrillation, Arrhythmias
Clinical Perspective
What Is New?
The present study demonstrated that chronic obstructive sleep apnea enhanced the profibrotic effect of epicardial adipose tissue on the neighboring atrial myocardium by stimulating the secretion of profibrotic adipokines such as activin A, transforming growth factor‐β1, matrix metalloproteinase‐9, tumor necrosis factor‐α, and interleukin‐6 from epicardial adipose tissue, which was significantly attenuated by metoprolol.
What Are the Clinical Implications?
These findings suggest that the autonomic nervous system plays an important role in chronic obstructive sleep apnea–induced atrial fibrillation, and modulation of the sympathetic nervous system might help to prevent atrial fibrillation in patients with obstructive sleep apnea.
Introduction
Obstructive sleep apnea (OSA), characterized by recurrent partial or complete collapse of the upper airway during sleep, is highly prevalent.1 Recently, OSA has been demonstrated to be an important risk factor for atrial fibrillation (AF) occurrence.2, 3 OSA can promote AF primarily by causing atrial structural and electrical remodeling.4, 5, 6, 7 Atrial fibrosis, the hallmark of structural remodeling–associated AF, has been found to be significantly increased in animal models with long‐term repetitive OSA and to play a prominent role in the occurrence of AF.5, 6, 7
Recently, the epicardial adipose tissue (EAT) has emerged as an important factor in the pathogenesis of cardiovascular diseases including AF. Several studies have shown a close association between EAT volume and the occurrence of AF, as reviewed by Wong et al.8 Previous studies demonstrated that EAT could contribute to the fibrosis of neighboring atrial myocardium by secreting profibrotic factors including inflammatory cytokines, growth factors, and matrix metalloproteinases (MMPs),9, 10 which may be the main mechanism underlying the association between EAT and AF.
Whether chronic OSA could promote EAT‐secreting profibrotic adipokines and thereby contribute to the fibrosis of neighboring atrial myocardium remains unknown. Increased sympathetic activity is 1 of the most important mechanisms underlying OSA‐related myocardial damage and OSA‐induced AF.11, 12 We previously found that the β‐adrenergic receptor antagonist metoprolol could inhibit atrial structural, autonomic, and metabolic remodeling and AF inducibility in a canine model of chronic OSA.13 However, the potential regulatory effects of metoprolol on the profibrotic adipokines secretion of EAT in chronic OSA are still unclear. Therefore, in the present study, we tested whether chronic OSA could promote EAT‐secreting profibrotic adipokines and thereby contribute to the fibrosis of neighboring atrial myocardium, and whether metoprolol could play a protective role in a canine model of chronic OSA.
Materials and Methods
The data that support the findings of this study are available from the corresponding authors on reasonable request.
Animals and Animal Models
Twenty‐one healthy male mongrel dogs (15‐20 kg, Experimental Animal Center of the First Affiliated Hospital of Harbin Medical University, Harbin, China) were used in the study. The present study was performed in accordance with the Guide for the Care and Use of Laboratory Animals and was approved by the Institutional Animal Care and Use Committee at the Harbin Medical University. The dogs were randomly divided into 3 groups: Sham group (n=7), OSA group (n=7), and OSA‐ and metoprolol‐treated group (OSA+Met group, n=7). The chronic OSA model was conducted as previously described.13 The dogs were anesthetized with intravenous ketamine (5.3 mg/kg), diazepam (0.25 mg/kg), and xylazine (1 mg/kg), and the adequacy of anesthesia was monitored on the basis of the disappearance of the corneal reflex and jaw tone. Then, tracheal intubation was performed, and the tracheal tubes were clamped to induce apnea at the end of the exhalation. In the first week the duration of the trachea blockage was 1 minute, and the duration of ventilation was 9 minutes. Thus, the apnea‐hypopnea index was set as 6. For the next 3 weeks, the duration of trachea blockage remained unchanged while the duration of ventilation was reduced by 1 minute every week. During the last 8 weeks the duration of trachea blockage was still 1 minute, and the duration of trachea ventilation was 5 minutes (apnea‐hypopnea index was 10). The procedure of trachea blockage and ventilation was performed for 4 hours every other day over 12 weeks. The dogs in sham group underwent anesthesia and tracheal intubation only. The dogs in OSA+Met group were administered metoprolol succinate (5 mg/kg per day) daily at the beginning of OSA simulation, and this regimen continued for 12 weeks.
Hematoxylin and Eosin and Masson Trichrome Staining
Atrial samples were collected in 4% paraformaldehyde and then embedded in paraffin. Left atrial (LA) EAT and its underlying atrial myocardium were stained with hematoxylin and eosin and Masson trichrome staining, respectively. Adipose tissue areas in the atrial myocardium at the junction between EAT and the atrial myocardium were measured on 3 randomly selected microscopic fields (×200), each slice analyzed with Image‐Pro Plus software (Version 4.0, Media Cybernetics, LP, Rockville, MD) and expressed as a mean percentage of total field area (adipose infiltration fraction). The collagen volume fractions of EAT and the adjacent atrial myocardium were calculated by averaging the percentage area of stained tissue within 3 randomly selected microscopic fields (×200) on each slice using Image‐Pro Plus software.
Immunohistochemical Studies
Activin A, transforming growth factor‐β1 (TGF‐β1), matrix metalloproteinase‐9 (MMP‐9), tumor necrosis factor‐α (TNF‐α), and interleukin‐6 (IL‐6) were stained on 5‐μm transmural sections of LA EAT. The sections were incubated overnight at 4°C with primary antibody (Biosynthesis Biotechnology Co Ltd, Beijing, China) and with peroxidase‐conjugated second antibody (Zhongshanjinqiao Biotech, Beijing, China) at 37°C for 20 minutes. Then, they were visualized with a DAB‐based colorimetric method.
Enzyme‐Linked Immunosorbent Assay
The activin A, TGF‐β1, MMP‐9, TNF‐α, and IL‐6 levels of LA EAT and the culture medium of 3T3‐L1 cells were measured using ELISA kits (BlueGene Biotech Co Ltd, Shanghai, China) according to the manufacturer's instructions.
3T3‐L1 Adipocyte Differentiation and Hypoxic Treatment
Murine preadipocytes (3T3‐L1 cells) were obtained from American Type Culture Collection (Manassas, VA) and cultured in DMEM containing 10% FBS and 1% penicillin/streptomycin (growth medium) in a 5% CO2 humidified atmosphere at 37°C. When the cells were subconfluenced at the culture bottle bottom, cell seeding was performed in 6‐well plates. 3T3‐L1 cells were differentiated into adipocytes according to previously described methods.14 Briefly, 3T3‐L1 cells were cultured to confluence completely, supplemented with 250 nmol/L dexamethasone, 25 nmol/L insulin, and 0.5 mmol/L isobutyl methylxanthine for 72 hours. Cells were then cultured for an additional 5 days in growth medium. Differentiation was determined microscopically with the criteria that fat droplets were observed in cytoplasm. Fully differentiated adipocytes were divided into 3 groups (Normoxia, Hypoxia, and Hypoxia+Metoprolol). In Hypoxia and Hypoxia+Metoprolol groups, cells were treated with vehicle and metoprolol (10 μmol/L), respectively, and then were incubated in a hypoxic chamber at an atmosphere of 1% O2, 95% N2, 5% CO2 and at 37°C for 12 hours. In the Normoxia group, cells were incubated in an atmosphere of 21% O2 and 5% CO2 at 37°C for 12 hours. Then the conditioned medium was collected to culture cardiac fibroblasts (CFs). Moreover, the cytokine concentration in the conditioned medium was measured by ELISA.
CF Isolation and Culture
Primary cultures of rat CFs were isolated from the hearts of 1‐ to 2‐day‐old Sprague‐Dawley rats using an enzymatic digestion solution containing 0.25% trypsin (Beyotime Institute of Biotechnology, Jiangsu, China) at 37°C. Cell cultures were incubated at 37°C in a humidified atmosphere of 5% CO2/95% air. CFs (second or third generation) were grown to subconfluence in serum‐containing medium and then growth arrested for 24 hours in serum‐free medium and were incubated with 3T3‐L1 adipocyte‐conditioned medium for 24 hours.
Western Blotting
Proteins were separated by electrophoresis on 8% to 12% SDS‐polyacrylamide gels and transferred moist to polyvinylidene difluoride membranes. Membranes were blocked by 5% nonfat dry milk in PBS and incubated overnight at 4°C. Membranes were washed with PBS containing 0.5% Tween 20 and incubated with primary antibodies overnight, then incubated with horseradish peroxidase‐conjugated secondary antibody (Zhongshanjinqiao Biotech, Beijing, China) for 1 hour. Antibodies against collagen I and collagen III were purchased from Biosynthesis Biotechnology Co, Ltd (Beijing, China). Antibodies against α‐smooth muscle actin was obtained from Abcam (Cambridge, MA). Antibody against TGF‐β1 was provided by Sigma‐Aldrich (St. Louis, MO). The images were captured on the Bio‐Rad system (Hercules, CA), and GAPDH (Kangcheng, Shanghai, China) was used as internal control.
Determination of Cell Viability
CF viability was detected using the CCK‐8 assay (Dojindo Molecular Technologies, Inc, Kumamoto, Japan). CFs were plated in 96‐well plates,with 5 duplicate wells in each group. When the cells had grown to ≈70% to 80% confluence, the cells were treated with conditioned medium as indicated. The CCK‐8 solution (10 μL) at a 1:10 dilution with FBS‐free DMEM (100 μL) was added to each well, and plates were incubated for 3 hours at 37°C. Absorbance was measured at 450 nm with a microplate reader (Molecular Devices, Sunnyvale, CA). The mean optical density (OD) of 5 wells in the indicated groups was used to calculate the percentage of cell viability as follows: percentage of cell viability=(ODtreatment group–ODblank group)/(ODcontrol group–ODblank group)×100%.
Statistical Methods
All data are presented as the mean±SD. Multiple group comparisons were performed via 1‐way ANOVA followed by Tukey tests. Natural logarithmic transformation was performed if the data did not satisfy statistical criteria for normal distribution, and the data were presented on the original scale. Analyses were conducted with GraphPad Prism version 5.0 (GraphPad Software, Inc, La Jolla, CA). P<0.05 was considered statistically significant.
Results
Metoprolol Attenuated OSA‐Induced Fibrosis and Infiltration of EAT
As shown in Figure 1, the fibrosis and the infiltration of EAT into atrial myocardium was significantly pronounced in chronic OSA dogs compared with those from the Sham group. Metoprolol significantly attenuated OSA‐induced fibrosis and infiltration of EAT.
Figure 1.
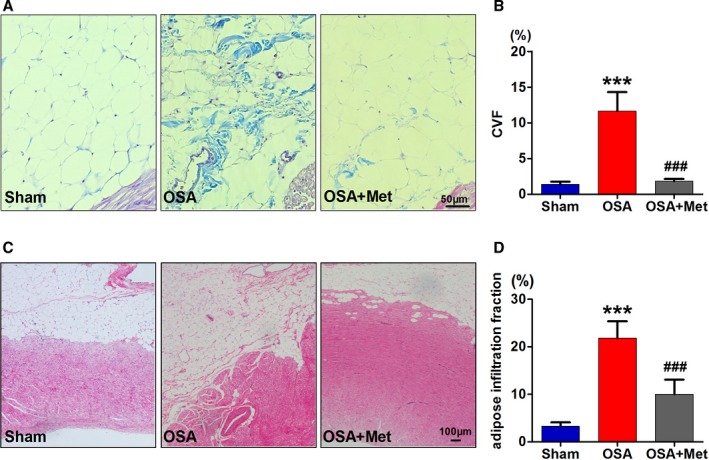
Metoprolol attenuated OSA‐induced fibrosis and infiltration of EAT. A, Representative examples of Masson trichrome staining of EAT. Scale bar=50 μm. B, Collagen volume fraction (CVF) of EAT. C, Representative examples of hematoxylin and eosin staining of epicardial fat layer and adipocytes infiltration. Scale bar=100 μm. D, Adipose infiltration fraction of each group, n=7 per group. ***P<0.001 vs the Sham group; ### P<0.001 vs the OSA group. EAT indicates epicardial adipose tissue; Met, metoprolol; OSA, obstructive sleep apnea.
Metoprolol Alleviated the Profibrotic Effect of EAT Induced by OSA
To investigate the effects of EAT on the fibrosis of the neighboring atrial myocardium, we examined the interstitial fibrosis of LA myocardium that was covered by EAT. As shown in Figure 2, Masson trichrome staining showed significantly more collagen deposition in the LA myocardium at the junction with EAT in OSA group than in that of Sham group, and the collagen deposition was observed around the infiltrated adipocytes. Treatment with metoprolol significantly attenuated the fibrosis of LA myocardium. These results suggested that OSA enhanced the profibrotic effect of EAT on the atrial myocardium beneath it, which was inhibited by metoprolol treatment.
Figure 2.
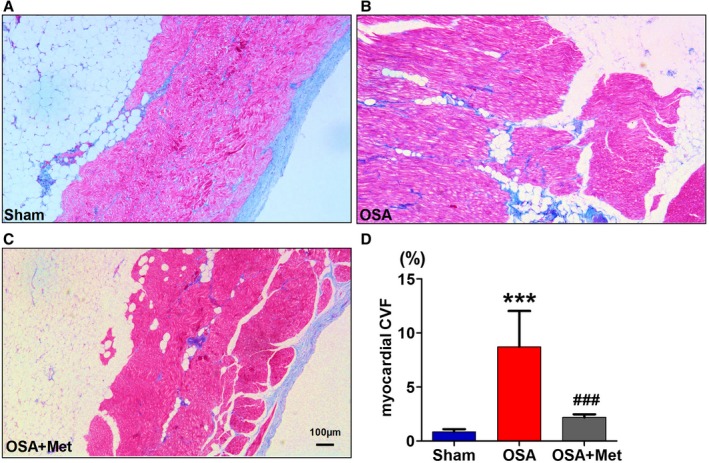
Metoprolol inhibited the profibrotic effects of EAT enhanced by OSA. A through C, Examples of Masson trichrome staining of the atrial myocardium with EAT covering of each group. D, Collagen volume fraction (CVF) of each group. n=7 per group. ***P<0.001 vs the Sham group; ### P<0.001 vs OSA group. Scale bar=100 μm. EAT indicates epicardial adipose tissue; Met, metoprolol; OSA, obstructive sleep apnea.
Metoprolol Inhibited Adipokine Secretion From EAT Induced by OSA
To illustrate the mechanism of the enhanced profibrotic effects of EAT during OSA, the expressions of main adipokines secreted by EAT were identified using immunohistochemistry and ELISA. The immunohistochemistry showed that the expressions of profibrotic and proinflammatory factors including activin A, TGF‐β1, MMP‐9, TNF‐α, and IL‐6 were significantly increased in EAT of OSA dogs over those in sham dogs (Figure 3), which was also confirmed by ELISA (Figure 4). OSA‐induced upregulation of these adipokines in EAT could be reduced by metoprolol treatment (Figures 3 and 4).
Figure 3.
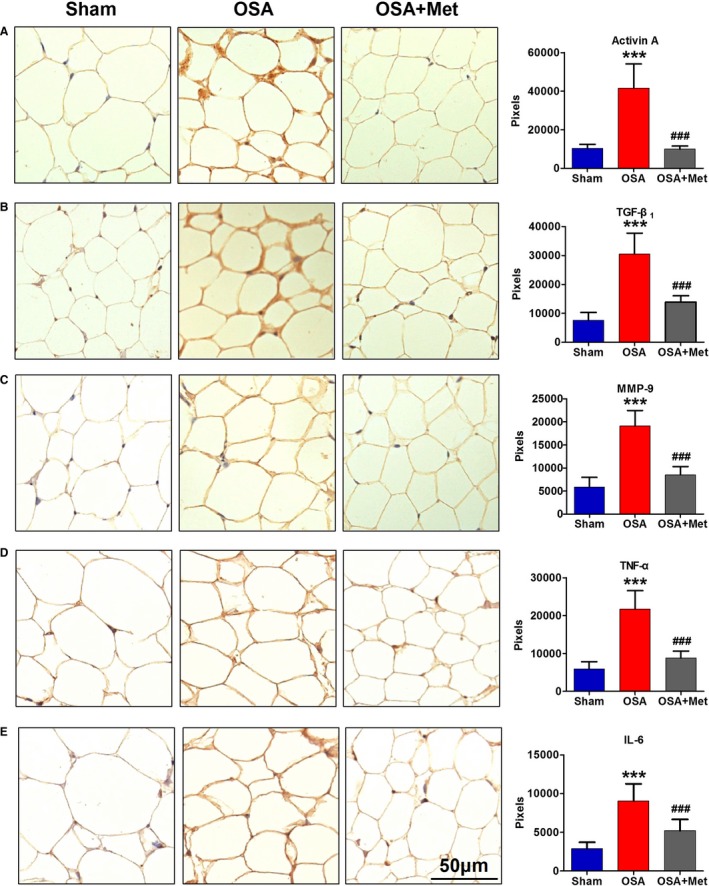
Immunohistochemistry result of adipokines in EAT. Representative immunohistochemical staining and quantification (signal intensity in pixels) of activin A (A), TGF‐β1 (B), MMP‐9 (C), TNF‐α (D), and IL‐6 (E) in EAT of each group. n=7 per group. ***P<0.001 vs the Sham group. ### P<0.001 vs OSA group. Scale bar=50 μm. EAT indicates epicardial adipose tissue; IL, interleukin; Met, metoprolol; MMP, matrix metalloproteinase; OSA, obstructive sleep apnea; TGF, transforming growth factor; TNF, tumor necrosis factor.
Figure 4.
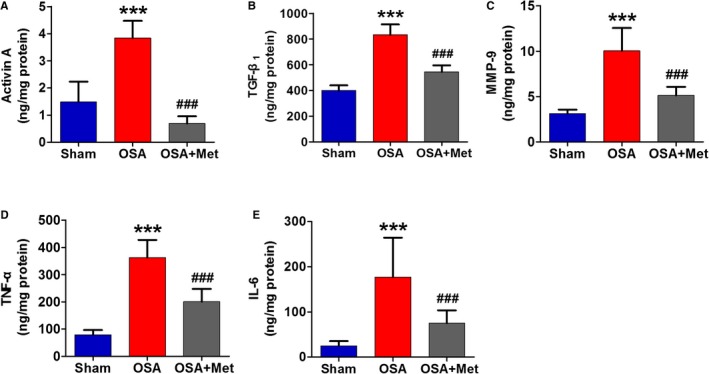
ELISA result of adipokines secreted by EAT. The content of activin A (A), TGF‐β1 (B), MMP‐9 (C), TNF‐α (D), and IL‐6 (E) secreted by EAT of each group. n=7 per group. ***P<0.001 vs the Sham group; ### P<0.001 vs the OSA group. EAT indicates epicardial adipose tissue; IL, interleukin; Met, metoprolol; MMP, matrix metalloproteinase; OSA, obstructive sleep apnea; TGF, transforming growth factor; TNF, tumor necrosis factor.
Metoprolol Inhibited Hypoxia‐Induced Adipokine Secretion in 3T3‐L1 Cells
To evaluate the effect of hypoxia on adipokine secretion, we measured the main adipokine concentration in the culture medium of 3T3‐L1 cells after 12 hours of hypoxia exposure using ELISA. The experiment was performed in quadruplicate. As shown in Figure 5, hypoxia significantly increased the secretion of activin A, TGF‐β1, and IL‐6 but had no significant effect on MMP‐9 or TNF‐α. Metoprolol treatment blocked hypoxia‐induced increase in activin A, TGF‐β1, and IL‐6.
Figure 5.
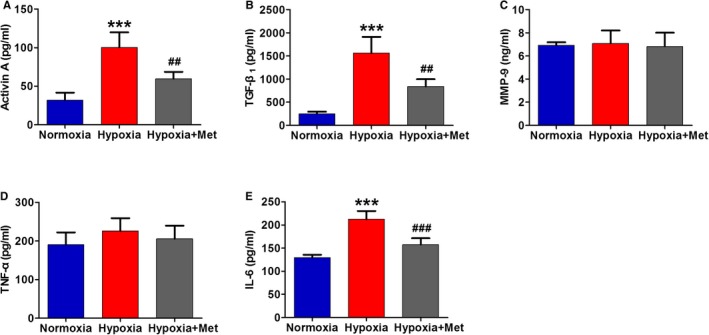
Metoprolol inhibited hypoxia‐induced adipokine secretion in 3T3‐L1 cells. The concentration of activin A (A), TGF‐β1 (B), MMP‐9 (C), TNF‐α (D), and IL‐6 (E) in the culture medium of 3T3‐L1 cells of each group. n=4 per group. ***P<0.001 vs the Normoxia group; ## P<0.05, ### P<0.001 vs the Hypoxia group. IL indicates interleukin; Met, metoprolol; MMP, matrix metalloproteinase; TGF, transforming growth factor; TNF, tumor necrosis factor.
Hypoxic 3T3‐L1 Adipocyte–Derived Conditioned Medium Increased CF Collagen Synthesis, TGF‐β1 Expression, and Cell Proliferation
We further observed the effect of the hypoxic 3T3‐L1 adipocyte–derived conditioned medium on collagen synthesis and TGF‐β1 expression in CFs. The experiment was performed in quadruplicate. The results showed that there was a significant increase in collagen I, collagen III, and TGF‐β1 protein expression in CFs treated with hypoxic 3T3‐L1 adipocyte–derived conditioned medium for 24 hours compared with cells subjected to normal culture medium (Figure 6A through 6C). We also observed the effect of the hypoxic 3T3‐L1 adipocyte–derived conditioned medium on cell differentiation and proliferation in CFs. As shown in Figure 6D, the differentiation marker α‐smooth muscle actin did not exhibit a statistical significance, although there was an increasing tendency. The hypoxic 3T3‐L1 adipocyte–derived conditioned medium led to an increase in cell viability determined by the CCK‐8 kit (Figure 6E), which indicated increased proliferation of CFs. Furthermore, compared with the adipocyte‐derived conditioned medium from the Hypoxia group, the adipocyte‐derived conditioned medium from the Hypoxia+Metoprolol group significantly inhibited collagen I, collagen III, and TGF‐β1 protein expression and cell proliferation of CFs.
Figure 6.
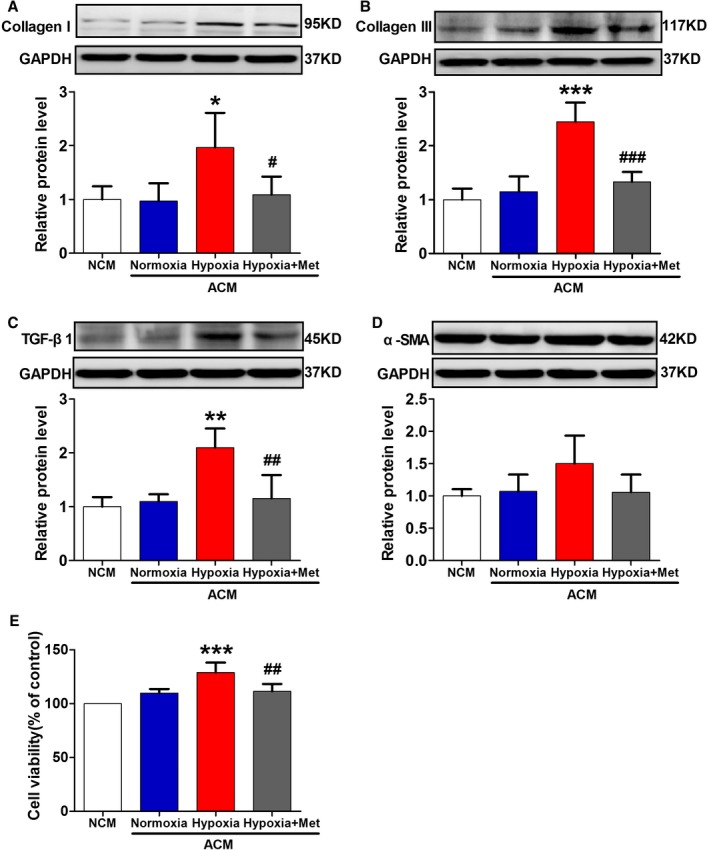
Effect of adipocyte‐derived conditioned medium on collagen synthesis, TGF‐β1 expression, and cell differentiation and proliferation in cardiac fibroblasts. Effect of the conditioned medium from the Normoxia, Hypoxia, and Hypoxia+Met groups of 3T3‐L1 cells on collagen I (A), collagen III (B), TGF‐β1 (C), and α‐SMA (D) protein expression and cell viability (E) in cardiac fibroblasts. n=4 per group. *P<0.05, **P<0.01, ***P<0.001 vs the normal culture medium (NCM) group; # P<0.05, ## P<0.001, ### P<0.001 vs the Hypoxia group. ACM indicates 3T3‐L1 adipocyte–derived conditioned medium; Met, metoprolol; SMA, smooth muscle actin; TGF, transforming growth factor.
Discussion
To our knowledge, the present study was the first experimental analysis of the contributory effect of EAT on the fibrosis of the underlying atrial myocardium, and the protective role of metoprolol during chronic OSA. We found that EAT promoted the fibrosis of the adjacent atrial myocardium, and the effect was enhanced by chronic OSA in a canine model. Moreover, chronic OSA induced profibrotic and proinflammatory cytokine secretion from EAT. We also found hypoxia‐induced adipokine secretion in cultured 3T3‐L1 adipocytes, and the hypoxic adipocyte–derived conditioned medium increased collagen and TGF‐β1 protein expression and cell proliferation of CFs. More importantly, the protective role of metoprolol was observed both in vivo and in vitro.
Previous studies showed a close relationship between the amount of LA EAT and AF independently of other AF risk factors as reviewed by Wong et al.8 Interestingly, a recent study demonstrated that epicardial fat thickness was associated with OSA severity.15 The promoting effect of EAT on atrial structural and electrical remodeling was thought to contribute to the association between EAT and AF.4, 5, 6, 7 Atrial fibrosis is the most prominent feature of clinical AF and a key process of arrhythmogenic structural remodeling, which provides a basis for unidirectional conduction block and macro reentry, resulting in an AF substrate.16 Studies showed that EAT could contribute to the fibrosis of neighboring atrial myocardium by secreting profibrotic factors including inflammatory cytokines, growth factors, and matrix metalloproteinases. Venteclef et al found the secretome from human EAT can promote myocardial fibrosis through the secretion of activin A.9 Recently, Abe et al demonstrated that, in AF patients, the total collagen in the LA myocardium was positively correlated with proinflammatory and profibrotic cytokines/chemokines, including IL‐6, monocyte chemoattractant protein‐1, TNF‐α, vascular endothelial growth factor, MMP‐2, and MMP‐9 in EAT.10 Consistent with the above studies, our results showed that the expressions of profibrotic and proinflammatory factors including activin A, TGF‐β1, MMP‐9, TNF‐α, and IL‐6 were significantly increased in EAT of OSA dogs. In addition to the well‐known profibrotic cytokines such as activin A,9 TGF‐β1,17 and MMP‐9,18 the proinflammatory factors TNF‐α19 and IL‐620 have been proven to induce atrial fibrosis. Therefore, the OSA‐induced upregulation of profibrotic and proinflammatory factors may be the main mechanism of the profibrotic effect of EAT. In this study we found that OSA induced epicardial adipocyte infiltration into atrial myocardium, which might facilitate the diffusion of adipokines into atrial myocardium and thereby contribute to the profibrotic effect of these adipokines.
Several pathophysiological consequences of OSA, including intermittent hypoxia, excessive arousals, increased sympathetic activation, and large negative intrathoracic pressure swings may provoke cardiovascular injury. Among the pathophysiological triggering factors, hypoxia has been suggested to play a key role in the pathophysiology of morphological and functional changes of the adipose tissue in OSA.21 Almendros et al found that changes in visceral adipose tissue partial pressure of oxygen in lean rats during intermittent hypoxia or obstructive apnea cycles of comparable duration were blunted.22 Therefore, to recapitulate the profibrotic remodeling of EAT induced by OSA, we evaluated the effect of hypoxia on adipokine secretion and further observed the profibrotic effect of the secretome in vitro. As a result, we found that 12 hours of hypoxia induced a significant increase in the secretion of activin A, TGF‐β1, and IL‐6 in 3T3‐L1 adipocytes, and the hypoxic adipocyte–derived secretome exerted a profibrotic effect on CFs. Previous studies have demonstrated that hypoxia could modulate the expressions of classic adipokines such as adiponectin and leptin in rodent and human adipocytes.23, 24 A recent study showed that chronic intermittent hypoxia induced a proinflammatory phenotype of visceral adipose tissue with proinflammatory M1 macrophage polarization in a murine model of obstructive sleep apnea.25 In cultured 3T3‐L1 adipocytes26 and human adipocytes,27 increased expression of inflammation‐related adipokines such as IL‐6 and monocyte migration inhibitory factor was observed during hypoxia. Therefore, it is reasonable to conclude that hypoxia is the main cause of adipokine secretion from EAT induced by OSA. Of note, there was a discrepancy in MMP‐9 and TNF‐α expressions between EAT of dogs with OSA and 3T3‐L1 adipocytes with hypoxia treatment. There are 2 explanations for the discrepancy. First, it may be due to the difference in duration and degree of hypoxia. Second, the adipokine profile of epicardial adipocytes and 3T3‐L1 adipocytes may differ because it has been reported that there is a difference in concentration of several adipokines between EAT‐conditioned media and subcutaneous adipose tissue–conditioned media in patients.9 There is evidence suggesting that hypoxia is also a major initiating factor for fibrosis of adipose tissue and that hypoxia‐inducible factor‐1α plays a critical role in the fibrotic response as reviewed by Sun et al.28 Recently, the fibrotic response and overexpression of hypoxia‐inducible factor‐1α were also observed in LA EAT in AF patients.10 Therefore, the fibrosis of EAT induced by OSA in this study also could be attributed to hypoxia.
Several studies have reported that the sympathetic nervous system regulates the expression of several adipokines through adipocyte adrenergic receptors. The nonselective β‐adrenergic receptor agonist isoproterenol has been found to stimulate proinflammatory cytokines TNF‐α and IL‐6 expression in differentiated 3T3‐L1 adipocytes.29, 30 Consistently, the nonselective β‐adrenergic receptor blocker propranolol has been proven to block acute stroke‐induced TNF‐α upregulation of epididymal adipose.31 A previous study demonstrated that acute hypoxia in adipocytes caused dysregulation of adiponectin secretion, and this effect was accompanied by increased protein expression of β1 and β2 adrenergic receptors,32 which indicates that β‐adrenergic receptors may be involved in this process. In our present study we found that the β1‐adrenergic receptor blocker metoprolol inhibited chronic OSA‐ or hypoxia‐induced adipokine secretion in both EAT and 3T3‐L1 adipocytes. Our result suggests that the regulatory effect of hypoxia on the expression of adipokines seems to depend mainly on adipocyte adrenergic receptor activation.
The efficacy of β‐adrenergic receptor blockers in the prevention of AF after heart surgery in patients has been confirmed in several clinical trials and meta‐analyses.33 Moreover, metoprolol therapy has been found to antagonize atrial gap junction remodeling and conduction in human chronic AF.34 Despite the reported benefits, the use of β‐adrenergic receptor blockers in OSA patients may be limited by their potential influence on apnea‐induced bradycardia. However, a recent study showed that β‐adrenergic receptor blockers attenuated apnea‐induced increases in heart rate but did not potentiate apnea‐induced heart rate decreases in patients with hypertension and OSA.35 We previously found that metoprolol could inhibit atrial structural, autonomic, and metabolic remodeling and AF incidence in the canine model of chronic OSA.13 Consistent with that, in this study, we found that metoprolol inhibited the profibrotic effect of EAT enhanced by chronic OSA as well. The sympathetic nervous system can be modulated by β‐adrenergic receptor antagonists and renal denervation in humans.36 Previous studies showed that reducing sympathetic activity by renal denervation significantly attenuated atrial effective refractory period shortening and renin‐angiotensin‐aldosterone system activation, thereby decreasing AF inducibility in a pig model of sleep apnea induced by short‐term negative tracheal pressure.37, 38 These results, together with our own, demonstrated the importance of the autonomic nervous system in both short‐ and long‐term OSA‐induced AF. Therefore we speculate that modulation of the sympathetic nervous system might help to prevent AF in patients with OSA, which needs to be clarified in future clinical studies.
According to the apnea‐hypopnea index value, the severity of OSA in our present animal model is mild. Therefore, our findings suggest that exposure to mild chronic intermittent hypoxia could induce proarrhythmic cardiac remodeling. Our result is consistent with that of Ray et al,39 who reported that mild intermittent hypoxia for 2 weeks in Wistar rats evoked significant cardiovascular pathophysiology. Limited clinical data are available to assess the association of mild OSA with the risk of atrial fibrillation or other arrhythmias40. Further clinical investigation will be required to validate the applicability of our findings to humans.
Limitations
Some limitations should be acknowledged when considering the results of the present study. First, although our data indicated the inhibiting effect of metoprolol on adipokine secretion of EAT induced by OSA, the precise molecular mechanism is still unclear and needs further investigation. Second, 3T3‐L1 adipocytes were used to investigate the effect of hypoxia on adipokine secretion in this study. Although our findings from the 3T3‐L1 cells more closely mimic the changes observed in vivo, the potential difference in the characteristics between 3T3‐L1 cells and canine epicardial adipocytes should not be ignored. Third, the changes in hemodynamic parameters, AF inducibility, and electrophysiological parameters were not examined, and additional work is needed to clarify the influence of EAT on atrial electrophysiological properties.
Conclusions
In summary, our work provided convincing experimental evidence, for the first time, that chronic OSA enhanced the profibrotic effect of EAT on the neighboring atrial myocardium. Hypoxia‐induced profibrotic adipokine secretion from EAT might be the main mechanism of the fibrosis‐promoting effect. Metoprolol significantly attenuated hypoxia‐induced adipokine secretion and thereby inhibited the profibrotic effect of EAT enhanced by OSA. This study gives insights into mechanisms underlying OSA‐induced AF and also provides experimental evidence for the protective effects of metoprolol.
Sources of Funding
This work was supported by grants from National Natural Science Foundation of China (No. 81100121 to Gong; Nos. 81670297, 81470462, and 81830012 to Li; No. 81300133 to Sheng) and the Fundamental Research Funds for the Provincial Universities (No. 2017LCZX08 to Li).
Disclosures
None.
(J Am Heart Assoc. 2019;8:e011155 DOI: 10.1161/JAHA.118.011155.)
Contributor Information
Yongtai Gong, Email: gongth@126.com.
Yue Li, Email: ly99ly@vip.163.com.
References
- 1. Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep‐disordered breathing among middle‐aged adults. N Engl J Med. 1993;328:1230–1235. [DOI] [PubMed] [Google Scholar]
- 2. Gami AS, Pressman G, Caples SM, Kanagala R, Gard JJ, Davison DE, Malouf JF, Ammash NM, Friedman PA, Somers VK. Association of atrial fibrillation and obstructive sleep apnea. Circulation. 2004;110:364–367. [DOI] [PubMed] [Google Scholar]
- 3. Tung P, Levitzky YS, Wang R, Weng J, Quan SF, Gottlieb DJ, Rueschman M, Punjabi NM, Mehra R, Bertisch S, Benjamin EJ, Redline S. Obstructive and central sleep apnea and the risk of incident atrial fibrillation in a community cohort of men and women. J Am Heart Assoc. 2017;6:e004500 DOI: 10.1161/JAHA.116.004500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Neilan TG, Farhad H, Dodson JA, Shah RV, Abbasi SA, Bakker JP, Michaud GF, van der Geest R, Blankstein R, Steigner M, John RM, Jerosch‐Herold M, Malhotra A, Kwong RY. Effect of sleep apnea and continuous positive airway pressure on cardiac structure and recurrence of atrial fibrillation. J Am Heart Assoc. 2013;2:e000421 DOI: 10.1161/JAHA.113.000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Iwasaki YK, Kato T, Xiong F, Shi YF, Naud P, Maguy A, Mizuno K, Tardif JC, Comtois P, Nattel S. Atrial fibrillation promotion with long‐term repetitive obstructive sleep apnea in a rat model. J Am Coll Cardiol. 2014;64:2013–2023. [DOI] [PubMed] [Google Scholar]
- 6. Zhao J, Xu W, Yun F, Zhao H, Li W, Gong Y, Yuan Y, Yan S, Zhang S, Ding X, Wang D, Zhang C, Dong D, Xiu C, Yang N, Liu L, Xue J, Li Y. Chronic obstructive sleep apnea causes atrial remodeling in canines: mechanisms and implications. Basic Res Cardiol. 2014;109:427. [DOI] [PubMed] [Google Scholar]
- 7. Ramos P, Rubies C, Torres M, Batlle M, Farre R, Brugada J, Montserrat JM, Almendros I, Mont L. Atrial fibrosis in a chronic murine model of obstructive sleep apnea: mechanisms and prevention by mesenchymal stem cells. Respir Res. 2014;15:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wong CX, Ganesan AN, Selvanayagam JB. Epicardial fat and atrial fibrillation: current evidence, potential mechanisms, clinical implications, and future directions. Eur Heart J. 2017;38:1294–1302. [DOI] [PubMed] [Google Scholar]
- 9. Venteclef N, Guglielmi V, Balse E, Gaborit B, Cotillard A, Atassi F, Amour J, Leprince P, Dutour A, Clément K, Hatem SN. Human epicardial adipose tissue induces fibrosis of the atrial myocardium through the secretion of adipo‐fibrokines. Eur Heart J. 2015;36:795–805. [DOI] [PubMed] [Google Scholar]
- 10. Abe I, Teshima Y, Kondo H, Kaku H, Kira S, Ikebe Y, Saito S, Fukui A, Shinohara T, Yufu K, Nakagawa M, Hijiya N, Moriyama M, Shimada T, Miyamoto S, Takahashi N. Association of fibrotic remodeling and cytokines/chemokines content in epicardial adipose tissue with atrial myocardial fibrosis in patients with atrial fibrillation. Heart Rhythm. 2018;15:1717–1727. [DOI] [PubMed] [Google Scholar]
- 11. Baguet JP, Barone‐Rochette G, Tamisier R, Levy P, Pépin JL. Mechanisms of cardiac dysfunction in obstructive sleep apnea. Nat Rev Cardiol. 2012;9:679–688. [DOI] [PubMed] [Google Scholar]
- 12. Yu L, Li X, Huang B, Zhou X, Wang M, Zhou L, Meng G, Wang Y, Wang Z, Deng J, Jiang H. Atrial fibrillation in acute obstructive sleep apnea: autonomic nervous mechanism and modulation. J Am Heart Assoc. 2017;6:e006264 DOI: 10.1161/JAHA.117.006264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sun L, Yan S, Wang X, Zhao S, Li H, Wang Y, Lu S, Dong X, Zhao J, Yu S, Li M, Li Y. Metoprolol prevents chronic obstructive sleep apnea‐induced atrial fibrillation by inhibiting structural, sympathetic nervous and metabolic remodeling of the atria. Sci Rep. 2017;7:14941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim S, Whelan J, Claycombe K, Reath DB, Moustaid‐Moussa N. Angiotensin II increases leptin secretion by 3T3‐L1 and human adipocytes via a prostaglandin‐independent mechanism. J Nutr. 2002;132:1135–1140. [DOI] [PubMed] [Google Scholar]
- 15. Mariani S, Fiore D, Barbaro G, Basciani S, Saponara M, D'Arcangelo E, Ulisse S, Moretti C, Fabbri A, Gnessi L. Association of epicardial fat thickness with the severity of obstructive sleep apnea in obese patients. Int J Cardiol. 2013;167:2244–2249. [DOI] [PubMed] [Google Scholar]
- 16. Nattel S. Molecular and cellular mechanisms of atrial fibrosis in atrial fibrillation. JACC Clin Electrophysiol. 2017;3:425–435. [DOI] [PubMed] [Google Scholar]
- 17. Rahmutula D, Marcus GM, Wilson EE, Ding CH, Xiao Y, Paquet AC, Barbeau R, Barczak AJ, Erle DJ, Olgin JE. Molecular basis of selective atrial fibrosis due to overexpression of transforming growth factor‐β1. Cardiovasc Res. 2013;99:769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nakano Y, Niida S, Dote K, Takenaka S, Hirao H, Miura F, Ishida M, Shingu T, Sueda T, Yoshizumi M, Chayama K. Matrix metalloproteinase‐9 contributes to human atrial remodeling during atrial fibrillation. J Am Coll Cardiol. 2004;43:818–825. [DOI] [PubMed] [Google Scholar]
- 19. Aschar‐Sobbi R, Izaddoustdar F, Korogyi AS, Wang Q, Farman GP, Yang F, Yang W, Dorian D, Simpson JA, Tuomi JM, Jones DL, Nanthakumar K, Cox B, Wehrens XH, Dorian P, Backx PH. Increased atrial arrhythmia susceptibility induced by intense endurance exercise in mice requires TNFα. Nat Commun. 2015;6:6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang Z, Chen XJ, Qian C, Dong Q, Ding D, Wu QF, Li J, Wang HF, Li WH, Xie Q, Cheng X, Zhao N, Du YM, Liao YH. Signal transducer and activator of transcription 3/microRNA‐21 feedback loop contributes to atrial fibrillation by promoting atrial fibrosis in a rat sterile pericarditis model. Circ Arrhythm Electrophysiol. 2016;9:e003396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bonsignore MR, McNicholas WT, Montserrat JM, Eckel J. Adipose tissue in obesity and obstructive sleep apnoea. Eur Respir J. 2012;39:746–767. [DOI] [PubMed] [Google Scholar]
- 22. Almendros I, Farré R, Planas AM, Torres M, Bonsignore MR, Navajas D, Montserrat JM. Tissue oxygenation in brain, muscle and fat in a rat model of sleep apnea: differential effect of obstructive apneas and intermittent hypoxia. Sleep. 2011;34:1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, Furukawa S, Tochino Y, Komuro R, Matsuda M, Shimomura I. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007;56:901–911. [DOI] [PubMed] [Google Scholar]
- 24. Wang B, Wood IS, Trayhurn P. Hypoxia induces leptin gene expression and secretion in human preadipocytes: differential effects of hypoxia on adipokine expression by preadipocytes. J Endocrinol. 2008;198:127–134. [DOI] [PubMed] [Google Scholar]
- 25. Murphy AM, Thomas A, Crinion SJ, Kent BD, Tambuwala MM, Fabre A, Pepin JL, Roche HM, Arnaud C, Ryan S. Intermittent hypoxia in obstructive sleep apnoea mediates insulin resistance through adipose tissue inflammation. Eur Respir J. 2017;49:1601731. [DOI] [PubMed] [Google Scholar]
- 26. Priyanka A, Nisha VM, Anusree SS, Raghu KG. Bilobalide attenuates hypoxia induced oxidative stress, inflammation, and mitochondrial dysfunctions in 3T3‐L1 adipocytes via its antioxidant potential. Free Radic Res. 2014;48:1206–1217. [DOI] [PubMed] [Google Scholar]
- 27. Wang B, Wood IS, Trayhurn P. Dysregulation of the expression and secretion of inflammation‐related adipokines by hypoxia in human adipocytes. Pflugers Arch. 2007;455:479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sun K, Tordjman J, Clement K, Scherer PE. Fibrosis and adipose tissue dysfunction. Cell Metab. 2013;18:470–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fu L, Isobe K, Zeng Q, Suzukawa K, Takekoshi K, Kawakami Y. β‐Adrenoceptor agonists downregulate adiponectin, but upregulate adiponectin receptor 2 and tumor necrosis factor‐α expression in adipocytes. Eur J Pharmacol. 2007;569:155–162. [DOI] [PubMed] [Google Scholar]
- 30. Fasshauer M, Klein J, Lossner U, Paschke R. Interleukin (IL)‐6 mRNA expression is stimulated by insulin, isoproterenol, tumour necrosis factor alpha, growth hormone, and IL‐6 in 3T3‐L1 adipocytes. Horm Metab Res. 2003;35:147–152. [DOI] [PubMed] [Google Scholar]
- 31. Wang YY, Lin SY, Chuang YH, Chen CJ, Tung KC, Sheu WH. Adipose proinflammatory cytokine expression through sympathetic system is associated with hyperglycemia and insulin resistance in a rat ischemic stroke model. Am J Physiol Endocrinol Metab. 2011;300:E155–E163. [DOI] [PubMed] [Google Scholar]
- 32. Famulla S, Schlich R, Sell H, Eckel J. Differentiation of human adipocytes at physiological oxygen levels results in increased adiponectin secretion and isoproterenol‐stimulated lipolysis. Adipocyte. 2012;1:132–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Crystal E, Connolly SJ, Sleik K, Ginger TJ, Yusuf S. Interventions on prevention of postoperative atrial fibrillation in patients undergoing heart surgery: a meta‐analysis. Circulation. 2002;106:75–80. [DOI] [PubMed] [Google Scholar]
- 34. Dhein S, Rothe S, Busch A, Rojas Gomez DM, Boldt A, Reutemann A, Seidel T, Salameh A, Pfannmüller B, Rastan A, Kostelka M, Mohr FW. Effects of metoprolol therapy on cardiac gap junction remodelling and conduction in human chronic atrial fibrillation. Br J Pharmacol. 2011;164:607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wolf J, Drozdowski J, Czechowicz K, Winklewski PJ, Jassem E, Kara T, Somers VK, Narkiewicz K. Effect of beta‐blocker therapy on heart rate response in patients with hypertension and newly diagnosed untreated obstructive sleep apnea syndrome. Int J Cardiol. 2016;202:67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krum H, Schlaich M, Whitbourn R, Sobotka PA, Sadowski J, Bartus K, Walton A, Sievert H, Thambar S, Abraham WT, Esler M. Catheter‐based renal sympathetic denervation for resistant hypertension: a multicenter safety and proof‐of‐principle cohort study. Lancet. 2009;373:1275–1281. [DOI] [PubMed] [Google Scholar]
- 37. Linz D, Mahfoud F, Schotten U, Ukena C, Neuberger HR, Wirth K, Böhm M. Renal sympathetic denervation suppresses postapneic blood pressure rises and atrial fibrillation in a model for sleep apnea. Hypertension. 2012;60:172–178. [DOI] [PubMed] [Google Scholar]
- 38. Linz D, Hohl M, Nickel A, Mahfoud F, Wagner M, Ewen S, Schotten U, Maack C, Wirth K, Böhm M. Effect of renal denervation on neurohumoral activation triggering atrial fibrillation in obstructive sleep apnea. Hypertension. 2013;62:767–774. [DOI] [PubMed] [Google Scholar]
- 39. Ray CJ, Dow B, Kumar P, Coney AM. Mild chronic intermittent hypoxia in Wistar rats evokes significant cardiovascular pathophysiology but no overt changes in carotid body‐mediated respiratory responses. Adv Exp Med Biol. 2015;860:245–254. [DOI] [PubMed] [Google Scholar]
- 40. Chowdhuri S, Quan SF, Almeida F, Ayappa I, Batool‐Anwar S, Budhiraja R, Cruse PE, Drager LF, Griss B, Marshall N, Patel SR, Patil S, Knight SL, Rowley JA, Slyman A; ATS Ad Hoc Committee on Mild Obstructive Sleep Apnea . An official American Thoracic Society research statement: impact of mild obstructive sleep apnea in adults. Am J Respir Crit Care Med. 2016;193:e37–e54. [DOI] [PubMed] [Google Scholar]