

Abstract
Background
Mutations in αB‐crystallin result in proteotoxic cardiomyopathy with desmin mislocalization to protein aggregates. Intermittent fasting (IF) is a novel approach to activate transcription factor EB (TFEB), a master regulator of the autophagy‐lysosomal pathway, in the myocardium. We tested whether TFEB activation can be harnessed to treat advanced proteotoxic cardiomyopathy.
Methods and Results
Mice overexpressing the R120G mutant of αB‐crystallin in cardiomyocytes (Myh6‐CryABR120G) were subjected to IF or ad‐lib feeding, or transduced with adeno‐associated virus–TFEB or adeno‐associated virus—green fluorescent protein after development of advanced proteotoxic cardiomyopathy. Adeno‐associated virus–short hairpin RNA–mediated knockdown of TFEB and HSPB8 was performed simultaneously with IF. Myh6‐CryABR120G mice demonstrated impaired autophagic flux, reduced lysosome abundance, and mammalian target of rapamycin activation in the myocardium. IF resulted in mammalian target of rapamycin inhibition and nuclear translocation of TFEB with restored lysosome abundance and autophagic flux; and reduced aggregates with normalized desmin localization. IF also attenuated left ventricular dilation and myocardial hypertrophy, increased percentage fractional shortening, and increased survival. Adeno‐associated virus–TFEB transduction was sufficient to rescue cardiomyopathic manifestations, and resulted in reduced aggregates and normalized desmin localization in Myh6‐CryABR120G mice. CryABR120G‐expressing hearts demonstrated increased interaction of desmin with αB‐crystallin and reduced interaction with chaperone protein, HSPB8, compared with wild type, which was reversed by both IF and TFEB transduction. TFEB stimulated autophagic flux to remove protein aggregates and transcriptionally upregulated HSPB8, to restore normal desmin localization in CryABR120G‐expressing cardiomyocytes. Short hairpin RNA–mediated knockdown of TFEB and HSPB8 abrogated IF effects, in vivo.
Conclusions
IF and TFEB activation are clinically relevant therapeutic strategies to rescue advanced R120G αB‐crystallin mutant‐induced cardiomyopathy by normalizing desmin localization via autophagy‐dependent and autophagy‐independent mechanisms.
Keywords: cardiomyopathy, HspB8, intermittent fasting, protein aggregates, TFEB, αB‐crystallin
Subject Categories: Animal Models of Human Disease, Basic Science Research, Cell Biology/Structural Biology, Myocardial Biology
Short abstract
See Editorial by Mukai et al
Clinical Perspective
What Is New?
Advanced cardiomyopathy, induced by expression of the R120G mutant of the small heat shock protein, αB‐crystallin, can be rescued by intermittent fasting and adeno‐associated virus–mediated gene therapy to transduce transcription factor EB, a master regulator of autophagy‐lysosome pathway.
Transcription factor EB activation by intermittent fasting and adeno‐associated virus–mediated gene delivery accelerates protein aggregate removal, and permits physiologic relocalization of desmin away from the aggregates to the Z‐discs and intercalated discs by transcriptional induction of HSPB8, another member of the small heat shock protein family.
What Are the Clinical Implications?
Intermittent fasting and adeno‐associated virus–mediated transcription factor EB gene delivery are promising translational strategies to abrogate cardiomyopathic manifestations with protein‐aggregate pathologic features in the myocardium.
These paradigms may also be broadly applicable to prevention and/or treatment of cardiomyopathies characterized by desmin mislocalization and related “desminopathies.”
Mutations in sarcomeric and cardiac cytoskeletal proteins are being increasingly ascribed causative roles in engendering cardiomyopathy spanning the spectrum of dilated, hypertrophic, restrictive, and arrythmogenic right ventricular dysplasia phenotypes.1 One such genetically heterogeneous group of disorders, termed “desminopathies,” is characterized by a deficiency or mislocalization of desmin, a muscle‐specific type III intermediate filament that serves a critical scaffolding role in linking the sarcomere to other intracellular structures.2 A cardiac‐enriched small heat shock family protein, αB‐crystallin, binds desmin; and a missense mutation that leads to an arginine‐to‐glycine change at position 120 (R120G) results in loss of its chaperone function,3 with formation of protein aggregates that manifest as an autosomal dominant cardiomyopathy, cataracts, and skeletal myopathy in humans.4 Analogously, mutations in desmin also provoke cardiomyopathy, and formation of amyloid protein aggregates in cardiac myocytes is a hallmark of disease pathogenesis.5 Whether facilitating removal of protein aggregates can reverse established cardiomyopathy in patients presenting with a full‐blown disease process has not been experimentally evaluated.
Protein aggregation leading to myofibrillar degeneration and sarcomere dysfunction,6, 7, 8 as well as cardiac myocyte apoptosis and interstitial fibrosis,9, 10 have been implicated as proximate mechanisms for development of the progressive cardiomyopathy in mice with cardiomyocyte‐specific expression of the R120G αB‐crystallin mutant (CryABR120G) protein. Although cardiomyocyte protein quality‐control pathways (namely, the heat shock family of chaperones,8 the ubiquitin‐proteasome pathway,11 and autophagy)7 are induced in these models, further upregulation of small heat shock proteins,12, 13 overexpression of an 11S subunit to activate proteasomes,14 and stimulation of autophagy with ATG7 overexpression or exercise15, 16 are individually sufficient to prevent development of aggregates and attenuate cardiomyopathy. Whether targeting these pathways individually or combinatorially rescues advanced cardiomyopathy with CryABR120G expression has not been evaluated.
Mislocalization of desmin from its physiologic location in association with Z‐discs between sarcomeres and intercalated discs at intercellular junctions to protein aggregates is a common feature of CryABR120G‐induced cardiomyopathy and desmin‐related cardiomyopathies in the human myocardium and in animal models.6, 17 CryABR120G‐expressing mice as well as mice with desmin mutations resulting in loss of its sarcomeric localization demonstrate impaired cardiac myocyte contractility9, 17 and mitochondrial abnormalities9, 18; and desmin null mice demonstrate myofibrillar breakdown with accelerated cardiomyocyte death and cardiomyopathy,19, 20 pointing to a critical role for desmin in cardiomyocyte homeostasis. The CryABR120G mutant protein binds desmin with increased affinity compared with wild‐type αB‐crystallin protein,21 is defective in chaperone function,3 and gets ubiquitinated to be sequestered in aggregates6, 7 or targeted for proteolytic degradation. Formation of protein aggregates is protective because strategies to reduce ubiquitination of CryABR120G by targeting the COP9 (Constitutive photomorphogenesis 9) signalosome22 or preventing its aggregation by depletion of p62,23 an adaptor protein that sequesters ubiquitinated proteins in aggregates, enhance CryABR120G‐induced cytotoxicity. However, despite accelerating protein aggregate pathologic features with age,10 cardiac function progressively declines in CryABR120G‐expressing hearts,7 suggesting the hypothesis that sequestration of other proteins, such as desmin, within aggregates may play a pathologic role in the progressive cardiomyopathy. Whether strategies to correct desmin localization will be synergistic with those targeting removal of protein aggregates remains to be explored.
Cardiomyocyte autophagy plays a critical role in slowing progression of CryABR120G‐induced cardiomyopathy because concomitant haploinsufficiency of Beclin‐1 resulted in accelerated disease progression in a mouse model of human CryABR120G protein expression.7 In studies presented herein, we have uncovered impairment in autophagic flux at an advanced, preterminal stage of cardiomyopathy, associated with inactivation of transcription factor EB (TFEB), the master regulator of autophagy and lysosomal machinery.24 Concurrently, we found hyperactivation of mammalian target of rapamycin (mTOR), the kinase that phosphorylates and inactivates TFEB, thus keeping it sequestered and inactive in the cytosol.25 We have observed that fasting is a potent stimulus for TFEB activation in the myocardium and that intermittent fasting (IF) facilitates mitochondrial quality control and preconditions the myocardium to ischemia‐reperfusion injury.26 Herein, we have evaluated the feasibility of IF and TFEB gene therapy, as a strategy to rescue advanced cardiomyopathy resulting from expression of the R120G αB‐crystallin mutant protein. Our findings reveal that TFEB activation coordinately drives protein quality control pathways in the myocardium to restore normal desmin localization via accelerating autophagic removal of protein aggregates and transcriptional induction of HSPB8 (Heat Shock Protein Family B (Small) Member 8), to attenuate established cardiomyopathy.
Methods
The authors declare that all supporting data are available within the article. The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedures reported in this article.
Studies in Mice
Transgenic mice with cardiomyocyte‐specific expression of R120G mutant of human αB‐crystallin protein (ie, Myh6‐CryABR120G transgenic mice described previously)8 and age‐ and sex‐matched littermate controls were used for all the studies. CryAB null mice27 were generously provided by Dr Eric Wawrousek. Echocardiographic studies and IF were performed as previously described (also see Data S1 for details).26 Terminal studies on mice were initiated between 8 and 10 am after an overnight period of feeding (ie, on a fed day). All animal studies were approved by the Institutional Animal Care and Use Committees at Washington University School of Medicine and at the John Cochran VA Medical Center. Assessment of autophagic flux, in vivo, was performed with injection of chloroquine (40 mg/kg IP, Sigma catalog No. C6628) 4 hours before euthanasia, as previously described.26
Ultrastructural Examination
Transmission electron microscopy was performed on mice hearts fixed in a modified Karnovsky's fixative, followed by imaging with a JEOL model 1200 EX electron microscope (JEOL, Tokyo, Japan), as previously described.26
Quantitative Polymerase Chain Reaction Analysis
Total RNA was extracted from mouse hearts or neonatal rat cardiac myocytes (NRCMs) using RNAeasy Fibrous Tissue Kit (Qiagen, 74704) or the Mini kit (Qiagen, 74104), respectively. Quantitative polymerase chain reaction analysis was performed as described.26 Primers used are shown in Table S1.
Assessment of Mitochondrial DNA Content
DNA was extracted from NRCMs modeled with adenoviral gain‐of function and loss‐of‐function approach pellets, and real‐time quantitative polymerase chain reaction was performed for the mitochondrial gene NADH2 (with primers 5′‐ATCACCACCATTCTCGCAAT‐3′ and 5′‐TCCTATGTGGGCAATTGATG‐3′) and the nuclear gene RCAN1 (with primers 5′‐GGTTTGCTGAGCCTCGAAG‐3′ and 5′‐CTTCATCCCTCCTTTGTAAC‐3′). The ratio of copies of mitochondrial/nuclear genes was calculated to represent the relative mitochondrial DNA content, as we have previously described.28
Generation of Adenoviral Constructs
Adenoviral particles coding for LacZ and human TFEB with N‐terminal FLAG have been described previously.29 Adenoviral particles for expression of short hairpin RNA (shRNA) targeting rat hspb8 and a LacZ shRNA control were generated with BLOCKiT adenoviral system (Invitrogen, K4941–00). Specific oligonucleotide sequences used were as follows: shRNA targeting rat hspb8–top strand oligonucleotide, 5′‐ CACCGTCAAAGTATTGGGAGGTTCGAAAACCTCCCAATACTTTGAC‐3′; bottom strand oligonucleotide, 5′‐AAAAGTCAAAGTATTGGGAGGTTTTCGAACCTCCCAATACTTTGAC‐3′. Adenoviruses were generated in HEK293A cells and titered per the manufacturer's instructions.
Generation of Adeno‐Associated Virus Constructs
Mouse TFEB cDNA was cloned into a phosphorylated adeno‐associated virus (AAV9) vector containing the CMV (cytomegalovirus) promoter (phosphorylated AAV9–CMV–FLAG–TFEB), as we have previously described.30 Coding sequences for human CRYAB and CRYAB protein with the R120G mutation were similarly cloned into a phosphorylated AAV9 vector containing the cardiac‐specific troponin T promoter.31 Constructs for in vivo knockdown of mouse endogenous TFEB and HSPB8 were generated using the specific oligonucleotide sequences targeting murine TFEB (5′‐CGGCAGTACTATGACTATGAT‐3′) and HSPB8 (5′‐GCCTTGGAAAGTGTGTGTCAA‐3′); and compared with a scrambled control sequence (with no complementarity to murine genome) (5′‐CCTAAGGTTAAGTCGCCCTCG‐3′) by cloning into an AAV9 vector under the control of a U6 promoter. AAV9 particles were generated at the Hope Center Viral Vectors Core at Washington University School of Medicine. A 1‐time dose of 3.5×1011 particles was administered to the mice by tail vein injection.
Subcellular Fractionation Into Cytoplasmic and Nuclear Fractions
Hearts were fractionated into nucleus‐enriched and cytoplasmic samples by using a CelLytic NuCLEAR extraction kit (Sigma), as previously described.26
Isolation of Soluble and Insoluble Fractions
Heart tissue was fractionated into soluble and insoluble fractions, as previously described (also see Data S1 for details).7
Immunoblotting
Crude cardiac or NRCM extracts were prepared, followed by immunoblotting, as described.26 Antibodies used were as follows: LAMP2 (Lysosomal Associated Membrane Protein 2), mouse monoclonal (Developmental Studies Hybridoma Bank, ABL‐93); LAMP1 (Santa Cruz Biotechnology, sc‐19992); anti‐LC3 (encoding for MAP1LC3B (Microtubule Associated Protein 1 Light Chain 3 Beta) subunit; Novus Biologicals, NB100–2220); SQSTM1 (Sequestosome 1) (Abcam, ab5416); TFEB (Bethyl Labs, A303–673A); HA (H6908; Sigma), p70S6K (phosphorylated Ribosomal protein S6 kinase beta‐1) (2708; Cell Signaling); phosphorylated p70S6K (9234; Cell Signaling); 4‐EBP1 (Eukaryotic translation initiation factor 4E‐binding protein 1) (9644; Cell Signaling); phosphorylated 4EBP1 (2855; Cell Signaling); phosphorylated mTOR (2974; Cell Signaling); mTOR (2983; Cell Signaling); histone H3 (9715, Cell Signaling); GAPDH (ab22555; Abcam); Hspb8 (3059S, Cell signaling); and ACTA1/α‐sarcomeric actin (Abcam, ab7799) or actin (Sigma, A2066). Protein abundance was normalized to actin or GAPDH protein expression and reported as fold change versus control.
Immunofluorescence Analysis
Primary cultures of NRCMs26 were fixed in 100% cold methanol for 20 minutes, followed by blocking with 1% normal serum in PBS for 1 hour at room temperature. Primary antibodies used were as follows: anti‐desmin (Santa Cruz Biotechnology, Inc, SC‐7559), anti–αB‐crystallin polyclonal antibody (ENZO Life, ADI‐SPA‐223‐F), anti‐p62/SQSTM1 (PROGEN Biotechnik, GP62‐C), anti–ubiquitinated FK‐2 (Millipore; 04‐263), and TFEB (Bethyl Labs, A303–673A) with overnight incubation at 4°C. Paraffin‐embedded heart sections (10 µm thick) were subjected to heat‐induced epitope retrieval, followed by blocking, and incubated overnight with primary antibody against desmin (Santa Cruz Biotechnology, SC‐7559). After serial washes, samples were stained with AlexaFluor488 (Invitrogen) and mounted with fluorescent 4’,6‐diamidino‐2‐phenylindole mounting medium (Vector Labs, H‐1200). Confocal imaging was performed on a Zeiss confocal LSM‐700 laser scanning confocal microscope using 63× Zeiss Plan‐Neofluar 40×/1.3 and 63×/1.4 oil immersion objectives, and images were acquired using Zen 2010 software. Assessment of terminal deoxynucleotidyl transferase‐mediated dUTP nick‐end labeling positivity in myocardial samples was performed as previously described.26 Intracellular protein aggregates in cardiac myocytes were quantitated on ×20 images of paraffin‐fixed tissue immunostained for desmin. Aggregates were identified using the ImageJ software by counting particles greater than a predefined threshold size and assessing an automated aggregate area as a percentage of the total area. A minimum of 10 images per heart were counted to obtain the average aggregate area percentage. At least 3 biological replicates were assessed for each group.
Flow Cytometry
NRCMs were incubated with JC‐1 (10 mg/mL for 10 minutes) at 37°C in 5% CO2, followed by trypsinization and flow cytometry on a FACScan instrument (Becton‐Dickinson), as previously described.26 Cyflogic software was used to analyze 20 000 events per run.
Assessment of Cell Death
Cell death assays were performed in 96‐well plates with a Live‐Dead cytotoxicity viability kit for mammalian cells (Invitrogen), as described previously.26
Coimmunoprecipitation Assays
One milligram of protein from the mouse hearts was incubated with anti‐desmin (SC7559; Santa Cruz Biotechnology), anti‐mouse FLAG (F3165; Sigma), and normal goat IgG (SC2028, Santa Cruz Biotechnology) or normal mouse IgG (SC2025, Santa Cruz Biotechnology); immunoprecipitation was performed using Dynabeads Protein G Immunoprecipitation Kit (Thermo Fisher Scientific, 10007D). The eluted samples are analyzed by SDS‐PAGE, followed by Western blot, to detect desmin (anti‐desmin, D1033, Sigma), αB‐crystallin (anti–αB‐crystallin, ADI‐SPA‐223‐F, Enzo Life), and HSPB8/HSP22 (anti‐HSPB8/HSP22, 3059S, Cell Signaling).
Statistical Analysis
Results are expressed as mean±SEM. Statistical differences were assessed with the unpaired Student t test for 2 independent groups and ANOVA for comparing results across multiple groups with GraphPad Prism software. Tukey's post hoc test was used after ANOVA for testing all pair‐wise comparisons. To overcome the limitations with large‐sample approximations using the above approaches, we redid the analysis using 2 additional statistical software programs for comparing results: R and SAS. With R, we performed t tests with permutation correction for P values by using permTS function of library “perm” of R, selecting method=“exact.ce” in permTS for a complete enumeration. We also tested such analyses via MULTTEST of SAS with mean t test for continuous variables by selecting “pooled” degrees of freedom method, “2‐tailed” tails for continuous tests, and “permutation” P‐value adjustment by selecting “20 000” number of resamples and a random seed. Application of the permTS (R) and MULTTEST (SAS) produced identical results. We have reported the R permTS function P value in Data S1 for quantitative analyses presented in Figures 1, 2, 3, 4, 5, 6, 7 through 8. Log‐rank statistic was used to assess differences in survival. A 2‐tailed value of P<0.05 was considered statistically significant.
Figure 1.
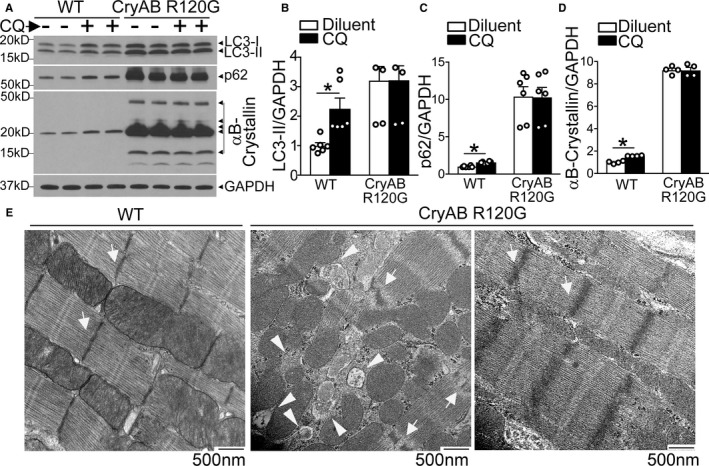
Autophagic flux is impaired in αB‐crystallin R120G mutant transgenic mice with onset of heart failure. A through D, Representative immunoblots (A) demonstrating expression of LC3 with quantitation of LC3‐II (B), p62 (C) and αB‐crystallin (D) in Myh6‐CryABR120G transgenic mice or littermate wild‐type (WT) controls at 40 weeks of age, injected with chloroquine (40 mg/kg) or diluent to assess autophagic flux. N=4 to 6/group. E, Representative myocardial transmission electron micrographs from in Myh6‐CryABR120G transgenic mice or littermate WT controls at 40 weeks of age. Arrows point to Z‐discs, and arrowheads point to autophagic structures. Representative of N=2/group. *P<0.05.
Figure 2.
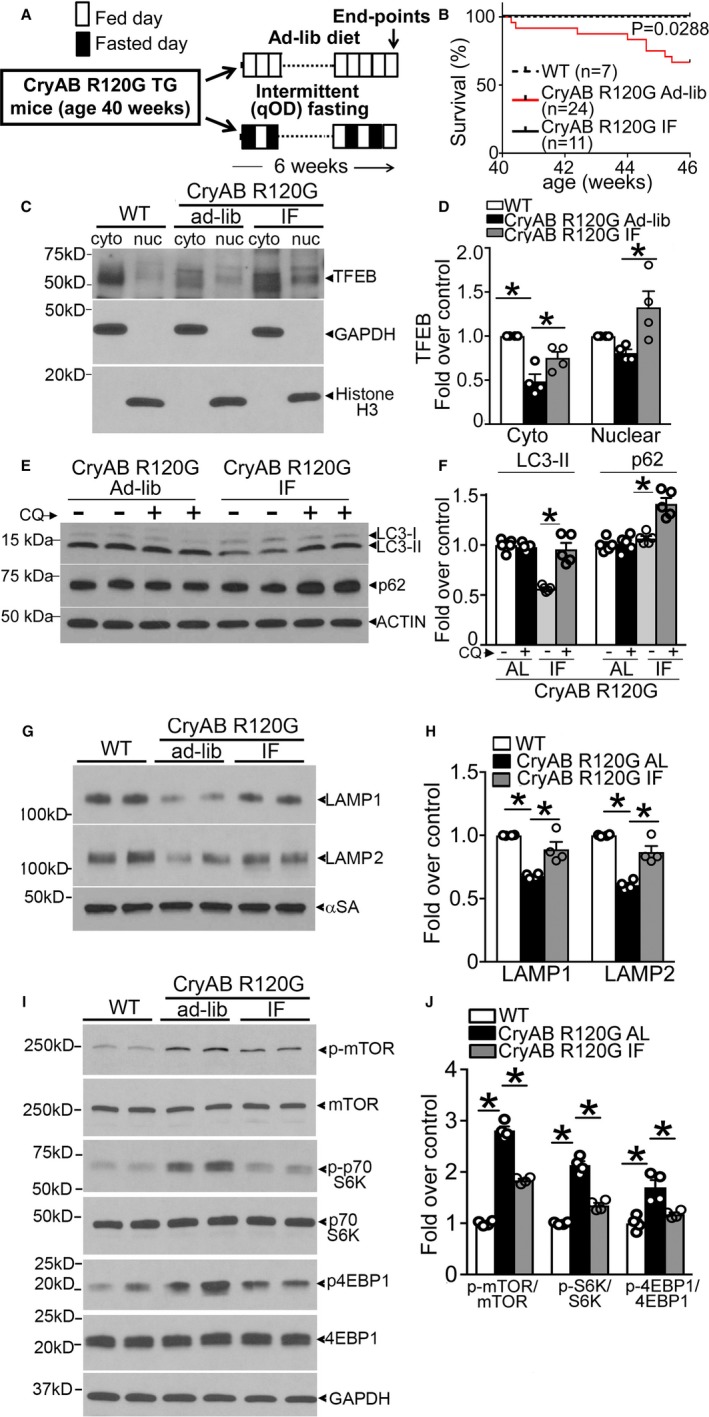
Intermittent fasting (IF) activates transcription factor EB (TFEB) and restores autophagic flux in αB‐crystallin R120G mutant transgenic mice. A, Schematic depicting experimental intervention with IF in Myh6‐CryABR120G transgenic mice. B, Survival curves in IF and ad‐lib (AL) fed Myh6‐CryABR120G transgenic mice and AL fed controls over the 6‐week experimental duration. P value depicted is by log‐rank test. C and D, Representative immunoblots (C) demonstrating expression of TFEB with quantitation (D) in the cytosolic and nuclear fractions from hearts of mice treated as in A (at 46 weeks) and subjected to biochemical fractionation. Expression of GAPDH and histone H3 is used to detect enrichment of cytosolic and nuclear proteins, respectively. N=4/group. E, Assessment of autophagic flux in mice Myh6‐CryABR120G transgenic mice subjected to IF or provided access to food AL as in A, and injected with chloroquine (40 mg/kg for 4 hours) or diluent with immunoblotting for LC3 and p62. F, Quantitation of LC3‐II and p62 in cardiac tissue from mice treated as in E. N=4/group. G and H, Representative immunoblot (G) with quantitation (H) of lysosome proteins LAMP1 and LAMP2 in total cardiac protein extracts from Myh6‐CryABR120G transgenic mice subjected to IF or AL feeding, or littermate wild type (WT) as in A. N=4/group. I and J, Representative immunoblots (I) demonstrating expression of mammalian target of rapamycin (mTOR) pathway proteins with quantitation (J) of phosphorylated mTOR (p‐mTOR), phosphorylated 70S6K (p‐S6K), and phosphorylated 4EBP1 (p‐4EBP1) in Myh6–αB‐crystallin R120G transgenic mice subjected to IF or AL feeding, and WT controls as in A. N=4/group. qOD (every other day). *P value by post hoc test after 1‐way ANOVA.
Figure 3.

Intermittent fasting (IF) promotes myocardial protein aggregate removal in αB‐crystallin R120G mutant transgenic mice. A through C, Representative immunoblot (A) with quantitation of αB‐crystallin in the soluble (B) and insoluble (C) myocardial fractions from Myh6‐CryABR120G transgenic mice subjected to IF or ad‐lib (AL) feeding, and littermate wild‐type (WT) controls at 46 weeks of age. N=4/group. P value is by t test. “Darker” and “lighter” indicate relative exposures of the same blot. D, Representative TEM (Transmission electron microscopy) images from Myh6‐CryABR120G transgenic mice subjected to IF or AL feeding and littermate WT mice. White arrows point to protein aggregates. Black arrows point to autolysosomes. Black arrowheads point to Z‐discs, where intracellular desmin is localized, pointing to partial restoration of Z‐disc architecture in intermittently fasted Myh6‐CryABR120G transgenic mice compared with AL fed controls. Representative of N=2/group.
Figure 4.
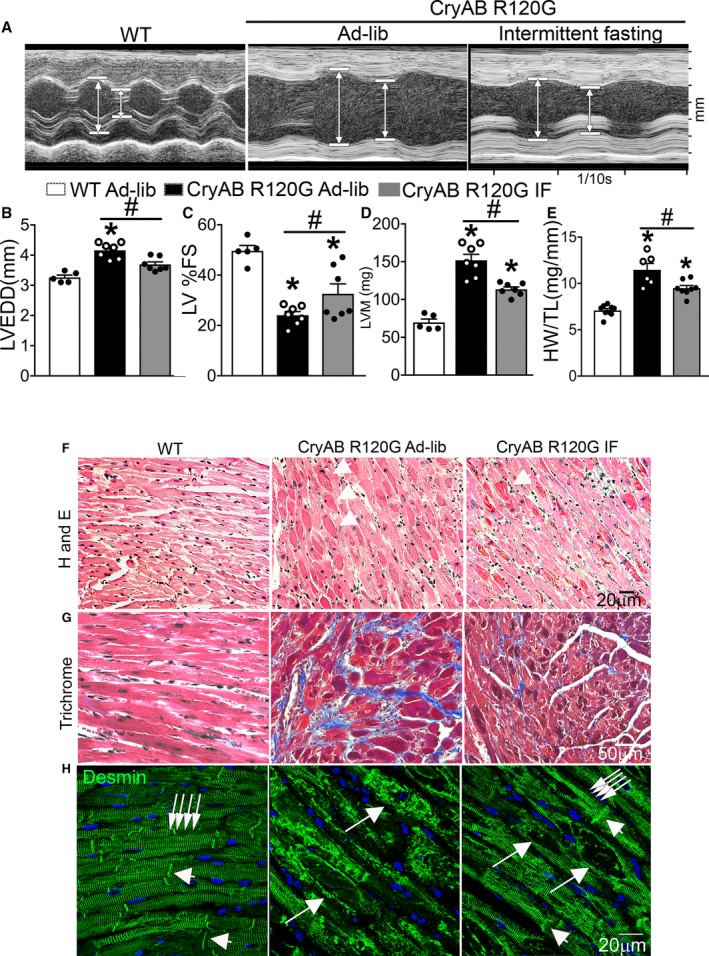
Intermittent fasting (IF) attenuates cardiomyopathy in αB‐crystallin R120G mutant transgenic mice and restores normal desmin localization. A, Representative 2‐dimensional–directed M‐mode echocardiographic images in Myh6‐CryABR120G transgenic mice subjected to IF or ad‐lib feeding for 6 weeks or ad‐lib fed littermate wild‐type (WT) mice at 46 weeks of age. B through E, Quantitation of left ventricular end‐diastolic diameter (LVEDD; B), left ventricular percentage endocardial fractional shortening (LV %FS; C), left ventricular mass (LVM; D), and heart weight normalized to tibial length (HW/TL; E) in mice treated as in A. N=5 to 7 mice/group. See Table S2 for additional echocardiographic data on these mice. F through H, Representative images demonstrating myocardial sections stained with hematoxylin‐eosin (H and E; F) and Masson's trichrome (G) and immunostained for desmin expression (H) in mice treated as in A. White arrows in F point to eosinophilic protein aggregates. In H, white arrows in groups point to Z‐discs and arrowheads indicate I lines to demonstrate desmin localization. Single long white arrows point to desmin localized in aggregates.
Figure 5.
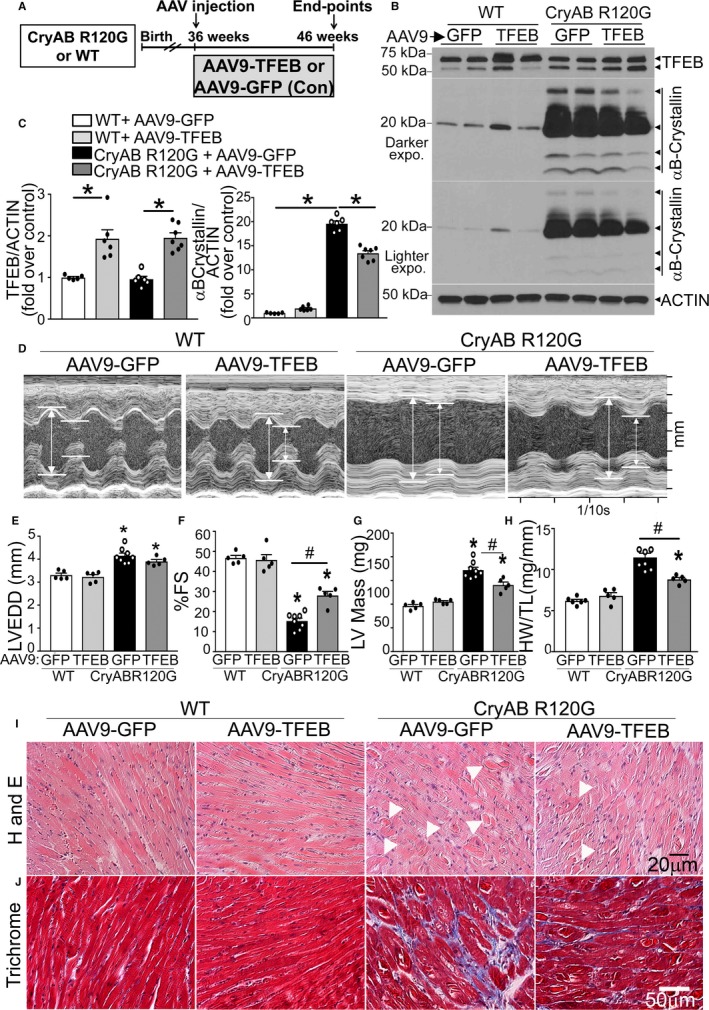
Adeno‐associated virus (AAV9)–mediated transcription factor EB (TFEB) transduction rescues cardiomyopathy in αB‐crystallin R120G mutant transgenic mice and promotes removal of protein aggregates. A, Schematic depicting experimental intervention with AAV9‐mediated TFEB transduction (or AAV9–green fluorescent protein [GFP] as control) in Myh6‐CryABR120G transgenic mice or littermate controls. B and C, Representative immunoblots demonstrating expression of TFEB in total protein extracts with quantitation (C, left) and αB‐crystallin (C, right) in hearts from Myh6‐CryABR120G transgenic mice or littermate wild‐type (WT) mice transduced with AAV9‐TFEB or AAV9‐GFP as in A, at 46 weeks of age. N=5 to 7/group. D through H, Representative 2‐dimensional–directed M‐mode echocardiograms (D) in Myh6‐CryABR120G transgenic mice or littermate WT mice transduced with AAV9‐TFEB or AAV9‐GFP as in A, at 46 weeks of age, with quantitation of left ventricular end‐diastolic diameter (LVEDD; E), left ventricular percentage endocardial fractional shortening (LV %FS; F), LV mass (G), and heart weight normalized to tibial length (HW/TL; H) in mice treated as in A. N=5 to 8/group. A total of 1 of 9 AAV9‐GFP–injected and 1 of 6 AAV9‐TFEB–injected Myh6‐CryABR120G transgenic mice died during the study. See Table S3 for additional echocardiographic data on these mice. I and J, Representative hematoxylin and eosin (H and E; I) and trichrome (J) stained myocardial sections from mice treated as in A. Arrowheads point to eosinophilic aggregates in I. *P<0.05 by post hoc test after 1‐way ANOVA.
Figure 6.
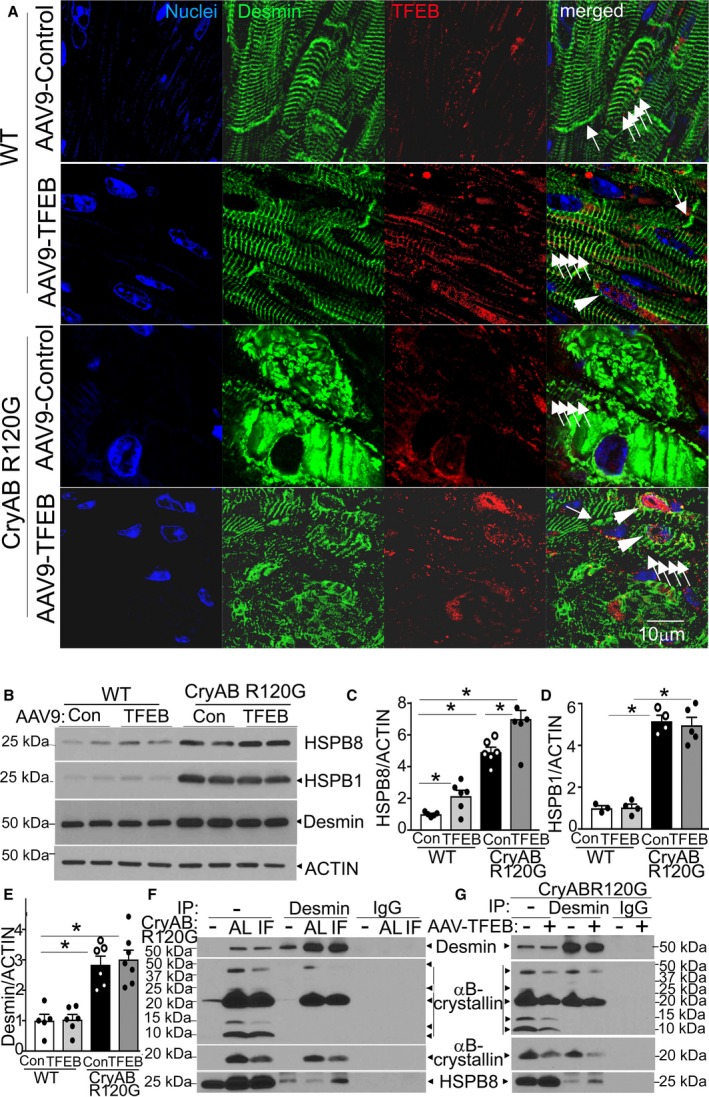
Adeno‐associated virus (AAV9)–mediated transcription factor EB (TFEB) transduction restores normal desmin localization in αB‐crystallin R120G mutant transgenic mice with upregulation of HSPB8. A, Representative images demonstrating immunolocalization of desmin (pseudocolored green) in 46‐week‐old Myh6‐CryABR120G transgenic mice or littermate controls transduced with AAV9‐TFEB or AAV9–green fluorescent protein (GFP) for 10 weeks. White arrowheads point to nuclear TFEB; white arrows in groups point to desmin associated with Z‐discs; single white arrows point to desmin associated with intercalated discs. B through E, Representative immunoblot (B) with quantitation of HSPB8 (C), HSPB1 (D), and desmin (E) in total cardiac protein extracts from mice treated as in A. N=3 to 7/group. F, Immunoblot demonstrating interaction of desmin with HSPB8 and αB‐crystallin in Myh6‐CryABR120G transgenic mice subjected to intermittent fasting (IF) or ad‐lib (AL) feeding and littermate control hearts (depicted as “‐”). Total protein (1 mg) from cardiac extracts was subjected to immunoprecipitation (IP) with anti‐desmin antibody or control IgG, followed by immunoblotting for HSPB8 and αB‐crystallin. At the exposure shown to demonstrate desmin in transgenic samples, a desmin signal was not evident in wild‐type (WT) hearts in the input lane (with 20 µg/sample) and was seen with longer exposure times (data not shown). G, Immunoblot demonstrating interaction of desmin with HSPB8 and αB‐crystallin in Myh6‐CryABR120G transgenic hearts treated with AAV9‐GFP or AAV9‐TFEB and subject to immunoprecipitation with anti‐desmin antibody (or IgG as control), as in G. For αB‐crystallin, both dark (top) and light (bottom) exposures are shown for F and G. *P<0.05 by post hoc test after 1‐way ANOVA.
Figure 7.
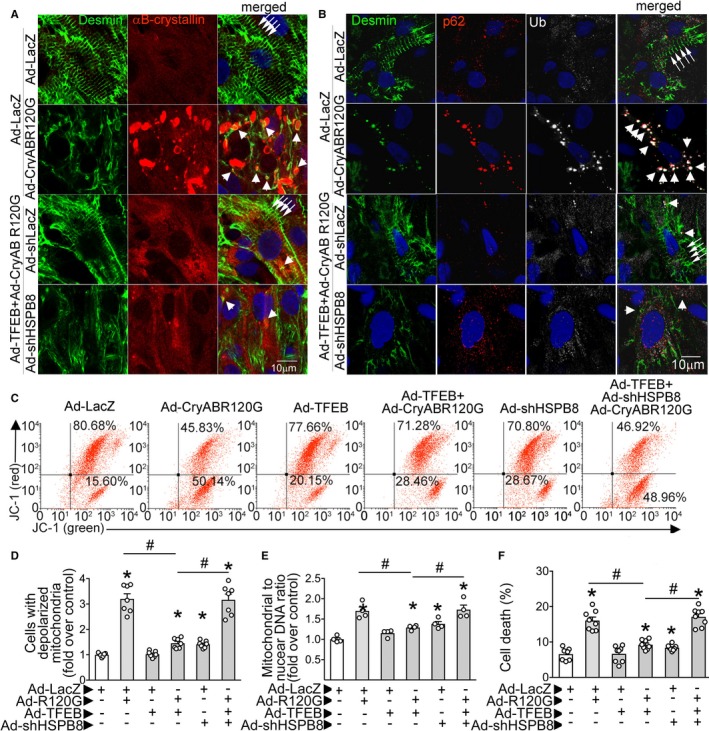
Transcription factor EB (TFEB) normalizes desmin localization via HSPB8 and promotes aggregate removal independent of HSPB8 expression. A, Representative confocal images demonstrating immunolocalization of desmin and αB‐crystallin in neonatal rat cardiac myocytes (NRCMs) adenovirally transduced with TFEB or control, with and without simultaneous knockdown of HSPB8 (ie, treated with adenoviral particles coding for R120G mutant of αB‐crystallin [multiplicity of infection {MOI}=10; for 48 hours] without or with HA‐tagged TFEB [MOI=10; for 24 hours] in the presence of adenoviral particles expressing short hairpin RNA targeting rat HSPB8 [short hairpin HSPB8 {shHSPB8}; MOI=100; for 72 hours]). Adenoviral particles coding for LacZ or short hairpin LacZ were added as controls simultaneously with Ad‐TFEB or Ad–αB‐crystallin overexpression and shHSPB8, respectively, to equalize the number of viral particles. Arrows point to desmin in Z‐discs, and arrowheads point to desmin colocalized with αB‐crystallin in aggregates. Representative of n=2 experiments. B, Representative confocal images demonstrating expression of desmin, ubiquitin, and p62 in NRCMs treated as in A. Arrows point to desmin in Z‐discs, and arrowheads point to desmin colocalized with p62 and ubiquitin in aggregates. Representative of n=2 experiments. C and D, Representative flow cytometric tracings demonstrating JC‐1 fluorescence (C) and quantitation of cells predominantly expressing JC‐1 monomers (lower right quadrants in C; D) for NRCMs treated as described for A. N=7/group. E, Mitochondrial DNA content indexed to nuclear DNA in cells treated as in A. N=4/group. F, Cell death in NRCMs treated as in A. N=7 to 8/group. *P<0.05 by post hoc test after 1‐way ANOVA.
Figure 8.
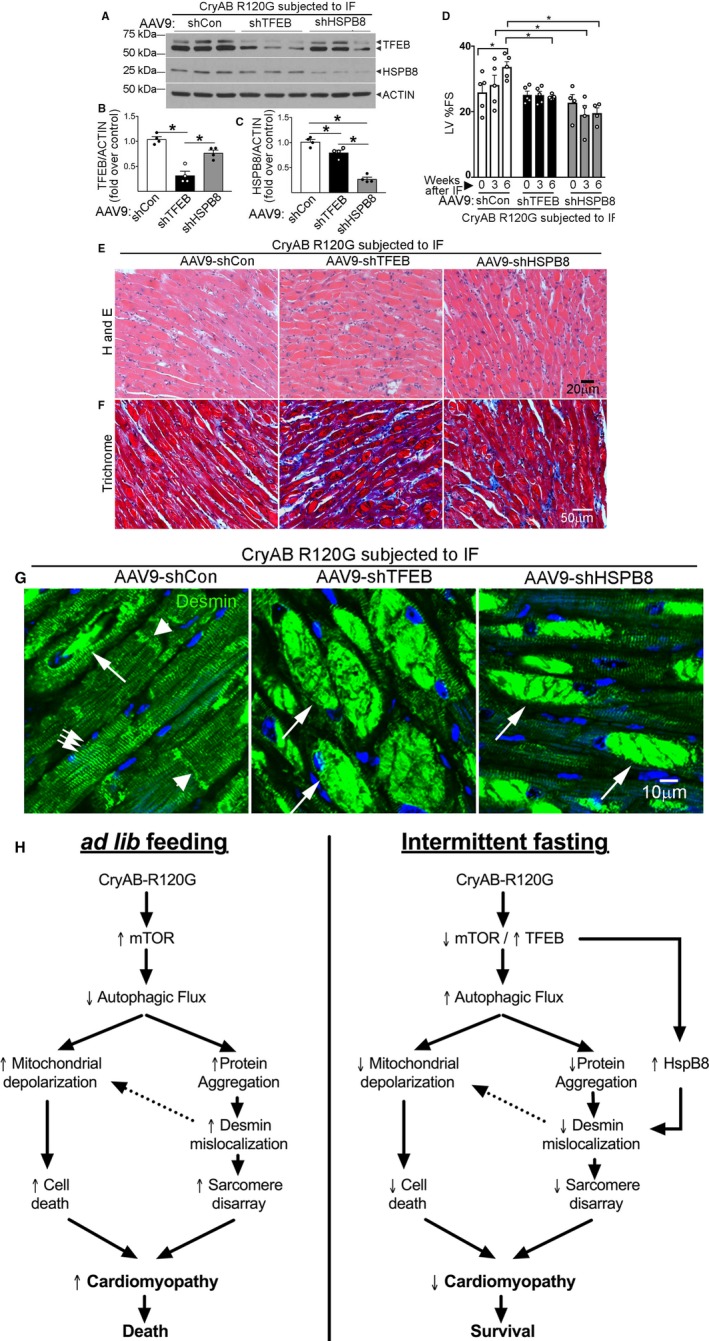
Knockdown of transcription factor EB (TFEB) and HSPB8 prevents intermittent fasting (IF)–mediated attenuation of proteotoxic cardiomyopathy with persistent desmin localization to aggregates. A through C, Representative immunoblot (A) with quantitation of TFEB expression (B) and HSPB8 abundance (C) in total cardiac protein extracts from Myh6‐CryABR120G transgenic mice transduced with adeno‐associated virus (AAV9)–short hairpin control (shCon), AAV9–short hairpin TFEB (shTFEB), or AAV9–short hairpin HSPB8 (shHSPB8) and subjected to IF, assessed at 46 weeks of age. N=4/group. *P<0.05 by post hoc test after 1‐way ANOVA. Please see schematic in Figure S12A for experimental design. D, Left ventricular percentage endocardial fractional shortening (LV %FS) in mice treated as in A and evaluated by echocardiography at baseline (4 weeks after injection of AAV9 particles and before IF, time=0 weeks) and 3 weeks and 6 weeks after initiating IF. N=4 to 5/group. *P<0.05 by post hoc test after 2‐way ANOVA. We did not observe mortality in these treatment groups. See Table S4 for additional echocardiographic data on these mice. E through G, Representative images demonstrating myocardial sections stained with hematoxylin‐eosin (H and E; E) and Masson's trichrome (F) and immunostained for desmin expression (G) in mice treated as in A. In G, white arrows in groups point to Z‐discs and arrowheads indicate intercalated discs to demonstrate desmin localization. Single long white arrows point to desmin localized in aggregates. H, Schematic depicting the mechanisms by which IF benefits advanced cardiomyopathy triggered by αB‐crystallin R120G mutation. Mutant R120G crystallin expression drives mammalian target of rapamycin (mTOR) activation with suppressed autophagic flux and aggregates sequestering desmin within the aggregates, thereby provoking mitochondrial abnormalities and cell death. IF suppresses mTOR activation, which activates TFEB to stimulate lysosome biogenesis and restores autophagic flux to remove mutant crystallin aggregates and relieve desmin sequestration. TFEB activation also stimulates HSPB8 expression, which chaperones desmin to its normal localization and restores mitochondrial quality to rescue cell death.
Results
Autophagic Flux Is Impaired in CryABR120G Transgenic Mice With Advanced Cardiomyopathy
To address the role of autophagy in treatment of established CryABR120G‐induced cardiomyopathy, as typically observed in individuals harboring this mutation when they first seek clinical attention,2 we examined autophagic flux in 40‐week‐old transgenic mice expressing the human R120G αB‐crystallin mutant protein in cardiac myocytes (Myh6‐CryABR120G). A sudden and marked increase in mortality is observed at this time in this model,8 accompanying widespread protein aggregate pathologic features in cardiomyocytes and an end‐stage cardiomyopathy phenotype. As shown in Figure 1A through 1C, short‐term inhibition of lysosomal acidification with chloroquine resulted in accumulation of LC3‐II (the autophagosome bound form of LC3, an index of autophagosome abundance) and p62 (SQSTM1, an adaptor protein that becomes degraded via autophagy) in wild‐type hearts compared with diluent‐treated counterparts. Myh6‐CryABR120G transgenic hearts demonstrated a marked increase in both LC3‐II and p62 compared with wild type, which did not change further with chloroquine treatment (Figure 1A through 1C). To confirm that chloroquine treatment was effective in impairing autophagosome processing in the Myh6‐CryABR120G transgenic mice, we also evaluated autophagic flux in skeletal muscle tissue. Unlike the decreased flux noted in cardiac tissue, chloroquine induced accumulation of LC3‐II and p62 in skeletal muscle, indicating a cardiac‐specific suppression of autophagic flux in this model (Figure S1A). Furthermore, in younger mice without evidence of heart failure, autophagic flux was preserved (Figure S1B and S1C). Taken together, these data indicate impaired flux through macroautophagy (henceforth termed “autophagy”) in Myh6CryABR120G transgenics with advanced cardiomyopathy compared with the preserved flux seen in wild‐type controls. Interestingly, αB‐crystallin levels are also upregulated in chloroquine‐ versus diluent‐treated wild‐type hearts (Figure 1A and 1D), indicating the normal turnover of this chaperone is, at least in part, via autophagy in mice at this age. Interestingly, cardiac extracts from Myh6‐CryABR120G transgenic mice demonstrated both slower and faster migrating bands on PAGE gels, which were confirmed to be αB‐crystallin protein (Figure S2) and likely represent ubiquitinated aggregates and cleavage products of the mutant crystallin protein, respectively, as previously described.22 The abundance of αB‐crystallin protein did not change with chloroquine treatment in the Myh6‐CryABR120G transgenic mice, consistent with impaired autophagic flux (Figure 1A and 1D). Ultrastructural examination of the cardiomyopathic Myh6‐CryABR120G myocardium in aged mice revealed accumulation of multiple autophagic structures (arrowheads, Figure 1E) as well as effaced Z‐discs (arrows, Figure 1E; with broad and often zig‐zag patterned Z‐discs in transgenics, as previously observed in a transgenic model with overexpression of mouse CryABR120G protein6 versus sharp Z‐discs in controls), consistent with the altered localization of desmin adjacent to the Z‐disc in this condition.
IF Restores Autophagic Flux and Rescues Lethality in CryABR120G Transgenic Mice With Advanced Cardiomyopathy
We have observed that IF transcriptionally upregulates the autophagy‐lysosome machinery and stimulates autophagic flux to precondition the heart to ischemia‐reperfusion injury and reduce cardiomyocyte cell death.26 More important, in this study, IF did not affect cardiac structure or function in wild‐type mice.26 Accordingly, we tested if IF could rescue the impaired autophagic flux in the immediate preterminal stage in Myh6‐CryABR120G transgenic mice with advanced cardiomyopathy (Figure 2A). Remarkably, IF over a period of 6 weeks completely prevented the mortality observed in Myh6‐CryABR120G mice over this period (Figure 2B). Examination of the state of activation of TFEB, the master regulator of the autophagy‐lysosome machinery, revealed that in comparison to age‐matched wild‐type controls, the abundance of nuclear TFEB (where it localizes to upon activation)25 was similar and cytosolic TFEB levels were reduced (Figure 2C and 2D). Because TFEB transcriptionally autoregulates its own expression,32 these data point to a failure of the transcriptional response that drives the autophagy‐lysosome pathway despite active protein aggregate pathologic features, consistent with the previous observation of reduced autophagy‐lysosome pathway gene transcripts in mice with transgenic expression of the murine CryABR120G mutant.15, 33, 34 Interestingly, IF significantly increased nuclear TFEB abundance in Myh6‐CryABR120G mice compared with their ad‐lib fed counterparts (Figure 2C and 2D), and this was associated with rescue of autophagic flux impairment, as indicated with chloroquine‐induced upregulation of LC3‐II and p62 levels versus respective diluent‐treated group (Figure 2E and 2F). The lack of TFEB activation was accompanied by reduction in LAMP1 and LAMP2 levels in ad‐lib fed Myh6‐CryABR120G mice (versus wild‐type mice, Figure 2G and 2H), suggestive of a reduction in lysosome abundance, which was restored to wild‐type levels with IF. To examine the mechanism for the lack of TFEB activation despite the advanced protein‐aggregate pathologic features in the Myh6‐CryABR120G myocardium, we assayed mTOR activity, which phosphorylates TFEB to prevent its nuclear translocation.25 Aged Myh6‐CryABR120G mouse hearts demonstrated increased levels of phosphorylated mTOR and its substrates, p70S6 kinase and 4EBP1, compared with wild‐type hearts, indicating increased mTOR activity, as observed previously in mice with transgenic expression of the murine CryABR120G mutant,34 which was attenuated with IF (Figure 2I and 2J).
IF Accelerates Protein Aggregate Removal in CryABR120G Transgenic Hearts
Prior studies have demonstrated that activation of autophagy is both necessary and sufficient to prevent formation of protein aggregates in cardiac myocytes expressing the CryABR120G protein.7, 33 To determine if restoration of autophagic flux was sufficient to remove protein aggregates, we examined αB‐crystallin protein expression in Myh6‐CryABR120G transgenic hearts fractionated into soluble and insoluble fractions.7 As shown in Figure 3A through 3C, intermittently fasted mice demonstrated reduced levels of soluble αB‐crystallin protein, as well as insoluble αB‐crystallin either in monomeric or aggregate form, compared with ad‐lib fed counterparts. Examination of myocardial ultrastructure demonstrated qualitatively fewer protein aggregates (Figure 3D, white arrows), with more abundant autolysosomes (black arrows), and reappearance of sharp Z‐discs, as seen in wild‐type controls, indicating partial restoration of Z‐disc ultrastructure in Myh6‐CryABR120G transgenic hearts with advanced cardiomyopathy (black arrowheads, compare with broad Z‐discs in ad‐lib fed Myh6‐CryABR120G myocardium).
IF Attenuates Cardiomyopathy and Restores Desmin Localization in CryABR120G Transgenic Mice
Echocardiographic characterization of intermittently fasted and ad‐lib fed Myh6‐CryABR120G mice revealed attenuation of left ventricular (LV) dilation (Figure 4A and 4B, Table S2), improved ejection performance (percentage LV endocardial fractional shortening, Figure 4A and 4C, Table S2), and attenuated hypertrophy (with reduced LV mass [Figure 4A and 4D] and heart weight [Figure 4A and 4E]) with IF, indicating that restoration of autophagic flux with IF correlated with attenuation of typical progression of the fatal cardiomyopathic manifestations, otherwise observed with expression of CryABR120G protein.6, 8 Intermittently fasted Myh6‐CryABR120G myocardium demonstrated fewer eosinophilic aggregates (Figure 4F [arrows] and 4H, with quantification in Figure S3A) with reduction in myocardial fibrosis (Figure 4G) and cell death (Figure S3B and S3C) compared with ad‐lib counterparts. We also observed that the normal distribution of desmin associated with Z‐discs (multiple arrows in groups) and intercalated discs (single arrow) seen in wild‐type cardiomyocytes was largely lost in Myh6‐CryABR120G transgenic mice and replaced by desmin localized to aggregates (Figure 4H, with appearance of a hollow core on immunohistochemistry secondary to lack of antibody penetration into aggregates, as observed previously6). Intriguingly, IF restored the expression of desmin in association with Z‐discs and intercalated discs (Figure 4H; multiple arrows in groups and single arrows, respectively) in the Myh6‐CryABR120G myocardium. To determine if this was secondary to synthesis of new desmin, we measured its transcripts. Our data demonstrate that although desmin transcripts are induced in Myh6‐CryABR120G hearts (versus wild type, Figure S4A), presumably to compensate for reduced desmin localized to Z‐discs and intercalated discs, we did not see further upregulation with IF, which suggests that IF restored localization of preexisting desmin associated with removal of the αB‐crystallin aggregates.
AAV9‐Mediated TFEB Transduction Rescues Advanced Cardiomyopathy in CryABR120G Transgenic Mice
We next tested if TFEB activation was sufficient to attenuate the cardiomyopathic manifestations in Myh6‐CryABR120G mice at an advanced stage of disease. To do so in a clinically relevant manner, we performed AAV9‐mediated gene transfer of mouse TFEB via intravenous injection (versus AAV9–green fluorescent protein as control) at 36 weeks of age and confirmed cardiac myocyte targeting at 40 weeks of age (Figure 5A; data not shown and vide infra). AAV9‐mediated TFEB transduction resulted in modest upregulation of myocardial TFEB abundance (≈2‐fold over control, see Figure 5B and 5C [left]) with increased abundance of nuclear TFEB (Figure S5A) and of TFEB target proteins (Figure S5B through S5F). This was sufficient to result in reduced abundance of αB‐crystallin, including the higher‐molecular‐weight forms versus AAV9–green fluorescent protein transduced myocardium (Figure 5B and 5C [right]) without affecting human CryABR120G transgene transcript levels (Figure S6A), generalized protein synthesis (Figure S6B and S6C), or mTOR activity (Figure S6D). More important, a modest upregulation of myocardial TFEB levels did not adversely affect left ventricular structure or function or myocardial architecture and fibrosis compared with AAV9–green fluorescent protein transduced controls (Figure 5D through 5J). AAV9‐mediated TFEB transduction improved LV ejection performance (LV percentage fractional shortening; Figure 5D and 5F, Table S3), significantly reduced LV end‐systolic diameter (Table S3), and attenuated hypertrophy (with reduced LV mass [Figure 5D and 5G, Table S3] and heart weight [Figure 5D and 5H]) without an effect on LV dilation (Figure 5D and 5E, Table S3) in Myh6‐CryABR120G mice with advanced cardiomyopathy. This was associated with reduced aggregates in the myocardium (Figure 5I, Figure S7A), reduced fibrosis (Figure 5J), and reduced myocardial cell death (Figure S7B and S7C), consistent with the notion that exogenous TFEB was sufficient to remove protein aggregates and attenuate cardiomyopathic manifestations at an advanced stage of pathology in mice expressing CryABR120G, mimicking the benefits observed with IF (Figure 4).
TFEB Activation Alters Desmin Interaction With αB‐Crystallin and HSPB8 to Restore Desmin Localization in CryABR120G Cardiomyopathic Hearts
We next examined if TFEB transduction altered desmin localization. AAV9‐mediated TFEB transduction did not affect desmin localization associated with the Z‐discs (multiple arrows in groups, Figure 6A) or the intercalated discs (single arrow, Figure 6A) in wild‐type mice. More important, AAV9‐TFEB transduction restored the normal pattern of desmin localization associated with the Z‐discs and intercalated discs in the Myh6‐CryABR120G myocardium, in cardiomyocytes that demonstrate nuclear TFEB expression (Figure 6A). This suggested the hypothesis that TFEB transcriptionally upregulates other members of the heat shock protein family to chaperone desmin to its normal localization. Prior studies have suggested that expression of recombinant HSPB1 (HSP22) and HSPB8 (HSP25), 2 members of the non–ATPase‐dependent heat shock protein family that αB‐crystallin belongs to, are sufficient to prevent CryABR120G amyloid oligomer formation.12 In addition, induction of HSPB8 either genetically or pharmacologically attenuated the development of cardiomyopathic manifestations in mice expressing CryABR120G in cardiac myocytes.35 We examined the protein levels of these 2 small heat shock family proteins in the TFEB (or green fluorescent protein)–transduced Myh6‐CryABR120G transgenic mice and wild‐type controls, and found that both are increased in Myh6‐CryABR120G transgenic myocardium versus control (Figure 6B through 6D). Interestingly, TFEB transduction resulted in further increase in protein levels of HSPB8 (Figure 6B and 6C), but not HSPB1 (Figure 6B and 6D), in both wild‐type and Myh6‐CryABR120G transgenic mice, consistent with the previously described presence of TFEB‐binding CLEAR (Coordinated Lysosomal Expression and Regulation) sites in the HSPB8 promoter.36 Indeed, exogenous TFEB was sufficient to selectively upregulate HSPB8 transcripts in NRCMs after short‐term transduction (Figure S8). Interestingly, myocardial HSPB8 transcript and protein levels were also upregulated with fasting (Figure S9A through S9C) and return to baseline levels with refeeding (Figure S9B and S9C), tracking the pulsed activation of myocardial TFEB with each episode that we have described previously,26 without an increase on a fed day noted at the termination of the 6 weeks of IF (Figure S9D and S9E).
The mechanism for TFEB‐induced normalization of desmin localization could be increased degradation of desmin from the protein aggregates and resynthesis of new desmin that gets chaperoned to its right location. We determined this to be less likely because we did not detect a change in total desmin protein abundance (Figure 6B and 6E) or in desmin transcripts in the AAV9‐TFEB treated samples (Figure S4B). The alternative explanation is that desmin protein is held in a strong interaction with the mutant αB‐crystallin protein,21 which is targeted to the aggregates. Indeed, immunoprecipitation studies with AAV9‐based cardiac‐myocyte specific expression of R120G mutant of CryAB and wild‐type CryAB proteins, driven by the cardiac troponin T promoter,31 demonstrate that desmin interacts more avidly with the mutant protein, in vivo (Figure S10A through S10D). TFEB‐mediated autophagic removal of the ubiquitinated mutant αB‐crystallin protein then frees up desmin, which binds to HSPB8 and is chaperoned back to its normal location in association with the Z‐discs and intercalated discs. Indeed, coimmunoprecipitation experiments demonstrated that desmin interaction with αB‐crystallin is markedly upregulated in the Myh6‐CryABR120G hearts, whereas its interaction with HSPB8 is concurrently diminished compared with wild type (Figure 6F). Interestingly, both IF and AAV9‐TFEB transduction resulted in a downregulation of desmin's interaction with αB‐crystallin with a simultaneous increase in HSPB8 protein that was immunoprecipitated with desmin (Figure 6F and 6G); and IF did not alter ubiquitination of desmin (Figure S11). Taken together, these data suggest that TFEB accelerates clearance of the mutant αB‐crystallin protein that sequesters desmin, whereby desmin is freed up to interact with HSPB8 and is, therefore, able to return to its correct location adjacent to the Z‐discs and intercalated discs.
TFEB Increases Autophagic Flux to Drive Degradation of Mutant αB‐Crystallin Independent of HSPB8 Induction
To examine the effect of targeting TFEB activation and the role of HSPB8 in autophagic removal of protein aggregates, we adenovirally transduced CryABR120G in NRCMs together with exogenous TFEB, with and without concomitant shRNA‐mediated HSPB8 knockdown (Figure S12A). TFEB transduction upregulated HSPB8 protein abundance (Figure S12A and S12B) and induced autophagic flux in control‐adenovirus transduced myocytes, with accumulation of LC3‐II and p62 in cells treated with bafilomycin A1 to inhibit lysosomal acidification versus diluent‐treated controls (Figure S12A, S12C, and S12D). Expression of R120G αB‐crystallin mutant resulted in impaired autophagic flux (lack of bafilomycin A1–induced accumulation of LC3‐II and p62 [Figure S12A, S12C, and S12D]), whereas concomitant TFEB transduction restored autophagic flux. More important, knockdown of HSPB8 did not prevent TFEB‐induced upregulation of autophagic flux (Figure S12A, S12C, and S12D) in cardiomyocytes expressing either CryABR120G or control. TFEB transduction resulted in reduced expression of αB‐crystallin protein (Figure S12A and S12E), including the high‐molecular‐weight aggregates specifically from the insoluble fractions (Figure S12A and S13), and knockdown of HSPB8 did not affect this decline (Figure S12E). Taken together, these data indicate that TFEB drives removal of CryABR120G protein aggregates via induction of autophagic flux independent of HSPB8 induction.
TFEB Induces Normalization of Desmin Expression, Preserves Mitochondrial Polarization, and Attenuates Cell Death in Cardiomyocytes via HSPB8 Induction
We next evaluated the effect of TFEB on desmin localization in NRCMs, cultured to induce sarcomere formation, which resulted in desmin localization to the Z‐discs (see arrows in groups in top row, in Figure 7A). Adenoviral expression of CryABR120G induced formation of aggregates and relocalization of desmin from the Z‐discs to aggregates (see single arrows in second row, Figure 7A). Overexpression of TFEB resulted in marked reduction of protein aggregates and reappearance of desmin at the Z‐discs (third row, Figure 7A). Remarkably, concomitant knockdown of HSPB8 completely prevented TFEB‐induced relocalization of desmin to the Z‐discs, without affecting TFEB‐induced reduction of aggregates in cardiomyocytes expressing CryABR120G (bottom row, Figure 7A). To confirm the presence of protein aggregates and their removal by TFEB expression, we evaluated the localization of p62, an adaptor protein that binds to ubiquitinated proteins to facilitate aggregate formation. Both αB‐crystallin and desmin colocalized with p62 and ubiquitin in puncta in CryABR120G‐transduced cardiac myocytes (see single arrows, second row, Figure 7B), and desmin was no longer localized to Z‐discs, as observed in controls (arrows in groups, top row, Figure 7B). TFEB transduction in CryABR120G‐expressing cardiac myocytes resulted in removal of p62 and ubiquitin‐positive aggregates in an HSPB8‐independent manner and normalization of desmin localization in an HSPB‐dependent manner (compare third and fourth rows, Figure 7B). Given the recently elucidated role for Z‐disc–associated desmin as a scaffold for mitochondria and maintaining their polarization,37 we examined the effect of TFEB in CryABR120G‐transduced cells on mitochondrial membrane potential assessed with a ratiometric dye, JC‐1. Cells transduced with CryABR120G demonstrated a marked increase in prevalence of depolarized mitochondria (Figure 7C and 7D), as well as increase in mitochondrial DNA content compared with controls (Figure 7E), indicating accumulation of damaged mitochondria in the setting of impaired autophagic flux (Figure S12A, S12C, and S12D). This was accompanied by increased cell death (Figure 7F), as described previously.37 Concomitant TFEB transduction markedly attenuated the CryABR120G‐induced increase in mitochondrial depolarization (Figure 7C and 7D) and reduced mitochondrial DNA content (Figure 7E), indicating removal of damaged mitochondria by TFEB‐induced restoration of autophagic flux in this setting (Figure S12A, S12C, and S12D). This was accompanied by a reduction in cell death (Figure 7F), in an HSPB8‐dependent manner. Intriguingly, shRNA‐mediated HSPB8 knockdown was sufficient to modestly increase the prevalence of depolarized mitochondria and cell death, suggesting that it plays a critical role in cardiac myocytes, in vitro, in a resting state. These findings indicate that TFEB activation drives normalization of desmin localization via both autophagy‐dependent and autophagy‐independent (HSPB8‐mediated) mechanisms, thereby preserving mitochondrial quality and cell viability as the likely mechanism in the attenuation of CryABR120G‐induced cardiomyopathy.
TFEB and HSPB8 Are Essential for IF‐Mediated Attenuation of Cardiomyopathy in CryABR120G Transgenic Mice
To determine the mechanistic role of TFEB and HSPB8 in the beneficial response to IF, we transduced 36‐week‐old mice with AAV9 particles encoding shRNA constructs targeting TFEB and HSPB8 or a nontargeting scrambled control. Baseline echocardiography was performed after 4 weeks, and mice were subjected to IF with repeated echocardiographic evaluation 3 and 6 weeks later, followed by postmortem analysis (Figure S13A). Viral transduction with the shRNA constructs results in ≈65% reduction in TFEB protein expression (with reduced levels of nuclear TFEB, Figure S14) and ≈70% reduction in HSPB8 protein compared with the control (Figure 8A through 8C). Interestingly, basal HSPB8 levels were also significantly reduced in AAV9–short hairpin TFEB (shTFEB) treated samples (compared with controls), confirming that HSPB8 is a TFEB target, in vivo (Figure 8A and 8C). We did not observe an effect of TFEB and HSPB8 knockdown on cardiac structure and function in the Myh6‐CryABR120G mice at 4 weeks after AAV injections to transduce shRNAs targeting TFEB and HSPB8 compared with control shRNA during a period of ad‐lib feeding (Figure 8D, Table S4). IF resulted in statistically significant improvement in LV ejection performance (LV percentage fractional shortening, Figure 8D, Table S4) in the Myh6‐CryABR120G mice transduced with the control shRNA and abrogation of LV dilation (Figure S14B, Table S4), similar to prior observations with IF in non–AAV9‐transduced mice (Figure 4C). By contrast, mice transduced with AAV9‐shTFEB as well as with AAV9–short hairpin HSPB8 (shHSPB8) did not demonstrate abrogation of progressive dilation and decline in LV systolic function compared with baseline, despite undergoing IF (Figure 8D, Figure S14B, Table S4). There was no change in body weight within any treatment group over this period (Figure S14C, Table S4). As observed in non–AAV9‐treated group (Figure 4F through 4H), IF resulted in reduced eosinophilic aggregates and fibrosis as well as restoration of desmin localization in Myh6‐CryABR120G mice treated with AAV9–short hairpin control (Figure 8E through 8G). By contrast, AAV9‐shTFEB and AAV9‐shHSPB8 transduced Myh6‐CryABR120G transgenic mice showed increased aggregate pathologic features and fibrosis as well as persistent abnormal desmin localization away from Z‐discs and intercalated discs, and in the aggregates, despite IF (Figure 8E through 8G). Quantitation of aggregates revealed a significantly increased aggregate load in both shTFEB‐ and shHSPB8‐transduced groups compared with short hairpin control (Figure S15A) and increased abundance of αB‐crystallin, including the higher‐molecular‐weight forms in shTFEB‐ and shHSPB8‐ versus short hairpin control–transduced myocardium (Figure S15B). Interestingly, shHSPB8 transduction appeared to partially affect autophagic flux, which appeared preserved in short hairpin control group and abrogated with shTFEB transduction in intermittently fasted Myh6‐CryABR120G transgenic mice (Figure S14D), indicating a role for HSPB8 in facilitating autophagic flux, in vivo, over a prolonged duration of observation. Taken together, these data indicate IF benefits require both TFEB and HSPB8 in the setting of advanced αB‐crystallin R120G mutation‐induced cardiomyopathy, consistent with the observation that TFEB activation is sufficient, per se, in transducing these benefits via mechanisms dependent on autophagy and HSPB8 induction.
Discussion
Myofibrillar cardiomyopathies such as observed with autosomal dominant inheritance of the R120G αB‐crystallin mutant protein increase mortality and morbidity attributable to heart failure, necessitating development of therapeutic approaches targeting specific pathobiologic mechanisms.2 Although prior studies have examined the efficacy of interventions to prevent and/or delay the onset of cardiomyopathy,15, 33, 38 the current study is the first to demonstrate rescue of advanced cardiomyopathy at a preterminal stage characterized by signs of heart failure.7, 8 Furthermore, we establish IF and TFEB‐gene therapy as promising strategies that merit exploration as therapies to treat human cardiomyopathy, resulting from this mutation. Expression of the human CryABR120G protein in cardiomyocytes impairs TFEB activation with marked impairment of autophagic flux. In contrast, IF and TFEB activation restore autophagic flux to remove protein aggregates, and simultaneously drive transcriptional upregulation of HSPB8, which binds to desmin and facilitates its physiologic localization to the intercalated discs and Z‐lines (Figure 8H). This results in attenuation of cardiomyopathy.
Contemporary literature has fostered a paradigm shift in understanding the role of lysosomes as active cellular managers that drive catabolism via autophagy and other degradative pathways and couple substrate supply to intracellular metabolism.39 In contrast to the increased autophagic flux observed at early stages of the disease,7 we observe decreased autophagic flux accompanying advanced CryABR120G‐induced cardiomyopathy. More important, although our experiments may have been underpowered to detect a difference between completely impaired versus reduced autophagic flux in CryABR120G mice at this advanced stage of cardiomyopathy, our results point to insufficient lysosome function in the setting of advanced protein aggregate pathologic features. Conceivably, the long‐term increase in protein turnover upregulates amino acid availability, mimicking an “overfed” state, which drives an increase mTOR complex 1 activity.40 This results in phosphorylation and cytosolic sequestration of TFEB, the master regulator of the autophagy‐lysosome machinery,25 with suppression of autophagic flux. Conversely, when these mice are subjected to nutrient deprivation by IF, this effect is overcome to inhibit mTOR complex 1 activity, resulting in TFEB activation.26 These effects are independent on an effect on body weight, which is not affected by IF in mice on standard chow26 (Figure S14C) and therefore points to tissue‐specific signaling effects. Although prior studies have examined the notion that TFEB stimulates autophagic flux to remove R120G mutant of αB‐crystallin,34 our data set is the first to comprehensively examine a clinically relevant paradigm for TFEB activation in vivo and explore relevant mechanisms. Intriguingly, TFEB transduction was sufficient to drive protein‐aggregate removal via enhancing autophagic flux and restore normal desmin localization via HSPB8 induction, pointing to a broader role for the postlysosomal TFEB‐mediated transcriptional program in facilitating protein quality control in cardiomyocytes. Furthermore, our data demonstrate that TFEB and HSPB8 play a critical mechanistic role in the IF‐mediated rescue of cardiomyopathy and normalization of desmin localization. Whether the activation of TFEB prevents the development and progression of cardiomyopathy in this model remains to be explored.
Our studies suggest a model in which the relative abundance and competitive‐binding affinities of CryABR120G and HSPB8 proteins to desmin dictate where desmin is localized to within the cardiomyocyte. Accordingly, in Myh6‐CryABR120G hearts, despite a compensatory transcriptional upregulation of desmin protein levels, a large fraction of desmin is bound to the mutant αB‐crystallin protein, thus localizing to aggregates rather than in association with the Z‐discs and intercalated discs. In contrast, the fraction of desmin bound to HSPB8 dramatically increases with a concomitant decline in mutant αB‐crystallin binding with IF and TFEB transduction, a scenario that favors physiologic desmin localization with attenuation of cardiomyopathic manifestations. Although we have not directly examined desmin degradation rates in vivo, our data demonstrating a lack of effect of IF on desmin transcripts, protein, and ubiquitination state support this model.
Prior studies have shown that CryABR120G protein aggregates into amyloid oligomers,41 a process that can be prevented by overexpression of the chaperones, HSPB8 (HSP22) and HSPB1 (HSP25)12; and transgenic expression of HSPB8 in cardiomyocytes prevents development of amyloid oligomers and cardiomyopathy.35 Our observations suggest that the desmin bound to the mutant crystallin protein is released on TFEB‐induced protein‐aggregate removal in the Myh6‐CryABR120G hearts and is chaperoned by HSPB8 to its normal localization. Intriguingly, induction of HSPB8 promotes autophagic clearance of aggregate‐prone proteins, such as mutant SOD1 (superoxide dismutase 1) and TDP43 (TAR DNA‐binding protein 43) (implicated in amyotrophic lateral sclerosis) and polyglutamine repeat‐bearing mutant androgen receptor (in spinal bulbar atrophy),42, 43 and mutations in HSPB8 implicated in causing Charcot Marie tooth disease and distal hereditary motor neuropathy 2A impair autophagosome maturation in neuronal cell types.44 Our findings demonstrate that shRNA‐mediated knockdown of HSPB8 had a modest effect on autophagic flux and autophagic clearance of αB‐crystallin aggregates, in vivo; and HSPB8 expression was essential to restore desmin localization with TFEB expression in CryABR120G‐expressing cardiomyocytes.
Normal localization of desmin in association with the Z‐discs and intercalated discs plays a critical scaffolding role in maintaining mitochondrial polarization and function18, 37 and sarcomere integrity.19 Mutations in the desmin tail that selectively impair its localization to the Z‐disc45 cause cardiomyopathy in humans,46 whereas experimental ablation of desmin in mice induces sarcomeric degeneration19 and a dilated cardiomyopathy with systolic dysfunction.20 Similarly, both missense mutations in desmin47 as well as a 7–amino acid deletion48 that impairs desmin assembly into intermediate filaments are sufficient to provoke an autosomal‐dominant cardiomyopathy in humans.47, 48 Reexpression of desmin via AAV9 transduction restored cardiac function and attenuated cardiomyopathy in desmin null mice.49 Indeed, the dominant‐negative effect of CryABR120G protein may be to sequester desmin (and other sarcomeric proteins and chaperones) away from their physiologic intracellular localization sites.
Reduced myocardial cell death and fibrosis with IF and TFEB activation, in vivo, are consistent with the restoration of polarized mitochondria and attenuated cell death with TFEB expression in CryABR120G‐expressing cardiomyocytes, in in vitro studies. Indeed, mitochondrial abnormalities are observed in the desmin null myocardium as well as CryABR120G transgenic hearts, that result in mitochondrial permeabilization and cell death,9, 33 which could be rescued by overexpression of an antiapoptotic protein, Bcl2.10, 50 Our data suggest that IF or TFEB‐induced normalization of desmin localization attenuates mitochondrial abnormalities as the proximate mechanism for attenuation of cardiomyopathy. Moreover, we have demonstrated that TFEB activation facilitates mitochondrial quality control via coordinated removal of damaged mitochondria and biogenesis of normal mitochondria under stress in cardiomyocytes, and this mechanism also needs to be formally evaluated in future studies.28
Our key finding, that IF and TFEB activation were sufficient to rescue established cardiomyopathy despite the markedly increased cell death9 and fibrosis7, 15 seen at this advanced stage of pathogenesis,8 underscores the potential for restoring desmin to favorably remodel sarcomere in failing cardiomyocytes to treat desmin‐related cardiomyopathies.2 Furthermore, this paradigm may also be broadly relevant to cardiomyopathy and heart failure of diverse causes, in which abnormal desmin localization and protein aggregation are observed, such as in hearts from patients with hypertrophic cardiomyopathy with reduced VPS34 expression51 and from patients with dilated cardiomyopathy.41, 52, 53 More important, these observations raise hopes for a therapeutic strategy targeting TFEB activation to slow or reverse disease pathogenesis, at a clinically relevant stage when most patients receive their initial diagnosis; and set the stage for experimental evaluation of therapies such as trehalose, a safe nonreducing disaccharide,39 or small molecules targeting mTOR25 that activate TFEB.
Author Contributions
Ma, Liu, Murphy, Foroughi, Kovacs, and Weinheimer performed experiments and analyzed the data; French, Benjamin, and Hill provided critical scientific reagents and scientific input; Kraja assisted with statistical analyses; Mani, Javaheri, and Diwan conceived the experiments; and Mani and Diwan wrote the article.
Sources of Funding
This study was supported by grants from the National Institutes of Health (HL107594) and the Department of Veterans Affairs (I01BX000448, 1I01BX001969, I01BX004235) to Diwan. Mani was supported by a Seed Grant from the St Louis VA Medical Center. Javaheri was supported by 5‐T32‐HL07081‐40 and K08‐HL138262 from the National Heart, Lung, and Blood Institute.
Disclosures
None.
Supporting information
Data S1. Supplementary Methods.
Table S1. List of Primers for Quantitative PCR Analysis
Table S2. Evaluation of Cardiac Function by M‐Mode Echocardiography in Wild Type ad lib Fed Mice and Myh6CryaABR120G Mice ‐ Either Treated With ad lib Feeding or Intermittent Fasting
Table S3. Evaluation of Cardiac Function by M‐Mode Echocardiography in Wild Type Mice and Myh6CryaABR120G Mice Treated With AAV9‐GFP or AAV9‐TFEB
Table S4. Evaluation of Cardiac Function by M‐Mode Echocardiography in Myh6CryaABR120G Mice Treated With AAV9 Control, AAV9 shTFEB, or AAV9shHspB8 and Subjected to Intermittent Fasting (IF)
Figure S1. Autophagic flux assessment reveals preserved autophagic flux in skeletal muscle from Myh6‐R120G αB‐Crystallin TG mice and in the transgenic mouse hearts at a young age. (A) lmmuno‐blot demonstrating LC3 and p62 expression in heart and skeletal muscle (gastrocnemius) tissue from 40 week old Myh6‐R120G αB‐Crystallin TG mice treated with CQ or diluent for autophagic flux assessment (representative of n=2 experiments). B and C, Representative immuno‐blot (B) depicting LC3‐II and p62 expression with quantitation (C) in young (8 week) Myh6‐R120G αB‐Crystallin TG mice treated with CQ or diluent for autophagic flux assessment. N=3/group. *Indicates P<0.05 by t test.
Figure S2. Expression of αB‐Crystallin in Myh6‐R120G αB‐Crystallin TG mice and CryAB null mice demonstrates specificity for immuno‐detection of αB‐Crystallin. lmmunoblot demonstrating expression of αB‐Crystallin in hearts from Myh6‐R120G αB‐Crystallin TG mice, CryAB null mice and wild‐type controls. Arrows point to various electrophoretic bands detected with the polyclonal αB‐crystallin antibody employed herein.
Figure S3. Intermittent fasting reduces aggregate pathology and myocardial cell death in Myh6‐R120G αB‐Crystallin TG Mice. A, Quantitation of aggregates in the myocardium (as % area). B and C, Representative TUNEL stained images (with DAPI‐stained nuclei, B) with quantitation of TUNEL + nuclei (arrows in B, quantified in C) in hearts from Myh6‐R120G αB‐Crystallin TG mice subjected to intermittent fasting (IF) or ad‐lib feeding for 6 weeks beginning at 40 weeks of age (see Figure 2A for schematic) and WT controls (N=3–5/group). *Indicates P<0.05 by t test for (A) and post‐hoc test after one‐way ANOVA for (C).
Figure S4. Intermittent fasting and TFEB activation do not alter desmin transcription. A, Transcript abundance for desmin in hearts from Myh6‐R120G αB‐Crystallin TG mice subjected to intermittent fasting or ad‐lib feeding, and littermate wild‐type controls. N=4/group. *Indicates P<0.05 by post‐hoc test after one‐way ANOVA. B, Transcript abundance for desmin in hearts from Myh6‐R120G αB‐Crystallin TG mice and littermate wild‐type control mice transduced with AAV‐TFEB or AAV‐GFP. N=3 to 6/group. *Indicates P<0.05 by post‐hoc test after one‐way ANOVA.
Figure S5. AAV‐mediated TFEB transduction upregulates abundance of candidate autophagy‐lysosome target proteins. A, lmmunoblot depicting nuclear TFEB abudance in cardiac tissue from CryABR120G mice transduced with AAV‐GFP or AAV‐TFEB at 36 weeks of age, and sacrificed 10 weeks later. B through F, Representative immuno‐blot with quantification of protein abundance for LC3‐II (C), LAMP1 (D), p62 (E) and LAMP2 (F) in cardiac tissue from wild‐type mice transduced with AAV‐GFP or AAV‐TFEB at 36 weeks of age, and sacrificed 10 weeks later. *Indicates P value by t test. N=3/group.
Figure S6. TFEB transduction does not alter transcript levels of the human CryAB R120G transgene and does not affect cardiac protein synthesis. A, Quantitative PCR analysis for detecting human CRYAB transgene expression with specific primer set (see Table S1) in Myh6‐R120G aB‐Crystallin TG mice transduced with AAV‐TFEB or AAV‐GFP. N=4/group. B and C, Wild‐type male C57bl6 mice (8 weeks old) were transduced with AAV‐GFP or AAV‐TFEB for 4 weeks and SunSET assay performed for assessing cardiac protein synthesis. Representative immuno‐blot (B) with quantitation of puromycin incorporation (C) in cardiac extracts from a non‐injected control mouse and AAV transduced mice. D, Representative immuno‐blots for detecting mTOR activation state in mice treated as in (B). P values were assessed by t test in (A and C).
Figure S7. TFEB transduction reduces aggregate pathology and myocardial cell death in Myh6‐R120G αB‐Crystallin TG mice. A, Quantitation of aggregates as% area of the myocaraium in AAV‐GFP and AAV‐TFEB transduced mice at 46 weeks of age (see Figure 6A for images). N=3/grouQ. *Indicates P<0.05 by t test. B and C, Representative TUNEL stained images (with DAPl‐stained nuclei, B) with quantitation of TUNE L+ nuclei (pseudo‐colored green, arrows in (C) in hearts from Myh6‐R120G αB‐Crystallin TG mice and WT controls transduced with AAV9‐TFEB or AAV9‐GFP for 10 weeks beginning at 36 weeks of age (see Figure 5A for schematic) and WT controls (N=3–4/group). *Indicates P<0.05 by post‐hoc test after one‐way ANOVA.
Figure S8. TFEB induces HSPB8 expression. Transcript abundance of various heat shock protein family members in NRCMs adenovirally transduced with HA‐tagged TFEB or Ad‐Lacz (MOI=10 each) for 24 h. N=3 to 4/group. *Indicates P<0.05 by t test.
Figure S9. Fasting increases HSPB8 transcript and protein levels in the myocardium. A, HSPB8 transcript levels in cardiac tissue from C57bl6 wild‐type mice fasted for various durations as shown. N=4/group. *Indicates P<0.05 by post‐hoc analysis after one way ANOVA (B and C) Representative immunoblot (B) and quantitation of HSPB8 protein levels (C) in the myocardium from fed wild‐type mice, mice fasted for 24 h, and mice fasted for 24 h followed by re‐feeding for 24 h. N=3/group.*Indicates P<0.05. D and E, Representative immunoblot (D) and guantitation of HSPB8 protein levels (E) in the myocardium from Myh6‐R120G αB‐Crystallin TG mice subjected to intermittent fasting (IF) or ad‐lib feeding for 6 weeks beginning at 40 weeks of age (see Figure 2A for schematic) and WT controls (N=4/group). *Indicates P<0.05 by post‐hoc test after one‐way ANOVA.
Figure S10. R120G mutant CRYAB Protein interacts more strongly with desmin than the wild‐type CRYAB protein, in vivo. A, Male C57bl6 mice at 8 weeks of age were transduced with AAV9 particles with cardiac troponin T promoter driven expression of FLAG‐tagged human CRYAB protein or its R120G mutant for 4 weeks. Representative immuno‐blot demonstrating expression of FLAG‐tagged CRYAB proteins is shown in (A). Mice not treated with AAV9 particles are shown as control; “ns” indicates non‐specific band. B, Quantitation of FLAG‐tagged CRYAB protein and its R120G mutant in cardiac extracts from mice treated as in (A). N=3/group. NS indicates not‐significant for P value by t test. C, lmmuno‐precipitation was performed with anti‐FLAG antibody (or lgG confrol) in cardiac extracts from mice treated as in (A) and immuno‐blot performed for desmin and CRYAB. Representative immuno‐blot is shown. (D) Quantitation of desmin expression in after immuno‐precipitation with anti‐FLAG antibody as shown in (C). N=3/group. *Indicates P<0.05 by t test.
Figure S11. Intermittent fasting does not alter desmin ubiquitination. lmmunoblot demonstrating poly‐ubiquitinated desmin in Myh6‐CryABR120G TG mice subjected to intermittent fasting (IF) or ad‐lib (AL) feeding and littermate control hearts (depicted as “‐”). One mg of total protein from cardiac extracts was subjected to immunoprecipitation with anti‐desmin antibody or control lgG, followed by immuno‐blotting for ubiquitin. The immunoblot for desmin is reproduced from the blot shown in Figure 6F. At the exposure shown to demonstrate desmin in TG samples, a desmin signal was not evident in WT hearts in the input lane (with 20 μg/sample; and was seen with longer exposure times [data not shown]).
Figure S12. TFEB promotes clearance of αB‐crystallin via induction of autophagic flux, independent of HSPB8 in NRCMs. A, Representative immunoblot demonstrating assessment of autophagic flux and HSPB8 expresion in NRCMs adenovirally transducesd with R120G mutant of αB‐crystallin (MOI=10; for 48 h) without or with HA‐tagged TFEB (MOI=10; for 24 h) to inhibi lysosomal acidification. Adenoviral particles coding for LacZ or shLacZ were added as controls simultaneously with Ad‐TFEB or Ad‐αB‐crystallin overexpression and sh‐HSPB8, respectively to equalize the number of viral particles. (B through D) Quantitation of HSPB8 (B), LC3‐II (C) and p62 (D) expression in NRCMs treated as in (A). N=3/group. *Indicates P<0.05 by t test within BfA and diluent treated groups; and # indicates P<0.05 by post‐hoc test after one‐way ANOVA for comparisons within the diluent treated group only. E, Quantitation of αB‐crystallin expression in the non‐BfA treated samples from A. *Indicates P<0.05 by one‐way ANOVA.
Figure S13. TFEB promotes removal of αB‐Crystallin aggregates in NRCMs. lmmunoblot demonstrating αB‐Crystallin expression in soluble and insoluble fractions from NRCMs adenovirally transduced with R 120G mutant of αB‐Crystallin (MOl=10; for 48 h) without or with HA‐tagged TFEB (MOl=10; for 24 h). Adenoviral particles coding for LacZ were added as controls simultaneously to equalize the number of viral particles.
Figure S14. Knockdown of TFEB and HSPB8 prevents intermittent fasting‐mediated attenuation of proteotoxic cardiomyopathy. A, Schematic depicting experimental intervention with AAV9‐ mediated shRNA transduction (directed to either TFEB or HSPB8 versus control sh RNA) in 36 week old Myh6‐ CryABR120G TG mice. Four weeks after AAV injection, baseline echocardiograms were obtained followed by 6 weeks of intermittent fasting (termed T=0 time point). Serial echocardiograms were then obtained at 3 weeks and 6 weeks (termed T=3 and T=6 time points, respectively), followed by sacrifice and post mortem biochemical, microscopic and immuno‐histochemical analysis. B, lmmunoblot depicting levels of nuclear TFEB after transduction with shTFEB versus shCon. C, Left ventricular end‐diastolic diameter assessed as fold‐change compared to baseline (T=0 weeks) data‐point in the control group, in mice treated as in (A). N=4 to 5/group. *Indicates P<0.05 by post‐hoc test after two‐way ANOVA. D, Body weight over the duration of the study in mice treated as in (A). N=4 to 5/group. *Indicates P<0.05 by post‐hoc test after two‐way ANOVA. D, Representative immuno‐blot demonstrating expression of LC3 and p62 in mice modeled as in (A) and injected with chloroquine (40 mg/kg) 4 h prior to sacrifice to assess autophagic flux. Representative of n=2 experiments.
Figure S15. Knockdown of TFEB and HSPB8 prevents intermittent fastingmediated removal of aggregates with persistent desmin localization to aggregates. A, Quantitation of aggregates as % area of the myocardium in Myh6‐CryABR120G TG mice transduced with AAV9‐shControl, AAV9‐shTFEB or AAV9‐shHSPB8 for 4 weeks at age of 36 weeks and subjected to intermittent fasting, assessed at 46 weeks of age. See Figure 8G for representative images. N=3/group. P value is by post‐hoc test after one‐way ANOVA. B and C, Representative immuno‐blot (B) with quantitation of αB‐crystallin in insoluble (C) myocardial fractions from Myh6‐CryABR120G TG mice at 46 weeks of age injected with AAV9 particles coding for shRNAs targeting TFEB or HSPB8, or control for 4 weeks and then subjected to intermittent fasting for 6 weeks. N=3/group. P value is by post‐hoc test after one‐way ANOVA.
Acknowledgments
The authors thank Dr Wandy Beatty from Department of Molecular Microbiology, Washington University School of Medicine, St. Louis, MO for performance of electron microscopy; and Joan Avery from the Center for Cardiovascular Research, Washington University School of Medicine, St. Louis, MO for technical assistance.
(J Am Heart Assoc. 2019;8:e010866 DOI: 10.1161/JAHA.118.010866.)
References
- 1. Burke MA, Cook SA, Seidman JG, Seidman CE. Clinical and mechanistic insights into the genetics of cardiomyopathy. J Am Coll Cardiol. 2016;68:2871–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goldfarb LG, Dalakas MC. Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest. 2009;119:1806–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bova MP, Yaron O, Huang Q, Ding L, Haley DA, Stewart PL, Horwitz J. Mutation R120G in alphaB‐crystallin, which is linked to a desmin‐related myopathy, results in an irregular structure and defective chaperone‐like function. Proc Natl Acad Sci USA. 1999;96:6137–6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vicart P, Caron A, Guicheney P, Li Z, Prevost MC, Faure A, Chateau D, Chapon F, Tome F, Dupret JM, Paulin D, Fardeau M. A missense mutation in the alphaB‐crystallin chaperone gene causes a desmin‐related myopathy. Nat Genet. 1998;20:92–95. [DOI] [PubMed] [Google Scholar]
- 5. Goldfarb LG, Park KY, Cervenakova L, Gorokhova S, Lee HS, Vasconcelos O, Nagle JW, Semino‐Mora C, Sivakumar K, Dalakas MC. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet. 1998;19:402–403. [DOI] [PubMed] [Google Scholar]
- 6. Wang X, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, Hewett T, Robbins J. Expression of R120G‐alphaB‐crystallin causes aberrant desmin and alphaB‐crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89:84–91. [DOI] [PubMed] [Google Scholar]
- 7. Tannous P, Zhu H, Johnstone JL, Shelton JM, Rajasekaran NS, Benjamin IJ, Nguyen L, Gerard RD, Levine B, Rothermel BA, Hill JA. Autophagy is an adaptive response in desmin‐related cardiomyopathy. Proc Natl Acad Sci USA. 2008;105:9745–9750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rajasekaran NS, Connell P, Christians ES, Yan LJ, Taylor RP, Orosz A, Zhang XQ, Stevenson TJ, Peshock RM, Leopold JA, Barry WH, Loscalzo J, Odelberg SJ, Benjamin IJ. Human alpha B‐crystallin mutation causes oxido‐reductive stress and protein aggregation cardiomyopathy in mice. Cell. 2007;130:427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maloyan A, Sanbe A, Osinska H, Westfall M, Robinson D, Imahashi K, Murphy E, Robbins J. Mitochondrial dysfunction and apoptosis underlie the pathogenic process in alpha‐B‐crystallin desmin‐related cardiomyopathy. Circulation. 2005;112:3451–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maloyan A, Sayegh J, Osinska H, Chua BH, Robbins J. Manipulation of death pathways in desmin‐related cardiomyopathy. Circ Res. 2010;106:1524–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu J, Chen Q, Huang W, Horak KM, Zheng H, Mestril R, Wang X. Impairment of the ubiquitin‐proteasome system in desminopathy mouse hearts. FASEB J. 2006;20:362–364. [DOI] [PubMed] [Google Scholar]
- 12. Sanbe A, Yamauchi J, Miyamoto Y, Fujiwara Y, Murabe M, Tanoue A. Interruption of CryAB‐amyloid oligomer formation by HSP22. J Biol Chem. 2007;282:555–563. [DOI] [PubMed] [Google Scholar]
- 13. Zhang H, Rajasekaran NS, Orosz A, Xiao X, Rechsteiner M, Benjamin IJ. Selective degradation of aggregate‐prone CryAB mutants by HSPB1 is mediated by ubiquitin‐proteasome pathways. J Mol Cell Cardiol. 2010;49:918–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li J, Horak KM, Su H, Sanbe A, Robbins J, Wang X. Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J Clin Invest. 2011;121:3689–3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, Hill JA, Sadoshima J, Robbins J. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123:5284–5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pattison JS, Osinska H, Robbins J. Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ Res. 2011;109:151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang X, Osinska H, Dorn GW, Nieman M, Lorenz JN, Gerdes AM, Witt S, Kimball T, Gulick J, Robbins J. Mouse model of desmin‐related cardiomyopathy. Circulation. 2001;103:2402–2407. [DOI] [PubMed] [Google Scholar]
- 18. Milner DJ, Mavroidis M, Weisleder N, Capetanaki Y. Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J Cell Biol. 2000;150:1283–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Milner DJ, Weitzer G, Tran D, Bradley A, Capetanaki Y. Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J Cell Biol. 1996;134:1255–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Milner DJ, Taffet GE, Wang X, Pham T, Tamura T, Hartley C, Gerdes AM, Capetanaki Y. The absence of desmin leads to cardiomyocyte hypertrophy and cardiac dilation with compromised systolic function. J Mol Cell Cardiol. 1999;31:2063–2076. [DOI] [PubMed] [Google Scholar]
- 21. Elliott JL, Der Perng M, Prescott AR, Jansen KA, Koenderink GH, Quinlan RA. The specificity of the interaction between alphaB‐crystallin and desmin filaments and its impact on filament aggregation and cell viability. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Su H, Li J, Zhang H, Ma W, Wei N, Liu J, Wang X. COP9 signalosome controls the degradation of cytosolic misfolded proteins and protects against cardiac proteotoxicity. Circ Res. 2015;117:956–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zheng Q, Su H, Ranek MJ, Wang X. Autophagy and p62 in cardiac proteinopathy. Circ Res. 2011;109:296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Settembre C, Di MC, Polito VA, Garcia AM, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A. A lysosome‐to‐nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Godar RJ, Ma X, Liu H, Murphy JT, Weinheimer CJ, Kovacs A, Crosby SD, Saftig P, Diwan A. Repetitive stimulation of autophagy‐lysosome machinery by intermittent fasting preconditions the myocardium to ischemia‐reperfusion injury. Autophagy. 2015;11:1537–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brady JP, Garland DL, Green DE, Tamm ER, Giblin FJ, Wawrousek EF. AlphaB‐crystallin in lens development and muscle integrity: a gene knockout approach. Invest Ophthalmol Vis Sci. 2001;42:2924–2934. [PubMed] [Google Scholar]
- 28. Ma X, Liu H, Murphy JT, Foyil SR, Godar RJ, Abuirqeba H, Weinheimer CJ, Barger PM, Diwan A. Regulation of the transcription factor EB‐PGC1alpha axis by beclin‐1 controls mitochondrial quality and cardiomyocyte death under stress. Mol Cell Biol. 2015;35:956–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ma X, Godar RJ, Liu H, Diwan A. Enhancing lysosome biogenesis attenuates BNIP3‐induced cardiomyocyte death. Autophagy. 2012;8:297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, Gonzales E, Tripoli DD, Czerniewski L, Ballabio A, Cirrito JR, Diwan A, Lee JM. Neuronal‐targeted TFEB accelerates lysosomal degradation of APP, reducing Aβ generation and amyloid plaque pathogenesis. J Neurosci. 2015;35:12137–12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Konkalmatt PR, Beyers RJ, O'Connor DM, Xu Y, Seaman ME, French BA. Cardiac‐selective expression of extracellular superoxide dismutase after systemic injection of adeno‐associated virus 9 protects the heart against post‐myocardial infarction left ventricular remodeling. Circ Cardiovasc Imaging. 2013;6:478–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ, Wollenberg AC, Di Bernardo D, Chan L, Irazoqui JE, Ballabio A. TFEB controls cellular lipid metabolism through a starvation‐induced autoregulatory loop. Nat Cell Biol. 2013;15:647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maloyan A, Gulick J, Glabe CG, Kayed R, Robbins J. Exercise reverses preamyloid oligomer and prolongs survival in alphaB‐crystallin‐based desmin‐related cardiomyopathy. Proc Natl Acad Sci USA. 2007;104:5995–6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pan B, Zhang H, Cui T, Wang X. TFEB activation protects against cardiac proteotoxicity via increasing autophagic flux. J Mol Cell Cardiol. 2017;113:51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sanbe A, Daicho T, Mizutani R, Endo T, Miyauchi N, Yamauchi J, Tanonaka K, Glabe C, Tanoue A. Protective effect of geranylgeranylacetone via enhancement of HSPB8 induction in desmin‐related cardiomyopathy. PLoS One. 2009;4:e5351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Palmieri M, Impey S, Kang H, di RA, Pelz C, Sardiello M, Ballabio A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011;20:3852–3866. [DOI] [PubMed] [Google Scholar]
- 37. Diokmetzidou A, Soumaka E, Kloukina I, Tsikitis M, Makridakis M, Varela A, Davos CH, Georgopoulos S, Anesti V, Vlahou A, Capetanaki Y. Desmin and alphaB‐crystallin interplay in the maintenance of mitochondrial homeostasis and cardiomyocyte survival. J Cell Sci. 2016;129:3705–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sanbe A, Osinska H, Villa C, Gulick J, Klevitsky R, Glabe CG, Kayed R, Robbins J. Reversal of amyloid‐induced heart disease in desmin‐related cardiomyopathy. Proc Natl Acad Sci USA. 2005;102:13592–13597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14:283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zoncu R, Bar‐Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside‐out mechanism that requires the vacuolar H(+)‐ATPase. Science. 2011;334:678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sanbe A, Osinska H, Saffitz JE, Glabe CG, Kayed R, Maloyan A, Robbins J. Desmin‐related cardiomyopathy in transgenic mice: a cardiac amyloidosis. Proc Natl Acad Sci USA. 2004;101:10132–10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Crippa V, D'Agostino VG, Cristofani R, Rusmini P, Cicardi ME, Messi E, Loffredo R, Pancher M, Piccolella M, Galbiati M, Meroni M, Cereda C, Carra S, Provenzani A, Poletti A. Transcriptional induction of the heat shock protein B8 mediates the clearance of misfolded proteins responsible for motor neuron diseases. Sci Rep. 2016;6:22827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Crippa V, Sau D, Rusmini P, Boncoraglio A, Onesto E, Bolzoni E, Galbiati M, Fontana E, Marino M, Carra S, Bendotti C, De Biasi S, Poletti A. The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum Mol Genet. 2010;19:3440–3456. [DOI] [PubMed] [Google Scholar]
- 44. Kwok AS, Phadwal K, Turner BJ, Oliver PL, Raw A, Simon AK, Talbot K, Agashe VR. HspB8 mutation causing hereditary distal motor neuropathy impairs lysosomal delivery of autophagosomes. J Neurochem. 2011;119:1155–1161. [DOI] [PubMed] [Google Scholar]
- 45. Mavroidis M, Panagopoulou P, Kostavasili I, Weisleder N, Capetanaki Y. A missense mutation in desmin tail domain linked to human dilated cardiomyopathy promotes cleavage of the head domain and abolishes its Z‐disc localization. FASEB J. 2008;22:3318–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li D, Tapscoft T, Gonzalez O, Burch PE, Quinones MA, Zoghbi WA, Hill R, Bachinski LL, Mann DL, Roberts R. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation. 1999;100:461–464. [DOI] [PubMed] [Google Scholar]
- 47. Sjoberg G, Saavedra‐Matiz CA, Rosen DR, Wijsman EM, Borg K, Horowitz SH, Sejersen T. A missense mutation in the desmin rod domain is associated with autosomal dominant distal myopathy, and exerts a dominant negative effect on filament formation. Hum Mol Genet. 1999;8:2191–2198. [DOI] [PubMed] [Google Scholar]
- 48. Munoz‐Marmol AM, Strasser G, Isamat M, Coulombe PA, Yang Y, Roca X, Vela E, Mate JL, Coll J, Fernandez‐Figueras MT, Navas‐Palacios JJ, Ariza A, Fuchs E. A dysfunctional desmin mutation in a patient with severe generalized myopathy. Proc Natl Acad Sci USA. 1998;95:11312–11317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Heckmann MB, Bauer R, Jungmann A, Winter L, Rapti K, Strucksberg KH, Clemen CS, Li Z, Schroder R, Katus HA, Muller OJ. AAV9‐mediated gene transfer of desmin ameliorates cardiomyopathy in desmin‐deficient mice. Gene Ther. 2016;23:673–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Weisleder N, Taffet GE, Capetanaki Y. Bcl‐2 overexpression corrects mitochondrial defects and ameliorates inherited desmin null cardiomyopathy. Proc Natl Acad Sci USA. 2004;101:769–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kimura H, Eguchi S, Sasaki J, Kuba K, Nakanishi H, Takasuga S, Yamazaki M, Goto A, Watanabe H, Itoh H, Imai Y, Suzuki A, Mizushima N, Sasaki T. Vps34 regulates myofibril proteostasis to prevent hypertrophic cardiomyopathy. JCI Insight. 2017;2:e89462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Heling A, Zimmermann R, Kostin S, Maeno Y, Hein S, Devaux B, Bauer E, Klovekorn WP, Schlepper M, Schaper W, Schaper J. Increased expression of cytoskeletal, linkage, and extracellular proteins in failing human myocardium. Circ Res. 2000;86:846–853. [DOI] [PubMed] [Google Scholar]
- 53. Di Somma S, Marotta M, Salvatore G, Cudemo G, Cuda G, De Vivo F, Di Benedetto MP, Ciaramella F, Caputo G, de Divitiis O. Changes in myocardial cytoskeletal intermediate filaments and myocyte contractile dysfunction in dilated cardiomyopathy: an in vivo study in humans. Heart. 2000;84:659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplementary Methods.
Table S1. List of Primers for Quantitative PCR Analysis
Table S2. Evaluation of Cardiac Function by M‐Mode Echocardiography in Wild Type ad lib Fed Mice and Myh6CryaABR120G Mice ‐ Either Treated With ad lib Feeding or Intermittent Fasting
Table S3. Evaluation of Cardiac Function by M‐Mode Echocardiography in Wild Type Mice and Myh6CryaABR120G Mice Treated With AAV9‐GFP or AAV9‐TFEB
Table S4. Evaluation of Cardiac Function by M‐Mode Echocardiography in Myh6CryaABR120G Mice Treated With AAV9 Control, AAV9 shTFEB, or AAV9shHspB8 and Subjected to Intermittent Fasting (IF)
Figure S1. Autophagic flux assessment reveals preserved autophagic flux in skeletal muscle from Myh6‐R120G αB‐Crystallin TG mice and in the transgenic mouse hearts at a young age. (A) lmmuno‐blot demonstrating LC3 and p62 expression in heart and skeletal muscle (gastrocnemius) tissue from 40 week old Myh6‐R120G αB‐Crystallin TG mice treated with CQ or diluent for autophagic flux assessment (representative of n=2 experiments). B and C, Representative immuno‐blot (B) depicting LC3‐II and p62 expression with quantitation (C) in young (8 week) Myh6‐R120G αB‐Crystallin TG mice treated with CQ or diluent for autophagic flux assessment. N=3/group. *Indicates P<0.05 by t test.
Figure S2. Expression of αB‐Crystallin in Myh6‐R120G αB‐Crystallin TG mice and CryAB null mice demonstrates specificity for immuno‐detection of αB‐Crystallin. lmmunoblot demonstrating expression of αB‐Crystallin in hearts from Myh6‐R120G αB‐Crystallin TG mice, CryAB null mice and wild‐type controls. Arrows point to various electrophoretic bands detected with the polyclonal αB‐crystallin antibody employed herein.
Figure S3. Intermittent fasting reduces aggregate pathology and myocardial cell death in Myh6‐R120G αB‐Crystallin TG Mice. A, Quantitation of aggregates in the myocardium (as % area). B and C, Representative TUNEL stained images (with DAPI‐stained nuclei, B) with quantitation of TUNEL + nuclei (arrows in B, quantified in C) in hearts from Myh6‐R120G αB‐Crystallin TG mice subjected to intermittent fasting (IF) or ad‐lib feeding for 6 weeks beginning at 40 weeks of age (see Figure 2A for schematic) and WT controls (N=3–5/group). *Indicates P<0.05 by t test for (A) and post‐hoc test after one‐way ANOVA for (C).
Figure S4. Intermittent fasting and TFEB activation do not alter desmin transcription. A, Transcript abundance for desmin in hearts from Myh6‐R120G αB‐Crystallin TG mice subjected to intermittent fasting or ad‐lib feeding, and littermate wild‐type controls. N=4/group. *Indicates P<0.05 by post‐hoc test after one‐way ANOVA. B, Transcript abundance for desmin in hearts from Myh6‐R120G αB‐Crystallin TG mice and littermate wild‐type control mice transduced with AAV‐TFEB or AAV‐GFP. N=3 to 6/group. *Indicates P<0.05 by post‐hoc test after one‐way ANOVA.
Figure S5. AAV‐mediated TFEB transduction upregulates abundance of candidate autophagy‐lysosome target proteins. A, lmmunoblot depicting nuclear TFEB abudance in cardiac tissue from CryABR120G mice transduced with AAV‐GFP or AAV‐TFEB at 36 weeks of age, and sacrificed 10 weeks later. B through F, Representative immuno‐blot with quantification of protein abundance for LC3‐II (C), LAMP1 (D), p62 (E) and LAMP2 (F) in cardiac tissue from wild‐type mice transduced with AAV‐GFP or AAV‐TFEB at 36 weeks of age, and sacrificed 10 weeks later. *Indicates P value by t test. N=3/group.
Figure S6. TFEB transduction does not alter transcript levels of the human CryAB R120G transgene and does not affect cardiac protein synthesis. A, Quantitative PCR analysis for detecting human CRYAB transgene expression with specific primer set (see Table S1) in Myh6‐R120G aB‐Crystallin TG mice transduced with AAV‐TFEB or AAV‐GFP. N=4/group. B and C, Wild‐type male C57bl6 mice (8 weeks old) were transduced with AAV‐GFP or AAV‐TFEB for 4 weeks and SunSET assay performed for assessing cardiac protein synthesis. Representative immuno‐blot (B) with quantitation of puromycin incorporation (C) in cardiac extracts from a non‐injected control mouse and AAV transduced mice. D, Representative immuno‐blots for detecting mTOR activation state in mice treated as in (B). P values were assessed by t test in (A and C).
Figure S7. TFEB transduction reduces aggregate pathology and myocardial cell death in Myh6‐R120G αB‐Crystallin TG mice. A, Quantitation of aggregates as% area of the myocaraium in AAV‐GFP and AAV‐TFEB transduced mice at 46 weeks of age (see Figure 6A for images). N=3/grouQ. *Indicates P<0.05 by t test. B and C, Representative TUNEL stained images (with DAPl‐stained nuclei, B) with quantitation of TUNE L+ nuclei (pseudo‐colored green, arrows in (C) in hearts from Myh6‐R120G αB‐Crystallin TG mice and WT controls transduced with AAV9‐TFEB or AAV9‐GFP for 10 weeks beginning at 36 weeks of age (see Figure 5A for schematic) and WT controls (N=3–4/group). *Indicates P<0.05 by post‐hoc test after one‐way ANOVA.
Figure S8. TFEB induces HSPB8 expression. Transcript abundance of various heat shock protein family members in NRCMs adenovirally transduced with HA‐tagged TFEB or Ad‐Lacz (MOI=10 each) for 24 h. N=3 to 4/group. *Indicates P<0.05 by t test.
Figure S9. Fasting increases HSPB8 transcript and protein levels in the myocardium. A, HSPB8 transcript levels in cardiac tissue from C57bl6 wild‐type mice fasted for various durations as shown. N=4/group. *Indicates P<0.05 by post‐hoc analysis after one way ANOVA (B and C) Representative immunoblot (B) and quantitation of HSPB8 protein levels (C) in the myocardium from fed wild‐type mice, mice fasted for 24 h, and mice fasted for 24 h followed by re‐feeding for 24 h. N=3/group.*Indicates P<0.05. D and E, Representative immunoblot (D) and guantitation of HSPB8 protein levels (E) in the myocardium from Myh6‐R120G αB‐Crystallin TG mice subjected to intermittent fasting (IF) or ad‐lib feeding for 6 weeks beginning at 40 weeks of age (see Figure 2A for schematic) and WT controls (N=4/group). *Indicates P<0.05 by post‐hoc test after one‐way ANOVA.
Figure S10. R120G mutant CRYAB Protein interacts more strongly with desmin than the wild‐type CRYAB protein, in vivo. A, Male C57bl6 mice at 8 weeks of age were transduced with AAV9 particles with cardiac troponin T promoter driven expression of FLAG‐tagged human CRYAB protein or its R120G mutant for 4 weeks. Representative immuno‐blot demonstrating expression of FLAG‐tagged CRYAB proteins is shown in (A). Mice not treated with AAV9 particles are shown as control; “ns” indicates non‐specific band. B, Quantitation of FLAG‐tagged CRYAB protein and its R120G mutant in cardiac extracts from mice treated as in (A). N=3/group. NS indicates not‐significant for P value by t test. C, lmmuno‐precipitation was performed with anti‐FLAG antibody (or lgG confrol) in cardiac extracts from mice treated as in (A) and immuno‐blot performed for desmin and CRYAB. Representative immuno‐blot is shown. (D) Quantitation of desmin expression in after immuno‐precipitation with anti‐FLAG antibody as shown in (C). N=3/group. *Indicates P<0.05 by t test.
Figure S11. Intermittent fasting does not alter desmin ubiquitination. lmmunoblot demonstrating poly‐ubiquitinated desmin in Myh6‐CryABR120G TG mice subjected to intermittent fasting (IF) or ad‐lib (AL) feeding and littermate control hearts (depicted as “‐”). One mg of total protein from cardiac extracts was subjected to immunoprecipitation with anti‐desmin antibody or control lgG, followed by immuno‐blotting for ubiquitin. The immunoblot for desmin is reproduced from the blot shown in Figure 6F. At the exposure shown to demonstrate desmin in TG samples, a desmin signal was not evident in WT hearts in the input lane (with 20 μg/sample; and was seen with longer exposure times [data not shown]).
Figure S12. TFEB promotes clearance of αB‐crystallin via induction of autophagic flux, independent of HSPB8 in NRCMs. A, Representative immunoblot demonstrating assessment of autophagic flux and HSPB8 expresion in NRCMs adenovirally transducesd with R120G mutant of αB‐crystallin (MOI=10; for 48 h) without or with HA‐tagged TFEB (MOI=10; for 24 h) to inhibi lysosomal acidification. Adenoviral particles coding for LacZ or shLacZ were added as controls simultaneously with Ad‐TFEB or Ad‐αB‐crystallin overexpression and sh‐HSPB8, respectively to equalize the number of viral particles. (B through D) Quantitation of HSPB8 (B), LC3‐II (C) and p62 (D) expression in NRCMs treated as in (A). N=3/group. *Indicates P<0.05 by t test within BfA and diluent treated groups; and # indicates P<0.05 by post‐hoc test after one‐way ANOVA for comparisons within the diluent treated group only. E, Quantitation of αB‐crystallin expression in the non‐BfA treated samples from A. *Indicates P<0.05 by one‐way ANOVA.
Figure S13. TFEB promotes removal of αB‐Crystallin aggregates in NRCMs. lmmunoblot demonstrating αB‐Crystallin expression in soluble and insoluble fractions from NRCMs adenovirally transduced with R 120G mutant of αB‐Crystallin (MOl=10; for 48 h) without or with HA‐tagged TFEB (MOl=10; for 24 h). Adenoviral particles coding for LacZ were added as controls simultaneously to equalize the number of viral particles.
Figure S14. Knockdown of TFEB and HSPB8 prevents intermittent fasting‐mediated attenuation of proteotoxic cardiomyopathy. A, Schematic depicting experimental intervention with AAV9‐ mediated shRNA transduction (directed to either TFEB or HSPB8 versus control sh RNA) in 36 week old Myh6‐ CryABR120G TG mice. Four weeks after AAV injection, baseline echocardiograms were obtained followed by 6 weeks of intermittent fasting (termed T=0 time point). Serial echocardiograms were then obtained at 3 weeks and 6 weeks (termed T=3 and T=6 time points, respectively), followed by sacrifice and post mortem biochemical, microscopic and immuno‐histochemical analysis. B, lmmunoblot depicting levels of nuclear TFEB after transduction with shTFEB versus shCon. C, Left ventricular end‐diastolic diameter assessed as fold‐change compared to baseline (T=0 weeks) data‐point in the control group, in mice treated as in (A). N=4 to 5/group. *Indicates P<0.05 by post‐hoc test after two‐way ANOVA. D, Body weight over the duration of the study in mice treated as in (A). N=4 to 5/group. *Indicates P<0.05 by post‐hoc test after two‐way ANOVA. D, Representative immuno‐blot demonstrating expression of LC3 and p62 in mice modeled as in (A) and injected with chloroquine (40 mg/kg) 4 h prior to sacrifice to assess autophagic flux. Representative of n=2 experiments.
Figure S15. Knockdown of TFEB and HSPB8 prevents intermittent fastingmediated removal of aggregates with persistent desmin localization to aggregates. A, Quantitation of aggregates as % area of the myocardium in Myh6‐CryABR120G TG mice transduced with AAV9‐shControl, AAV9‐shTFEB or AAV9‐shHSPB8 for 4 weeks at age of 36 weeks and subjected to intermittent fasting, assessed at 46 weeks of age. See Figure 8G for representative images. N=3/group. P value is by post‐hoc test after one‐way ANOVA. B and C, Representative immuno‐blot (B) with quantitation of αB‐crystallin in insoluble (C) myocardial fractions from Myh6‐CryABR120G TG mice at 46 weeks of age injected with AAV9 particles coding for shRNAs targeting TFEB or HSPB8, or control for 4 weeks and then subjected to intermittent fasting for 6 weeks. N=3/group. P value is by post‐hoc test after one‐way ANOVA.