

Abstract
Background
Prostaglandin E2 (PGE 2) is a major prostanoid with multiple actions that potentially affect blood pressure (BP). PGE 2 acts through 4 distinct E‐prostanoid (EP) receptor isoforms: EP1 to EP4. The EP4 receptor (EP4R) promotes PGE 2‐dependent vasodilation, but its role in the pathogenesis of hypertension is not clear.
Methods and Results
To address this issue, we studied mice after temporal‐ and cell‐specific deletion of EP4R. First, using a mouse line with loss of EP4 expression induced universally after birth, we confirm that EP4R mediates a major portion of the acute vasodilatory effects of infused PGE 2. In addition, EP4 contributes to control of resting BP, which was increased by 5±1 mm Hg in animals with generalized deficiency of this receptor. We also show that EP4 is critical for limiting elevations in BP caused by high salt feeding and long‐term infusion of angiotensin II. To more precisely identify the mechanism for these actions, we generated mice in which EP4R loss is induced after birth and is limited to smooth muscle. In these mice, acute PGE 2‐dependent vasodilation was attenuated, indicating that this response is mediated by EP4R in vascular smooth muscle cells. However, absence of EP4R only in this vascular compartment had a paradoxical effect of lowering resting BP, whereas the protective effect of EP4R on limiting angiotensin II–dependent hypertension was unaffected.
Conclusions
Taken together, our findings support a complex role for EP4R in regulation of BP and in hypertension, which appears to involve actions of the EP4R in tissues beyond vascular smooth muscle cells.
Keywords: angiotensin II, blood pressure, E‐prostanoid 4 receptor, hypertension, prostaglandin E2, renin, vascular smooth muscle cells
Subject Categories: Basic Science Research, Mechanisms, High Blood Pressure, Hypertension
Clinical Perspective
What Is New?
We used novel mouse lines with temporal‐ and cell‐specific deletion of the E‐prostanoid 4 receptor (EP4R) to investigate its role in blood pressure control and in the pathogenesis of hypertension.
This study, for the first time, clearly demonstrates a role for EP4R in baseline control of blood pressure and as a mitigating factor in the development of hypertension.
We also show that EP4R in vascular smooth muscle cells mediates the vasodilator actions of prostaglandin E2 and serves as a key trigger for renin release, influencing the level of baseline blood pressure.
Our data indicate that the effects EP4R in blood pressure control are complex. Its effects to attenuate hypertension are not attributable to compensatory vasodilation mediated by receptors in vascular smooth muscle cells and likely involve receptor actions in other cell lineages.
What Are the Clinical Implications?
Powerful protective actions of prostanoids in hypertension and cardiovascular disease have been revealed through clinical studies of NSAIDs, which act by specific inhibition of cyclooxygenase enzymes, reducing production of prostanoids.
In hypertension, characterizing the mechanisms underlying the beneficial effects of prostanoids, such as those mediated by the EP4R, can inform pathogenesis, uncover therapeutic targets, and suggest approaches for developing safer analgesic agents.
Introduction
Prostanoids, generated through the cyclooxygenase pathway of arachidonic acid metabolism, have a wide range of biological functions. Because of their relatively short half‐lives, these compounds typically act locally in the tissues where they are synthesized and their actions are mediated by specific receptors belonging to the large family of G‐protein–coupled, 7‐transmembrane receptors.1 NSAIDs, among the most widely used medications worldwide, act by inhibiting cyclooxygenase enzymes, thereby broadly attenuating the production of all prostanoids.2 NSAIDs are effective analgesic agents, but their use has been associated with exacerbation of hypertension3, 4 and increased cardiovascular risk,5, 6 suggesting that ≥1 prostanoids have beneficial actions to prevent hypertension and cardiovascular disease.
Prostaglandin E2 (PGE2) is a highly abundant prostanoid with several actions relevant to blood pressure (BP) control and cardiovascular functions.7, 8, 9, 10, 11 For example, it is a potent vasodilator12 and can induce natriuresis.13 Moreover, PGE2 influences immune responses and inflammation,14, 15, 16 which have recently been shown to contribute to the pathogenesis of hypertension17, 18 and cardiovascular diseases.18, 19 The actions of PGE2 are mediated by a series of E‐prostanoid (EP) receptors, which are divided into 4 pharmacological classes, EP1 to EP4, with differing tissue distributions and varied coupling to intracellular signaling pathways.20, 21, 22 Among the EP receptor isoforms, EP2 and EP4 have been classically considered as “relaxant” receptors1, 23, 24, 25 based on their preferential coupling to Gs proteins, linked to enhanced formation of cAMP and relaxation of bronchial and vascular smooth muscle.26
Our group27 and others28 have shown that genetic deficiency of microsomal PGE synthase 1, the major downstream PGE synthase enzyme responsible for synthesis of PGE2, is associated with elevated baseline BP and exaggerated angiotensin II– or deoxycorticosterone acetate (DOCA)‐salt–dependent hypertension in mice. Although the EP4 receptor (EP4R) contributes to PGE2‐dependent vasodilation,22, 29 the role of the EP4R in hypertension is not clear. In studies described herein, we find that inducible deletion of EP4R across all tissues is associated with elevated BP and exaggerated response to long‐term angiotensin II infusion, consistent with a role for EP4R to mitigate hypertension. However, cell‐specific deletion of EP4R from vascular smooth muscle cells (VSMCs) does not reproduce this prohypertensive phenotype, indicating that resistance to hypertension is not mediated by vasodilatory actions of EP4R.
Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure.30 Mouse lines are available on request from the authors.
Experimental Animals
A mouse line with a conditional Ptger4 flox allele was generated using homologous recombination in embryonic stem cells.31 To delete EP4R in the whole body, or specifically in VSMCs, we crossed the Ptger4 flox/flox mice with mouse lines expressing tamoxifen‐inducible Cre recombinase transgenes under control of β‐actin (ESR1‐Cre 32) or smooth muscle myosin heavy chain (SMMHC 33) promoters. After serial crosses, ESR1‐Cre + Ptger4 flox/flox total‐body knockout (TBKO; n=18) and SMMHC‐Cre + Ptger4 flox/flox smooth muscle knockout (SMKO; n=44) mouse lines were generated on an inbred C57BL/6 background. To induce expression of the Cre recombinase transgenes, tamoxifen (75 mg/kg per day; Sigma, St Louis, MO) dissolved in corn oil was administered by IP injection to individual mice for 5 consecutive days. Experiments were then initiated 2 weeks after tamoxifen dosing was completed.
Mouse lines were bred in Association for Assessment and Accreditation of Laboratory Animal Care International–accredited animal facilities at the Durham VA Medical Center. All studies were approved by Duke University and Durham Veterans’ Affairs Medical Center Institutional Animal Care and Use Committees and conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animals were housed in temperature‐controlled rooms with 12‐hour light/dark cycles and had free access to standard rodent chow and water unless specified.
BP Measurements in Conscious Mice
BP was measured continuously in conscious mice using radio telemetry, as described previously.34 After telemetry implantation, mice were allowed to recover for 7 days before BP recording started. A 7‐day baseline period during which the mice were fed with regular chow (0.4% NaCl) was followed by low‐salt (<0.01% NaCl) and high‐salt (6% NaCl) diets for 7 days, respectively. Finally, animals were returned to the regular chow for 3 days, and angiotensin II (Sigma Aldrich, St Louis, MO) was then infused using an osmotic minipump (1000 ng/kg per minute; Alzet, Cupertino, CA), as described previously.35 BP was continuously monitored for up to 3 weeks after angiotensin II infusion was initiated.
Assessment of Acute Vascular Responses to Agonists
We examined acute vascular responses to short‐term intravenous infusions of PGE2 (Cayman Chemicals, MI) and angiotensin II in mice anesthetized with 2% isoflurane, as described previously.35 A catheter (PE‐50) was inserted into the left jugular vein for administration of basal fluids and vasoconstrictors. A second catheter (pulled‐down PE‐50) attached to a pressure transducer (model MLT844, ADInstruments, Colorado Springs, CO) was placed in the left carotid artery. Intra‐arterial BP was recorded continuously through the carotid catheter using the PowerLab data acquisition system and LabChart software (ADInstruments). PGE2 (100 μg/kg) was injected intravenously and followed by 1 μg/kg of angiotensin II 10 minutes later, while intra‐arterial pressures were continuously monitored.
Furosemide‐Stimulated Renin mRNA Expression
To assess capacity for furosemide‐stimulated renin expression, separate cohorts of control and SMKO (n=9 per group) mice were treated with furosemide (2.28 mmol/L in drinking water; Sigma) or vehicle for 5 days. Kidneys were harvested, and RNA was extracted from the renal cortex for further analysis, as described previously.36
Quantitative Reverse Transcription–Polymerase Chain Reaction
Quantitative reverse transcription–polymerase chain reaction (PCR) was used to assess mRNA expression levels. Isoflurane‐anesthetized mice were perfused with ice‐cold PBS, and tissues were snap frozen in liquid nitrogen and stored in −80°C for RNA isolation. Total RNA was extracted using the RNeasy Mini or Micro Kit (Qiagen). Reverse transcription was performed using qScript cDNA Supermix (Quanta Biosciences, Gaithersburg, MD), and quantitative PCR was performed using the SYBR Green PCR Master Mix (Applied Biosystems, Carlsbad, CA). The amount of target gene relative to endogenous control (GAPDH) was determined by the Δ cycle threshold (Ct) method. The primer sequences are provided in the Table.
Table 1.
Sequences of Primers for RT‐PCR
| Variable | Forward Primers (5′→3′) | Reverse Primers (5′→3′) |
|---|---|---|
| EP4 | TCTCTGGTGGTGCTCATCTG | TGCAAATCTGGGTTTCTGCT |
| Renin‐1 | ATGAAGGGGGTGTCTGTGGGGTC | ATGCGGGGAGGGTGGGCACCTG |
| GAPDH | TCACCACCATGGAGAAGGC | GCTAAGCAGTTGGTGGTGCA |
EP indicates E‐prostanoid; RT‐PCR, reverse transcription–polymerase chain reaction.
Statistical Analysis
GraphPad v7.0 (GraphPad Software, San Diego, CA) was used for statistical analysis. Normality was determined using the Shapiro‐Wilk test. For comparisons between 2 groups in which normal distribution of the data could not be established, statistical significance was assessed using the Mann‐Whitney test. In these cases, data are expressed as median with interquartile range (IQR). For comparisons between 2 groups with normally distributed data, statistical significance was assessed using the unpaired t test and data are expressed as mean±SEM. Multiple comparisons between groups were assessed by 1‐way ANOVA, followed by Bonferroni post hoc test. Comparisons within groups were assessed by paired t test. Probability value for significance was defined as P<0.05.
Results
Characterization of Mouse Line With Tamoxifen‐Induced Deletion of EP4R in All Tissues
To generate mice lacking EP4 expression in all tissues (TBKO), we used a tamoxifen‐inducible Cre recombinase transgene driven by the β‐actin promoter (ESR‐Cre). To document the degree of EP4R excision, EP4R expression was quantified by real‐time PCR in tissues from ESR‐Cre + Ptger4 flox/flox (TBKO) and ESR‐Cre − Ptger4 flox/flox (control) mice (Figure 1). Two weeks after tamoxifen administration, mean±SEM EP4 mRNA levels were significantly reduced in all tissues examined from TBKO mice by 96.3±3.1% in kidney (n=6 and n=5; P=0.004), 95.0±4.3% in thymus (n=4; P=0.029), 70.5±4.4% in aorta (n=4; P=0.057), and 99.7±2.2% in heart (n=5; P=0.008). These data suggest efficient deletion of EP4R is achieved in ESR‐Cre + Ptger4 flox/flox adult mice after induction of the transgene with tamoxifen.
Figure 1.
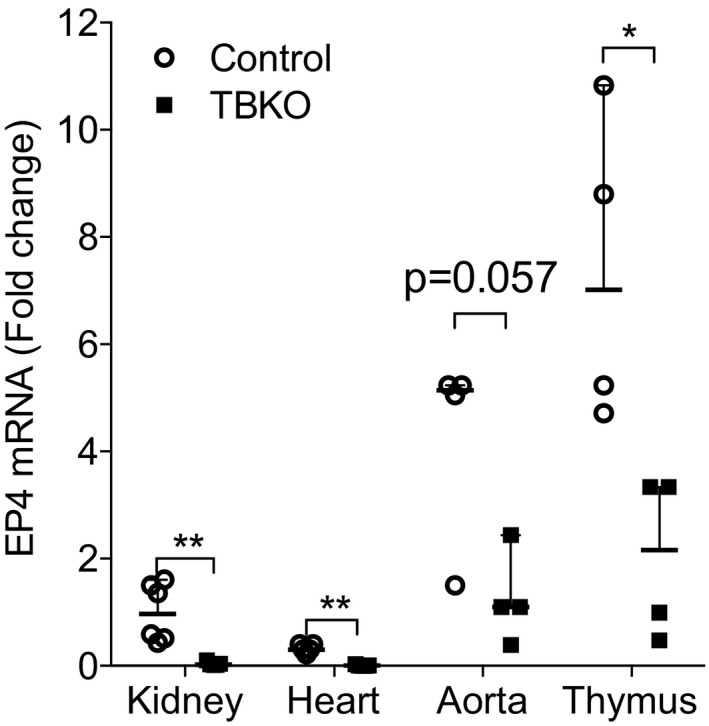
Verification of E‐prostanoid 4 receptor (EP4R) deletion in total‐body knockout (TBKO) mice. EP4 mRNA expression was measured in total RNA extracted from kidney, heart, aorta, and thymus. In all tissues tested, EP4R expression was significantly reduced in TBKO compared with control mice. n=4 to 6 per group. Data are expressed as median with range. *P<0.05, **P<0.01.
Acute Vasodilatory Responses to PGE2 Are Attenuated in Mice Lacking EP4R in All Tissues
To determine the impact of the complete absence of EP4R on PGE2‐dependent vasodilation, we measured hemodynamic responses to short‐term bolus infusions of PGE2 in TBKO and control mice. As shown in Figure 2, administration of PGE2 to control mice caused a rapid decrease in BP to a nadir of −11 mm Hg (IQR, −14 to −9 mm Hg; n=5). This response was significantly attenuated in TBKOs, in which maximal change in BP was −2 mm Hg (IQR, −6 to −1 mm Hg; n=4; P=0.016 versus controls). By contrast, short‐term administration of the vasoconstrictor angiotensin II caused rapid increases in BP that were similar in controls (18 mm Hg; IQR, 15–24 mm Hg) and TBKOs (21 mm Hg; IQR, 20–22 mm Hg) (P=0.73). These findings suggest that EP4R is the major mediator of vasodilation induced by PGE2. Moreover, the absence of EP4R does not affect the extent of vasoconstriction induced by angiotensin II.
Figure 2.
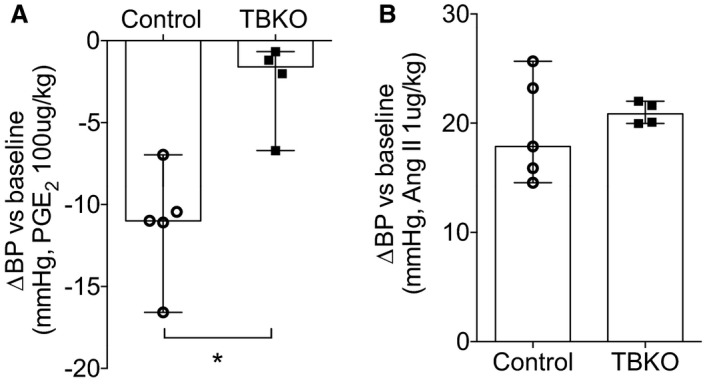
Eliminating E‐prostanoid 4 receptor from all tissues leads to reduced acute vasodilator responses to prostaglandin E2 (PGE 2). Acute blood pressure (BP) changes to PGE 2 and angiotensin II administration were measured through the carotid artery. A, The PGE 2‐induced acute BP decrease was significantly reduced in total‐body knockout (TBKO) mice. B, Short‐term administration of angiotensin II caused similar acute BP increases in both genotypes. n=4 and n=5 per group. Data are expressed as median with range. *P<0.05.
Elevated BPs and Enhanced Salt Sensitivity in TBKO Mice Lacking EP4R in All Tissues
BP in TBKO and control mice was measured using radio telemetry at baseline on a standard (0.4% NaCl) diet and during sequential feeding of low‐sodium (<0.01% NaCl) and high‐sodium (6% NaCl) chow. As shown in Figure 3A, mean arterial pressure (MAP) in control animals under baseline conditions of a standard 0.4% diet was 105±2 mm Hg (n=9). Under similar conditions, the absence of EP4R in the TBKOs was associated with a significant elevation in baseline MAP to 110±1 mm Hg (n=8; P=0.041 versus control). BP in TBKOs remained higher than controls throughout the periods of low‐ and high‐salt feeding (P=0.038 and P=0.004 versus control, respectively; Figure 3A). Within groups, the low‐salt diet did not significantly affect MAP in either mouse line compared with baseline. However, MAP increased significantly with high‐salt feeding in both controls (mean±SEM, 108±2 mm Hg [P=0.014] versus 0.4% NaCl; n=9) and the TBKOs (mean±SEM, 118±2 mm Hg [P=0.007] versus 0.4% NaCl; n=6). In addition, the change of MAP from low‐ to high‐salt feeding period was significantly higher in TBKO mice (5 mm Hg; IQR, 2–7 mm Hg) compared with controls (1 mm Hg; IQR, 0–2 mm Hg; P=0.026; Figure 3B). Thus, the absence of EP4R in TBKOs caused elevated baseline BP and enhanced salt sensitivity.
Figure 3.
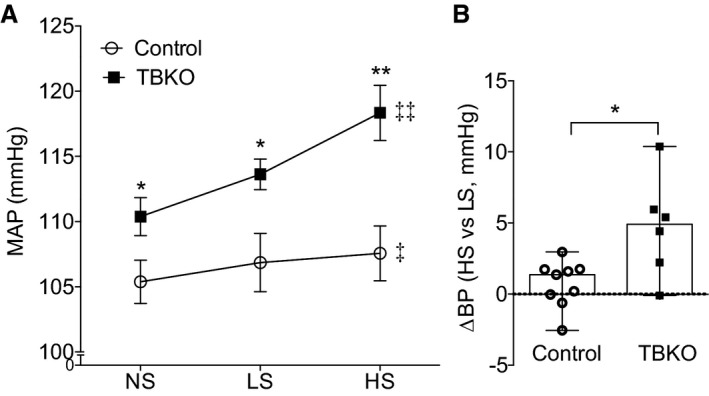
Total body deletion of E‐prostanoid 4 receptor leads to elevated baseline blood pressure (BP) and salt sensitivity. A, Mean arterial pressure (MAP), measured by radio telemetry, was significantly higher in total‐body knockout (TBKO) mice compared with controls at baseline and during low‐salt (LS) or high‐salt (HS) feeding. Feeding an LS diet had no effect on BP in the control mice; however, MAP increased significantly in both groups with HS feeding. B, The BP change from LS to HS feeding period was significantly elevated in TBKOs compared with the control group. n=6 to 9 per group. Data are expressed as mean±SEM (A) and median with range (B). NS indicates normal salt. *P<0.05, **P<0.01 vs control; ‡ P<0.05, ‡‡ P<0.01 vs baseline with NS feeding.
Reduced Expression of EP4R in All Tissues Causes Exaggerated Angiotensin II–Dependent Hypertension
To induce hypertension in TBKOs and controls, we infused angiotensin II subcutaneously via osmotic minipump at a dose of 1000 ng/kg per minute for 3 weeks. As shown in Figure 4A, angiotensin II caused rapid increases in BP in both groups, and the BP in TBKOs remained elevated (mean±SEM, 146±3 mm Hg; n=7) compared with controls (mean±SEM, 132±3 mm Hg; n=8; P=0.008) throughout the period of angiotensin II infusion (Figure 4A). Even considering the lower baseline BP in the control group, the change in MAP from baseline to the averaged entire period of angiotensin II infusion was significantly higher in TBKOs (33 mm Hg; IQR, 27–36 mm Hg) than in controls (22 mm Hg; IQR, 14–30 mm Hg; P=0.029; Figure 4B).
Figure 4.
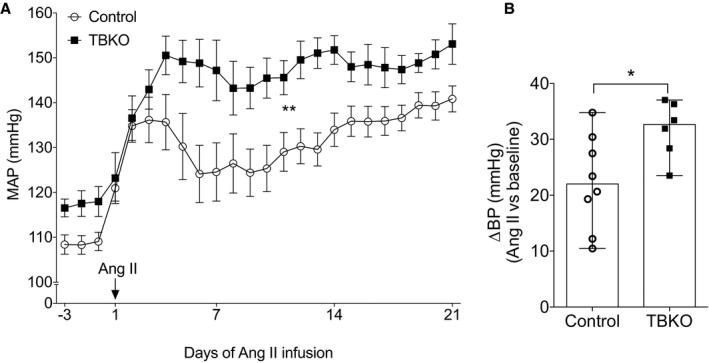
Exaggerated angiotensin II–dependent hypertension in mice lacking E‐prostanoid 4 receptor. A, Daily comparisons of mean arterial pressure (MAP) between the control and total‐body knockout (TBKO) groups at baseline and during angiotensin II infusion. B, Net change in MAP compared with baseline (change in blood pressure [∆BP]) for TBKO and control mice during the 21 days of angiotensin II infusion. The ∆BP with angiotensin II infusion was significantly higher in TBKOs (32±2 mm Hg) than controls (22±3 mm Hg). n=7 to 8 per group. Data are expressed as mean±SEM (A) and median with range (B). *P<0.05, **P<0.01 vs control.
Generation of Mice Lacking EP4R in Smooth Muscle
Because actions of the EP4R to promote vasodilation could abrogate BP elevation, we hypothesized that the protective effect of EP4R in angiotensin II–dependent hypertension is attributable to signaling and relaxation of VSMCs. To test this hypothesis, we generated mice lacking EP4R only in VSMCs by crossing a tamoxifen‐inducible Cre transgene driven by the SMMHC promoter with the C57BL/6 Ptger4 flox/flox mouse line. We have previously documented specificity of SMMC‐Cre + expression in smooth muscle cell lineages using the mTmG reporter mouse line.35 We found diminished EP4R expression in VSMCs by measuring the EP4 mRNA expression in whole aorta tissue using quantitative reverse transcription–PCR (P=0.007 versus control; Figure 5). In addition, as shown in Figure 6, acute vasodilatory responses to PGE2 were significantly attenuated in SMKOs (P=0.012), whereas acute angiotensin II–induced vasoconstriction was significantly increased in SMKOs compared with controls (P=0.028; n=8 and n=9). These data suggest that the acute vascular response to PGE2 stimulation is primarily mediated by EP4R in VSMCs.
Figure 5.
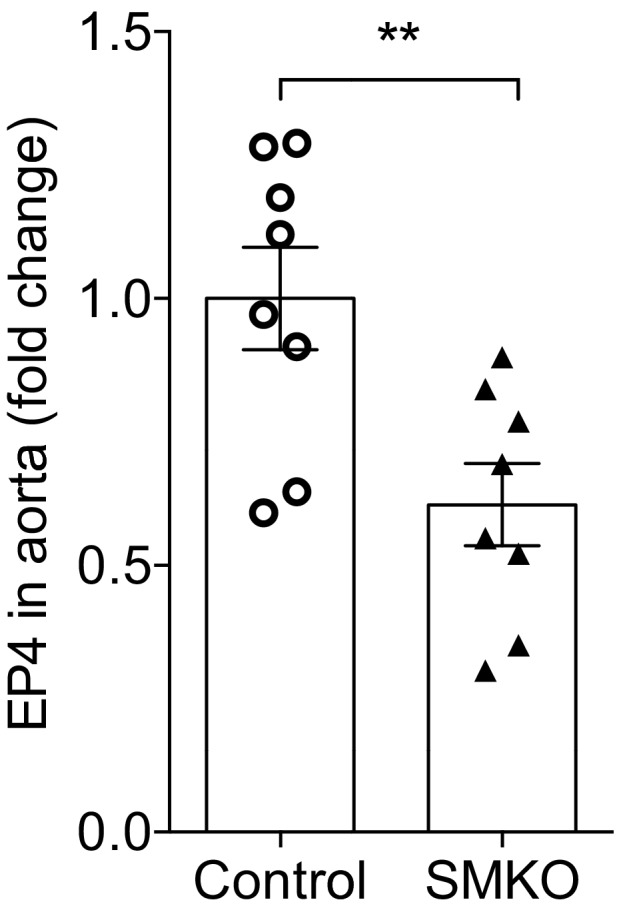
Verification of E‐prostanoid 4 (EP4) receptor deletion in vascular tissue of smooth muscle knockout (SMKO) mice. EP4 mRNA expression was significantly reduced in aortae from SMKO mice compared with controls. n=8 per group. Data are expressed as mean±SEM. **P<0.01.
Figure 6.
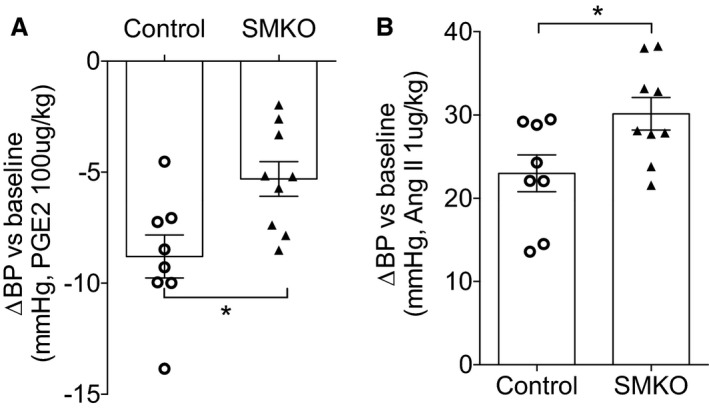
E‐prostanoid 4 receptor in vascular smooth muscle cells regulates acute vascular responses to prostaglandin E2 (PGE 2) and angiotensin II. A, The decrease in blood pressure (BP) after short‐term administration of PGE 2 was significantly attenuated in smooth muscle knockout (SMKO) mice. B, Acute pressor responses to angiotensin II were significantly enhanced in SMKO mice. n=8 to 9 per group. Data are expressed as mean±SEM. *P<0.05.
Reduced BP in Mice Lacking EP4R in VSMCs
We measured BP in SMKOs and controls using radio telemetry. As shown in Figure 7A, BP at baseline was unexpectedly and significantly reduced in SMKO mice (mean±SEM, 104±2 mm Hg; n=10) compared with controls (mean±SEM, 109±2 mm Hg; n=11; P=0.041). We also assessed the impact of altered dietary sodium intake on BP in the experimental groups. As shown in Figure 7A, compared with controls, BP remained lower in SMKOs during both low‐ and high‐salt feeding. However, the BP change from low‐ to high‐salt feeding period was similar in SMKOs and controls (Figure 7B). Thus, the absence of EP4R in VSMCs was associated with lower baseline BP without any significant effect on sodium‐dependent BP responses.
Figure 7.
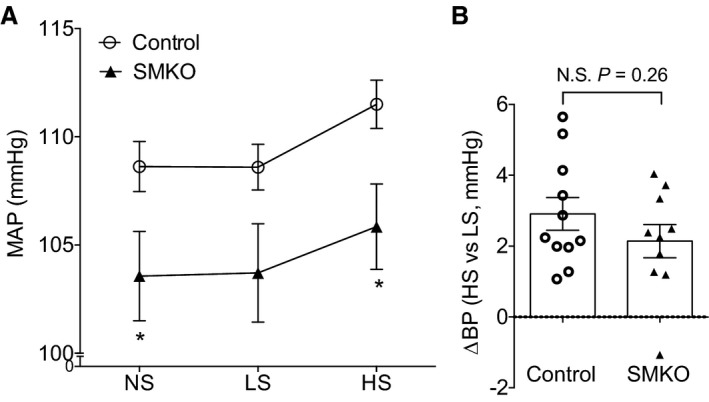
Mice lacking E‐prostanoid 4 receptor in vascular smooth muscle cells have reduced baseline blood pressure (BP). A, The mean arterial pressures (MAPs) during baseline and the high‐salt (HS) feeding period were significantly lower in smooth muscle knockout (SMKO) mice compared with control. B, The MAP change from low‐salt (LS) to HS feeding was similar between the 2 groups. n=10 to 11 per group. Data are expressed as mean±SEM. NS indicates normal salt; N.S., not significant. *P<0.05 vs control.
Angiotensin II–Dependent Hypertension Is Unaffected in SMKOs
To determine the impact of eliminating EP4R from VSMCs on the development of hypertension, we performed long‐term infusions of angiotensin II in SMKOs and controls, as described above. As shown in Figure 8A, the BP responses to angiotensin II infusion were indistinguishable between SMKOs and controls, with MAPs of 138±3 and 139±3 mm Hg, respectively (n=10 per group). Similarly, the changes in BP with angiotensin II compared with baseline were not significantly different between SMKOs (mean±SEM, 32±3 mm Hg) and controls (mean±SEM, 27±3 mm Hg; Figure 8B).
Figure 8.
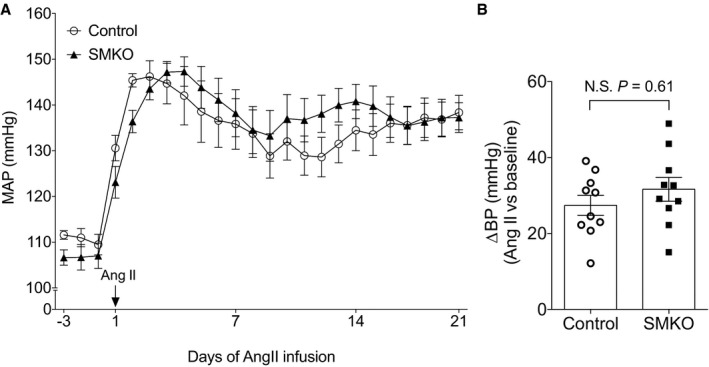
Absence of E‐prostanoid 4 receptor from vascular smooth muscle cells has no effect on angiotensin II–dependent hypertension. A, There were no differences in daily mean arterial pressure (MAP) between smooth muscle knockout (SMKO) mice and control during angiotensin II infusion. B, Net change in MAP during the 21 days of angiotensin II infusion compared with baseline (change in blood pressure [∆BP]) was similar in control and SMKO mice. n=10 per group. Data are expressed as mean±SEM. N.S. indicates not significant.
Furosemide‐Induced Renin Expression Was Reduced in SMKOs
We have previously shown that renin responses are impaired in mice completely lacking EP4R,36 implicating EP4R as a key trigger for renin regulation. In view of the reduced baseline BP in SMKO mice, we hypothesized that reduced renin expression might be contributing to their low BP. Accordingly, we analyzed the renin mRNA expression in the renal cortex after administration of furosemide, which blocks sodium reabsorption by Na‐K‐Cl cotransporter (NKCC) 2 and thereby stimulates renin expression and release.37, 38, 39 After administration of furosemide in drinking water for 5 days, renin expression was significantly lower in SMKO mice compared with controls (mean±SEM fold change, 8.6±1.1 versus 11.7±0.9; P=0.02; n=8 and n=9). This indicates that EP4R signaling in VSMCs is required for appropriate regulation of renin expression.
Discussion
A series of clinical and epidemiological studies have identified associations between the use of NSAIDs and risk for hypertension and cardiovascular disease.3, 4, 5, 40, 41, 42, 43, 44 Because the pharmacological actions of NSAIDs are mediated by specific inhibition of cyclooxygenase enzymes and reduced production of prostanoids, these clinical studies highlight protective actions of prostanoids in these disorders. Characterizing the mechanisms underlying the beneficial effects of prostanoids in cardiovascular diseases can inform pathogenesis, uncover therapeutic targets, and suggest approaches for developing safer analgesic agents. Previous studies have demonstrated powerful actions of prostacyclin to prevent hypertension, coronary artery disease, and cardiac fibrosis.45, 46, 47, 48 Similarly, a protective role for PGE2 in hypertension has also been well documented.27, 49 For PGE2, it has been suggested that mitigating actions in hypertension depend on vasodilation of both the systemic and renal circulations, reducing peripheral vascular resistance and promoting sodium excretion by the kidney in the face of hypertensive challenge.50, 51, 52, 53, 54, 55, 56, 57
The vasodilator actions of PGE2 are presumed to be mediated by its EP2 receptor and EP4R, which both signal by activating adenylyl cyclase and generating cAMP, a well‐known stimulus for vascular relaxation.1, 20, 25, 58 In this regard, previous work suggested that the EP2 receptor contributes to PGE2‐induced vasodilation49, 59 and also mediates protective actions of PGE2 in hypertension.59, 60 However, effects of the EP2 receptor to prevent development of hypertension have not been universally observed.49 Moreover, a role for the alternative vasodilator receptor, EP4R, in BP regulation and hypertension has not been clearly defined. Herein, using conditional gene targeting, we find that PGE2 elicits vasodilation via EP4R in VSMCs. We also show that the predominant role of EP4R in hypertension is to attenuate BP increases triggered by high‐salt feeding or elevated levels of angiotensin II. However, the effects of EP4R to resist the development of hypertension are not mediated through its actions in VSMCs.
Among its vascular actions, the EP4R is essential for normal remodeling and closure of the ductus arteriosus after birth.61, 62 Specifically, ductus arteriosus closure is triggered by reduced EP4R signaling coincident with abrupt decrease in PGE2 in the circulation immediately after delivery61; these actions are mediated by EP4R expressed in VSMCs.63 Accordingly, complete elimination of EP4R before birth causes perinatal mortality attributable to patent ductus arteriosus.61 To circumvent this complication, we used tamoxifen‐inducible Cre transgenes to delete the EP4R gene in adult mice. In TBKOs, this system achieved efficient diminution of EP4R expression in all tissues tested, and there were no other apparent untoward effects of deleting EP4R in adult animals. Moreover, elimination of EP4R resulted in substantial attenuation of PGE2‐dependent vasodilation, indicating that EP4R is the predominant mediator of vascular responses to PGE2.
Our studies in TBKOs, with reduced expression of EP4R across all tissues, identified a role for EP4R to resist the development of hypertension. Specifically, global removal of EP4R from adult mice caused a significant ≈5–mm Hg increase in resting BP along with salt sensitivity, characterized by exaggerated increases in BP during high‐salt feeding. In addition, angiotensin II–dependent hypertension was significantly augmented in TBKOs. These observations are in line with previous studies from our laboratory27 and others28 showing that absence of microsomal PGE synthase 1, the major synthetic enzyme for PGE2 downstream of cyclooxygenase, also produces exaggerated hypertension.27 Taken together, these data indicate that the PGE2/EP4R axis functions to oppose elevations in BP triggered by increased dietary salt intake or activation of the renin‐angiotensin system.
On the basis of the critical role of EP4R in PGE2‐dependent vasodilation, we posited that impaired vasodilation in TBKOs would eliminate an important compensatory pathway for buffering elevations in BP. To test whether this mechanism explained exaggerated hypertension in the TBKOs, we evaluated mice with cell‐specific deletion of EP4R from smooth muscle (SMKO). As in the TBKOs, acute vascular responses to PGE2 were largely abrogated in SMKOs, confirming that EP4R signaling in VSMCs mediates PGE2‐induced vasodilation. However, in contrast to TBKOs, we found that baseline BP was significantly reduced in SMKOs. Moreover, the phenotypes of enhanced salt sensitivity and accelerated angiotensin II–dependent hypertension seen in the TBKOs were not present in the SMKOs. Thus, eliminating EP4R‐dependent vasodilatory responses in isolation did not lead to further increases in BP, suggesting that other functions of EP4R are responsible for its protective actions in hypertension. In this regard, Harris and associates recently showed that cyclooxygenase‐2 expressed in macrophages prevents salt‐sensitive hypertension through a pathway involving PGE2 and EP4R, by regulating sodium deposition in skin.64 These findings are in line with work by Machnik et al65 showing that the skin is an important reservoir for sodium storage during high‐salt feeding, triggering macrophages to produce vascular endothelial growth factor‐C, inducing lymphangiogenesis, and buffering the propensity of a high‐salt diet to increase BP.
Along with the minimal impact of deleting EP4R from VSMCs to development of hypertension, the frank reduction in baseline BP in the SMKOs was also unexpected. However, in previous studies, we demonstrated a key role for the EP4R in regulating renin release and activating the renin‐angiotensin system.36 In this prior work, performed in a unique line of recombinant inbred EP4R‐deficient mice, we showed that renin expression and release were significantly diminished in the absence of EP4R signaling.36 But because these mice lacked EP4R expression in all tissues, the key cellular site of EP4R action controlling renin could not be determined. Herein, we find significant attenuation of furosemide‐stimulated renin expression in SMKOs, suggesting a mechanism for reduced baseline BP in SMKOs and pinpointing EP4R expression in smooth muscle lineages as a critical control point for renin. These findings are consistent with previous work showing that renin‐producing cells in the juxtaglomerular apparatus of the kidney are contiguous with the glomerular afferent arteriole, and they have ultrastructural features and expression markers consistent with smooth muscle cell lineage.66, 67
Taken together, our study reveals complex roles for the EP4R in BP regulation. EP4R in VSMCs mediates acute vasodilator responses to PGE2, while also acting as a control point for activation of the renin‐angiotensin system. Although the absence of EP4R‐dependent regulation of renin in isolation is sufficient to lower BP, this phenotype is supplanted by hypertension when expression of EP4R is more broadly reduced. Thus, the dominant physiological impact of EP4R in BP homeostasis is to oppose the development of hypertension.
Sources of Funding
These studies were supported by the National Institutes of Health Grant DK105049 (to Coffman), the National Heart, Lung, and Blood Institute Grant K99‐HL‐109167 (to Herrera), the Career Development Award from the Biomedical Laboratory Research and Development Service of the Department of Veterans Affairs Office of Research and Development IK2BX002240 (to Sparks), the Edna and Fred L. Mandel Center for Hypertension and Atherosclerosis Research, the James R. Clapp Distinguished Professorship Fund (to Coffman), and the Duke O'Brien Center for Kidney Research.
Disclosures
None.
(J Am Heart Assoc. 2019;8:e010745 DOI: 10.1161/JAHA.118.010745.)
References
- 1. Narumiya S, FitzGerald GA. Genetic and pharmacological analysis of prostanoid receptor function. J Clin Invest. 2001;108:25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vane J, Botting R. Inflammation and the mechanism of action of anti‐inflammatory drugs. FASEB J. 1987;1:89–96. [PubMed] [Google Scholar]
- 3. Solomon DH, Schneeweiss S, Levin R, Avorn J. Relationship between COX‐2 specific inhibitors and hypertension. Hypertension. 2004;44:140–145. [DOI] [PubMed] [Google Scholar]
- 4. Dedier J, Stampfer MJ, Hankinson SE, Willett WC, Speizer FE, Curhan GC. Nonnarcotic analgesic use and the risk of hypertension in us women. Hypertension. 2002;40:604–608; discussion 601–603. [DOI] [PubMed] [Google Scholar]
- 5. Solomon DH, Schneeweiss S, Glynn RJ, Kiyota Y, Levin R, Mogun H, Avorn J. Relationship between selective cyclooxygenase‐2 inhibitors and acute myocardial infarction in older adults. Circulation. 2004;109:2068–2073. [DOI] [PubMed] [Google Scholar]
- 6. Yu Y, Ricciotti E, Scalia R, Tang SY, Grant G, Yu Z, Landesberg G, Crichton I, Wu W, Pure E, Funk CD, FitzGerald GA. Vascular COX‐2 modulates blood pressure and thrombosis in mice. Sci Transl Med. 2012;4:132ra154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Okuno T, Lindheimer MD, Oparil S. Central effects of prostaglandin E2 on blood pressure and plasma renin activity in rats: role of the sympathoadrenal system and vasopressin. Hypertension. 1982;4:809–816. [DOI] [PubMed] [Google Scholar]
- 8. Colina‐Chourio JA, Godoy‐Godoy N, Avila‐Hernandez RM. Role of prostaglandins in hypertension. J Hum Hypertens. 2000;14(suppl 1):S16–S19. [DOI] [PubMed] [Google Scholar]
- 9. Smith MC, Dunn MJ. The role of prostaglandins in human hypertension. Am J Kidney Dis. 1985;5:A32–A39. [DOI] [PubMed] [Google Scholar]
- 10. Sun H, Wang Y. Prostaglandin E2 in remote control of myocardial remodeling. Circulation. 2012;125:2818–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yuhki K, Kojima F, Kashiwagi H, Kawabe J, Fujino T, Narumiya S, Ushikubi F. Roles of prostanoids in the pathogenesis of cardiovascular diseases: novel insights from knockout mouse studies. Pharmacol Ther. 2011;129:195–205. [DOI] [PubMed] [Google Scholar]
- 12. Tang L, Loutzenhiser K, Loutzenhiser R. Biphasic actions of prostaglandin E(2) on the renal afferent arteriole: role of EP(3) and EP(4) receptors. Circ Res. 2000;86:663–670. [DOI] [PubMed] [Google Scholar]
- 13. Villa E, Garcia‐Robles R, Haas J, Romero JC. Comparative effect of PGE2 and PGI2 on renal function. Hypertension. 1997;30:664–666. [DOI] [PubMed] [Google Scholar]
- 14. Chizzolini C, Chicheportiche R, Alvarez M, de Rham C, Roux‐Lombard P, Ferrari‐Lacraz S, Dayer JM. Prostaglandin E2 synergistically with interleukin‐23 favors human Th17 expansion. Blood. 2008;112:3696–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boniface K, Bak‐Jensen KS, Li Y, Blumenschein WM, McGeachy MJ, McClanahan TK, McKenzie BS, Kastelein RA, Cua DJ, de Waal Malefyt R. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J Exp Med. 2009;206:535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Napolitani G, Acosta‐Rodriguez EV, Lanzavecchia A, Sallusto F. Prostaglandin E2 enhances Th17 responses via modulation of IL‐17 and IFN‐gamma production by memory CD4+ T cells. Eur J Immunol. 2009;39:1301–1312. [DOI] [PubMed] [Google Scholar]
- 17. Coffman TM. Under pressure: the search for the essential mechanisms of hypertension. Nat Med. 2011;17:1402–1409. [DOI] [PubMed] [Google Scholar]
- 18. Meng X, Yang J, Dong M, Zhang K, Tu E, Gao Q, Chen W, Zhang C, Zhang Y. Regulatory T cells in cardiovascular diseases. Nat Rev Cardiol. 2016;13:167–179. [DOI] [PubMed] [Google Scholar]
- 19. Chinetti‐Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. 2015;12:10–17. [DOI] [PubMed] [Google Scholar]
- 20. Coleman RA, Smith WL, Narumiya S. International union of pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev. 1994;46:205–229. [PubMed] [Google Scholar]
- 21. Tsuboi K, Sugimoto Y, Ichikawa A. Prostanoid receptor subtypes. Prostaglandins Other Lipid Mediat. 2002;68–69:535–556. [DOI] [PubMed] [Google Scholar]
- 22. Yokoyama U, Iwatsubo K, Umemura M, Fujita T, Ishikawa Y. The prostanoid EP4 receptor and its signaling pathway. Pharmacol Rev. 2013;65:1010–1052. [DOI] [PubMed] [Google Scholar]
- 23. Tilley SL, Coffman TM, Koller BH. Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. J Clin Invest. 2001;108:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenases)‐1 and ‐2. J Biol Chem. 1996;271:33157–33160. [DOI] [PubMed] [Google Scholar]
- 25. Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–1226. [DOI] [PubMed] [Google Scholar]
- 26. Breyer MD, Breyer RM. G protein‐coupled prostanoid receptors and the kidney. Annu Rev Physiol. 2001;63:579–605. [DOI] [PubMed] [Google Scholar]
- 27. Facemire CS, Griffiths R, Audoly LP, Koller BH, Coffman TM. The impact of microsomal prostaglandin E synthase 1 on blood pressure is determined by genetic background. Hypertension. 2010;55:531–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jia Z, Aoyagi T, Yang T. mPGES‐1 protects against DOCA‐salt hypertension via inhibition of oxidative stress or stimulation of NO/cGMP. Hypertension. 2010;55:539–546. [DOI] [PubMed] [Google Scholar]
- 29. Abran D, Dumont I, Hardy P, Peri K, Li DY, Molotchnikoff S, Varma DR, Chemtob S. Characterization and regulation of prostaglandin E2 receptor and receptor‐coupled functions in the choroidal vasculature of the pig during development. Circ Res. 1997;80:463–472. [PubMed] [Google Scholar]
- 30. Yang TY. Complex role for E‐Prostanoid (EP) 4 receptors in hypertension. Dans. 2018. Available at: 10.17026/dans-28u-529p. Accessed January 31, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schneider A, Guan Y, Zhang Y, Magnuson MA, Pettepher C, Loftin CD, Langenbach R, Breyer RM, Breyer MD. Generation of a conditional allele of the mouse prostaglandin EP4 receptor. Genesis. 2004;40:7–14. [DOI] [PubMed] [Google Scholar]
- 32. Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen‐inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. [DOI] [PubMed] [Google Scholar]
- 33. Nelson RD, Stricklett P, Gustafson C, Stevens A, Ausiello D, Brown D, Kohan DE. Expression of an AQP2 Cre recombinase transgene in kidney and male reproductive system of transgenic mice. Am J Physiol. 1998;275:C216–C226. [DOI] [PubMed] [Google Scholar]
- 34. Crowley SD, Gurley SB, Oliverio MI, Pazmino AK, Griffiths R, Flannery PJ, Spurney RF, Kim HS, Smithies O, Le TH, Coffman TM. Distinct roles for the kidney and systemic tissues in blood pressure regulation by the renin‐angiotensin system. J Clin Invest. 2005;115:1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sparks MA, Stegbauer J, Chen D, Gomez JA, Griffiths RC, Azad HA, Herrera M, Gurley SB, Coffman TM. Vascular type 1A angiotensin II receptors control BP by regulating renal blood flow and urinary sodium excretion. J Am Soc Nephrol. 2015;26:2953–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Facemire CS, Nguyen M, Jania L, Beierwaltes WH, Kim HS, Koller BH, Coffman TM. A major role for the EP4 receptor in regulation of renin. Am J Physiol Renal Physiol. 2011;301:F1035–F1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Plata C, Meade P, Hall A, Welch RC, Vazquez N, Hebert SC, Gamba G. Alternatively spliced isoform of apical Na(+)‐K(+)‐Cl(‐) cotransporter gene encodes a furosemide‐sensitive Na(+)‐Cl(‐)cotransporter. Am J Physiol Renal Physiol. 2001;280:F574–F582. [DOI] [PubMed] [Google Scholar]
- 38. Limmer F, Schinner E, Castrop H, Vitzthum H, Hofmann F, Schlossmann J. Regulation of the Na(+)‐K(+)‐2 cl(‐) cotransporter by cGMP/cGMP‐dependent protein kinase I after furosemide administration. FEBS J. 2015;282:3786–3798. [DOI] [PubMed] [Google Scholar]
- 39. Mutig K, Kahl T, Saritas T, Godes M, Persson P, Bates J, Raffi H, Rampoldi L, Uchida S, Hille C, Dosche C, Kumar S, Castaneda‐Bueno M, Gamba G, Bachmann S. Activation of the bumetanide‐sensitive Na+, K+,2Cl‐ cotransporter (NKCC2) is facilitated by Tamm‐Horsfall protein in a chloride‐sensitive manner. J Biol Chem. 2011;286:30200–30210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Graham DJ, Campen D, Hui R, Spence M, Cheetham C, Levy G, Shoor S, Ray WA. Risk of acute myocardial infarction and sudden cardiac death in patients treated with cyclo‐oxygenase 2 selective and non‐selective non‐steroidal anti‐inflammatory drugs: nested case‐control study. Lancet. 2005;365:475–481. [DOI] [PubMed] [Google Scholar]
- 41. Johnsen SP, Larsson H, Tarone RE, McLaughlin JK, Norgard B, Friis S, Sorensen HT. Risk of hospitalization for myocardial infarction among users of rofecoxib, celecoxib, and other NSAIDs: a population‐based case‐control study. Arch Intern Med. 2005;165:978–984. [DOI] [PubMed] [Google Scholar]
- 42. Helin‐Salmivaara A, Virtanen A, Vesalainen R, Gronroos JM, Klaukka T, Idanpaan‐Heikkila JE, Huupponen R. NSAID use and the risk of hospitalization for first myocardial infarction in the general population: a nationwide case‐control study from Finland. Eur Heart J. 2006;27:1657–1663. [DOI] [PubMed] [Google Scholar]
- 43. Haag MD, Bos MJ, Hofman A, Koudstaal PJ, Breteler MM, Stricker BH. Cyclooxygenase selectivity of nonsteroidal anti‐inflammatory drugs and risk of stroke. Arch Intern Med. 2008;168:1219–1224. [DOI] [PubMed] [Google Scholar]
- 44. Chan AT, Manson JE, Albert CM, Chae CU, Rexrode KM, Curhan GC, Rimm EB, Willett WC, Fuchs CS. Nonsteroidal antiinflammatory drugs, acetaminophen, and the risk of cardiovascular events. Circulation. 2006;113:1578–1587. [DOI] [PubMed] [Google Scholar]
- 45. Francois H, Athirakul K, Howell D, Dash R, Mao L, Kim HS, Rockman HA, Fitzgerald GA, Koller BH, Coffman TM. Prostacyclin protects against elevated blood pressure and cardiac fibrosis. Cell Metab. 2005;2:201–207. [DOI] [PubMed] [Google Scholar]
- 46. Cheng Y, Austin SC, Rocca B, Koller BH, Coffman TM, Grosser T, Lawson JA, FitzGerald GA. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science. 2002;296:539–541. [DOI] [PubMed] [Google Scholar]
- 47. Kobayashi T, Tahara Y, Matsumoto M, Iguchi M, Sano H, Murayama T, Arai H, Oida H, Yurugi‐Kobayashi T, Yamashita JK, Katagiri H, Majima M, Yokode M, Kita T, Narumiya S. Roles of thromboxane A(2) and prostacyclin in the development of atherosclerosis in apoE‐deficient mice. J Clin Invest. 2004;114:784–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xiao CY, Hara A, Yuhki K, Fujino T, Ma H, Okada Y, Takahata O, Yamada T, Murata T, Narumiya S, Ushikubi F. Roles of prostaglandin I(2) and thromboxane A(2) in cardiac ischemia‐reperfusion injury: a study using mice lacking their respective receptors. Circulation. 2001;104:2210–2215. [DOI] [PubMed] [Google Scholar]
- 49. Tilley SL, Audoly LP, Hicks EH, Kim HS, Flannery PJ, Coffman TM, Koller BH. Reproductive failure and reduced blood pressure in mice lacking the EP2 prostaglandin E2 receptor. J Clin Invest. 1999;103:1539–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nasrallah R, Hassouneh R, Hebert RL. PGE2, kidney disease, and cardiovascular risk: beyond hypertension and diabetes. J Am Soc Nephrol. 2016;27:666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Silldorff EP, Yang S, Pallone TL. Prostaglandin E2 abrogates endothelin‐induced vasoconstriction in renal outer medullary descending vasa recta of the rat. J Clin Invest. 1995;95:2734–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen J, Zhao M, He W, Milne GL, Howard JR, Morrow J, Hebert RL, Breyer RM, Chen J, Hao CM. Increased dietary NaCl induces renal medullary PGE2 production and natriuresis via the EP2 receptor. Am J Physiol Renal Physiol. 2008;295:F818–F825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang Y, Guan Y, Schneider A, Brandon S, Breyer RM, Breyer MD. Characterization of murine vasopressor and vasodepressor prostaglandin E(2) receptors. Hypertension. 2000;35:1129–1134. [DOI] [PubMed] [Google Scholar]
- 54. Ren Y, D'Ambrosio MA, Garvin JL, Wang H, Carretero OA. Response to prostaglandin E2 mediates connecting tubule glomerular feedback. Hypertension. 2014;63:e20. [DOI] [PubMed] [Google Scholar]
- 55. Hristovska AM, Rasmussen LE, Hansen PB, Nielsen SS, Nusing RM, Narumiya S, Vanhoutte P, Skott O, Jensen BL. Prostaglandin E2 induces vascular relaxation by E‐prostanoid 4 receptor‐mediated activation of endothelial nitric oxide synthase. Hypertension. 2007;50:525–530. [DOI] [PubMed] [Google Scholar]
- 56. Foudi N, Kotelevets L, Louedec L, Leseche G, Henin D, Chastre E, Norel X. Vasorelaxation induced by prostaglandin E2 in human pulmonary vein: role of the EP4 receptor subtype. Br J Pharmacol. 2008;154:1631–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ren Y, D'Ambrosio MA, Garvin JL, Wang H, Carretero OA. Prostaglandin E2 mediates connecting tubule glomerular feedback. Hypertension. 2013;62:1123–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Audoly LP, Tilley SL, Goulet J, Key M, Nguyen M, Stock JL, McNeish JD, Koller BH , Coffman TM. Identification of specific EP receptors responsible for the hemodynamic effects of PGE2. Am J Physiol. 1999;277:H924–H930. [DOI] [PubMed] [Google Scholar]
- 59. Audoly LP, Ruan X, Wagner VA, Goulet JL, Tilley SL, Koller BH, Coffman TM, Arendshorst WJ. Role of EP(2) and EP(3) PGE(2) receptors in control of murine renal hemodynamics. Am J Physiol Heart Circ Physiol. 2001;280:H327–H333. [DOI] [PubMed] [Google Scholar]
- 60. Kennedy CR, Zhang Y, Brandon S, Guan Y, Coffee K, Funk CD, Magnuson MA, Oates JA, Breyer MD, Breyer RM. Salt‐sensitive hypertension and reduced fertility in mice lacking the prostaglandin EP2 receptor. Nat Med. 1999;5:217–220. [DOI] [PubMed] [Google Scholar]
- 61. Nguyen M, Camenisch T, Snouwaert JN, Hicks E, Coffman TM, Anderson PA, Malouf NN, Koller BH. The prostaglandin receptor EP4 triggers remodelling of the cardiovascular system at birth. Nature. 1997;390:78–81. [DOI] [PubMed] [Google Scholar]
- 62. Yokoyama U, Minamisawa S, Shioda A, Ishiwata R, Jin MH, Masuda M, Asou T, Sugimoto Y, Aoki H, Nakamura T, Ishikawa Y. Prostaglandin E2 inhibits elastogenesis in the ductus arteriosus via EP4 signaling. Circulation. 2014;129:487–496. [DOI] [PubMed] [Google Scholar]
- 63. Gruzdev A, Nguyen M, Kovarova M, Koller BH. PGE2 through the EP4 receptor controls smooth muscle gene expression patterns in the ductus arteriosus critical for remodeling at birth. Prostaglandins Other Lipid Mediat. 2012;97:109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang MZ, Yao B, Wang Y, Yang S, Wang S, Fan X, Harris RC. Inhibition of cyclooxygenase‐2 in hematopoietic cells results in salt‐sensitive hypertension. J Clin Invest. 2015;125:4281–4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, Park JK , Beck FX, Muller DN, Derer W, Goss J, Ziomber A, Dietsch P, Wagner H, van Rooijen N, Kurtz A, Hilgers KF, Alitalo K, Eckardt KU, Luft FC, Kerjaschki D, Titze J. Macrophages regulate salt‐dependent volume and blood pressure by a vascular endothelial growth factor‐C‐dependent buffering mechanism. Nat Med. 2009;15:545–552. [DOI] [PubMed] [Google Scholar]
- 66. Sequeira Lopez ML, Pentz ES, Robert B, Abrahamson DR, Gomez RA. Embryonic origin and lineage of juxtaglomerular cells. Am J Physiol Renal Physiol. 2001;281:F345–F356. [DOI] [PubMed] [Google Scholar]
- 67. Brunskill EW, Sequeira‐Lopez ML, Pentz ES, Lin E, Yu J, Aronow BJ, Potter SS, Gomez RA. Genes that confer the identity of the renin cell. J Am Soc Nephrol. 2011;22:2213–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]