

Abstract
Background
Endothelial NO synthase plays a central role in regulating vasodilation and blood pressure. Intracellular Ca2+ mobilization is a critical modulator of endothelial NO synthase function, and increased cytosolic Ca2+ concentration in endothelial cells is able to induce endothelial NO synthase phosphorylation. Ca2+ release mediated by 3 subtypes of inositol 1,4,5‐trisphosphate receptors (IP 3Rs) from the endoplasmic reticulum and subsequent Ca2+ entry after endoplasmic reticulum Ca2+ store depletion has been proposed to be the major pathway to mobilize Ca2+ in endothelial cells. However, the physiological role of IP 3Rs in regulating blood pressure remains largely unclear.
Methods and Results
To investigate the role of endothelial IP 3Rs in blood pressure regulation, we first generated an inducible endothelial cell–specific IP 3R1 knockout mouse model and found that deletion of IP 3R1 in adult endothelial cells did not affect vasodilation and blood pressure. Considering all 3 subtypes of IP 3Rs are expressed in mouse endothelial cells, we further generated inducible endothelial cell–specific IP 3R triple knockout mice and found that deletion of all 3 IP 3R subtypes decreased plasma NO concentration and increased basal blood pressure. Furthermore, IP 3R deficiency reduced acetylcholine‐induced vasodilation and endothelial NO synthase phosphorylation at Ser1177.
Conclusions
Our results reveal that IP 3R‐mediated Ca2+ release in vascular endothelial cells plays an important role in regulating vasodilation and physiological blood pressure.
Keywords: blood pressure, calcium, calcium signaling, endothelial cell, hypertension
Subject Categories: Basic Science Research, Vascular Biology, Hypertension, Genetically Altered and Transgenic Models
Clinical Perspective
What Is New?
We described the role of inositol 1,4,5‐trisphosphate receptors, a family of intracellular Ca2+ release channels localized in the membrane of the endoplasmic reticulum, in regulating vasodilation and blood pressure using genetically engineered mouse models.
We show that deletion of all 3 inositol 1,4,5‐trisphosphate receptor subtypes in mouse endothelial cells leads to hypertension, with reductions in both plasma NO concentration and acetylcholine‐induced vasodilation.
What Are the Clinical Implications?
Given that genome‐wide association studies suggested that variants in inositol 1,4,5‐trisphosphate receptors are associated with hypertension in humans, inositol 1,4,5‐trisphosphate receptor deficiency in endothelial cells may exacerbate human hypertension through reducing NO production and vasodilation.
Introduction
Hypertension or high blood pressure is characterized as a sustained elevated level of systolic and/or diastolic blood pressure and is a major risk factor for cardiovascular morbidity and mortality.1 Accumulating evidence suggests that hypertension could also be linked to noncardiovascular diseases, including dementia, cancer, oral health disorders, and osteoporosis.2 Affecting ≈1 in 4 adults, hypertension is one of the most important preventable causes of premature death worldwide. However, despite the availability of various types of treatments, optimal blood pressure control in patients with hypertension is hard to achieve. One reason is that the pathogenesis of essential or primary hypertension is multifactorial and extremely complex. In fact, the origin of essential hypertension is generally unclear, and the pathogenesis of hypertension, as well as the molecular mechanism of blood pressure control, is still not well understood.
The endothelium is located at the interface between the vessel wall and lumen and has been recognized for decades as a critical mediator in controlling blood pressure, primarily via producing a variety of vasoactive substances, such as NO, thromboxane, and endothelin‐1.3 NO is one of the most well‐known vessel‐relaxing factors and is produced by a NO synthase (NOS) from l‐arginine in the presence of oxygen and the cofactors Ca2+, calmodulin, reduced nicotinamide adenine dinucleotide phosphate, and tetrahydrobiopterin.4 Intracellular Ca2+ has long been proposed as a critical factor regulating the activity of endothelial NOS (eNOS). Numerous stimuli, including hormonal and chemical signals, but also mechanical changes such as shear stress, can elicit rapid increases in intracellular Ca2+ in endothelial cells, resulting in Ca2+‐dependent activation of eNOS and subsequent NO release.5, 6, 7, 8, 9 Genetically deficient eNOS mice are hypertensive, with lower circulating nitrite levels, indicating the importance of eNOS and NO in blood pressure regulation.10, 11 However, how endothelial Ca2+ mobilization via plasma membrane Ca2+ entry and endoplasmic reticulum (ER) Ca2+ release regulates eNOS in vivo is not clear.
Inositol 1,4,5‐trisphosphate receptors (IP3Rs) are a family of intracellular Ca2+ release channels located on the ER membrane, which in mammals consists of 3 different subtypes (IP3R1, IP3R2, and IP3R3) encoded by 3 genes, Itpr1, Itpr2, and Itpr3, respectively.12 All 3 subtypes of IP3Rs are expressed in endothelial cells.13, 14 However, it remains unclear whether the different subtypes of IP3Rs in endothelial cells have distinct or redundant functions. A recent study found that IP3R1 deletion by the endothelial‐specific receptor tyrosine kinase‐Cre (Tie2‐Cre) in mice reduced acetylcholine‐induced vasodilation and increased basal blood pressure.15 However, Tie2‐Cre is expressed early during embryonic development. Furthermore, Tie2‐Cre is expressed not only in endothelial cells but also in blood cell lineages.16 Therefore, Tie2‐Cre is not the ideal tool to use when generating endothelial cell–specific gene deletion mouse models, especially for physiological studies. To investigate the specific roles of IP3Rs in adult endothelial cells, we used an inducible endothelial cell–specific platelet‐derived growth factor receptor‐β–Cre (iCre+) to delete IP3R1 alone as well as all 3 IP3R subtypes together in mouse endothelial cells. Our results demonstrate that deletion of IP3R1 in adult endothelial cells does not affect vascular reactivity or blood pressure. In fact, the different IP3R subtypes in adult endothelial cells function redundantly because deletion of all 3 IP3R subtypes reduced plasma NO concentration and acetylcholine‐induced eNOS phosphorylation, resulting in less vasodilation and hypertension. Our study reveals an essential role of IP3R‐mediated Ca2+ signaling in regulating vascular function and blood pressure with a mechanism of functional redundancy among the different IP3R subtypes.
Methods
The data, analytical methods, and study materials are available to other researchers for purposes of reproducing results or replicating procedures, as described in this article or by contacting corresponding authors.
Mice
The generation of floxed mice for each IP3R subtype has been described previously.17, 18 The iCre+ mice expressing an inducible Cre recombinase under the control of the platelet‐derived growth factor receptor‐β promoter has also been described previously.19 All IP3R floxed mice and the iCre+ mice were backcrossed with C57BL/6 mice for >8 generations. The B6.Cg‐Tg (Tek‐cre)1Ywa/J (Tie2‐Cre+) mice and the Gt(ROSA)26Sortm4(ACTB‐tdTomato,‐EGFP)Luo/J (ROSAmT/mG) reporter mice carrying membrane‐targeted, 2‐color fluorescent Cre‐reporter allele were purchased from the Jackson Laboratory.
To generate endothelial cell–specific single IP3R1 knockout and triple IP3R knockout mice, the 7‐ to 8‐week‐old iCre+ IP3R1 floxed (IP3R1f/f) and iCre+IP3R1f/fIP3R2f/fIP3R3f/f mice were intraperitoneally injected with tamoxifen (50 mg/kg per day; Sigma‐Aldrich) for 5 consecutive days, and they were considered as endothelial cell–specific IP 3R1 knockout (ECR1KO) and endothelial cell–specific IP 3R triple knockout (ECTKO) mice, respectively. The littermate iCre‐IP3R1f/f and iCre‐IP3R1f/fIP3R2f/fIP3R3f/f mice also treated with tamoxifen using the same protocol were considered as control mice. Only male mice were used in all experiments. All mice were housed under a 12‐hour day/night cycle at a temperature of 25.0°C, with free access to a standard diet and clean water.
Endothelial Cell Isolation
Endothelial cells were freshly isolated by enzymatic digestion. After euthanasia, the lungs were perfused with ice‐cold Hank's balanced salt solution (8 g/L NaCl, 0.4 g/L KCl, 1 g/L glucose, 60 mg/L KH2PO4, and 47.5 mg/L Na2HPO4, pH 7.4) via the right ventricle to remove blood cells from inside the tissue. The lung was then cut into small pieces and digested in serum‐free M199 medium containing 1.5 mg/mL collagenase II (Worthington) and 0.5607 mg/mL dispase II (Roche) for 1 hour at 37°C. After digestion, the cells were filtrated through a 70‐μm cell strainer and precipitated by centrifugation at 225 g for 10 minutes. The cells were resuspended and incubated with magnetic beads (sheep anti‐rat; Invitrogen) linked to anti‐CD31 antibody (rat anti‐mouse; BD Pharmingen) for 30 minutes at 4°C, and endothelial cells were harvested by magnetic separation.
Ca2+ Imaging
Ca2+ imaging was performed and analyzed, as previously described.20 Briefly, isolated endothelial cells from individual mice (3 mice per group) were washed with physiological saline solution (in mmol/L: NaCl 137, KCl 5.4, MgSO4 1.0, glucose 10, CaCl2 1.8, and HEPES 10, pH 7.4) and then incubated with 5 μmol/L Fluo‐4‐AM (Invitrogen) for 30 minutes at 37°C. After that, cells were washed with physiological saline solution 3 times and imaged with a Zeiss 880 inverted confocal microscope. Acetylcholine (10 μmol/L) was then applied to elicit intracellular Ca2+ mobilization in endothelial cells. The fluorescence was excited with 488‐nm light, and emitted light >510 nm was collected.
Quantitative Real‐Time Polymerase Chain Reaction Analysis
Quantitative real‐time polymerase chain reaction (PCR) was performed and analyzed, as previously described.21 Briefly, total RNA was extracted from freshly isolated endothelial cells using the RNeasy MiniKit (Qiagen), and cDNA was synthesized using the TransScript One‐Step gDNA removal and cDNA Synthesis SuperMix Kit (TransGen Biotech). Quantitative real‐time PCR was performed using the TransStart Tip Green qPCR SuperMix (TransGen Biotech), according to the manufacturer's instructions. The sequences for primers of Itpr1‐3 and Gapdh have been previously described.22 The primer sequences for quantitative PCR are listed in Table S1. Endothelial cells isolated from 3 mice were pooled as 1 sample. At least 3 samples were prepared for quantitative real‐time PCR per group. Each sample was assayed in triplicate, and the transcript level of each target gene was normalized to Gapdh.
Immunoblotting
Thoracic aortas were isolated from individual mice (4 mice per group), cleaned from connective tissues and adipose, and equilibrated in Krebs solution (in mmol/L: NaCl 118, KCl 4.6, NaHCO3 25, CaCl2 2.5, MgSO4 1.2, KH2PO4 1.2, and glucose 12, pH 7.4) at 37°C for 1 hour. After equilibration, the arteries were stimulated with vehicle or acetylcholine (10 μmol/L; 10 minutes) and then were quickly frozen in liquid nitrogen and homogenized with protein lysis buffer consisting of 8 mol/L urea, 2 mol/L thiourea, 3% SDS, 75 mmol/L dithiothreitol, 0.05 mol/L Tris‐HCl (pH 6.8), and 0.03% Bromophenol Blue. Western blotting was then performed as previously described.23 The primary antibodies against mouse eNOS (Millipore, catalog No. 07–520, at 1:1000 dilution), phosphorylated eNOS at Ser1177 (Abcam, catalog No. ab195944, at 1:1000 dilution), phosphorylated eNOS at Thr495 (Cell Signaling Technology, catalog No. 9574s, at 1:1000 dilution), and GAPDH (Cell Signaling Technology, catalog No. 97166, at 1:1000 dilution) were commercially purchased.
Myography
To assess vascular reactivity, thoracic aortas and the second order of superior mesenteric arteries were isolated from individual mice (at least 6 mice per group) and quickly immersed in ice‐cold Krebs solution. After the connective tissues were carefully removed, the vessel rings were prepared and mounted in a myograph chamber (620 mol/L; Danish Myo Technology, Denmark), as previously described.22 Each chamber was filled with 5 mL Krebs solution aerated with 95% O2 to 5% CO2 and maintained at 37°C. Each vessel ring was stretched in a stepwise manner to the optimal resting tension (thoracic aortas to ≈9 mN; mesenteric arteries to ≈1.5 mN), equilibrated for 30 minutes, and then exposed to 100 mmol/L K+ Krebs solution to induce a reference contraction. Thereafter, after the application of phenylephrine (10 μmol/L; Sigma‐Aldrich) to elicit a preconstriction, cumulative doses of acetylcholine (1 nmol/L to 10 μmol/L) and sodium nitroprusside (1 nmol/L to 10 μmol/L; Sigma‐Aldrich) were then applied to elicit vasodilation. The vasodilation in response to the cumulatively administrated acetylcholine or sodium nitroprusside was expressed as decreases in force and as percentages of the peak of phenylephrine‐induced contraction.
Measurement of Blood Pressure by a Tail‐Cuff System
Systolic blood pressure was measured by a noninvasive tail‐cuff system (NIBP system, IN125/m; AD Instruments), as previously described.22 Briefly, the mice were placed in a restrainer with the caudal artery positioned right above the sensor. All the mice (11 control and 12 mutant mice for the IP3R1 single knockout studies; 13 control and 17 mutant mice for the IP3R triple knockout studies) were given at least 1 week to adapt to the system before blood pressure measurement. Systolic blood pressure was continuously measured at least 6 times and averaged for each mouse.
Telemetry Measurement
A total of 14 male mice at the age of 6 months (4 months after tamoxifen injection) were anesthetized with IP injection of a mixture of ketamine (100 mg/kg) and xylazine (5 mg/kg). Each mouse was implanted with a radiotelemetric transmitter (PA‐C10; Data Sciences International) and housed in a separated cage under a 12‐hour day/night cycle. Animals undergoing survival surgery were treated with carprofen for 2 to 5 days after operation to reduce discomfort. After a 1‐week recovery period from surgery, blood pressures and heart rates were recorded every 5 minutes for 3 consecutive days in conscious, freely moving mice, as previously described.22 After data acquisition, the mice will be euthanized by IP injection of nembutal (150 mg/kg), followed by cervical dislocation.
Histology
The second order of superior mesenteric arteries was isolated from 5 control and 6 ECTKO mice, fixed in 4% paraformaldehyde at 4°C overnight, and thereafter processed for frozen sections. Hematoxylin‐eosin staining was performed on 8‐μm sections, and images were acquired with light microscopy, as previously described.24 The wall thickness and the media/lumen ratio were quantified using Image J software.
Measurement of Serum Nitrite and Nitrate
NO in solution is rapidly oxidized into nitrite and nitrate, which can be used to quantitate NO production. Serum was collected from 6 control and 6 ECTKO mice 4 months after tamoxifen induction, and NO production was assessed using the Nitric Oxide Assay Kit (Nanjing Jiancheng Bioengineering Institute), which is designed to accurately measure NO production after reduction of nitrate to nitrite using the improved Griess method.
Statistical Analysis
Scatter diagram and box‐and‐whisker plots were drawn using GraphPad Prism 5. Statistical analysis was performed using 2‐tailed, unpaired, Student t test or 2‐way ANOVA with Bonferroni post hoc test for multiple comparisons. All data were presented as mean±SEM (error bars). P<0.05 was considered statistically significant (P<0.05 and P<0.01 versus control).
Study Approval
All animal care and use procedures in this study were approved by the Institutional Animal Care and Use Committee at UC San Diego (San Diego, CA; protocol reference No. S01049) and at Peking University Shenzhen Graduate School (Shenzhen, China; protocol reference No. AP0017).
Results
Single Deletion of IP3R1 in Endothelial Cells Does Not Affect Vascular Function and Blood Pressure
To investigate the specific role of IP3R1‐mediated Ca2+ release in endothelial cells of adult mice, we crossed IP3R1f/f mice with mice that express a tamoxifen‐inducible form of Cre recombinase in vascular endothelial cells using a phage artificial chromosome containing the platelet‐derived growth factor receptor‐β (Pdgfb) gene (iCre+).19 To confirm the efficiency and cell specificity of gene deletion by iCre+, we also crossed iCre+ mice with ROSAmT/mG reporter mice that express a cell membrane–targeted, 2‐color fluorescent Cre reporter allele.25
Administration of tamoxifen in adult iCre+ROSAmT/mG mice led to expression of cell membrane–localized enhanced green fluorescent protein in almost all endothelial cells of the mesenteric artery, whereas nonendothelial cells expressed cell membrane–localized tdTomato (Figure S1), suggesting that the Cre recombinase activity of iCre+ was specifically induced in endothelial cells. Furthermore, we found that Itpr1 mRNA levels were dramatically reduced in endothelial cells isolated from iCre+IP3R1f/f (ECR1KO) mice 4 months after tamoxifen injection compared with control cells, whereas Itpr2 and Itpr3 mRNA levels were comparable between control and ECR1KO cells (Figure 1A). However, single deletion of IP3R1 was not sufficient to block Ca2+ mobilization induced by the endothelium‐dependent vasodilator acetylcholine in endothelial cells (Figure 1B). In fact, the amplitude of Ca2+ transient induced by 10 μmol/L acetylcholine in ECR1KO cells was >80% of that in control cells (Figure 1C), implicating that the other 2 IP3R subtypes can compensate for the loss of IP3R1 in endothelial cells. Accordingly, systolic blood pressure measured by the tail‐cuff system was comparable between control and ECR1KO mice at 2, 3, and 4 months after tamoxifen injection (Figure 1D). In addition, vascular reactivity in response to either acetylcholine or the endothelium‐independent vasodilator sodium nitroprusside (a NO donor) in the thoracic aorta and mesenteric artery was not significantly altered in ECR1KO mice compared with control mice 4 months after tamoxifen injection (Figure 1E and 1F). Furthermore, the mRNA levels of muscarinic acetylcholine receptor M1 (CHRM1), M2 (CHRM2), M3 (CHRM3), M4 (CHRM4), and M5 (CHRM5), nicotinic acetylcholine receptor subunit α7 (CHRNA7), and eNOS (NOS3) were also comparable between control and ECR1KO mice (Figure 1G). Taken together, these results demonstrated that single deletion of IP3R1 in adult endothelial cells was not sufficient to alter vasodilation and cause hypertension, as reported in a recent study that found that IP3R1 deletion by Tie2‐Cre in mice reduced acetylcholine‐induced vasodilation and increased basal blood pressure.15
Figure 1.
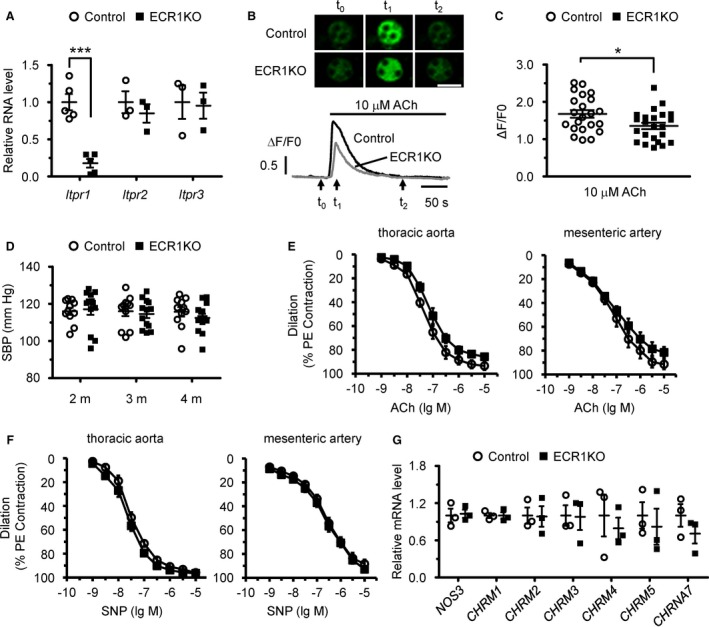
Endothelial cell–specific deletion of inositol 1,4,5‐trisphosphate receptor 1 (IP 3R1) in adult mice had no major effects on blood pressure and vasodilation. A, mRNA levels of IP 3Rs in isolated endothelial cells from control and endothelial cell–specific IP 3R1 knockout (ECR1KO) mice. n=3 to 5 (with endothelial cells from 3 mice pooled as 1 sample) per group. B, Confocal Ca2+ imaging of isolated endothelial cells using Fluo‐4‐AM. Intracellular Ca2+ mobilization was elicited by 10 μmol/L acetylcholine. Top, Sequential confocal images of endothelial cells at the time points of time 0, time 1, and time 2, as indicated by the arrows at the bottom. Bar=20 μm. Bottom, Representative traces of Ca2+ signals in control (black) and ECR1KO (gray) endothelial cells. F0, the Fluo‐4 fluorescence at rest. F/F0, normalized fluorescence. C, The amplitude of Ca2+ signals induced by 10 μmol/L acetylcholine. n=20 to 30 cells from 3 independent experiments per group. D, Systolic blood pressure (SBP) measured in control and ECR1KO mice at 2, 3, and 4 months after tamoxifen injection using the tail‐cuff system. n=11 to 12 mice per group. E, Vascular reactivity in response to endothelium‐dependent agonist acetylcholine in ECR1 aortas and mesenteric arteries showed a slight trend toward reduced vasodilation that was not statistically significant. F, Vascular reactivity in response to endothelium‐independent agonist sodium nitroprusside (SNP) in aortas and mesenteric arteries. The vessels were preconstricted by 10 μmol/L phenylephrine, and the vasorelaxing effects of acetylcholine and sodium nitroprusside (SNP) were presented as a percentage of phenylephrine‐induced contraction. n=6 mice per group. G, Quantitative real‐time polymerase chain reaction analysis of the expression of NOS3 and major acetylcholine receptors, including CHRM1,CHRM2,CHRM3,CHRM4,CHRM5, and CHRNA7, in control and ECR1KO endothelial cells. n=3 (with endothelial cells from 3 mice pooled as 1 sample) per group. Significance was determined using a 2‐tailed, unpaired, Student t test or 2‐way ANOVA analysis with Bonferroni post hoc test. Error bars represent mean±SEM. *P<0.05, ***P<0.001 vs control.
The difference between our ECR1KO mice and the recently published Tie2‐Cre‐IP3R1 knockout mice could be explained by the fact that Tie2‐Cre is expressed at early embryonic stages not only in endothelial cells but also in multiple blood cell lineages.16 To further confirm this possibility, we then generated the same Tie2‐Cre+IP3R1f/f mice (Figure S2A). However, no significant changes in systolic blood pressure were found between Tie2‐Cre‐IP3R1f/f and Tie2‐Cre+IP3R1f/f mice at the ages of 3 and 6 months, respectively (Figure S2B). Acetylcholine‐induced vasodilation in the thoracic aorta and mesenteric artery was also comparable between Tie2‐Cre‐IP3R1f/f and Tie2‐Cre+IP3R1f/f mice at the age of 6 months (Figure S2C). In addition, the ratio of heart weight/body weight was not significantly increased in Tie2‐Cre+IP3R1f/f mice at the age of 6 months when compared with Tie2‐Cre‐IP3R1f/f mice (Figure S2D). Thus, our data also do not support the published results that IP3R1 deletion by Tie2‐Cre in mice reduced acetylcholine‐induced vasodilation and increased basal blood pressure.15
Deletion of All 3 IP3R Subtypes in Adult Endothelial Cells Results in Hypertension
Because we did not observe any obvious functional deficits in the IP3R1 single knockout animals, we considered that the other 2 IP3R subtypes might play a compensatory function in the absence of IP3R1 because both our data (Figure 1A) and others showed that all 3 IP3R subtypes are expressed in endothelial cells.13, 14 Therefore, we generated iCre+IP3R1f/fIP3R2f/fIP3R3f/f (ECTKO) mice (Figure S3A). In isolated endothelial cells from control and ECTKO mice 4 months after tamoxifen injection, mRNA levels of all 3 IP3R subtypes were dramatically reduced in ECTKO cells compared with control cells (Figure 2A), suggesting that all IP3Rs were efficiently deleted by tamoxifen injection. Furthermore, Ca2+ transients induced by 10 μmol/L acetylcholine were almost abolished in ECTKO endothelial cells (Figure 2B and 2C). Considering the effect of single deletion of IP3R1 on acetylcholine‐induced Ca2+ transient in endothelial cells (Figure 1B and 1C), our data demonstrate that the different IP3R subtypes in mouse endothelial cells have redundant roles in acetylcholine‐induced Ca2+ mobilization.
Figure 2.
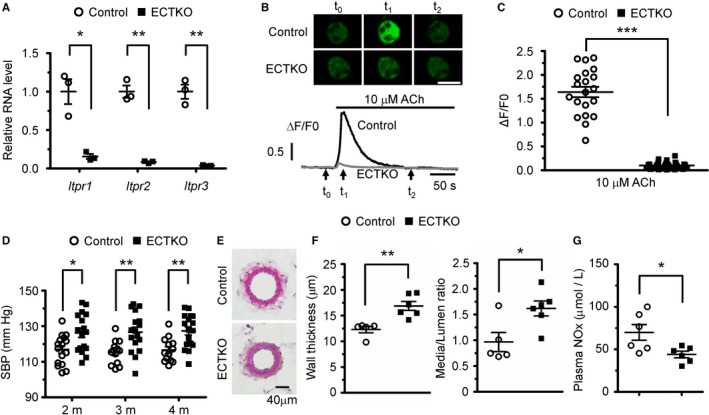
Endothelial cell–specific deletion of all 3 subtypes of inositol 1,4,5‐trisphosphate receptors (IP 3Rs) in adult mice increased systolic blood pressure (SBP). A, Quantitative real‐time polymerase chain reaction analysis of the 3 IP 3R subtypes in isolated endothelial cells from control and endothelial cell–specific IP 3R triple knockout (ECTKO) mice. n=3 (with endothelial cells from 3 mice pooled as 1 sample) per group. B, Confocal Ca2+ imaging of endothelial cells isolated from control and ECTKO mice. Top, Sequential confocal images of endothelial cells at the time point of time 0, time 1, and time 2, as indicated by the arrows at the bottom. Bar=20 μm. Bottom, Representative traces of Ca2+ signals in control (black) and ECTKO (gray) endothelial cells. F0, the Fluo‐4 fluorescence at rest. F/F0, normalized fluorescence. C, The amplitude of Ca2+ signals induced by 10 μmol/L acetylcholine in control and ECTKO endothelial cells. n=20 to 30 cells from 3 independent experiments per group. D, SBP measured in control and ECTKO mice at 2, 3, and 4 months after tamoxifen administration using the tail‐cuff system. n=13 to 17 mice per group. E, Representative hematoxylin and eosin–stained sections of the second‐order mesenteric arteries isolated from control and ECTKO mice 4 months after tamoxifen administration. Bar=40 μm. F, The wall thickness and the ratio of medial/luminal area were calculated in control and ECTKO mice. n=5 to 6 mice per group. G, The concentration of nitrite and nitrate (NOx) in the serum measured in control and ECTKO mice 4 months after tamoxifen administration. n=6 mice per group. Significance was determined using a 2‐tailed, unpaired, Student t test. Error bars represent mean±SEM. *P<0.05, **P<0.01, ***P<0.001 vs control.
We next investigated whether deletion of all 3 subtypes of IP3Rs in endothelial cells affects blood pressure regulation. We found that systolic blood pressure, measured by the tail‐cuff system, was significantly increased in ECTKO mice compared with control mice at 2, 3, and 4 months after tamoxifen injection (Figure 2D). Consistently, we also found that the wall thickness and the media/lumen ratio of mesenteric arteries were both significantly increased in ECTKO mice 4 months after tamoxifen injection when compared with control mice (Figure 2E and 2F). The importance of the vasodilator, NO, in regulating blood pressure has been well recognized, and lower circulating NO levels have been highlighted in numerous animal models as well as in patients with hypertension.10, 11, 26 Therefore, we investigated whether loss of endothelial IP3Rs in mice affects NO production by measuring the concentration of nitrite and nitrate (NOx) in the plasma. At 4 months after tamoxifen injection, the concentration of NOx in the plasma of ECTKO mice at baseline was significantly reduced compared with control mice (Figure 2G), indicating that deletion of IP3Rs in adult mouse endothelial cells decreases NO production.
We further used a telemetry system to analyze the hemodynamics of control and ECTKO mice under conscious and unrestrained conditions. The averaged results from data collected continuously over 24 hours showed that all control and ECTKO mice 4 months after tamoxifen injection displayed characteristic diurnal variations in blood pressure (Figure 3A). Systolic blood pressure, diastolic blood pressure, and mean blood pressure measured at every time point, or averaged days or nights, are all significantly increased in ECTKO mice compared with control mice (Figure 3A and 3B), consistent with what was observed by the tail‐cuff system. Taken together, all these data strongly suggest that IP3R‐mediated Ca2+ release in mouse endothelial cells is required for normal blood pressure regulation, and IP3R deficiency in endothelial cells is sufficient to cause hypertension.
Figure 3.
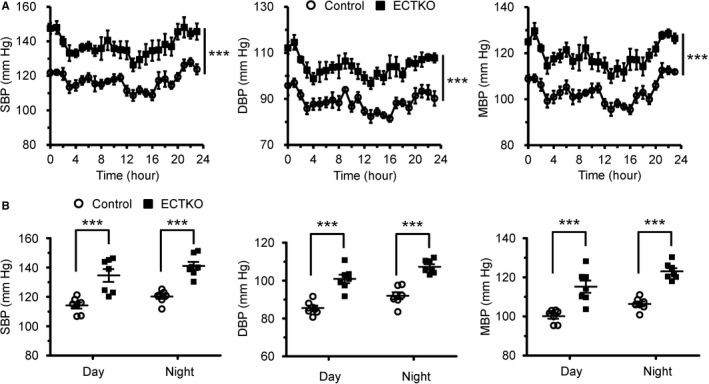
Basal blood pressure was significantly elevated in endothelial cell–specific inositol 1,4,5‐trisphosphate receptor triple knockout (ECTKO) mice. Blood pressures were measured in unstrained mice using an implantable telemetry system. A, Systolic blood pressure (SBP), diastolic blood pressure (DBP), and mean blood pressure (MBP) in control and ECTKO mice. The data were presented at times indicated on a 24‐hour scale, and data shown at each point represent 1‐hour rolling averages of data sampled each minute in 3 consecutive days. n=7 mice per group. B, Mean values for SBP, DBP, and MBP were calculated during the day (8 am–8 pm) and night (8 pm–8 am). n=7 mice per group. Significance was determined using a 2‐tailed, unpaired, Student t test or 2‐way ANOVA analysis with Bonferroni post hoc test. Error bars represent mean±SEM. ***P<0.001 vs control.
IP3R Deletion in Endothelial Cells Caused Vascular Dysfunction and Defective eNOS Activity
Next, we investigated whether IP3R deficiency influences vascular reactivity in response to vasoconstrictor and vasodilator treatment. Using wire myography, we measured vascular contractility and dilation in control and ECTKO mice 4 months after tamoxifen injection. Both the contraction induced by 100 mmol/L potassium and the dose‐dependent response to phenylephrine in the aorta were not significantly altered in ECTKO mice relative to their controls (Figure S3B). The contraction of the mesenteric artery induced by 100 mmol/L potassium was also not significantly changed in ECTKO mice (Figure S3C). However, the dose‐dependent response to phenylephrine in the ECTKO mesenteric arteries displayed a small leftward shift compared with control samples (Figure S3C), implicating that endothelial dysfunction resulting from IP3R deficiency might also affect the basal vascular tone toward increased constriction. Consistently, vasodilation induced by acetylcholine in both the aorta and mesenteric artery was markedly reduced in ECTKO mice compared with control mice (Figure 4A). In contrast, vasodilation induced by the endothelium‐independent vasodilator sodium nitroprusside (a NO donor) in both the aorta and the mesenteric artery was not significantly changed between control and ECTKO mice (Figure 4B). Considering that the plasma NOx concentration at baseline was reduced in ECTKO mice (Figure 2G), we next investigated whether the observed blunted vasodilation was attributable to the defective response of eNOS to acetylcholine because eNOS is the primary enzyme to produce NO in endothelial cells. Therefore, we isolated aortas from control and ECTKO mice and measured the protein and phosphorylation levels of eNOS in response to acetylcholine stimulation. We found that the protein level of eNOS was not significantly altered in ECTKO aortas (Figure 4C and 4D). However, the increase in the phosphorylation level of eNOS at Ser1177 in response to acetylcholine stimulation was blunted in ECTKO aortas compared with control aortas (Figure 4C and 4D), which is consistent with the blunted vasodilation observed in ECTKO arteries. On the other hand, reduction of eNOS phosphorylation at Thr495 in response to acetylcholine stimulation was comparable between control and ECTKO aortas (Figure 4C and 4D). Taken together, our data strongly suggest that IP3R‐mediated Ca2+ release plays an essential role in regulating vasodilation via modulating eNOS phosphorylation.
Figure 4.
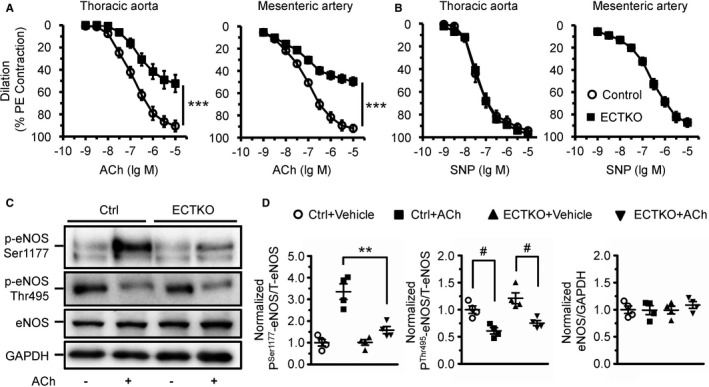
Deficiency of inositol 1,4,5‐trisphosphate receptors (IP 3Rs) in endothelial cells affected acetylcholine‐induced vasodilation and endothelial NO synthase (eNOS) phosphorylation. A and B, Vascular reactivity in response to endothelium‐dependent agonist acetylcholine (A) and endothelium‐independent agonist sodium nitroprusside (SNP) (B) in aortas and mesenteric arteries. The vessels were preconstricted by 10 μmol/L phenylephrine, and the vasorelaxing effects of acetylcholine and SNP were presented as a percentage of phenylephrine‐induced contraction. n=6 to 12 mice per group. C, Expression and phosphorylation of eNOS measured by Western blot. Thoracic aortas isolated from control and endothelial cell–specific IP 3R triple knockout (ECTKO) mice were treated with or without 10 μmol/L acetylcholine for 10 minutes. D, The levels of phosphorylated eNOS at Ser1177 and Thr495 were normalized to total eNOS. The levels of total eNOS were normalized to GAPDH. n=4 mice per group. Significance was determined using a 2‐tailed, unpaired, Student t test or 2‐way ANOVA analysis with Bonferroni post hoc test. Error bars represent mean±SEM. **P<0.01, ***P<0.001 vs control; # P<0.05 vs vehicle.
Discussion
In this study, we demonstrated that IP3Rs play an essential role in regulating vasodilation and blood pressure, with the different IP3R subtypes displaying functional redundancy. The IP3R is a ubiquitously expressed ER Ca2+ release channel, which has 3 different subtypes in mammals encoded by 3 distinct genes. The 3 full‐length amino acid sequences are 60% to 80% homologous overall, with regions, including the ligand‐binding and pore domains, having much higher homology.27, 28 Furthermore, multiple IP3R subtypes are coexpressed in most mammalian cell types outside the central nervous system,12 suggesting a functional redundancy between different subtypes. Indeed, various studies using genetically engineered mouse models have demonstrated that there is functional redundancy among the IP3R subtypes in cells that express >1. Deletion of both IP3R2 and IP3R3 was required to create a pancreatic acinar cell secretion phenotype.29 In T lymphocytes, deletion of all 3 IP3R subtypes was required to induce a developmental defect in double‐negative to double‐positive transition and T‐cell acute lymphoblastic leukemia.17 In B cells, different IP3R subtypes also play a redundant role in regulating both B‐cell receptor–mediated Ca2+ release and B‐cell development.20
In contrast to our current experiments, a recent publication performed concomitantly to our study reported that single deletion of IP3R1 by Tie2‐Cre in mice impaired endothelium‐dependent vasodilation, elevated the basal blood pressure, and eventually increased the ratio of heart weight/body weight,15 suggesting that IP3R1‐mediated Ca2+ release plays a dominant role in regulating endothelial cell function. In contrast with these findings, we found that single deletion of IP3R1 in endothelial cells by the same Tie2‐Cre was not able to cause the same phenotype in mice. The parameters, including blood pressure, vascular reactivity in response to acetylcholine, and the ratio of heart weight/body weight in our Tie2‐Cre+R1f/f mice, are all comparable with control mice. The difference between our current study and the previous report may result from different gene‐targeting strategies and/or mouse genetic backgrounds. In our IP3R1 floxed mouse model, the exon 5 was flanked, whereas the exon 4 was targeted in the mouse model of Yuan et al.15 In addition, the mice reported by Yuan et al15 were backcrossed with C57BL/6 2 more generations than ours. It is possible that these differences could contribute, in part, to the discrepancy in the phenotypes observed in our and their mouse models. However, our results showed that deletion of IP3R1 alone has minimal effects on the amplitudes of acetylcholine‐induced Ca2+ signals in isolated endothelial cells. Furthermore, the 3 IP3R subtypes are all expressed in endothelial cells, as confirmed by quantitative real‐time PCR in this study and also reported by others.13, 14 In addition, our previous work demonstrated that deletion of all 3 IP3R subtypes by Tie2‐Cre resulted in T‐cell acute lymphoblastic leukemia and mouse lethality from 2 months of age.17 Tie2‐Cre is not an ideal tool to study the role of endothelial cells in vascular biology because Tie2‐Cre is also expressed in hematopoietic cells.16, 30 Another constitutively active panendothelial Cre, VE‐cadherin‐Cre, which has also been widely used for endothelium‐specific gene deletion, is also expressed in hematopoietic cells.30, 31 Therefore, we used the iCre+, an inducible panendothelial Cre, to target IP3R genes in adult vascular endothelial cells, and we found that deletion of all IP3R genes in endothelial cells was efficient after tamoxifen administration. Accordingly, deletion of all 3 IP3R subtypes was able to block acetylcholine‐induced Ca2+ signals in isolated endothelial cells. Consistently, deletion of all 3 IP3R subtypes in endothelial cells by iCre+ reduced acetylcholine‐induced vasodilation and increased the basal blood pressure. Therefore, our study strongly suggests there is functional redundancy between the different IP3R subtypes in endothelial cells.
In contrast to the vasodilatory effects of IP3R‐mediated Ca2+ release in endothelial cells, it has been well recognized that Ca2+ release from ER via IP3Rs in smooth muscle cells leads to vasoconstriction via activation of myosin light chain kinase that subsequently phosphorylates myosin light chain, enabling molecular cross‐bridge formation between myosin and actin filaments.32, 33 Deletion of all IP3R subtypes in smooth muscle cells has been shown to reduce vascular contraction and myosin light chain 20 phosphorylation in response to vasoconstrictor, and it alleviated angiotensin II–induced hypertension.22 Therefore, IP3R‐mediated Ca2+ mobilization in vascular endothelial cells and smooth muscle cells plays an opposing role in regulating vascular contractility, which might provide the basis for paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries.34, 35 On the other hand, endothelial cells from different tissues or organs have distinct morphological, structural, and gene‐expression profiles.36, 37, 38 In additional to vascular tone, endothelial cells may also play essential roles in regulating vascular permeability, angiogenesis, inflammation, and vascular remodeling.26, 37 Thus, it will be worthy to examine diverse tissues or organs to determine whether and how IP3R‐mediated intracellular Ca2+ mobilization could mediate different functions.
Among the various vasoactive factors released by vascular endothelial cells, NO is definitely one of the most well‐characterized molecules. NO regulates vascular tone and physiological blood pressure, and it participates in the pathogenesis of many cardiovascular diseases, including atherosclerosis, myocardial infarction, diabetes mellitus, and hypertension.39, 40, 41, 42, 43 In endothelial cells, the production of NO is controlled by eNOS, an enzyme highly dependent on Ca2+/calmodulin‐mediated activation. Active calmodulin binds to eNOS, leading to caveolin detaching from eNOS, and transforms eNOS into its active form.44, 45 Previous studies have shown that removal of extracellular Ca2+, treatment with calmodulin antagonist, or addition of IP3R blockers, such as heparin, dramatically reduces NO production and endothelium‐dependent artery dilation.45, 46, 47 In addition, genetic depletion of stromal interaction molecule 1 from endothelial cells also significantly impairs the endothelium‐dependent vasorelaxation.48 Consistent with these findings, our data also demonstrated that deficiency of IP3R‐mediated Ca2+ release in endothelial cells reduces endothelium‐dependent vasodilation and plasma NOx levels. Considering that shear stress has been proposed to play a critical role in regulating NO production and intracellular Ca2+ homeostasis in endothelial cells,44, 49, 50, 51 it remains to be determined whether loss of IP3R‐mediated Ca2+ release could also affect NO production and Ca2+ signals induced by shear stress.
In addition to the classic Ca2+/calmodulin‐dependent activation, eNOS can also be activated via phosphorylation at multiple sites.52, 53 The most extensively studied eNOS phosphorylation site is Ser1177, which is located in the reductase domain close to the carboxy‐terminus. Phosphorylation of this site has been associated with eNOS activation in response to numerous stimuli, including mechanical factors, such as shear stress,54 and humoral factors, such as bradykinin55 and insulin.56 Up to now, it has been proposed that kinases, such as 5’ AMP‐activated protein kinase, protein kinase B, protein kinase A, calmodulin‐dependent protein kinase II, and protein kinase G, as well as phosphatase protein phosphatase 2A were able to regulate the phosphorylation of eNOS at Ser1177.52 Overall, Ser1177 has been generally considered the most important of the regulatory eNOS phosphorylation sites. Phosphorylation of eNOS at Ser1177 increases eNOS activation mediated by Ca2+/calmodulin binding, and it can also lead to activation of eNOS at resting levels of [Ca2+]. Consistently, deficiency of IP3R‐mediated Ca2+ release in endothelial cells dramatically reduced acetylcholine‐induced phosphorylation of eNOS at Ser1177. We assume that this could be, at least in part, a consequence of insufficient activation of Ca2+/calmodulin‐dependent protein kinase II because intracellular Ca2+ signals were almost eliminated in IP3R‐deficient endothelial cells. On the other hand, activity of eNOS is reduced by phosphorylation of eNOS at Thr495 in the Ca2+/calmodulin binding domain.57, 58 Protein kinase C has been suggested to phosphorylate eNOS at Thr495,59 whereas protein phosphatase 1, protein phosphatase 2A, and calcineurin dephosphorylate eNOS at Thr495.52, 53 However, dephosphorylation of eNOS at Thr495 induced by acetylcholine was not significantly altered in ECTKO arteries. Furthermore, in contrast to results reported by Yuan et al,15 we found that protein levels of aortic eNOS were not significantly changed between control and ECTKO mice, suggesting that IP3R‐mediated Ca2+ release is not required for maintaining eNOS expression in endothelial cells.
Taken together, we demonstrated that IP3R‐mediated Ca2+ mobilization plays an essential role in regulating vascular dilation and basal blood pressure. In endothelial cells, all 3 IP3R subtypes exist and may function redundantly. Deletion of all 3 IP3R subtypes in mouse endothelial cells impaired acetylcholine‐induced Ca2+ signals, eNOS phosphorylation at Ser1177, and vasodilation. Deficiency of all IP3Rs also reduced plasma NOx levels and increased basal blood pressure. Considering that IP3Rs have been strongly implicated in hypertension in humans, as revealed by genome‐wide associated studies, our study provides mechanistic insight into the relationship between IP3R function and human hypertension.
Author Contributions
Lin, Zhao, Jing, Li, Tang, Huang, Zhang, and Wang performed research; Ouyang, Fang, Liu, Jia, and Chen designed the research; Ouyang, Fang, Trexler, and Chen wrote the manuscript. Liu, and Jia provided materials.
Sources of Funding
The work was supported by the National Science Foundation of China (31370823, 91439130, 81700289, 31800767), the Guangdong Province Basic Research Foundation (2018A030310012), the Shenzhen Basic Research Foundation (KCYJ20160428154108239, KQJSCX20170330155020267, KCYJ20170818090044949, KQTD2015032709315529), and the National Institutes of Health (NIH) (Chen). Chen is the American Heart Association Endowed Chair in Research. Fang is also supported by NIH grant K99HL143210.
Disclosures
None.
Supporting information
Table S1. Primers Sequences for RT‐PCR
Figure S1. Endolthelial cell‐specific gene deletion in adult mice by Pdgfb‐iCreER. The Pdgfb‐iCreER (iCre+) mice were crossed with ROSAmT/mG reporter mice to generate the iCre+ROSAmT/mG mice. Administration of tamoxifen in adult iCre+ROSAmT/mG mice led to expression of cell membrane‐localized EGFP in endothelial cells, while non‐endothelial cells expressed cell membrane‐localized tdTomato. A, Schematic diagram demonstrating the crossing strategy and the induction of EGFP expression. B, Confocal fluorescent imaging showing the localization of EGFP and tdTomato in the cross‐section of the mesenteric artery isolated from iCre+ROSAmT/mG mice after tamoxifen administration. Scale bar, 40 μm.
Figure S2. Deletion of IP3R1 by Tie2‐Cre in mice did not alter blood pressure and vasodilation. The constitutively active Tie2‐Cre instead of the inducible Pdgfb‐iCreER was used to delete IP3R1 in endothelial cells from as early as the embryonic stage. A, Quantitative RT‐PCR analysis of the expression of 3 IP3R subtypes in isolated endothelial cells from Tie2‐Cre−R1f/f and Tie2‐Cre+R1f/f mice. n=3 (with endothelial cells from 3 mice pooled as one sample) per group. Significance was determined by the 2‐tailed, unpaired student t test. **P<0.0 vs Tie2‐Cre−R1f/f mice. Data are presented as mean±SEM. B, Systolic blood pressure (SBP) were measured at the ages of 3 and 6 months using the tail cuff system, respectively. n=5 to 8 mice per group. Significance was determined by 2‐way ANOVA analysis with Bonferroni post‐hoc test. C, Vascular reactivity in response to ACh in aortas and mesenteric arteries. The vessels were pre‐constricted by 10 μmol/L phenylephrine (PE) and the vasorelaxing effects of ACh were presented as a percentage of PE‐induced contractions. n=6 mice per group. Significance was determined by 2‐way ANOVA analysis with Bonferroni post‐hoc test. D, The ratio of ventricle weight to body weight was comparable between Tie2‐Cre−R1f/f and Tie2‐Cre+R1f/f mice at the age of 6 months. n=9 mice per group. Significance was determined by the 2‐tailed, unpaired student t test.
Figure S3. Mouse breeding strategy and measurement of vascular contractility in control and ECTKO mice. A, Schematic diagram showing the mouse breeding strategy of generate iCre+IP3R1f/f IP3R2f/f IP3R3f/f mice. B, Reference contraction induced by high potassium (100 mmol/L) and the dose‐dependent contractile response to phenylephrine (PE) in control and ECTKO aortas. n=6 per group. C, Reference contraction induced by high potassium (100 mmol/L) and the dose‐dependent contractile response to phenylephrine (PE) in control and ECTKO mesenteric arteries. n=6 per group. For all dose‐response curves, data were expressed as a percentage of the peak of K+‐induced contraction, and significance was determined by 2‐tailed, unpaired student t test or 2‐way ANOVA analysis with Bonferroni post‐hoc test. **P<0.0 vs control. Error bars represent mean±SEM.
Acknowledgments
The authors would like to thank Marcus Fruttiger at UCL Institute of Ophthalmology (London, UK) for providing us the inducible platelet‐derived growth factor receptor‐β–Cre mice.
(J Am Heart Assoc. 2019;8:e011704 DOI: 10.1161/JAHA.118.011704.)
Contributor Information
Ju Chen, Email: juchen@ucsd.edu.
Kunfu Ouyang, Email: ouyangkunfu@pkusz.edu.cn.
References
- 1. Blacher J, Levy BI, Mourad JJ, Safar ME, Bakris G. From epidemiological transition to modern cardiovascular epidemiology: hypertension in the 21st century. Lancet. 2016;388:530–532. [DOI] [PubMed] [Google Scholar]
- 2. Kokubo Y, Iwashima Y. Higher blood pressure as a risk factor for diseases other than stroke and ischemic heart disease. Hypertension. 2015;66:254–259. [DOI] [PubMed] [Google Scholar]
- 3. Feletou M, Kohler R, Vanhoutte PM. Endothelium‐derived vasoactive factors and hypertension: possible roles in pathogenesis and as treatment targets. Curr Hypertens Rep. 2010;12:267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Balligand JL, Feron O, Dessy C. eNOS activation by physical forces: from short‐term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol Rev. 2009;89:481–534. [DOI] [PubMed] [Google Scholar]
- 5. Palmer RM, Ashton DS, Moncada S. Vascular endothelial cells synthesize nitric oxide from L‐arginine. Nature. 1988;333:664–666. [DOI] [PubMed] [Google Scholar]
- 6. Luksha L, Agewall S, Kublickiene K. Endothelium‐derived hyperpolarizing factor in vascular physiology and cardiovascular disease. Atherosclerosis. 2009;202:330–344. [DOI] [PubMed] [Google Scholar]
- 7. Santulli G, Cipolletta E, Sorriento D, Del Giudice C, Anastasio A, Monaco S, Maione AS, Condorelli G, Puca A, Trimarco B, Illario M, Iaccarino G. CaMK4 gene deletion induces hypertension. J Am Heart Assoc. 2012;1:e001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chataigneau T, Feletou M, Huang PL, Fishman MC, Duhault J, Vanhoutte PM. Acetylcholine‐induced relaxation in blood vessels from endothelial nitric oxide synthase knockout mice. Br J Pharmacol. 1999;126:219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K, Kanaide H, Takeshita A. Hydrogen peroxide is an endothelium‐derived hyperpolarizing factor in mice. J Clin Invest. 2000;106:1521–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, Smithies O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 1996;93:13176–13181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. [DOI] [PubMed] [Google Scholar]
- 12. Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev. 2007;87:593–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mountian I, Manolopoulos VG, De Smedt H, Parys JB, Missiaen L, Wuytack F. Expression patterns of sarco/endoplasmic reticulum Ca(2+)‐ATPase and inositol 1,4,5‐trisphosphate receptor isoforms in vascular endothelial cells. Cell Calcium. 1999;25:371–380. [DOI] [PubMed] [Google Scholar]
- 14. Grayson TH, Haddock RE, Murray TP, Wojcikiewicz RJ, Hill CE. Inositol 1,4,5‐trisphosphate receptor subtypes are differentially distributed between smooth muscle and endothelial layers of rat arteries. Cell Calcium. 2004;36:447–458. [DOI] [PubMed] [Google Scholar]
- 15. Yuan Q, Yang J, Santulli G, Reiken SR, Wronska A, Kim MM, Osborne BW, Lacampagne A, Yin Y, Marks AR. Maintenance of normal blood pressure is dependent on IP3R1‐mediated regulation of eNOS. Proc Natl Acad Sci USA. 2016;113:8532–8537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schlaeger TM, Mikkola HK, Gekas C, Helgadottir HB, Orkin SH. Tie2Cre‐mediated gene ablation defines the stem‐cell leukemia gene (SCL/tal1)‐dependent window during hematopoietic stem‐cell development. Blood. 2005;105:3871–3874. [DOI] [PubMed] [Google Scholar]
- 17. Ouyang K, Leandro Gomez‐Amaro R, Stachura DL, Tang H, Peng X, Fang X, Traver D, Evans SM, Chen J. Loss of IP3R‐dependent Ca2+ signalling in thymocytes leads to aberrant development and acute lymphoblastic leukemia. Nat Commun. 2014;5:4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li X, Zima AV, Sheikh F, Blatter LA, Chen J. Endothelin‐1‐induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol‐1,4,5‐trisphosphate(IP3)‐receptor type 2‐deficient mice. Circ Res. 2005;96:1274–1281. [DOI] [PubMed] [Google Scholar]
- 19. Claxton S, Kostourou V, Jadeja S, Chambon P, Hodivala‐Dilke K, Fruttiger M. Efficient, inducible Cre‐recombinase activation in vascular endothelium. Genesis. 2008;46:74–80. [DOI] [PubMed] [Google Scholar]
- 20. Tang H, Wang H, Lin Q, Fan F, Zhang F, Peng X, Fang X, Liu J, Ouyang K. Loss of IP3 receptor‐mediated Ca(2+) release in mouse B cells results in abnormal B cell development and function. J Immunol. 2017;199:570–580. [DOI] [PubMed] [Google Scholar]
- 21. Wang H, Jing R, Trexler C, Li Y, Tang H, Pan Z, Zhu S, Zhao B, Fang X, Liu J, Chen J, Ouyang K. Deletion of IP3R1 by Pdgfrb‐Cre in mice results in intestinal pseudo‐obstruction and lethality. J Gastroenterol. 2018. Available at: https://link.springer.com/article/10.1007%2Fs00535-018-1522-7. Accessed January 30, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin Q, Zhao G, Fang X, Peng X, Tang H, Wang H, Jing R, Liu J, Lederer WJ, Chen J, Ouyang K. IP3 receptors regulate vascular smooth muscle contractility and hypertension. JCI Insight. 2016;1:e89402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fang X, Bogomolovas J, Wu T, Zhang W, Liu C, Veevers J, Stroud MJ, Zhang Z, Ma X, Mu Y, Lao DH, Dalton ND, Gu Y, Wang C, Wang M, Liang Y, Lange S, Ouyang K, Peterson KL, Evans SM, Chen J. Loss‐of‐function mutations in co‐chaperone BAG3 destabilize small HSPs and cause cardiomyopathy. J Clin Invest. 2017;127:3189–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fang X, Stroud MJ, Ouyang K, Fang L, Zhang J, Dalton ND, Gu Y, Wu T, Peterson KL, Huang HD, Chen J, Wang N. Adipocyte‐specific loss of PPARgamma attenuates cardiac hypertrophy. JCI Insight. 2016;1:e89908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double‐fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. [DOI] [PubMed] [Google Scholar]
- 26. Widlansky ME, Gokce N, Keaney JF Jr, Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol. 2003;42:1149–1160. [DOI] [PubMed] [Google Scholar]
- 27. Patel S, Joseph SK, Thomas AP. Molecular properties of inositol 1,4,5‐trisphosphate receptors. Cell Calcium. 1999;25:247–264. [DOI] [PubMed] [Google Scholar]
- 28. Taylor CW, Genazzani AA, Morris SA. Expression of inositol trisphosphate receptors. Cell Calcium. 1999;26:237–251. [DOI] [PubMed] [Google Scholar]
- 29. Futatsugi A, Nakamura T, Yamada MK, Ebisui E, Nakamura K, Uchida K, Kitaguchi T, Takahashi‐Iwanaga H, Noda T, Aruga J, Mikoshiba K. IP3 receptor types 2 and 3 mediate exocrine secretion underlying energy metabolism. Science. 2005;309:2232–2234. [DOI] [PubMed] [Google Scholar]
- 30. Payne S, De Val S, Neal A. Endothelial‐specific Cre mouse models. Arterioscler Thromb Vasc Biol. 2018;38:2550–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen MJ, Yokomizo T, Zeigler BM, Dzierzak E, Speck NA. Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature. 2009;457:887–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sanders KM. Invited review: mechanisms of calcium handling in smooth muscles. J Appl Physiol (1985). 2001;91:1438–1449. [DOI] [PubMed] [Google Scholar]
- 33. Ogut O, Brozovich FV. Regulation of force in vascular smooth muscle. J Mol Cell Cardiol. 2003;35:347–355. [DOI] [PubMed] [Google Scholar]
- 34. Ludmer PL, Selwyn AP, Shook TL, Wayne RR, Mudge GH, Alexander RW, Ganz P. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N Engl J Med. 1986;315:1046–1051. [DOI] [PubMed] [Google Scholar]
- 35. el‐Tamimi H, Mansour M, Wargovich TJ, Hill JA, Kerensky RA, Conti CR, Pepine CJ. Constrictor and dilator responses to intracoronary acetylcholine in adjacent segments of the same coronary artery in patients with coronary artery disease: endothelial function revisited. Circulation. 1994;89:45–51. [DOI] [PubMed] [Google Scholar]
- 36. Chi JT, Chang HY, Haraldsen G, Jahnsen FL, Troyanskaya OG, Chang DS, Wang Z, Rockson SG, van de Rijn M, Botstein D, Brown PO. Endothelial cell diversity revealed by global expression profiling. Proc Natl Acad Sci USA. 2003;100:10623–10628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Conway EM, Carmeliet P. The diversity of endothelial cells: a challenge for therapeutic angiogenesis. Genome Biol. 2004;5:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Marcu R, Choi YJ, Xue J, Fortin CL, Wang Y, Nagao RJ, Xu J, MacDonald JW, Bammler TK, Murry CE, Muczynski K, Stevens KR, Himmelfarb J, Schwartz SM, Zheng Y. Human organ‐specific endothelial cell heterogeneity. iScience. 2018;4:20–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Atochin DN, Wang A, Liu VW, Critchlow JD, Dantas AP, Looft‐Wilson R, Murata T, Salomone S, Shin HK, Ayata C, Moskowitz MA, Michel T, Sessa WC, Huang PL. The phosphorylation state of eNOS modulates vascular reactivity and outcome of cerebral ischemia in vivo. J Clin Invest. 2007;117:1961–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Venugopal SK, Devaraj S, Yuhanna I, Shaul P, Jialal I. Demonstration that C‐reactive protein decreases eNOS expression and bioactivity in human aortic endothelial cells. Circulation. 2002;106:1439–1441. [DOI] [PubMed] [Google Scholar]
- 41. Scherrer‐Crosbie M, Ullrich R, Bloch KD, Nakajima H, Nasseri B, Aretz HT, Lindsey ML, Vancon AC, Huang PL, Lee RT, Zapol WM, Picard MH. Endothelial nitric oxide synthase limits left ventricular remodeling after myocardial infarction in mice. Circulation. 2001;104:1286–1291. [DOI] [PubMed] [Google Scholar]
- 42. Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J Clin Invest. 2001;108:1341–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pasceri V, Willerson JT, Yeh ET. Direct proinflammatory effect of C‐reactive protein on human endothelial cells. Circulation. 2000;102:2165–2168. [DOI] [PubMed] [Google Scholar]
- 44. Kuchan MJ, Frangos JA. Role of calcium and calmodulin in flow‐induced nitric oxide production in endothelial cells. Am J Physiol. 1994;266:C628–C636. [DOI] [PubMed] [Google Scholar]
- 45. Busse R, Mulsch A. Calcium‐dependent nitric oxide synthesis in endothelial cytosol is mediated by calmodulin. FEBS Lett. 1990;265:133–136. [DOI] [PubMed] [Google Scholar]
- 46. Luckhoff A, Pohl U, Mulsch A, Busse R. Differential role of extra‐ and intracellular calcium in the release of EDRF and prostacyclin from cultured endothelial cells. Br J Pharmacol. 1988;95:189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang B, Liu D, Wang C, Wang Q, Zhang H, Liu G, Tao X, Zhang L. Mechanism of endothelial nitric oxide synthase phosphorylation and activation by tentacle extract from the jellyfish Cyanea capillata . PeerJ. 2017;5:e3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kassan M, Zhang W, Aissa KA, Stolwijk J, Trebak M, Matrougui K. Differential role for stromal interacting molecule 1 in the regulation of vascular function. Pflugers Arch. 2015;467:1195–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schwarz G, Droogmans G, Nilius B. Shear stress induced membrane currents and calcium transients in human vascular endothelial cells. Pflugers Arch. 1992;421:394–396. [DOI] [PubMed] [Google Scholar]
- 50. Shen J, Luscinskas FW, Connolly A, Dewey CF Jr, Gimbrone MA Jr. Fluid shear stress modulates cytosolic free calcium in vascular endothelial cells. Am J Physiol. 1992;262:C384–C390. [DOI] [PubMed] [Google Scholar]
- 51. Ayajiki K, Kindermann M, Hecker M, Fleming I, Busse R. Intracellular pH and tyrosine phosphorylation but not calcium determine shear stress‐induced nitric oxide production in native endothelial cells. Circ Res. 1996;78:750–758. [DOI] [PubMed] [Google Scholar]
- 52. Mount PF, Kemp BE, Power DA. Regulation of endothelial and myocardial NO synthesis by multi‐site eNOS phosphorylation. J Mol Cell Cardiol. 2007;42:271–279. [DOI] [PubMed] [Google Scholar]
- 53. Dudzinski DM, Michel T. Life history of eNOS: partners and pathways. Cardiovasc Res. 2007;75:247–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Boo YC, Sorescu G, Boyd N, Shiojima I, Walsh K, Du J, Jo H. Shear stress stimulates phosphorylation of endothelial nitric‐oxide synthase at Ser1179 by Akt‐independent mechanisms: role of protein kinase A. J Biol Chem. 2002;277:3388–3396. [DOI] [PubMed] [Google Scholar]
- 55. Harris MB, Ju H, Venema VJ, Liang H, Zou R, Michell BJ, Chen ZP, Kemp BE, Venema RC. Reciprocal phosphorylation and regulation of endothelial nitric‐oxide synthase in response to bradykinin stimulation. J Biol Chem. 2001;276:16587–16591. [DOI] [PubMed] [Google Scholar]
- 56. Montagnani M, Chen H, Barr VA, Quon MJ. Insulin‐stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser(1179). J Biol Chem. 2001;276:30392–30398. [DOI] [PubMed] [Google Scholar]
- 57. Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin‐dependent endothelial nitric oxide synthase activity. Circ Res. 2001;88:E68–E75. [DOI] [PubMed] [Google Scholar]
- 58. Greif DM, Kou R, Michel T. Site‐specific dephosphorylation of endothelial nitric oxide synthase by protein phosphatase 2A: evidence for crosstalk between phosphorylation sites. Biochemistry. 2002;41:15845–15853. [DOI] [PubMed] [Google Scholar]
- 59. Matsubara M, Hayashi N, Jing T, Titani K. Regulation of endothelial nitric oxide synthase by protein kinase C. J Biochem. 2003;133:773–781. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers Sequences for RT‐PCR
Figure S1. Endolthelial cell‐specific gene deletion in adult mice by Pdgfb‐iCreER. The Pdgfb‐iCreER (iCre+) mice were crossed with ROSAmT/mG reporter mice to generate the iCre+ROSAmT/mG mice. Administration of tamoxifen in adult iCre+ROSAmT/mG mice led to expression of cell membrane‐localized EGFP in endothelial cells, while non‐endothelial cells expressed cell membrane‐localized tdTomato. A, Schematic diagram demonstrating the crossing strategy and the induction of EGFP expression. B, Confocal fluorescent imaging showing the localization of EGFP and tdTomato in the cross‐section of the mesenteric artery isolated from iCre+ROSAmT/mG mice after tamoxifen administration. Scale bar, 40 μm.
Figure S2. Deletion of IP3R1 by Tie2‐Cre in mice did not alter blood pressure and vasodilation. The constitutively active Tie2‐Cre instead of the inducible Pdgfb‐iCreER was used to delete IP3R1 in endothelial cells from as early as the embryonic stage. A, Quantitative RT‐PCR analysis of the expression of 3 IP3R subtypes in isolated endothelial cells from Tie2‐Cre−R1f/f and Tie2‐Cre+R1f/f mice. n=3 (with endothelial cells from 3 mice pooled as one sample) per group. Significance was determined by the 2‐tailed, unpaired student t test. **P<0.0 vs Tie2‐Cre−R1f/f mice. Data are presented as mean±SEM. B, Systolic blood pressure (SBP) were measured at the ages of 3 and 6 months using the tail cuff system, respectively. n=5 to 8 mice per group. Significance was determined by 2‐way ANOVA analysis with Bonferroni post‐hoc test. C, Vascular reactivity in response to ACh in aortas and mesenteric arteries. The vessels were pre‐constricted by 10 μmol/L phenylephrine (PE) and the vasorelaxing effects of ACh were presented as a percentage of PE‐induced contractions. n=6 mice per group. Significance was determined by 2‐way ANOVA analysis with Bonferroni post‐hoc test. D, The ratio of ventricle weight to body weight was comparable between Tie2‐Cre−R1f/f and Tie2‐Cre+R1f/f mice at the age of 6 months. n=9 mice per group. Significance was determined by the 2‐tailed, unpaired student t test.
Figure S3. Mouse breeding strategy and measurement of vascular contractility in control and ECTKO mice. A, Schematic diagram showing the mouse breeding strategy of generate iCre+IP3R1f/f IP3R2f/f IP3R3f/f mice. B, Reference contraction induced by high potassium (100 mmol/L) and the dose‐dependent contractile response to phenylephrine (PE) in control and ECTKO aortas. n=6 per group. C, Reference contraction induced by high potassium (100 mmol/L) and the dose‐dependent contractile response to phenylephrine (PE) in control and ECTKO mesenteric arteries. n=6 per group. For all dose‐response curves, data were expressed as a percentage of the peak of K+‐induced contraction, and significance was determined by 2‐tailed, unpaired student t test or 2‐way ANOVA analysis with Bonferroni post‐hoc test. **P<0.0 vs control. Error bars represent mean±SEM.