

Abstract
Background
The aortic valve of the heart experiences constant mechanical stress under physiological conditions. Maladaptive valve injury responses contribute to the development of valvular heart disease. Here, we test the hypothesis that MG53 (mitsugumin 53), an essential cell membrane repair protein, can protect valvular cells from injury and fibrocalcific remodeling processes associated with valvular heart disease.
Methods and Results
We found that MG53 is expressed in pig and human patient aortic valves and observed aortic valve disease in aged Mg53−/− mice. Aortic valves of Mg53−/− mice showed compromised cell membrane integrity. In vitro studies demonstrated that recombinant human MG53 protein protects primary valve interstitial cells from mechanical injury and that, in addition to mediating membrane repair, recombinant human MG53 can enter valve interstitial cells and suppress transforming growth factor‐β‐dependent activation of fibrocalcific signaling.
Conclusions
Together, our data characterize valve interstitial cell membrane repair as a novel mechanism of protection against valvular remodeling and assess potential in vivo roles of MG53 in preventing valvular heart disease.
Keywords: cell membrane repair, fibrosis, heart valve, transforming growth factor‐β, valvular heart disease
Subject Categories: Valvular Heart Disease, Basic Science Research, Fibrosis
Clinical Perspective
What Is New?
MG53, an essential cell membrane repair protein, is expressed in aortic valves.
Mg53−/− mice display signs of aortic valve disease.
Recombinant human MG53 protects aortic valve interstitial cells from membrane injury and reduces fibrocalcific signaling.
What Are the Clinical Implications?
Targeting valvular cell membrane repair represents a potential novel mechanism to treat valvular heart disease.
Introduction
Valvular heart disease (VHD) is a common cause of cardiovascular disease, afflicting over 5 million patients in North America alone.1, 2 These numbers are rapidly increasing because of aging populations. VHD leads to maladaptive cardiac remodeling and heart failure without surgical valve replacement. There are currently no pharmacological options to specifically treat valve disease.
The 4 heart valves open and close with every cardiac cycle, playing an integral role in regulating blood flow throughout the heart chambers. The aortic valve separates the left ventricle from the aorta, is exposed to the highest cardiac pressures, and is the most common valve implicated in disease. Valve leaflets are composed of endothelial and interstitial cells, the latter of which are the most prevalent cell type and proposed to play critical roles in tissue repair.3, 4, 5, 6, 7, 8, 9 Quiescent aortic valve interstitial cells (VICs) become “activated” in response to injury, experiencing a fibroblast‐to‐myofibroblast‐like transition, and later “osteoblastic” in nature, cumulatively resulting in valvular fibrocalcific changes hallmarked by extracellular matrix remodeling and calcium deposition.8, 10 Physiologically, these valve leaflet changes result in narrowing of the valve lumen, termed aortic stenosis, and progression of cardiac disease.
Our laboratory has identified MG53 (mitsugumin 53), a 477‐amino acid TRIM (tripartite motif‐containing) protein, as an essential component of the cell membrane repair machinery.11, 12, 13, 14, 15, 16, 17, 18, 19 In response to injury, MG53 acts as a sensor of the extracellular oxidative environment to nucleate recruitment of intracellular vesicles to damaged sites for membrane patch formation. MG53 is highly expressed in mechanically‐active tissues such as cardiac and skeletal muscle and can protect these cells from injury secondary to various pathophysiological stresses.
Given the tremendous stress experienced by heart valves and the crucial contributions of fibrocalcific signaling to valve disease, we hypothesized that MG53 can both facilitate repair of acute membrane injury to VICs and modulate the fibrocalcific responses that contribute to the development of VHD. We present data to show that MG53 is expressed in aortic valves and that aged Mg53−/− mice develop aortic valve disease. Additionally, we observed that MG53 protects against both VIC membrane damage and transforming growth factor (TGF)‐ß‐induced VIC fibrocalcific changes. Together, these findings support the therapeutic potential for MG53 in modulating VHD.
Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedures upon reasonable request to the corresponding author.
Porcine Aortic Valve Tissue and Cell Culture
Immediately after euthanasia of adult pigs by The Ohio State University Laboratory Animal Resources, hearts were excised, and aortic valves were dissected. For tissue western blotting, valve leaflets were immediately washed with phosphate‐buffered saline (PBS), frozen, and processed in radio‐immunoprecipitation assay lysis buffer as described below. For primary VIC isolation, valve leaflets were immediately washed with PBS and then incubated in standard media (10% fetal bovine serum, 1% antibiotic‐antimycotic [penicillin, streptomycin, amphotericin B], Dulbecco's modified Eagle's medium) with 600 U/mL collagenase II at 37°C for 18 hours.20 For all cell culture experiments, VICs were plated in plastic, tissue culture‐treated cell culture flasks and maintained in standard media through a maximum of 8 passages.
Human Patient Aortic Valve Tissue
Human patient aortic valve tissues were obtained from 1 explanted heart of a patient undergoing cardiac transplantation and from donor hearts obtained in collaboration with the Lifeline of Ohio Organ Procurement Program. All human tissues were experimented on with approval from the Institutional Review Board of The Ohio State University (Study Number 2012H0197) and conformed to the Declaration of Helsinki. Informed consent was acquired from the cardiac transplant patient. Frozen leaflets were processed in radio‐immunoprecipitation assay lysis buffer as described below.
Western Blotting
Cell and tissue protein was extracted in radio‐immunoprecipitation assay lysis buffer with protease and phosphatase inhibitor cocktails. Protein concentrations were determined via Bradford assays. Prepared samples were separated via sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE) and transferred to polyvinylidene fluoride membranes. Amongst the different experiments, membranes were incubated with antibodies against MG53 (custom‐made rabbit monoclonal antibody), glyceraldehyde 3‐phosphate dehydrogenase (GAPDH; Santa Cruz, sc‐32233), phospho‐Smad2 (Cell Signaling Technology, #3108), Smad2/3 (Cell Signaling Technology, #8685), and fibronectin (Sigma‐Aldrich, F3648) and imaged on film or an Odyssey Fc Imaging System (LI‐COR Biosciences [Lincoln, Nebraska, USA]). Densitometric quantification of detected signals was performed on the LI‐COR Biosciences Image Studio Lite version 5.2 imaging software.
The custom made monoclonal antibody can recognize MG53 in mouse, rat, pig, dog, and human tissues. Compared with the available commercial antibodies, this antibody showed high specificity and affinity for MG53. In particular, it can resolve up to 10 picograms of the recombinant human MG53 (rhMG53) protein in western blot. For assessing MG53 expression in mouse myocardial tissue, only 0.06 μg of total protein was required. This antibody has been used previously for IHC staining to resolve low levels of MG53 expression in the mouse lung15 and kidney18 tissues. Specificity of the antibody is shown in the present study with heart muscle derived from Mg53−/− mice.
Mice
Mg53−/− mice were first generated in the 129S1/SvImJ strain as described in Cai et al, 11 have been maintained and utilized in several studies of tissue injury, and have been backcrossed for more than thirty generations.11, 12, 13, 14, 15, 18 Here, wild type and Mg53−/− mice derived from the original strain were used in all studies. In echocardiography and histopathology experiments, we were able to use wild‐type littermates for all the 10‐month‐old Mg53−/− mice and for 2 of the 24‐month‐old Mg53−/− mice. The additional mice in these studies were age‐matched and of the same 129S1/SvImJ genetic background. In Evans blue dye uptake experiments, we used littermate control mice. Based on our previous studies, we did not identify sex differences in cardiac function with the Mg53−/− mice. However, for economic use of the animal resources, we employed the male wild‐type and Mg53−/− mice for the echocardiography studies and histology studies that required paraffin embedding. The female mice were used for Evans blue dye uptake experiments that required cryo‐embedding. All mice were housed in The Ohio State University Laboratory Animal Resources vivarium, and all procedures and experiments were approved by The Ohio State University Institutional Animal Care and Use Committee and performed in accord with National Institutes of Health guidelines.
Echocardiography
In echocardiographic studies, mice were anesthetized with 2% isoflurane and imaged using a Vevo 2100 Imaging System (FUJIFILM VisualSonics, Inc [Toronto, Ontario, Canada]) and the MX400 22 to 55 MHz ultrasound transducer. For aortic valve velocity measurements, the parasternal long axis view was used to visualize the left ventricular outflow tract with color Doppler. Mice were tilted to reverse Trendelenburg positions to obtain flows of optimal clarity, and pulse‐wave velocity measurements were then obtained in blood flow past the aortic valve. Continuous‐wave velocity measurements could not be performed as the Vevo 2100 Imaging System does not have these technical capabilities. Peak velocities represent the mean of the highest 10% of collected velocities for each mouse.
Histopathology and Immunohistochemistry
Male mouse hearts were perfused with 4% paraformaldehyde and excised for histopathology and immunohistochemistry. Hearts were fixed in 4% paraformaldehyde for at least 24 hours, dehydrated in 70% ethanol for 24 hours, and transferred to The Ohio State University Comparative Pathology and Mouse Phenotyping Core for paraffin‐embedding. Paraffin blocks were then sectioned to 4 μm at The Ohio State University Pathology Core Facility and mounted on Fisherbrand Superfrost Plus microscope slides. Sections with aortic valve leaflets were used for all histopathological and immunohistochemical analyses. Hematoxylin and eosin and Masson trichrome staining was performed by the Pathology Core Facility, and Alizarin Red S staining was performed by the Comparative Pathology and Mouse Phenotyping Core. Immunohistochemistry was performed for fibronectin (Abcam, ab2413; 1:200).
Pig aortic valve sections were subjected to immunohistochemical staining with MG53 antibody (1:200). Histopathology sections were imaged on an Axiovert 200 microscope (Zeiss [Oberkochen, Germany]) with cellSens Dimension 1.18 imaging software (OLYMPUS [Tokyo, Japan]). Immunohistochemistry sections were imaged on a Zeiss LSM 780 confocal microscope with Zeiss ZEN 2011 imaging software.
Microelectrode Needle‐Induced Cell Membrane Injury
VIC membrane injury assays were performed as previously described for other cell types.11, 14, 15, 16, 18, 19 For microelectrode needle penetration, VICs were transfected with GFP (green fluorescent protein)‐human MG53 and then transferred to a Delta T glass‐bottomed dish. Single cells were visualized on an Eclipse TE2000‐S microscope (Nikon [Tokyo, Japan]) with a Radiance 2100 Laser Scanning System (BIO‐RAD [Hercules, California, USA]) and BIO‐RAD LaserSharp2000 5.1 imaging software. Under fluorescence and live cell recording, a microelectrode needle was used to penetrate the VIC membrane.
Glass Bead Cell Injury Assay
Glass bead damage assays used VICs cultured in 75 mm2 cell culture flasks to 70% to 90% confluency. VICs were collected and concentrated in PBS to 1.5×106 cells per milliliter. VICs (50 μL [≈75 000 cells]) and glass microbeads (50 μL) were then aliquoted on 96‐well plates and treated in duplicate with varying doses of rhMG53; (0, 12.5, 25, 50, 100, and 200 μg/mL). Immediately after treatments, plates were secured on a benchtop shaker and vigorously shaken for 15 minutes. Plates were spun down, and supernatants were collected for LDH (lactate dehydrogenase) spectrophotometric measurements (Takara, Cat. #MK401) to assess VIC injury.14, 15, 16, 18
Evans Blue Dye Uptake
One percent EBD was administered to littermate wild‐type and Mg53−/− mice via intraperitoneal injection of 10 μL EBD per gram of mouse body weight. Mice were euthanized 24 hours post‐injection, and hearts were excised and OCT‐embedded for cryosectioning. Hearts were sectioned to 8 μm, post‐fixed in 4% paraformaldehyde for 10 minutes at room temperature, washed in PBS, and mounted on Fisherbrand Superfrost Plus microscope slides.21 EBD fluorescence was imaged on a Zeiss Axiovert 200 microscope with cellSens Dimension 1.18 imaging software. EBD uptake for individual aortic valve leaflets was measured on Fiji (ImageJ) 2.0.0 as Corrected Total fluorescence=integrated density of selected leaflet−(area of selected leaflet×mean fluorescence of background readings).
Conjugation of rhMG53 to Fluorescent Probe and Live Cell Imaging
rhMG53 and bovine serum albumin (BSA) were diluted in distilled water to a final concentration of 2 mg/mL. rhMG53 (1 mg) and BSA (1 mg) were conjugated to Alexa 647‐NHS using an Alexa Fluor 647 Protein Labeling Kit according to manufacturer's protocol (Invitrogen, Cat. #A20173). Equal concentrations of either rhMG53‐Alexa647 or BSA‐Alexa647 were added to VICs that were 50% confluent on glass‐bottomed cell culture dishes (35 mm diameter with a 15‐mm internal glass diameter).14 Cells were then incubated at 37°C for 30 minutes and imaged immediately thereafter to assess intracellular fluorescent intensity on a Zeiss LSM 780 confocal microscope with Zeiss ZEN 2011 imaging software.
TGF‐ß Signaling Studies
The role of MG53 in modulating VIC TGF‐ß signaling was investigated by culturing VICs in 25 cm2 cell culture flasks to 70% to 90% confluency and then conditioning them overnight in either serum‐free medium (to assess changes in protein phosphorylation) or 1% fetal bovine serum medium (to assess changes in protein expression). Cells were pretreated for 30 minutes with either rhMG53 (50 μg/mL) or equimolar BSA and then treated with TGF‐ß (1 ng/mL) for 20 minutes (to assess changes in protein phosphorylation) or 24 hours (to assess changes in protein expression). For western blotting, cells were processed in radio‐immunoprecipitation assay lysis buffer as described above.
Statistical Analyses
All statistical analyses were performed with GraphPad Prism 7.0b with data points denoting mean±SD. Shapiro–Wilk normality tests (α=0.05) were used to test normality. For normally‐distributed data, statistical significance is shown as P values obtained via a 2‐tailed Student t test when comparing 2 experimental groups and ANOVA with multiple comparisons when comparing >2 groups. Ordinary, 1‐way ANOVA tests with Tukey multiple comparison testing were generally used; 2‐way ANOVA tests were used when an experiment had 2 independent variables; and Dunnett multiple comparison testing was used when comparing multiple experimental groups to a single control group. When data were not normally distributed, a Mann–Whitney non‐parametric test was used to obtain P values. A P value of <0.05 was considered statistically significant.
Results
MG53 is Expressed in Pig and Human Patient Aortic Valve Tissue
In mice, MG53 is predominantly expressed in mechanically‐active cardiac and skeletal muscles.11, 12 More recent studies have established expression of low levels of MG53 in non‐muscle tissues as well.15, 18 MG53 expression in large animal and human tissues is less well‐characterized.22, 23, 24 Given that the aortic valve experiences tremendous mechanical stress and that large animals share similar cardiovascular physiological environments with human patients, we first performed western blotting with pig tissue lysates to determine MG53 protein expression in pig aortic valves (Figure 1A). We then confirmed MG53 expression in aortic valves from human donor hearts and from 1 patient with heart failure and VHD (Figure 1B). De‐identified patient details are presented in Table S1. The human heart samples were derived from The Ohio State University Heart Bank. Most of the samples in the Heart Bank are from patients with ischemic cardiomyopathies, end‐stage heart failure, or healthy donor hearts that failed to be transplanted. In the future, once we are able to obtain a reasonable number of human diseased valve samples, we could then design studies to examine if there are any changes in MG53 protein expression, subcellular distribution, or genetic mutations in human patients with VHD.
Figure 1.
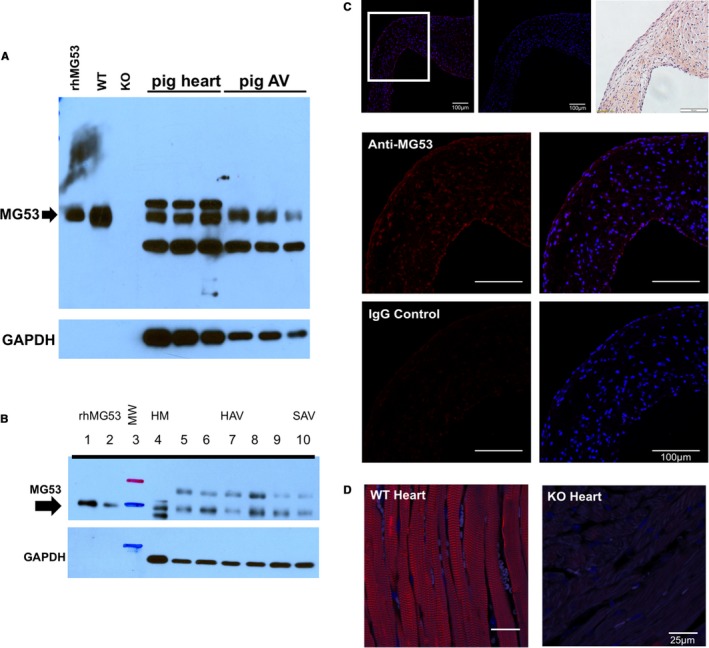
MG53 is expressed in pig and human patient aortic valve tissue. A, Western blotting shows that MG53 (53 kDa, arrow) is expressed in pig myocardium and aortic valves. rhMG53 (0.03 ng) and wild‐type mouse myocardium (0.06 μg) were used as positive controls and Mg53−/− myocardium (0.06 μg) as a negative control. 10 μg of lysates from pig tissues were loaded from 3 different animals. The apparent lack of GAPDH expression in the mouse myocardial tissue reflects the ≈150‐fold less loading of mouse vs pig protein lysates. B, Western blotting shows that MG53 (53 kDa, arrow) is expressed in human myocardium and aortic valves. rhMG53 was loaded in lanes 1 to 2 (0.05 and 0.02 ng, respectively); ladder in lane 3; non‐failing myocardium (2.5 μg) in lane 4; non‐diseased valves (10 μg) in lanes 5 to 9; and a stenotic valve (10 μg) in lane 10. C, Immunohistochemistry of axial sections of pig aortic valve shows that MG53 (red) is expressed in the valve interstitial layers as well as its endothelial linings. In the top‐most row, ×20 magnification images are shown of (left to right) MG53 staining, rabbit immunoglobulin G control staining, and hematoxylin and eosin staining. In the lower rows and from the ×20 boxed area, ×40 magnification images are shown of MG53 and rabbit immunoglobulin G control staining (red) and overlaid DAPI (4′,6‐diamidino‐2‐phenylindole) staining (blue). D, MG53 staining (red) of wild‐type and Mg53−/− mouse myocardium are positive and negative controls, respectively. AV indicates aortic valve; HAV, healthy aortic valves; HM, healthy myocardium; IgG, immunoglobulin G; KO, knockout (Mg53−/−); MW, molecular weight; SAV, stenotic aortic valve; rh, recombinant human; WT, wild‐type.
We conducted immunohistochemistry to determine the tissue distribution of MG53 in the pig aortic valves (Figure 1C). Clearly, the monoclonal antibody against MG53 detected signals in both interstitial and endothelial cell layers, whereas the control immunoglobulin G showed negative staining. Specificity of the antibody against MG53 used in the immunohistochemistry experiments was confirmed with wild‐type and Mg53−/− mouse myocardium (Figure 1D).
Aged Mg53−/− Mice Develop Aortic Valve Disease
The physiological role of MG53 in modulating aortic valve function was investigated with in vivo assessments of wild‐type and Mg53−/− mice. Given the marked and well‐established contributions of aging to VHD,25, 26, 27, 28 we screened 10‐and 24‐month‐old wild‐type versus Mg53−/− mice for aortic valve disease via echocardiography and histology.
In clinical echocardiography, color Doppler imaging and peak aortic valve outflow velocity are the most robust tools to assess aortic valve dysfunction.29 Stenotic valves have more turbulent flow and increased peak aortic valve velocities attributable to the fluid dynamics of blood flowing past fibrocalcific leaflets that narrow the valve lumen. Such environments contribute to continued cycles of pathological valve remodeling. In murine echocardiography, numerous studies have used peak aortic valve outflow velocity as a metric to evaluate aortic stenosis.30, 31, 32, 33, 34 Some studies have used echocardiographic calculations of aortic valve cusp separation distance to quantify the dimensions of the aortic valve lumen.33, 34, 35, 36, 37 While cusp separation distance provides a unidimensional assessment of VHD, it is often less sensitive than peak aortic valve velocity measurement in screening for aortic valve dysfunction in rodents.
Echocardiographic analysis of mice at 10 months showed no differences in peak aortic valve velocity between wild‐type and Mg53−/− mice (1335±317.5 mm/s versus 1274±66.3 mm/s, P=0.9962) (Figure 2A). Wild‐type mice did not show apparent changes in peak aortic valve velocity from 10 to 24 months (1335±317.5 mm/s versus 1516±414.3 mm/s, P=0.8578). However, 24‐month‐old Mg53−/− mice developed significant increases in peak aortic valve velocity compared with those at 10 months, presenting hemodynamic evidence of aortic valve disease. Figure 2A shows the mean peak aortic valve velocity of Mg53−/− mice increased from 1274±66.3 mm/s (at 10 months) to 2929±1073 mm/s (at 24 months, P<0.0001) and rose significantly higher at 24 months than that of age‐matched wild‐type mice (1516±414.3 mm/s, P<0.0001). Representative color Doppler images and pulse wave aortic valve jets are shown (Figure 2B), and representative videos are provided (Videos S1 and S2).
Figure 2.
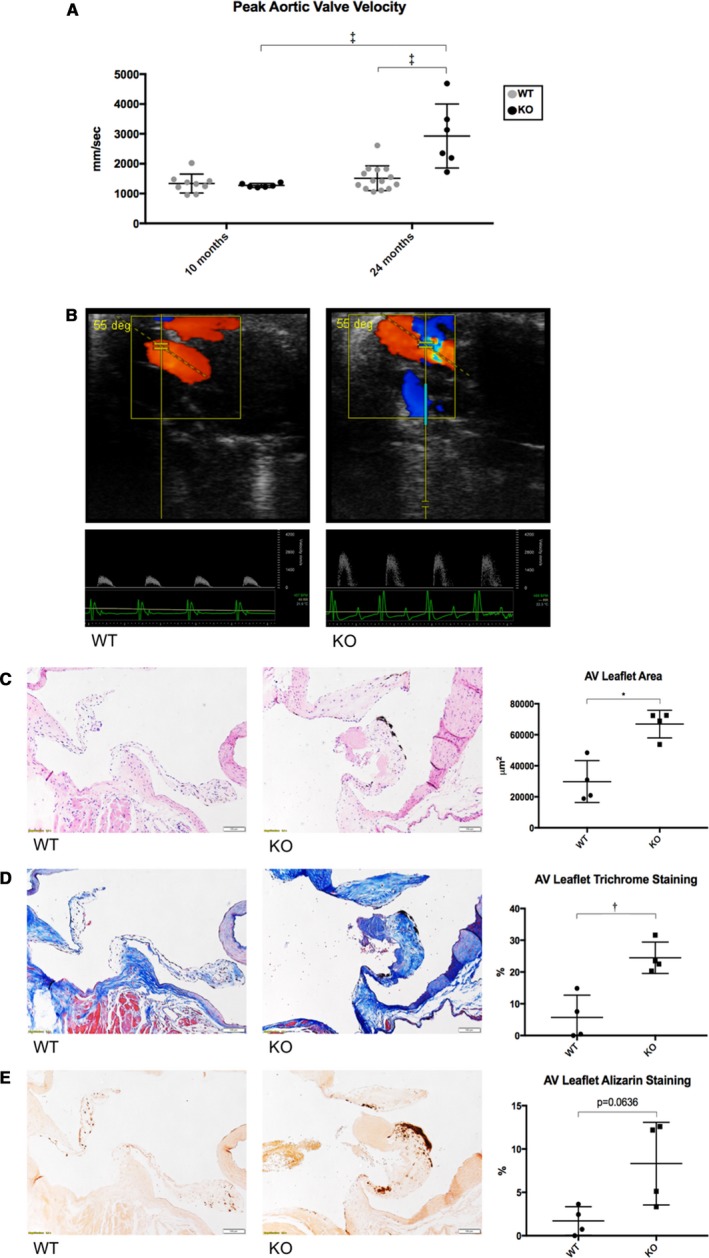
Welcome3jah3Aged Mg53−/− mice develop aortic valve disease. A, Peak aortic valve outflow velocities were measured via echocardiography with higher velocities associated with presence of aortic stenosis. Ten‐ and 24‐month‐old wild‐type mice had no statistical difference in peak aortic valve velocities. Twenty‐four‐month‐old Mg53−/− mice showed statistically significant higher velocities compared with both 10‐month‐old Mg53−/− mice and 24‐month‐old wild‐type mice. A 2‐way ANOVA test with Tukey multiple comparison testing (α=0.05) was used to obtain P values. n=9 ten‐month‐old wild‐type mice; n=6 ten‐month‐old Mg53−/− mice; n=14 twenty‐four‐month‐old wild‐type mice; n=6 twenty‐four‐month‐old Mg53−/− mice. B, Representative color Doppler images and pulse‐wave aortic valve jets are shown for wild‐type (left) and Mg53−/− (right) mice. Aortic valve disease is associated with more turbulent color images and higher velocities as measured by the y‐axis of the pulse‐wave jets. C, Coronal sections of aortic valve leaflets are shown opening upwards, left ventricle below and aorta above. Histologic analyses of aged wild‐type (left) and Mg53−/− (right) mouse hearts using hematoxylin and eosin staining showed increased area; (D) Masson trichrome staining showed increased fibrosis; and (E) Alizarin Red S staining showed increased calcification in Mg53−/− aortic valve leaflets (n=4) compared with those from wild‐type control mice (n=4). Mann–Whitney test was used to obtain the P value for (C), and 2‐tailed Student t tests were used to obtain P values for (D and E). AV indicates aortic valve; KO, knockout (Mg53−/−); WT, wild‐ type. *P<0.05, **P<0.01, ****P<0.0001.
Histologic analysis of mouse hearts with hematoxylin and eosin staining showed that aged Mg53−/− mice have thickened aortic valve leaflets compared with age‐matched wild‐type controls, pathology consistent with the development of aortic valve disease (Figure 2C). Furthermore, Masson trichrome staining showed increased valvular fibrosis (Figure 2D, see also Figure S1). Alizarin Red S staining showed increased valvular calcification (Figure 2E). Together, these stainings provide histologic evidence of fibrocalcific aortic valve remodeling in aged Mg53−/− mice. Interestingly, similar histologic changes were also observed in mitral valves of aged Mg53−/− mice (Figure S2).
Echocardiographic assessment previously showed that Mg53−/− mice up to 10 months of age display normal cardiac function under basal conditions.12, 13, 38 Here, we also found no significant differences between wild‐type and Mg53−/− mice in left ventricular ejection fraction, fractional shortening, heart rate, and volume, even at 24 months of age. Cardiac morphology measurements (heart weight and heart weight/body weight) similarly showed no statistical differences (Figure S3). These results demonstrate that valve dysfunction observed in Mg53−/− mice is not a consequence of more general cardiac dysfunction or disease. Further, other investigators have also observed that mice with significant aortic stenosis may have well‐preserved ventricular function.34 Such preserved physiology may be a result of adaptive changes in Mg53−/− mouse hearts or reflective of mice that have not yet progressed to heart failure.
MG53 Protects Against VIC Membrane Injury
MG53 orchestrates membrane repair patch formation in various cells, protecting them from membrane injury caused by various stimuli.11, 13, 14, 15, 16, 18, 19 Here, we used live cell imaging to demonstrate that GFP‐MG53 translocates to membrane injury sites following microelectrode needle penetration of primary cultured VICs (Figure 3A, Video S3). This is similar to our previous findings with skeletal muscle, cardiac muscle, and epithelial cells, where formation of a membrane repair patch is associated with rapid movement of GFP‐MG53 to injury sites.11, 13, 14, 15, 18, 19
Figure 3.
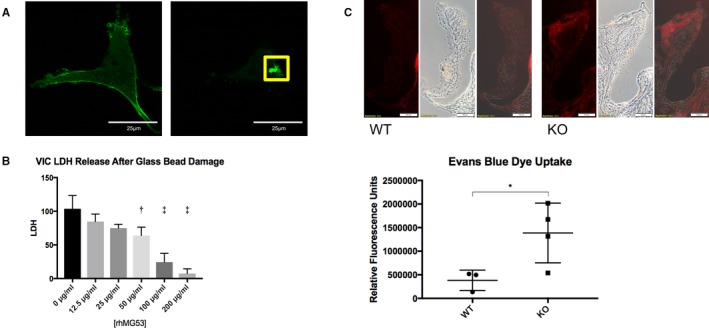
MG53 protects against valve interstitial cell (VIC) membrane injury. A, In pig VICs transfected with GFP‐MG53 (left), microelectrode needle penetration of the VIC membrane causes translocation of GFP‐MG53 to the membrane injury site (right, boxed; n=10 cells). B, VICs were exposed to physical damage via vigorous shaking with glass micro‐beads. rhMG53 reduces LDH release—an index of cell membrane injury—in a dose‐dependent manner (n=3 biological replicates). An ordinary, 1‐way ANOVA test with Dunnett multiple comparison testing (α=0.05) was used to obtain P values. The ANOVA P‐value was P<0.0001. Multiple comparison testing compared each rhMG53‐treatment group to the control, non‐treated group to obtain P values <0.05 for cells treated with 50, 100, and 200 μg/mL rhMG53. C, EBD is impermeable to cells with intact membranes. Aortic valve leaflets of 24‐month‐old Mg53−/− mice (n=4) show significantly increased EBD uptake compared with those of littermate wild‐type mice (n=3). Representative images are shown on top, and relative fluorescence quantification is shown below. A 2‐tailed Student t test was used to obtain the P‐value. These results suggest increased endothelial permeability in 24‐month‐old Mg53−/− compared with wild‐type controls. Further, given that EBD will not penetrate cells with intact plasma membranes, these results also suggest a greater in vivo burden of cell membrane injury in aortic valves from Mg53−/− mice. Imaging shows increased damage particularly in endothelial and subendothelial cell layers. KO indicates knockout (Mg53−/−); LDH, lactate dehydrogenase; rh, recombinant human; WT, wild‐type. *P<0.05, **P<0.01, ***P<0.001.
LDH release was additionally used as a marker of cell membrane injury.14, 15, 16, 18 Glass bead‐induced damage to the cell membrane leds to exposure of phosphatidylserine (PS) to the extracellular space. The rhMG53 protein can bind to the exposed PS, facilitating membrane patch formation. We have shown previously that incubation of cells with rhMG53 could reduce LDH release in multiple cell lines including HEK, C2C12, alveolar lung cells, keratinocytes, and proximal tubular epithelial cells.14, 15, 18, 39, 40 Here we found that rhMG53 applied to the culture medium protected VICs from glass bead‐induced membrane damage in a dose‐dependent manner (Figure 3B).
Recently, Anstine et al reported increased EBD accumulation in aortic valves from aged versus young mice.21 While Anstine et al studied EBD accumulation as a marker of valve endothelial permeability, EBD is a cell‐impermeable dye and will not enter cells with intact plasma membranes. We thus performed EBD uptake experiments in 24‐month‐old wild‐type versus Mg53−/− mice and found that Mg53−/− mice had more EBD uptake per aortic valve leaflet, indicating increased in vivo membrane damage to aged Mg53−/− valvular cells (Figure 3C). We also conducted experiments in younger Mg53−/− and wild‐type littermate mice and found no significant differences in EBD uptakes between the 2 groups (Figure S4).
We previously demonstrated that rhMG53 protein delivered via intravenous infusion could protect against ischemia‐reperfusion‐induced myocardial infarction in pigs.17 To test if systemic administration of rhMG53 could penetrate aortic valves, we performed intravenous injection of rhMG53 to an aged Mg53−/− mouse and found that at 4 hours after intravenous infusion, rhMG53 could be detected in the aortic valve but not in the myocardium (Figure S5). We have shown that the exogenous rhMG53 protein could recognize and accumulate in injured tissues but not in healthy tissues.14, 17 Thus, this finding provides additional evidence that Mg53−/− mice had aortic valve dysfunction despite preserved ventricular contractile function.
Together, these results demonstrate that MG53 maintains its ability to promote cell membrane repair in VICs and that membrane injury may represent a mechanism contributing to the development of age‐related VHD. Without MG53, the valvular cells show an increased burden of membrane injury. In time, accumulated cell injury may lead to remodeling of valve tissue and hemodynamic and histologic signs of valve disease as observed in Mg53−/− mice (Figure 2). The ability of MG53 to modulate VIC injury responses lays the foundation for our next set of studies to investigate MG53‐mediated protection against signaling processes that contribute to the development of VHD. Since systemically‐administered rhMG53 could access the injured valves, this finding supports the potential for the usage of rhMG53 protein to treat valvular disease.
rhMG53‐Mediated Suppression of TGF‐ß Signaling in Aortic VICs
One signature of VHD is the increased fibrotic remodeling in valve leaflets. Immunohistochemistry demonstrates significantly elevated fibronectin staining in aortic valves of aged Mg53−/− mice compared with age‐matched wild‐type controls (Figure 4A).41, 42, 43, 44, 45
Figure 4.
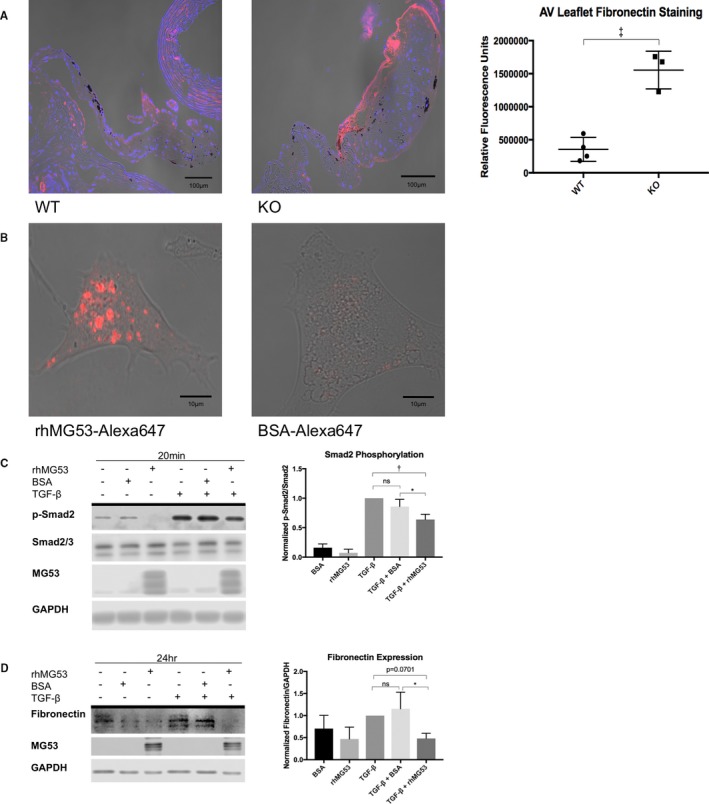
rhMG53 suppresses TGF‐β‐induced fibrocalcific signaling in valve interstitial cells. A, Immunohistochemistry of aged wild‐type (left) and Mg53−/− (right) mouse hearts show increased fibronectin staining in Mg53−/− aortic valve leaflets (n=3) compared with those from wild‐type control mice (n=4). A 2‐tailed Student t test was used to obtain the P value. In different tissues, fibronectin mediates both fibrosis and calcification such as that observed in histologic analysis of 24‐month‐old Mg53−/− mouse valves (Figure 2).41, 42, 43, 44 Fibronectin is a downstream product of the TGF‐ß pathway and has been associated with VIC injury, facilitating continued signaling events and VIC activation.7, 8, 45, 48, 49, 50, 57 Given the relationship between TGF‐ß and fibronectin and with increased fibronectin staining in Mg53−/− aortic valves, we aimed to target our in vitro studies through our histopathology findings. Specifically, we tested if rhMG53 treatment could modulate VIC TGF‐ß signaling and downstream fibronectin expression. B, To guide our mechanistic insights, we first wanted to determine if rhMG53 could enter pig aortic VICs. Here, we conjugated rhMG53 and BSA as a control to the Alexa‐647 fluorophore to allow visualization of rhMG53‐specific cell entry via live cell imaging (n=10 cells). Importantly, rhMG53 VIC entry was observed after treating cells for 30 minutes, thus our use of this pretreatment time in our in vitro signaling studies. C, Short time points to study cell signaling events in pig VICs showed that rhMG53 reduces TGF‐β‐induced Smad2 phosphorylation (n=3). D, Longer time points to study differences in protein expression in pig VICs demonstrated that rhMG53 reduces TGF‐β‐induced fibronectin expression (n=4). Ordinary, 1‐way ANOVA tests with Tukey multiple comparison testing (α=0.05) were used to obtain P‐values. AV indicates aortic valve; BSA, bovine serum albumin; KO, knockout (Mg53−/−); rh, recombinant human; TGF, transforming growth factor; WT, wild type. *P<0.05, **P<0.01, ***P=0.001.
Recent studies show that MG53 exerts protection against fibrosis, protecting tissues and organs beyond acute cell membrane repair.17, 18, 46 Given that fibrosis represents a critical process in the pathogenesis of VHD, we tested the ability of rhMG53 to modulate TGF‐ß signaling, a central signaling pathway associated with valvular injury response and fibrocalcific remodeling.36, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57
Canonical TGF‐ß signaling occurs via ligand‐induced TGF‐ß receptor II (TßRII) dimerization with and phosphorylation of TGF‐ß receptor I (TßRI) followed by TGF‐ß receptor I phosphorylation of Smad2. Smad2 then heterodimerizes with Smad3 and translocates to the nucleus where it acts as a transcription factor to increase expression of fibrotic genes.58, 59
For live cell imaging, we conjugated rhMG53 and BSA with Alexa647, a fluorescent probe with high quantum yield. Thirty‐minute incubation of VICs with rhMG53‐Alexa647 resulted in protein uptake into the cells, not observed with BSA‐Alexa647 (Figure 4B). Based on this time point, VICs were pretreated with rhMG53 for 30 minutes, followed by a 20‐minute treatment with TGF‐ß (1 ng/mL). Western blot confirmed the uptake of rhMG53 by the VICs (Figure 4C and 4D). Figure S6 demonstrated the expression of low level of native MG53 protein in VICs derived from the pigs. Figure 4C shows that TGF‐ß causes Smad2 activation, indicated by elevated Smad2 phosphorylation. Cells pretreated with rhMG53 showed significantly less TGF‐ß‐induced Smad2 phosphorylation as compared with BSA‐pretreated control cells. Moreover, in downstream studies, prolonged (24‐hour) treatment of VICs with TGF‐ß (1 ng/mL) increased fibronectin expression; this change was also mitigated by rhMG53 (Figure 4D).
Discussion
In this study, we demonstrate that MG53, an essential cell membrane repair protein, can facilitate repair of injury to VICs and modulate the fibrocalcific processes that contribute to the development of VHD. We show that MG53 is expressed in pig and human aortic valves. Aortic valves of aged Mg53−/− knockout mice had increased tissue injury and displayed signs of aortic valve dysfunction and disease. We found that MG53 protects aortic valve interstitial cells from both membrane injury and fibrocalcific remodeling. Given the preserved contractile function of Mg53−/− mouse hearts under physiological conditions, the observation that aged Mg53−/− mice display hemodynamic and histological signs of aortic valve disease highlights a critical in vivo mechanism of MG53‐mediated valvular protection (Figure 5).
Figure 5.
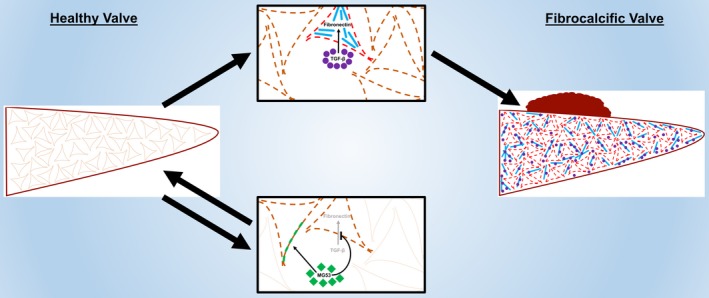
A suggested model summarizes our results. Accumulated membrane injury to VICs (dashed lines) is associated with increased TGF‐β‐induced fibronectin expression, allowing for pooled TGF‐β signaling, constitutive VIC activation (red lines), and downstream fibrocalcific changes. These remodeling processes result in VHD. MG53 protects VICs from both membrane injury (green patches) and fibrocalcific TGF‐β signaling to prevent such remodeling and maintain valvular homeostasis. Future studies will specify mechanisms of MG53‐mediated protection and will assess the translational potential for MG53‐based protein therapies or MG53‐coated cardiovascular materials in preventing or treating VHD.
Disruptions in cell membrane repair in response to injury contribute to a vast number of diseases in many different tissues.60, 61, 62, 63 However, valvular cell membrane injury and repair remain an understudied aspect of heart valve biology. Under physiologic conditions, heart valves experience tremendous mechanical stress from hemodynamic forces, and maladaptive valve injury responses are directly associated with VHD. VICs represent ≈80% of the cells in the valve and maintenance of their integrity is critical for the valve function. After damage to the valve endothelial cell layer, VICs are prone to continued injury from the maladaptive hemodynamic environment. In these processes, quiescent VICs become “activated”, experiencing a fibroblast‐to‐myofibroblast‐like transition, and later “osteoblastic” in nature, cumulatively resulting in valvular fibrocalcific changes and the development of VHD.
Many studies have characterized the vital roles for VICs in the valve injury response, with excessive TGF‐ß signaling activation as a result of VIC injury.3, 4, 5, 6, 7, 8, 9 The injury response and TGF‐β signaling contribute to the vicious cycle of valvular fibrotic remodeling and disease. We provide evidence to show that MG53‐mediated protection against VIC injury represents a promising means to maintain valve cell homeostasis and prevent progression of valvular pathology. Beyond membrane repair, our studies show that rhMG53 can mitigate TGF‐β‐induced fibrotic response in VICs. Through Smad2 phosphorylation, TGF‐ß drives pathologic remodeling of quiescent, fibroblast‐like VICs to an activated, myofibroblast‐like phenotype. Fibronectin is recognized as a potent downstream mediator of TGF‐ß activity, contributing to VIC activation and hence progression of valve disease.7, 8, 45, 48, 49, 57 We showed that rhMG53 interferes with TGF‐β signaling to reduce the Smad2 phosphorylation and fibronectin expression, mediators of fibrocalcific activation in VICs. Clearly, the anti‐fibrotic function of MG53 requires the entry of MG53 into the VICs; whether or not it involves the intrinsic E3 ubiquitin ligase activity will require further studies in the future.
The greatest limitation in the valve biology field remains the lack of suitable mouse models of acquired valve disease. Most mouse valve disease models involve single‐ or double‐knockouts of genes critical to cholesterol handling, still requiring a long‐term high‐fat diet or aging thereafter.35, 37, 64 Studies from Weiss et al demonstrated that aging and cholesterol handling may play a significant role in the pathogenesis of aortic valve disease.37 Despite these insights, clinical trials utilizing cholesterol‐lowering medications have yielded insignificant benefits, highlighting the need for a therapy more specifically targeting valvular cells.65 We found that the Mg53−/− mice displayed an age‐dependent phenotype of VHD, without baseline changes of ventricular contractile function. Thus, the Mg53−/− mice could potentially provide a useful model for VHD research. We previously demonstrated a cardioprotective function of rhMG53 in a porcine model of ischemia‐reperfusion‐induced myocardial infarction.17 Here, we provide important findings to show that systemically‐administered rhMG53 could penetrate the injured valve tissues, thus supporting the therapeutic potential for rhMG53 protein to treat valvular disease.
Since our discovery of MG53 in 2009,11 notable progress has been made in advancing the mechanistic action of this protein in the biology of tissue repair as well as in regulation of metabolic syndromes. MG53 belongs to the TRIM family of proteins that contain the conserved RING motif with E3‐ligase activity. Several studies have shown that insulin receptor substrate 1 66, 67, 68 and focal adhesion kinase69 are E3‐ligase substrates for MG53‐mediated ubiquitination and degradation. Song et al67 reported MG53 protein was increased in animal models of diabetes mellitus and proposed that MG53‐mediated downregulation of insulin receptor substrate 1 served as a causative factor for the development the diabetic phenotype. Yi et al68 and others70, 71, 72 observed normal MG53 expression in human diabetic muscle and murine models of diabetes mellitus. Early publications with mice lacking insulin receptor substrate 1 revealed no clear phenotype of type 2 diabetes mellitus.73, 74 Thus, MG53‐mediated down‐regulation of insulin receptor substrate 1 cannot be a causative factor for the development of type 2 diabetes mellitus. While this remains a controversial field of research, future studies need to be performed to determine the role of MG53 in modulating the metabolic aspect of VHDs.
Overall, this study contributes to our understandings of valve biology—an understudied field of cardiovascular biology despite valve disease representing a fast‐growing clinical burden in the United States and throughout the world. Our work demonstrated a dual function for MG53 in maintaining the integrity of the VICs and in controlling TGF‐β‐mediated fibrotic remodeling associated with VHD. Further understanding of the signaling components associated with MG53/TGF‐β crosstalk in modulation of the fibrocalcific response in the diseased valves could advance our basic understanding of cell membrane repair in valve physiology and disease, as well as facilitating the translation of therapeutic application of rhMG53 to alleviate and treat VHD or improve its surgical management. Such therapies may include an oral or injectable rhMG53‐based formulation that targets valve cells, rhMG53‐coated sutures or bioprosthetic valves that can be used during valve operations, or a topical rhMG53‐based formulation that can be applied during valve repairs.
Author Contributions
Adesanya, Russell, Park, Zhou performed experiments, and Sermersheim, Gumpper, Koenig, Lincoln, Tan, and Zhu assisted with experiments and analyses. Adesanya and Ma designed experiments and analyzed data; Whitson and Janssen developed protocols to procure and store human valve tissue. Adesanya and Ma wrote the manuscript, and all authors participated in revision of the manuscript.
Sources of Funding
This work was supported by National Institutes of Health grants awarded to Ma (AR061385, AR070752, and DK106394, AG056919) and Zhu (AR067766 and HL124122) and a National Institutes of Health training fellowship awarded to Adesanya (TL1TR001069); an American Medical Association Foundation Seed Grant to Adesanya; and a College of Medicine Physiology Training Program Fellowship awarded to Adesanya.
Disclosures
Ma is a founder of TRIM‐edicine, Inc, a biotechnology company developing MG53 for therapeutic applications in regenerative medicine. Intellectual property rights of MG53 are patented by Robert Wood Johnson Medical School, Rutgers University. The remaining authors have no disclosures to report.
Supporting information
Table S1. De‐Identified Patient Details are Provided for the Human Tissue Western Blot
Figure S1. Intensity of the trichrome staining in mouse aortic valve.
Figure S2. Coronal sections of mitral valve leaflets are shown opening downwards, left ventricle below and left atrium above.
Figure S3. Wild type and Mg53−/− mice show similar cardiac function.
Figure S4. Evans blue dye (EBD) uptake in a limited number of younger wild type and Mg53−/− littermate mice.
Figure S5. Systemically administered rhMG53 protein can penetrate murine aortic valve tissue.
Figure S6. Western blot of MG53 expression in the pig aortic valvular interstitial cells (AVICs).
Videos S1 and S2. Representative color Doppler videos from 24‐month‐old wild‐type (left) and Mg53−/− (right) mice. Best viewed with Windows Media Player.
Video S3. GFP‐MG53 translocates to VIC membrane injury site after microelectrode needle penetration. Best viewed with Windows Media Player.
Acknowledgments
We thank Matthew Joseph, Dr Dondrae Coble, Lori Mattox, and The Ohio State University Laboratory Animal Resources for large animal assistance; The Ohio State University Small Animal Imaging Core for echocardiography assistance; and Julie Rectenwald, Jania Johnson, Kaitlyn Thatcher, Denise Peterson, the Pathology Core Facility, and the Comparative Pathology and Mouse Phenotyping Core (supported by National Institutes of Health grant P30CA016058) for histology assistance. We thank Lifeline of Ohio for helping with procurement of the human donor hearts.
(J Am Heart Assoc. 2019;8:e009960 DOI: 10.1161/JAHA.118.009960.)
References
- 1. Thanassoulis G. Lipoprotein (a) in calcific aortic valve disease: from genomics to novel drug target for aortic stenosis. J Lipid Res. 2016;57:917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rajamannan NM, Gersh B, Bonow RO. Calcific aortic stenosis: from bench to the bedside—emerging clinical and cellular concepts. Heart. 2003;89:801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lester WM, Gotlieb AI. In vitro repair of the wounded porcine mitral valve. Circ Res. 1988;62:833–845. [DOI] [PubMed] [Google Scholar]
- 4. Lester WM, Damji AA, Tanaka M, Gedeon I. Bovine mitral valve organ culture: role of interstitial cells in repair of valvular injury. J Mol Cell Cardiol. 1992;24:43–53. [DOI] [PubMed] [Google Scholar]
- 5. Lester WM, Damji AA, Gedeon I, Tanaka M. Interstitial cells from the atrial and ventricular sides of the bovine mitral valve respond differently to denuding endocardial injury. In Vitro Cell Dev Biol. 1993;29A:41–50. [DOI] [PubMed] [Google Scholar]
- 6. Tamura K, Jones M, Yamada I, Ferrans VJ. Wound healing in the mitral valve. J Heart Valve Dis. 2000;9:53–63. [PubMed] [Google Scholar]
- 7. Fayet C, Bendeck MP, Gotlieb AI. Cardiac valve interstitial cells secrete fibronectin and form fibrillar adhesions in response to injury. Cardiovasc Pathol. 2007;16:203–211. [DOI] [PubMed] [Google Scholar]
- 8. Liu AC, Gotlieb AI. Transforming growth factor‐beta regulates in vitro heart valve repair by activated valve interstitial cells. Am J Pathol. 2008;173:1275–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Han L, Gotlieb AI. Fibroblast growth factor‐2 promotes in vitro mitral valve interstitial cell repair through transforming growth factor‐beta/Smad signaling. Am J Pathol. 2011;178:119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dweck MR, Boon NA, Newby DE. Calcific aortic stenosis: a disease of the valve and the myocardium. J Am Coll Cardiol. 2012;60:1854–1863. [DOI] [PubMed] [Google Scholar]
- 11. Cai C, Masumiya H, Weisleder N, Matsuda N, Nishi M, Hwang M, Ko JK, Lin P, Thornton A, Zhao X, Pan Z, Komazaki S, Brotto M, Takeshima H, Ma J. MG53 nucleates assembly of cell membrane repair machinery. Nat Cell Biol. 2009;11:56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cao CM, Zhang Y, Weisleder N, Ferrante C, Wang X, Lv F, Zhang Y, Song R, Hwang M, Jin L, Guo J, Peng W, Li G, Nishi M, Takeshima H, Ma J, Xiao RP. MG53 constitutes a primary determinant of cardiac ischemic preconditioning. Circulation. 2010;121:2565–2574. [DOI] [PubMed] [Google Scholar]
- 13. Wang X, Xie W, Zhang Y, Lin P, Han L, Han P, Wang Y, Chen Z, Ji G, Zheng M, Weisleder N, Xiao RP, Takeshima H, Ma J, Cheng H. Cardioprotection of ischemia/reperfusion injury by cholesterol‐dependent MG53‐mediated membrane repair. Circ Res. 2010;107:76–83. [DOI] [PubMed] [Google Scholar]
- 14. Weisleder N, Takizawa N, Lin P, Wang X, Cao C, Zhang Y, Tan T, Ferrante C, Zhu H, Chen PJ, Yan R, Sterling M, Zhao X, Hwang M, Takeshima M, Cai C, Cheng H, Takeshima H, Xiao RP, Ma J. Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci Transl Med. 2012;4:139ra185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jia Y, Chen K, Lin P, Lieber G, Nishi M, Yan R, Wang Z, Yao Y, Li Y, Whitson BA, Duann P, Li H, Zhou X, Zhu H, Takeshima H, Hunter JC, McLeod RL, Weisleder N, Zeng C, Ma J. Treatment of acute lung injury by targeting MG53‐mediated cell membrane repair. Nat Commun. 2014;5:4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li X, Xiao Y, Cui Y, Tan T, Narasimhulu CA, Hao H, Liu L, Zhang J, He G, Verfaillie CM, Lei M, Parthasarathy S, Ma J, Zhu H, Liu Z. Cell membrane damage is involved in the impaired survival of bone marrow stem cells by oxidized low‐density lipoprotein. J Cell Mol Med. 2014;18:2445–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu J, Zhu H, Zheng Y, Xu Z, Li L, Tan T, Park KH, Hou J, Zhang C, Li D, Li R, Liu Z, Weisleder N, Zhu D, Lin P, Ma J. Cardioprotection of recombinant human MG53 protein in a porcine model of ischemia and reperfusion injury. J Mol Cell Cardiol. 2015;80:10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Duann P, Li H, Lin P, Tan T, Wang Z, Chen K, Zhou X, Gumpper K, Zhu H, Ludwig T, Mohler PJ, Rovin B, Abraham WT, Zeng C, Ma J. MG53‐mediated cell membrane repair protects against acute kidney injury. Sci Transl Med. 2015;7:279ra236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yao Y, Zhang B, Zhu H, Li H, Han Y, Chen K, Wang Z, Zeng J, Liu Y, Wang X, Li Y, He D, Lin P, Zhou X, Park KH, Bian Z, Chen Z, Gong N, Tan T, Zhou J, Zhang M, Ma J, Zeng C. MG53 permeates through blood‐brain barrier to protect ischemic brain injury. Oncotarget. 2016;7:22474–22485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gould RA, Butcher JT. Isolation of valvular endothelial cells. J Vis Exp. 2010;46:2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Anstine LJ, Bobba C, Ghadiali S, Lincoln J. Growth and maturation of heart valves leads to changes in endothelial cell distribution, impaired function, decreased metabolism and reduced cell proliferation. J Mol Cell Cardiol. 2016;100:72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lemckert FA, Bournazos A, Eckert DM, Kenzler M, Hawkes JM, Butler TL, Ceely B, North KN, Winlaw DS, Egan JR, Cooper ST. Lack of MG53 in human heart precludes utility as a biomarker of myocardial injury or endogenous cardioprotective factor. Cardiovasc Res. 2016;110:178–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Touma M, Kang X, Zhao Y, Cass AA, Gao F, Biniwale R, Coppola G, Xiao X, Reemtsen B, Wang Y. Decoding the long noncoding RNA during cardiac maturation: a roadmap for functional discovery. Circ Cardiovasc Genet. 2016;9:395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guo J, Jia F, Jiang Y, Li Q, Yang Y, Xiao M, Xiao H. Potential role of MG53 in the regulation of transforming‐growth‐factor‐beta1‐induced atrial fibrosis and vulnerability to atrial fibrillation. Exp Cell Res. 2018;362:436–443. [DOI] [PubMed] [Google Scholar]
- 25. Nakao K, Mao P, Ghidoni J, Angrist A. An electron microscopic study of the aging process in the rat heart valve. J Gerontol. 1966;21:72–85. [DOI] [PubMed] [Google Scholar]
- 26. Kim KM, Valigorsky JM, Mergner WJ, Jones RT, Pendergrass RF, Trump BF. Aging changes in the human aortic valve in relation to dystrophic calcification. Hum Pathol. 1976;7:47–60. [DOI] [PubMed] [Google Scholar]
- 27. Kume T, Kawamoto T, Okura H, Watanabe N, Toyota E, Neishi Y, Okahashi N, Yamada R, Yoshida K. Rapid progression of mild to moderate aortic stenosis in patients older than 80 years. J Am Soc Echocardiogr. 2007;20:1243–1246. [DOI] [PubMed] [Google Scholar]
- 28. Bhatia N, Basra SS, Skolnick AH, Wenger NK. Aortic valve disease in the older adult. J Geriatr Cardiol. 2016;13:941–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baumgartner H, Hung J, Bermejo J, Chambers JB, Evangelista A, Griffin BP, Iung B, Otto CM, Pellikka PA, Quinones M; EAE/ASE . Echocardiographic assessment of valve stenosis: EAE/ASE recommendations for clinical practice. Eur J Echocardiogr. 2009;10:1–25. [DOI] [PubMed] [Google Scholar]
- 30. Nus M, MacGrogan D, Martinez‐Poveda B, Benito Y, Casanova JC, Fernandez‐Aviles F, Bermejo J, de la Pompa JL. Diet‐induced aortic valve disease in mice haploinsufficient for the Notch pathway effector RBPJK/CSL. Arterioscler Thromb Vasc Biol. 2011;31:1580–1588. [DOI] [PubMed] [Google Scholar]
- 31. Honda S, Miyamoto T, Watanabe T, Narumi T, Kadowaki S, Honda Y, Otaki Y, Hasegawa H, Netsu S, Funayama A, Ishino M, Nishiyama S, Takahashi H, Arimoto T, Shishido T, Miyashita T, Kubota I. A novel mouse model of aortic valve stenosis induced by direct wire injury. Arterioscler Thromb Vasc Biol. 2014;34:270–278. [DOI] [PubMed] [Google Scholar]
- 32. Huk DJ, Hammond HL, Kegechika H, Lincoln J. Increased dietary intake of vitamin A promotes aortic valve calcification in vivo. Arterioscler Thromb Vasc Biol. 2013;33:285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miller JD, Weiss RM, Heistad DD. Calcific aortic valve stenosis: methods, models, and mechanisms. Circ Res. 2011;108:1392–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Casaclang‐Verzosa G, Enriquez‐Sarano M, Villaraga HR, Miller JD. Echocardiographic approaches and protocols for comprehensive phenotypic characterization of valvular heart disease in mice. J Vis Exp. 2017;120:54110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Miller JD, Weiss RM, Serrano KM, Brooks RM II, Berry CJ, Zimmerman K, Young SG, Heistad DD. Lowering plasma cholesterol levels halts progression of aortic valve disease in mice. Circulation. 2009;119:2693–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miller JD, Weiss RM, Serrano KM, Castaneda LE, Brooks RM, Zimmerman K, Heistad DD. Evidence for active regulation of pro‐osteogenic signaling in advanced aortic valve disease. Arterioscler Thromb Vasc Biol. 2010;30:2482–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weiss RM, Ohashi M, Miller JD, Young SG, Heistad DD. Calcific aortic valve stenosis in old hypercholesterolemic mice. Circulation. 2006;114:2065–2069. [DOI] [PubMed] [Google Scholar]
- 38. Zhang C, Chen B, Wang Y, Guo A, Tang Y, Khataei T, Shi Y, Kutschke WJ, Zimmerman K, Weiss RM, Liu J, Benson CJ, Hong J, Ma J, Song LS. MG53 is dispensable for T‐tubule maturation but critical for maintaining T‐tubule integrity following cardiac stress. J Mol Cell Cardiol. 2017;112:123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhu H, Lin P, De G, Choi KH, Takeshima H, Weisleder N, Ma J. Polymerase transcriptase release factor (PTRF) anchors MG53 protein to cell injury site for initiation of membrane repair. J Biol Chem. 2011;286:12820–12824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lin P, Zhu H, Cai C, Wang X, Cao C, Xiao R, Pan Z, Weisleder N, Takeshima H, Ma J. Nonmuscle myosin IIA facilitates vesicle trafficking for MG53‐mediated cell membrane repair. FASEB J. 2012;26:1875–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Winnard RG, Gerstenfeld LC, Toma CD, Franceschi RT. Fibronectin gene expression, synthesis and accumulation during in vitro differentiation of chicken osteoblasts. J Bone Miner Res. 1995;10:1969–1977. [DOI] [PubMed] [Google Scholar]
- 42. Yoshiba K, Yoshiba N, Nakamura H, Iwaku M, Ozawa H. Immunolocalization of fibronectin during reparative dentinogenesis in human teeth after pulp capping with calcium hydroxide. J Dent Res. 1996;75:1590–1597. [DOI] [PubMed] [Google Scholar]
- 43. Watson KE, Parhami F, Shin V, Demer LL. Fibronectin and collagen I matrixes promote calcification of vascular cells in vitro, whereas collagen IV matrix is inhibitory. Arterioscler Thromb Vasc Biol. 1998;18:1964–1971. [DOI] [PubMed] [Google Scholar]
- 44. Altrock E, Sens C, Wuerfel C, Vasel M, Kawelke N, Dooley S, Sottile J, Nakchbandi IA. Inhibition of fibronectin deposition improves experimental liver fibrosis. J Hepatol. 2015;62:625–633. [DOI] [PubMed] [Google Scholar]
- 45. Messier RH Jr, Bass BL, Aly HM, Jones JL, Domkowski PW, Wallace RB, Hopkins RA. Dual structural and functional phenotypes of the porcine aortic valve interstitial population: characteristics of the leaflet myofibroblast. J Surg Res. 1994;57:1–21. [DOI] [PubMed] [Google Scholar]
- 46. He B, Tang RH, Weisleder N, Xiao B, Yuan Z, Cai C, Zhu H, Lin P, Qiao C, Li J, Mayer C, Li J, Ma J, Xiao X. Enhancing muscle membrane repair by gene delivery of MG53 ameliorates muscular dystrophy and heart failure in delta‐Sarcoglycan‐deficient hamsters. Mol Ther. 2012;20:727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jian B, Narula N, Li QY, Mohler ER III, Levy RJ. Progression of aortic valve stenosis: TGF‐beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003;75:457–465; discussion 465‐456. [DOI] [PubMed] [Google Scholar]
- 48. Walker GA, Masters KS, Shah DN, Anseth KS, Leinwand LA. Valvular myofibroblast activation by transforming growth factor‐beta: implications for pathological extracellular matrix remodeling in heart valve disease. Circ Res. 2004;95:253–260. [DOI] [PubMed] [Google Scholar]
- 49. Cushing MC, Liao JT, Anseth KS. Activation of valvular interstitial cells is mediated by transforming growth factor‐beta1 interactions with matrix molecules. Matrix Biol. 2005;24:428–437. [DOI] [PubMed] [Google Scholar]
- 50. Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol. 2007;171:1407–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kim L, Kim DK, Yang WI, Shin DH, Jung IM, Park HK, Chang BC. Overexpression of transforming growth factor‐beta 1 in the valvular fibrosis of chronic rheumatic heart disease. J Korean Med Sci. 2008;23:41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Doetschman T, Barnett JV, Runyan RB, Camenisch TD, Heimark RL, Granzier HL, Conway SJ, Azhar M. Transforming growth factor beta signaling in adult cardiovascular diseases and repair. Cell Tissue Res. 2012;347:203–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yip CY, Chen JH, Zhao R, Simmons CA. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arterioscler Thromb Vasc Biol. 2009;29:936–942. [DOI] [PubMed] [Google Scholar]
- 54. Chen JH, Chen WL, Sider KL, Yip CY, Simmons CA. Beta‐catenin mediates mechanically regulated, transforming growth factor‐beta1‐induced myofibroblast differentiation of aortic valve interstitial cells. Arterioscler Thromb Vasc Biol. 2011;31:590–597. [DOI] [PubMed] [Google Scholar]
- 55. Hagler MA, Hadley TM, Zhang H, Mehra K, Roos CM, Schaff HV, Suri RM, Miller JD . TGF‐beta signalling and reactive oxygen species drive fibrosis and matrix remodelling in myxomatous mitral valves. Cardiovasc Res. 2013;99:175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sritharen Y, Enriquez‐Sarano M, Schaff HV, Casaclang‐Verzosa G, Miller JD. Pathophysiology of aortic valve stenosis: is it both fibrocalcific and sex specific? Physiology (Bethesda). 2017;32:182–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ignotz RA, Massague J. Transforming growth factor‐beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 1986;261:4337–4345. [PubMed] [Google Scholar]
- 58. Meng XM, Nikolic‐Paterson DJ, Lan HY. TGF‐beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325–338. [DOI] [PubMed] [Google Scholar]
- 59. Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. McNeil PL, Steinhardt RA. Plasma membrane disruption: repair, prevention, adaptation. Annu Rev Cell Dev Biol. 2003;19:697–731. [DOI] [PubMed] [Google Scholar]
- 61. McNeil PL, Kirchhausen T. An emergency response team for membrane repair. Nat Rev Mol Cell Biol. 2005;6:499–505. [DOI] [PubMed] [Google Scholar]
- 62. Andrews NW. Membrane repair and immunological danger. EMBO Rep. 2005;6:826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wenzel K, Geier C, Qadri F, Hubner N, Schulz H, Erdmann B, Gross V, Bauer D, Dechend R, Dietz R, Osterziel KJ, Spuler S, Ozcelik C. Dysfunction of dysferlin‐deficient hearts. J Mol Med (Berl). 2007;85:1203–1214. [DOI] [PubMed] [Google Scholar]
- 64. Sider KL, Blaser MC, Simmons CA. Animal models of calcific aortic valve disease. Int J Inflam. 2011;2011:364310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Borer JS, Sharma A. Drug therapy for heart valve diseases. Circulation. 2015;132:1038–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lee CS, Yi JS, Jung SY, Kim BW, Lee NR, Choo HJ, Jang SY, Han J, Chi SG, Park M, Lee JH, Ko YG. TRIM72 negatively regulates myogenesis via targeting insulin receptor substrate‐1. Cell Death Differ. 2010;17:1254–1265. [DOI] [PubMed] [Google Scholar]
- 67. Song R, Peng W, Zhang Y, Lv F, Wu HK, Guo J, Cao Y, Pi Y, Zhang X, Jin L, Zhang M, Jiang P, Liu F, Meng S, Cao CM, Xiao RP. Central role of E3 ubiquitin ligase MG53 in insulin resistance and metabolic disorders. Nature. 2013;494:375–379. [DOI] [PubMed] [Google Scholar]
- 68. Yi JS, Park JS, Ham YM, Nguyen N, Lee NR, Hong J, Kim BW, Lee H, Lee CS, Jeong BC, Song HK, Cho H, Kim YK, Lee JS, Park KS, Shin H, Choi I, Lee SH, Park WJ, Park SY, Choi CS, Lin P, Karunasiri M, Tan T, Duann P, Zhu H, Ma J, Ko YG. MG53‐induced IRS‐1 ubiquitination negatively regulates skeletal myogenesis and insulin signalling. Nat Commun. 2013;4:2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nguyen N, Yi JS, Park H, Lee JS, Ko YG. Mitsugumin 53 (MG53) ligase ubiquitinates focal adhesion kinase during skeletal myogenesis. J Biol Chem. 2014;289:3209–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ma LL, Zhang FJ, Kong FJ, Qian LB, Ma H, Wang JA, Yan M. Hypertrophied myocardium is refractory to sevoflurane‐induced protection with alteration of reperfusion injury salvage kinase/glycogen synthase kinase 3beta signals. Shock. 2013;40:217–221. [DOI] [PubMed] [Google Scholar]
- 71. Xu Y, Ma LL, Zhou C, Zhang FJ, Kong FJ, Wang WN, Qian LB, Wang CC, Liu XB, Yan M, Wang JA. Hypercholesterolemic myocardium is vulnerable to ischemia‐reperfusion injury and refractory to sevoflurane‐induced protection. PLoS One. 2013;8:e76652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yuan H, Niu Y, Liu X, Yang F, Niu W, Fu L. Proteomic analysis of skeletal muscle in insulin‐resistant mice: response to 6‐week aerobic exercise. PLoS One. 2013;8:e53887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Terauchi Y, Iwamoto K, Tamemoto H, Komeda K, Ishii C, Kanazawa Y, Asanuma N, Aizawa T, Akanuma Y, Yasuda K, Kodama T, Tobe K, Yazaki Y, Kadowaki T. Development of non‐insulin‐dependent diabetes mellitus in the double knockout mice with disruption of insulin receptor substrate‐1 and beta cell glucokinase genes. Genetic reconstitution of diabetes as a polygenic disease. J Clin Invest. 1997;99:861–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tamemoto H, Kadowaki T, Tobe K, Yagi T, Sakura H, Hayakawa T, Terauchi Y, Ueki K, Kaburagi Y, Satoh S, Sekihara H, Yoshioka S, Horikoshi H, Furuta Y, Ikawa Y, Kasuga M, Yazaki Y, Aizawa S. Insulin resistance and growth retardation in mice lacking insulin receptor substrate‐1. Nature. 1994;372:182–186. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. De‐Identified Patient Details are Provided for the Human Tissue Western Blot
Figure S1. Intensity of the trichrome staining in mouse aortic valve.
Figure S2. Coronal sections of mitral valve leaflets are shown opening downwards, left ventricle below and left atrium above.
Figure S3. Wild type and Mg53−/− mice show similar cardiac function.
Figure S4. Evans blue dye (EBD) uptake in a limited number of younger wild type and Mg53−/− littermate mice.
Figure S5. Systemically administered rhMG53 protein can penetrate murine aortic valve tissue.
Figure S6. Western blot of MG53 expression in the pig aortic valvular interstitial cells (AVICs).
Videos S1 and S2. Representative color Doppler videos from 24‐month‐old wild‐type (left) and Mg53−/− (right) mice. Best viewed with Windows Media Player.
Video S3. GFP‐MG53 translocates to VIC membrane injury site after microelectrode needle penetration. Best viewed with Windows Media Player.