

Abstract
Background
Circulating microparticles have emerged as biomarkers and effectors of vascular disease. Elevated rates of cardiovascular disease are seen in HIV‐1–seropositive individuals. The aims of this study were to determine: (1) if circulating microparticles are elevated in antiretroviral therapy–treated HIV‐1–seropositive adults; and (2) the effects of microparticles isolated from antiretroviral therapy–treated HIV‐1–seropositive adults on endothelial cell function, in vitro.
Methods and Results
Circulating levels of endothelial‐, platelet‐, monocyte‐, and leukocyte‐derived microparticles were determined by flow cytometry in plasma from 15 healthy and 15 antiretroviral therapy–treated, virologically suppressed HIV‐1–seropositive men. Human umbilical vein endothelial cells were treated with microparticles from individual subjects for 24 hours; thereafter, endothelial cell inflammation, oxidative stress, senescence, and apoptosis were assessed. Circulating concentrations of endothelial‐, platelet‐, monocyte‐, and leukocyte‐derived microparticles were significantly higher (≈35%–225%) in the HIV‐1–seropositive compared with healthy men. Microparticles from HIV‐1–seropositive men induced significantly greater endothelial cell release of interleukin‐6 and interleukin‐8 (≈20% and ≈35%, respectively) and nuclear factor‐κB expression while suppressing anti‐inflammatory microRNAs (miR‐146a and miR‐181b). Intracellular reactive oxygen species production and expression of reactive oxygen species–related heat shock protein 70 were both higher in cells treated with microparticles from the HIV‐1–seropositive men. In addition, the percentage of senescent cells was significantly higher and sirtuin 1 expression lower in cells treated with HIV‐1–related microparticles. Finally, caspase‐3 was significantly elevated by microparticles from HIV‐1–seropositive men.
Conclusions
Circulating concentrations of endothelial‐, platelet‐, monocyte‐, and leukocyte‐derived microparticles were higher in antiretroviral therapy–treated HIV‐1–seropositive men and adversely affect endothelial cells promoting cellular inflammation, oxidative stress, senescence, and apoptosis. Circulating microparticles may contribute to the vascular risk associated with HIV‐1 infection.
Keywords: endothelial dysfunction, HIV‐1, inflammation, microparticles, microRNA
Subject Categories: Inflammation, Atherosclerosis, Biomarkers, Endothelium/Vascular Type/Nitric Oxide, Pathophysiology
Clinical Perspective
What Is New?
Circulating concentrations of endothelial‐, platelet‐, monocyte‐, and leukocyte‐derived microparticles are elevated in antiretroviral therapy–treated HIV‐1–seropositive men.
Microparticles from antiretroviral therapy–treated HIV‐1–seropositive men induce greater endothelial cell activation, inflammation, oxidative stress, senescence, and apoptotic susceptibility compared with microparticles from healthy men.
Endothelial cell NO synthase activity is also disrupted by circulating microparticles from antiretroviral therapy–treated HIV‐1–seropositive men.
What Are the Clinical Implications?
Cardiovascular disease, in particular atherosclerotic vascular disease, has emerged as a major cause of morbidity and mortality in HIV‐1–infected individuals.
Elevations in circulating microparticles, particularly endothelial‐, platelet‐, monocyte‐, and leukocyte‐derived microparticles, are associated with increased risk of atherosclerosis and atherothrombotic events.
Circulating microparticles may be an important contributor to the atherogenic vascular phenotype associated with HIV‐1. Microparticle number and phenotype represent potential novel targets for therapeutic intervention aimed at reducing cardiovascular risk in antiretroviral therapy–treated HIV‐1–seropositive adults.
Introduction
Since the introduction of effective antiretroviral therapy (ART) and consequent improvement in life expectancy, cardiovascular disease (CVD), in particular atherosclerotic CVD, has emerged as a major cause of morbidity and mortality in HIV‐1–infected individuals. HIV‐1–seropositive adults have nearly twice the risk for atherosclerosis and myocardial infarction than uninfected adults,1, 2 and the incidences of coronary heart disease, myocardial infarction, and stroke are expected to continue to increase with the aging of this population.3, 4 The mechanisms underlying the increased risk of atherosclerosis in HIV‐1–infected individuals are not completely understood. Traditional risk factors and exposure to ART do not fully account for the increased incidence and severity of vascular disease and events associated with HIV‐1.3 For example, autopsy findings from HIV‐1–seropositive children and young adults have reported clinically relevant atherosclerotic lesions and endovascular injury in peripheral, coronary, and cerebral arteries, in the absence of CVD risk factors.4
Endothelial cell dysfunction is a primary initiating event in the cause of atherosclerosis5 and is ubiquitous with HIV‐1 infection.4 Although not a target of the virus, HIV‐1 infection promotes a proatherogenic endothelial phenotype characterized by increased cellular inflammation, oxidative stress, and apoptosis and diminished NO production.6 However, the factors underlying endothelial cell dysfunction with HIV‐1 infection are not well defined.
The vascular endothelium is significantly influenced by circulating cell‐derived microparticles.7, 8, 9, 10 Microparticles are small (100–1000 nm) membrane vesicles that are released into the circulation by various cell types (eg, endothelial cells, platelets, leukocytes, and monocytes) in response to a myriad of physiologic and pathologic stimuli that induce cell activation, injury, and apoptosis.7, 10, 11 The initial notion that microparticles in the circulation were merely “inert cellular debris” rapidly changed when it became apparent that circulating microparticles can trigger a proatherogenic endothelial cell phenotype by disrupting endothelial NO production, inducing endothelial inflammation, oxidative stress, and senescence as well as accelerating apoptosis.12, 13, 14 In addition, microparticles have been shown to contribute to plaque development, thrombogenicity, and instability.15 More important, the number, size, composition, and biological effects of microparticles are largely dictated by the stimulus for release.10, 11 Under healthy conditions, cells constitutively release microparticles that aid in cell‐to‐cell communication, activate repair or defense mechanisms, and/or stimulate immune responses.11, 16 Under pathologic conditions, however, microparticles are released in greater number and their functional phenotype is more likely to evoke and perpetuate pathologic cellular responses.10, 17, 18 The numerical and functional phenotype of circulating microparticles in HIV‐1–seropositive adults may represent important mediating factors underlying HIV‐1–associated endothelial dysfunction.
Accordingly, the aims of the present study were to determine: (1) if circulating microparticles are elevated in ART‐treated HIV‐1–seropositive adults; and (2) the effects of microparticles isolated from ART‐treated HIV‐1–seropositive adults on endothelial cell function, in vitro. We hypothesized that circulating levels of endothelial (EMP)–, platelet (PMP)–, monocyte (MMP)–, and leukocyte (LMP)–derived microparticles are elevated in ART‐treated HIV‐1–seropositive adults; and that microparticles from ART‐treated HIV‐1–seropositive adults will induce greater endothelial cell activation, inflammation, oxidative stress, senescence, and apoptotic susceptibility than microparticles from healthy adults.
Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Subjects
Thirty sedentary, nonobese young and middle‐aged adult men (age, 24–64 years) were studied: 15 healthy and 15 HIV‐1 seropositive. All HIV‐1–seropositive men were receiving antiretroviral regimens in accordance with contemporary Department of Health and Human Services guidelines, including an integrase inhibitor (n=9), a nonnucleoside reverse transcriptase inhibitor (n=5), or a protease inhibitor (n=1) combined with 2 reverse transcriptase inhibitors. In addition, the HIV‐1–seropositive men had documented virologic suppression (<50 copies HIV‐1 RNA/mL) for at least 1 year. All subjects were normotensive and free of overt cardiovascular and metabolic disease assessed by medical history, resting and exercise ECGs, and fasting blood chemistry. All subjects were current nonsmokers and had not smoked for at least 7 years before enrollment in the study. Before participation in the study, all subjects provided written informed consent according to the guidelines of the University of Colorado Boulder. This study was approved by the University of Colorado Institutional Review Board.
Body Composition and Metabolic Measures
Body mass was measured to the nearest 0.1 kg using a medical beam balance. Percentage body fat was determined by dual‐energy X‐ray absorptiometry (Lunar Corp, Madison, WI). Body mass index was calculated as weight (kg) divided by height (m) squared. Minimal waist circumference was measured according to published guidelines.19 Fasting plasma lipid, lipoprotein, glucose, and insulin concentrations were determined using standard techniques.
Microparticle Identification and Isolation
Microparticle isolation and identification was performed as previously described.20 Venous blood from an antecubital vein was collected into tubes containing sodium citrate and centrifuged at 1500g for 10 minutes at room temperature. Plasma was collected and stored at −80°C for batch analysis and microparticle isolation. For the characterization and quantification of circulating microparticle subspecies, all plasma samples were centrifuged at 13 000g for 2 minutes and 200 μL was transferred to a TruCount tube (BD Biosciences, Franklin Lakes, NJ). Microparticle subspecies were determined using markers indicative of endothelial (EMP: CD62E+), platelet (PMP: CD62P+), monocyte (MMP: CD14+), and leukocyte (LMP: CD45+) cell lineage. Anti‐human CD62E/allophycocyainin (catalog No. 336012), CD62P/fluorescein isothiocyanate (catalog No. 304903), CD14/APC (catalog No. 367118), and CD45/fluorescein isothiocyanate (catalog No. 368508) antibodies were purchased from Biolegend (San Diego, CA). Samples were incubated with fluorochrome‐labeled antibodies for 20 minutes in the dark at room temperature. After incubation, samples were fixed with 2% paraformaldehyde (ChemCruz Biochemicals, Santa Cruz, CA) and diluted with PBS. Thereafter, all samples were analyzed using an FACSAria I flow cytometer (BD Biosciences). Microparticle size threshold was established using Megamix‐Plus SSC calibrator beads (Biocytex, Marseille, France), and only events >0.16 and <1 μm were counted. The concentration of microparticles was determined using the formula: [(number of events in region containing microparticles/number of events in absolute count bead region)×(total number of beads per test/total volume of sample)].
To isolate microparticles from each subject sample for use in cell experiments, 1 to 2 mL plasma from the sodium citrate tubes was centrifuged at 13 000g for 2 minutes to remove cellular debris and then recentrifuged at 20 500g for 30 minutes at 4°C to pellet microparticles.21 The pelleted microparticles were then resuspended in media, and the concentration of microparticles in the media was determined by fluorescence‐activated cell sorting.
Cell Culture and Microparticle Treatment
Human umbilical vein endothelial cells (HUVECs) (Life Technologies, ThermoFisher, Waltham, MA) were cultured in endothelial growth media (EBM‐2 BulletKit; Lonza, Basel, Switzerland) supplemented with 100 U/mL penicillin and 100 μg/mL streptomycin under standard cell culture conditions (37°C and 5% CO2). Growth medium was replaced 24 hours after initial culture and every 2 days thereafter. Cells were serially passaged after reaching 80% to 90% confluence, and cells were harvested for experimentation after reaching ≈90% confluence on passages 3 to 4. For experimentation, HUVECs were seeded into 6‐well tissue culture plates with media containing an equal concentration of microparticles from either an HIV‐1–seronegative or an HIV‐1–seropositive adult for 24 hours. Cells were treated with microparticles on a 2:1 microparticle/cell basis; this is equivalent to treating each cell with microparticles from 0.4 to 2 nL of plasma. After treatment, cells and media were harvested for the determination of cellular protein expression, microRNA (miR) expression, and soluble cytokine release.
Intracellular Protein Expression
Whole cell lysates were obtained from microparticle‐treated HUVECs for the quantification of intracellular proteins. HUVECs harvested after microparticle treatment were washed in ice‐cold PBS and incubated in ice‐cold radioimmunoprecipitation assay buffer containing protease and phosphatase inhibitors (ThermoFisher) for 15 minutes.22 Cell lysates were sonicated for 20 seconds (four 5‐second cycles spaced by 90 seconds between each cycle) and incubated on ice for an additional 15 minutes.22 Thereafter, cell lysates were centrifuged at 13 000g at 4°C for 7 minutes and the supernatant was collected. Protein concentration was determined using the Bio‐Rad DC protein assay (Bio‐Rad, Hercules, CA). Protein expression was measured by capillary electrophoresis immunoassay (Wes; ProteinSimple, Santa Clara, CA). Briefly, 2 to 3 ng of cell lysate was combined with a provided sample master mix (ProteinSimple) consisting of 1× sample buffer, 1× fluorescent molecular weight markers, and 40 mmol/L dithiothreitol. Samples were vortex mixed and heated at 95°C for 5 minutes before combining with blocking solution, primary antibodies, horseradish peroxidase–conjugated secondary antibody, chemiluminescent substrate, and separation and stacking matrices for automated electrophoresis (375 V for 25 minutes) and immunodetection using the Wes system. Protein expression was quantified as peak area for the protein of interest normalized to peak area of β‐actin in the sample. Rabbit primary antibodies against nuclear factor‐κB (NF‐κB) p65 (D14E12), phosphorylated NF‐κB p65 (Ser536) (93H1), caspase‐3 (8G10) and cleaved caspase‐3 (Asp175) (5A1E), heat shock protein 70 (Hsp70), and sirtuin 1 (D1D7) (diluted 1:50, 1:25, 1:25, 1:25, and 1:50, respectively) (Cell Signaling Technologies, Danvers, MA); endothelial NO synthase (eNOS) (No. PA1‐037), phosphorylated eNOS (Ser1177) (No. PA5‐35879, 1:25) and β‐actin (No. PA1‐16889), (diluted 1:25, 1:25 and 1:300, respectively) (ThermoFisher) were used. Initial titrations were performed to optimize antibody and total protein concentration for each protein.
Intracellular Oxidative Stress
For the determination of intracellular oxidative stress, HUVECs were seeded in 96‐well tissue culture plates (Thermo Scientific, Waltham, MA) and allowed to adhere overnight. Adherent cells were washed and treated with 2′,7′‐dichlorofluorescin diacetate (Abcam, Cambridge, MA) stain (25 μmol/L) for 45 minutes. After 2′,7′‐dichlorofluorescin diacetate treatment, cells were washed twice and stimulated with media or media containing microparticles for 3 hours. Immediately thereafter, fluorescence was measured using a GEMINI EM microplate reader (Molecular Devices, Sunnyvale, CA) and reported as the percentage relative to control.23
Senescence‐Associated β‐Galactosidase Assay
Cellular senescence was quantified using cytochemical senescence‐associated β‐galactosidase staining. After microparticle treatment, HUVECs were washed twice with PBS and incubated with 2 mL of fixative (2% formaldehyde and 0.2% glutaraldehyde) for 5 minutes. Fixed cells were washed twice with PBS and then incubated for 14 hours with 2 mL of freshly prepared staining solution (1 mg/mL 5‐bromo‐4‐chloro‐3‐indolyl‐β‐d‐galactopyranoside in dimethylformamide, 40 mmol/L citric acid/sodium phosphate, 5 mmol/L potassium ferrocyanide, 5 mmol/L potassium ferricyanide, 150 mmol/L NaCl, and 2 mmol/L MgCl2) (ThermoFisher). The staining solution was then removed, and cells were washed twice with PBS and once with methanol and allowed to air dry. Cells were visualized by light microscopy (Zeiss, Thornwood, NY) and quantified in 5 random image fields containing a minimum of 100 cells per field for each condition. All images were quantified by a blinded investigator (K.A.S). Cells with blue cytoplasmic staining were identified as senescent‐positive cells. Senescent cells (percentage) were determined as senescence‐associated β‐galactosidase–positive cells/the total number of cells counted.24
Intracellular miR Expression
Intracellular miR expression for specific miRs associated with cellular inflammation (miR‐126, miR‐146a, and miR‐181b), senescence (miR‐34a), apoptosis (miR‐126 and miR‐Let‐7a), and eNOS (miR‐21, miR‐126, and miR‐155) was determined by reverse transcription–polymerase chain reaction.24 After microparticle treatment, 1.0×105 cells were harvested and total cellular RNA was isolated using the miRVANA RNA isolation kit (Exiqon, Vedbake, Denmark). RNA concentration was determined using a Nanodrop Lite spectrophotometer (ThermoFisher). Thereafter, 150 ng of RNA was reverse transcribed using the miScript II Reverse Transcription Kit (Qiagen, Hilden, Germany).24 Reverse transcription–polymerase chain reaction was performed using the BioRad CFX96 reverse transcription–polymerase chain reaction platform with the miScript SYBR green PCR kit (Qiagen) and specific primers miR‐21, miR‐34a, miR‐92a, miR‐126, miR‐146a, miR‐155, miR‐181b, miR‐Let‐7a, and U6 (Qiagen).24 All samples were assayed in duplicate. miR expression was quantified using the comparative cycle threshold (Ct) method and normalized to U6.24 The relative expression of each transcript was calculated as the 2−ΔCt, where 2−(Ct[miR]−Ct[RNU6]), and presented as arbitrary units (AUs).
Cytokine Release
Concentration of interleukin‐6 and interleukin‐8 was quantified in the media from microparticle‐treated cells using chemiluminescent ELISA (R&D Systems, Minneapolis, MN).25 Intra‐assay coefficient of variation for the chemiluminescent ELISAs was <8% for each assay.
Statistical Analysis
The distribution of the data was assessed by the Shapiro‐Wilk test and the homogeneity of variances by the Levene test. Group differences in subject characteristics, circulating microparticles concentrations, cellular protein expression, miR expression, oxidative stress, and senescence were determined by independent Student t test or Mann‐Whitney U test. Data were presented as mean±SEM for normally distributed variables and as the median (interquartile range [IQR]) for nonnormally distributed variables. Pearson correlations were determined between variables of interest. Statistical significance was set a priori at P<0.05.
Results
Subject Characteristics
Selected subject characteristics are presented in the Table. There were no significant group differences in age, anthropometric variables, or hemodynamic variables. Indeed, body composition and blood pressure were similar between the healthy and seropositive men. Although plasma triglyceride concentrations were slightly, albeit significantly, higher in the seropositive men, there were no significant group differences in plasma cholesterol concentrations or glucose and insulin levels. HIV‐1–seropositive individuals had received ART for a mean of 93.5 months (range, 36–228 months).
Table 1.
Selected Subject Characteristics
| Characteristics | Healthy Subjects (n=15) | HIV‐1–Seropositive Subjects (n=15) |
|---|---|---|
| Age, y | 49±3 | 46±2 |
| Body mass, kg | 78.4±2.4 | 81.3±2.6 |
| BMI, kg/m2 | 25.1±0.7 | 26.3±0.7 |
| Body fat, % | 24.0±1.2 | 26.2±1.7 |
| Systolic blood pressure, mm Hg | 118±2 | 122±3 |
| Diastolic blood pressure, mm Hg | 71±3 | 76±2 |
| Total cholesterol, mg/dL | 186.9±7.8 | 174.0±8.1 |
| LDL‐C, mg/dL | 121.2±5.5 | 104.0±8.1 |
| HDL‐C, mg/dL | 46.3±2.2 | 43.2±2.7 |
| Triglycerides, mg/dL | 87.4±9.5 | 146.5±14.7a |
| Glucose, mg/dL | 88.4±1.8 | 89.8±2.4 |
| Insulin, μU/mL | 6.9±0.7 | 6.9±0.8 |
| Average sleep, h/night | 7.0±0.2 | 7.3±0.4 |
| CD4+ cell count, cells/mm3 | … | 615±82 |
| Viral load, copies/mL | … | <50 |
Values are expressed as mean±SEM. BMI indicates body mass index; HDL‐C, high‐density lipoprotein; LDL‐C, low‐density lipoprotein.
P<0.05.
Circulating Microparticles
Circulating levels of EMPs, PMPs, MMPs, and LMPs in the healthy and HIV‐1–seropositive men are shown in Figure 1. The concentration of each microparticle subspecies was markedly higher (P<0.05) in the HIV‐1–infected men: EMPs (≈35%: median [IQR] 105 [94–140] versus 79 [59–115] microparticles/μL); PMPs (≈225%: median [IQR] 124 [86–169] versus 38 [30–78] microparticles/μL); MMPs (≈70%; median [IQR] 127 [66–257] versus 75 [46–87] microparticles/μL); and LMPs (≈60%; median [IQR] 530 [423–664] versus 336 [129–462] microparticles/μL). There were no significant relations between EMP concentrations and number of months receiving ART or CD4+ cell count.
Figure 1.
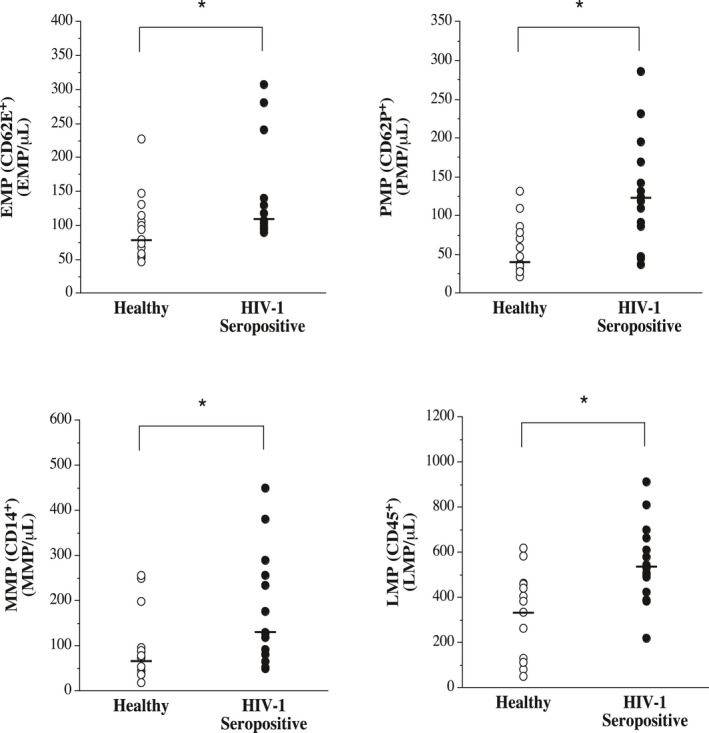
Circulating concentrations of endothelial (EMP)–, platelet (PMP)–, monocyte (MMP)–, and leukocyte (LMP)–derived microparticles in antiretroviral therapy–treated HIV‐1–seropositive men and healthy men. Values are mean±SEM. *P<0.05.
Endothelial Inflammation
The effect of microparticles on HUVEC cytokine release and expression of NF‐κB p65, phosphorylated NF‐κB p65 (Ser536), and miR‐146a is shown in Figure 2. Microparticles from HIV‐1–seropositive men induced greater (P<0.05) release of interleukin‐6 (mean±SEM, 121.4±6.3 versus 101.9±5.3 pg/mL) and interleukin‐8 (mean±SEM, 258.3±14.5 versus 191.4±12.5 pg/mL) from HUVECs into the media compared with the microparticles from healthy men. Intracellular expression of total NF‐κB p65 was not significantly different between the cells treated with microparticles from the HIV‐1–seropositive or healthy men (median [IQR] 2.18 [1.79–3.84] versus 3.06 [1.14–3.45] AUs). However, the level of phosphorylated NF‐κB p65 (Ser536) (active NF‐κB) was 25% higher (P<0.05) in HUVECs treated with microparticles from the HIV‐1–seropositive (median [IQR] 1.23 [1.07–1.33] AUs) compared with healthy (median [IQR] 1.03 [0.86–1.14] AUs) adults. Intracellular expression of miR‐146a, and miR involved in the inhibition of NF‐κB activation, was several‐fold lower (P<0.05) in cells treated with microparticles from HIV‐1–seropositive men than those from healthy controls (mean±SEM, 2.80±0.41 versus 6.59±0.72 AUs). Expression of miR‐181b (mean±SEM, 1.35±0.20 versus 3.18±0.63 AUs) was also lower in the cells treated with microparticles from the HIV‐1–infected men. In the overall study population, phosphorylated NF‐κB p65 (Ser536) expression was positively related to interleukin‐6 (r=0.42; P<0.05) and interleukin‐8 (r=0.41; P<0.05) release and inversely related to miR‐146a expression (r=−0.47; P<0.05) (Figure 2). There was no correlation between interleukin‐6 or interleukin‐8 concentrations and miR‐181b.
Figure 2.
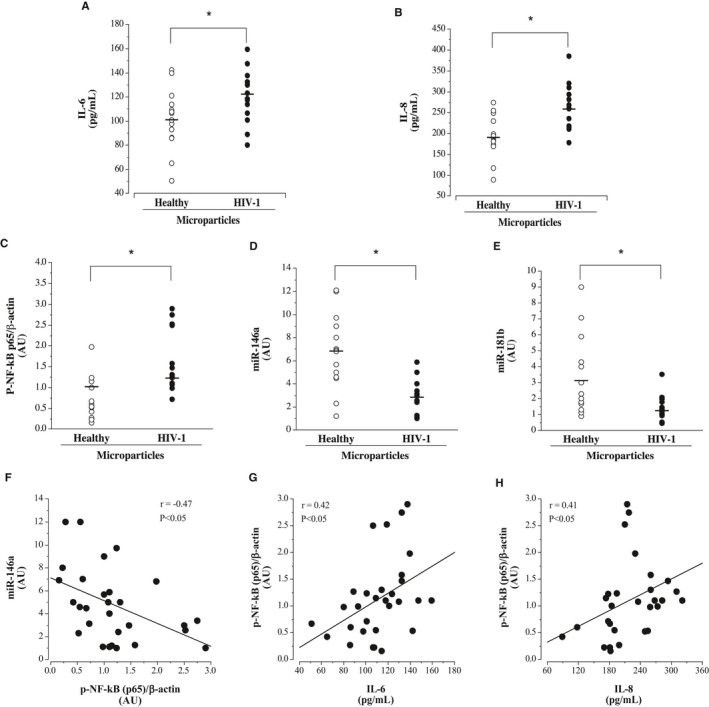
Endothelial cell release of interleukin‐6 (A) and interleukin‐8 (B) and intracellular expression of phosphorylated nuclear factor (NF)‐κB p65 (Ser536) (C), miR‐146a (D), and miR‐181b (E) in response to treatment with microparticles from antiretroviral therapy–treated HIV‐1–seropositive men and healthy men. F, Relation between miR‐146a and phosphorylated NF‐κB p65 in microparticle‐treated human umbilical vein endothelial cells (HUVECs). Relation between phosphorylated NF‐κB p65 and interleukin‐6 (G) and interleukin‐8 (H) concentrations from microparticle‐treated HUVECs. Values are mean±SEM. AU indicates arbitrary unit. *P<0.05.
Endothelial Oxidative Stress
Microparticles from HIV‐1–seropositive men prompted greater (≈40%; P<0.05) cellular reactive oxygen species (ROS) production compared with microparticles from healthy men (mean±SEM, 147±13% versus 107±9% of control) (Figure 3). Moreover, expression of the ROS‐induced chaperone protein, Hsp70, was also significantly higher in the HUVECs treated with the microparticles from HIV‐1–infected men (mean±SEM, 2.81±0.57 versus 1.36±0.13 AUs). ROS production was positively and significantly correlated with Hsp70 expression (r=0.37).
Figure 3.
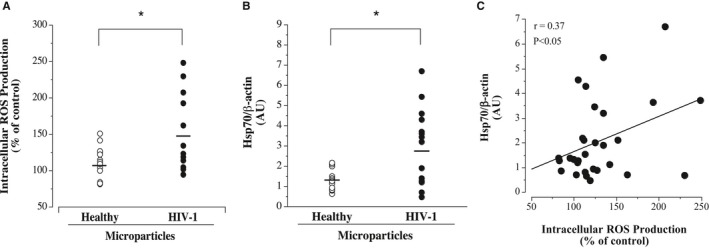
Endothelial cell reactive oxygen species (ROS) production (A) and intracellular heat shock protein 70 (Hsp70) expression (B) in response to microparticles from antiretroviral therapy–treated HIV‐1–seropositive men and healthy men. C, Relation between Hsp70 and ROS production in microparticle‐treated human umbilical vein endothelial cells. Values are mean±SEM. AU indicates arbitrary unit. *P<0.05.
Endothelial Senescence
Endothelial cell senescence as well as cell expression of sirtuin 1 and miR‐34a are shown in Figure 4. Microparticles from the ART‐treated HIV‐1–seropositive adults induced greater endothelial cell senescence. The percentage of senescence‐associated β‐galactosidase staining cells was significantly higher in HUVECs treated with microparticles from HIV‐1–seropositive (mean±SEM, 36±1%) compared with healthy (mean±SEM, 27±1%) adults. HUVEC expression of sirtuin 1 was ≈50% lower (mean±SEM, 4.17±0.54 versus 6.17±0.71 AUs; P<0.05), whereas the expression of miR‐34a was 190% higher (median [IQR] 2.80 [2.11–3.61] versus 0.96 [0.87–3.05] AUs) in cells treated with microparticles from HIV‐1–seropositive versus healthy men. Sirtuin 1 expression was strongly and inversely correlated (r=−0.75; P<0.05) with miR‐34a expression.
Figure 4.
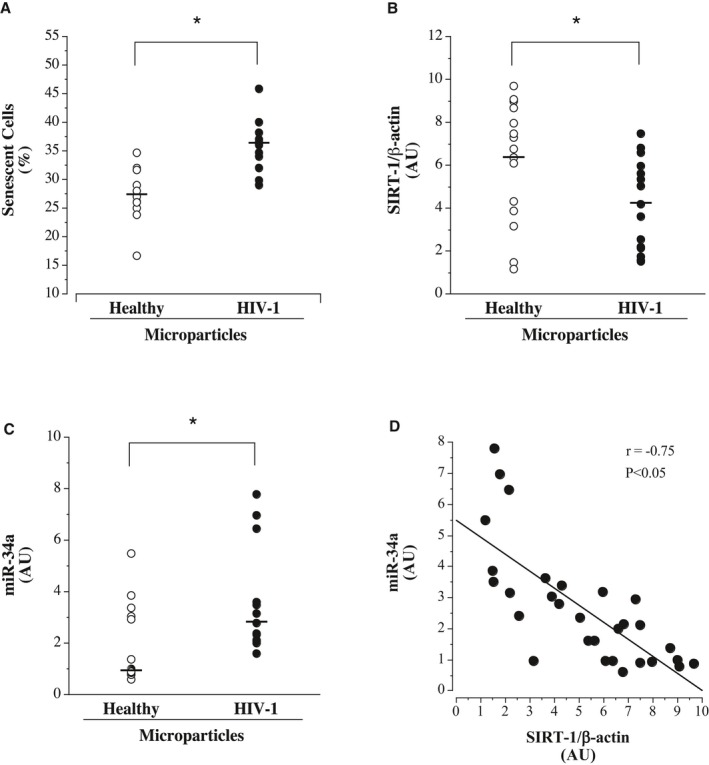
Endothelial cell senescence (A) and intracellular sirtuin 1 (B) and miR‐34a (C) expression in response to treatment with microparticles from antiretroviral therapy–treated HIV‐1–seropositive men and healthy men. D, Relation between cellular miR‐34a and sirtuin 1 expression in microparticle‐treated human umbilical vein endothelial cells. Values are mean±SEM. AU indicates arbitrary unit. *P<0.05.
Endothelial Apoptosis
Endothelial cell expression levels of caspase‐3, active caspase‐3, and miR‐Let‐7a are shown in Figure 5. Microparticles from HIV‐1–seropositive men induced significantly higher (≈50%) expression of total caspase‐3 (mean±SEM, 2.50±0.21 versus 1.66±0.13 AUs) and active caspase‐3 (mean±SEM, 0.92±0.09 versus 0.64±0.08 AUs) than microparticles from healthy men. Higher caspase‐3 expression was accompanied by reduced (P<0.05) expression of miR‐Let‐7a in the HIV‐1–seropositive (mean±SEM, 3.10±0.40 AUs) compared with healthy (mean±SEM, 7.5±1.1 AUs) microparticle‐treated cells. Total caspase‐3 levels were inversely related (r=−0.48; P<0.05) with miR‐Let‐7a expression.
Figure 5.
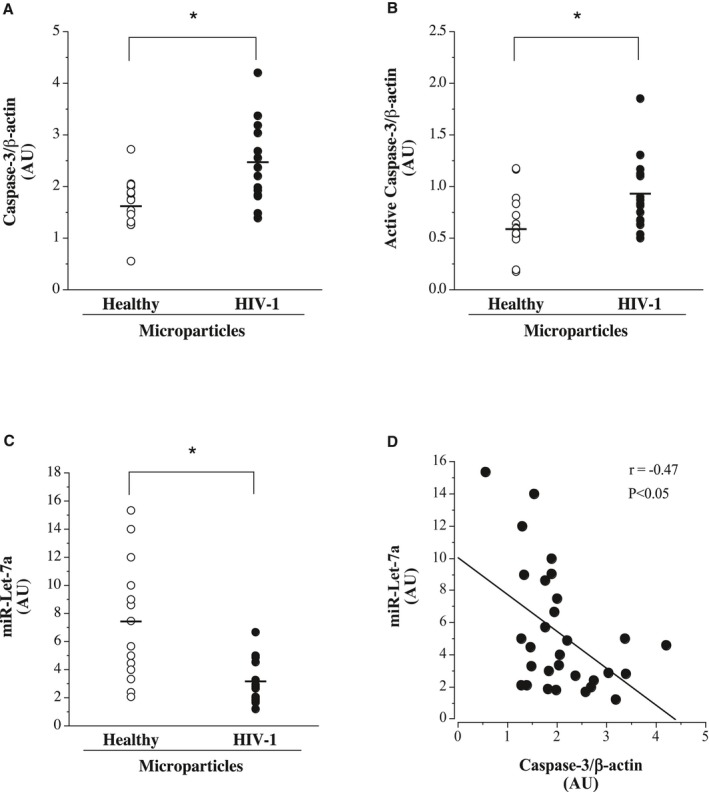
Endothelial cell expression of caspase‐3 (A), active caspase‐3 (Asp175) (B), and miR‐Let‐7a (C) in response to treatment with microparticles from antiretroviral therapy–treated HIV‐1–seropositive and healthy adult men. D, Relation between intracellular miR‐Let‐7a and caspase‐3 expression in microparticle‐treated human umbilical vein endothelial cells. Values are mean±SEM. AU indicates arbitrary unit. *P<0.05.
eNOS Expression
Cellular expression levels of eNOS, phosphorylated eNOS (Ser1177), miR‐21, miR‐126, and miR‐155 are shown in Figure 6. Total eNOS expression was not significantly different between cells treated with microparticles from HIV‐1–seropositive (median [IQR] 1.65 [0.99–2.11] AUs) and healthy (median [IQR] 1.48 [0.46–2.30] AUs) adults; however, phosphorylated eNOS (Ser1177) expression was ≈45% lower (P<0.05) in the cells treated with microparticles from HIV‐1–infected men (median [IQR] 1.40 [0.76–2.03] versus 2.50 [1.48–2.98] AUs). Along with eNOS activity, cellular expression of miR‐21 (median [IQR] 0.43 [0.26–1.02] versus 1.13 [0.31–1.32] AUs) and miR‐126 (median [IQR] 2.53 [0.76–3.29] versus 3.48 [2.90–4.53] AUs) was lower in cells treated with microparticles from HIV‐1–seropositive compared with healthy men. There was no difference in the cellular expression of miR‐155 between HIV‐1 (median [IQR] 0.30 [0.1–0.72] AUs) and healthy (median [IQR] 0.31 [0.14–0.56] AUs) microparticle‐treated cells. In the overall study population, phosphorylated eNOS (Ser1177) was positively associated with miR‐21 (r=0.46; P<0.05) and miR‐126 (r=0.40; P<0.05) expression.
Figure 6.
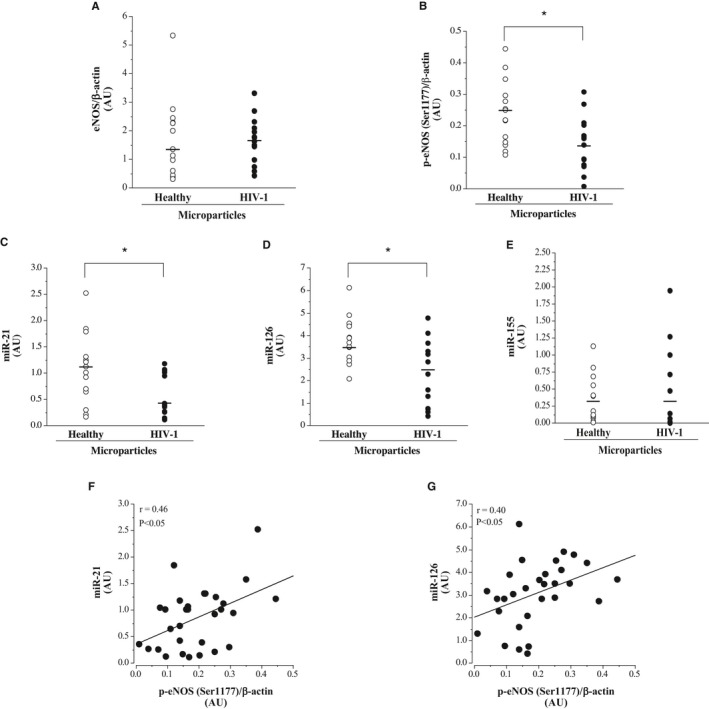
Endothelial cell expression of endothelial NO synthase (eNOS; A), phosphorylated eNOS (Ser1177) (B), miR‐21 (C), miR‐126 (D), and miR‐155 (E) in response to treatment with microparticles from antiretroviral therapy–treated HIV‐1–seropositive and healthy adult men. Relation between intracellular miR‐21 (F) and miR‐126 (G) and phosphorylated eNOS expression in microparticle‐treated human umbilical vein endothelial cells. Values are mean±SEM. AU indicates arbitrary unit. *P<0.05.
Discussion
The key findings of the present study are as follows: (1) circulating concentrations of EMPs, PMPs, MMPs, and LMPs are markedly higher in ART‐treated HIV‐1–seropositive men compared with healthy men; and (2) microparticles isolated from ART‐treated HIV‐1–seropositive men induce a proinflammatory, pro‐oxidative, prosenescent, and proapoptotic endothelial phenotype. To our knowledge, this is the first study to determine both the number and functional phenotype of circulating microparticles in ART‐treated HIV‐1–infected adults. Of note, all cells were treated with an equal number of microparticles from each subject. Therefore, all observed microparticle effects are attributable to differences in microparticle composition and not concentration. Elevations in circulating microparticles that are capable of causing deleterious, atherogenic effects on endothelial cell function represent novel factors that could contribute mechanistically to the increased risk and incidence of atherosclerotic vascular disease associated with HIV‐1.
Although most eukaryotic cells possess the capacity to release microparticles, clinical interest in circulating microparticles has focused primarily on EMPs, PMPs, LMPs, and MMPs because of their principal involvement in the initiation, development, and progression of atherosclerotic vascular disease and its clinical consequences.10, 26, 27 Elevations in these microparticles are associated with several cardiovascular pathologic conditions and risk factors, including atherothrombosis, myocardial infarction, ischemic left ventricular dysfunction, unstable angina, diabetes mellitus, hypertension, and metabolic syndrome.7, 18, 27 We demonstrate that the circulating levels of EMPs, PMPs, LMPs, and MMPs are significantly higher (35% to 225%) in ART‐treated HIV‐1–seropositive adults. Although we are the first to report elevations in each of these microparticle subtypes, this finding is not overly surprising considering HIV‐1 infection is known to cause activation of their parent cells, even in virologically suppressed individuals.28, 29 Cell activation is likely the primary mechanism underlying the elevated circulating concentrations of these specific microparticles.9, 10 Indeed, a ubiquitous cellular response to activation is rapid elevation in p38 activity, resulting in cytoskeletal rearrangement and membrane vesiculation.10 Regardless of mechanism of release, the importance of circulating microparticles may not simply be based on their concentration but, rather, their potential effects on the vasculature.7, 30, 31 For example, microparticles isolated from adults with coronary artery disease have been shown to suppress NO‐mediated endothelium‐dependent relaxation in isolated arteries.11 Considering that HIV‐1, regardless of ART and independent of other risk factors, is associated with vascular dysfunction, characterized by heightened endothelial inflammation, oxidative stress, and apoptotic burden, it is possible that circulating microparticles, both in terms of their elevated number and functional phenotype, may be a contributing factor.4, 6, 9
Endothelial cell release of the proinflammatory cytokines interleukin‐6 and interleukin‐8 is a precipitating event in atherogenesis.32 In the present study, microparticles from ART‐treated HIV‐1–seropositive adults, despite being similar in number, induced significantly more endothelial cell release of interleukin‐6 and interleukin‐8, in vitro, compared with microparticles from healthy adults. Cellular NF‐κB activation was markedly higher in cells treated with microparticles from the HIV‐1–infected individuals. NF‐κB is the principal transcription factor regulating both interleukin‐6 and interleukin‐8 production and release.32 Thus, the increase in endothelial cell NF‐κB activation in response to the HIV‐1–related microparticles is consistent with, and likely underlies, the enhanced release of interleukin‐6 and interleukin‐8. Moreover, the changes in NF‐κB activity in response to microparticles from the HIV‐1–seropositive men were directionally aligned with observed changes in cellular expression of miR‐146a and miR‐181b, 2 miRs involved in NF‐κB regulation.33, 34 Both miR‐146a and miR‐181b were 2‐fold lower in cells treated with microparticles from the ART‐treated HIV‐1–seropositive adults. miR‐146a quells NF‐κB activation by targeting several upstream activators of NF‐κB, such as interleukin‐1 receptor–associated kinase 1 and tumor necrosis factor receptor–associated factor 6.33 miR‐181b suppresses NF‐κB–mediated gene transcription by inhibiting importin‐ɑ3, a protein involved in nuclear translocation of NF‐κB.33, 34 Reduced expression of miR‐146a and miR‐181b has been directly linked with increased NF‐κB–mediated endothelial inflammation.33, 34 In addition to inflammation, elevated NF‐κB activation has been shown to contribute to the pathogenesis of atherosclerosis and thrombosis endothelial dysfunction, atherogenesis, and heart failure.35, 36, 37, 38 Collectively, these findings demonstrate the profound effects of circulating microparticles from HIV‐1–infected individuals on endothelial cell inflammatory processes and outline novel mechanisms for the increase in vascular inflammation associated with HIV‐1.
Oxidative stress often accompanies inflammation and is central to the development of atherosclerosis.39 Although ROS production is an unavoidable consequence of cellular metabolism and is important for cellular signaling, aberrant ROS production results in DNA and organelle damage as well as protein dysregulation and dysfunction, rendering the cell susceptible to disease.39 Herein, we demonstrate that microparticles from ART‐treated HIV‐1–seropositive adults increased endothelial ROS production significantly more (≈40%) than microparticles from healthy controls. Consistent with greater ROS production, cellular expression of Hsp70 was significantly higher in the cells treated with microparticles from HIV‐1–seropositive adults. Hsp70 is a chaperone protein that is expressed constitutively at low levels but is promptly upregulated in response to ROS‐induced cellular stress.40 Hsp70 is a sensitive and specific indicator of cellular oxidative stress and damage,40 providing additional evidence of increased endothelial oxidative burden. Intracellular ROS is known to cause endothelial activation, damage, and dysfunction, leading to senescence, apoptosis, atherosclerosis, and major adverse cardiac events.41, 42, 43 Thus, circulating microparticles may play an important role in mediating and promoting the pro‐oxidative endothelial phenotype that is known to occur with HIV‐1 infection.6
Endothelial cell senescence and apoptosis renders the vasculature highly susceptible to atherosclerosis. Senescent‐prone endothelial cells exhibit diminished regenerative, angiogenic, and vasomotor function.44 In addition, senescent endothelial cells develop a proinflammatory senescence‐associated secretory phenotype prone to production and release of proinflammatory cytokines and signaling molecules, such as interleukin‐6 and interleukin‐8.45 In the present study, microparticles from the HIV‐1–infected men induced an increase in endothelial cell senescence. The percentage of senescence‐associated β‐galactosidase–stained cells was markedly higher in cells exposed to microparticles from the HIV‐1–seropositive men than those from the healthy men. The mechanisms underlying the prosenescent effects of HIV‐1–related microparticles appear to involve downregulation of key protein and miR critical to antisenescent cellular signaling.45, 46 Indeed, sirtuin 1 expression, an NAD+‐dependent histone deacetylase that inhibits the activity and expression of proteins associated with cellular senescence and apoptosis,46, 47 was significantly lower in the cells treated with microparticles from the HIV‐1–seropositive men compared with microparticles from the healthy men. Reduced cellular expression of sirtuin 1 is recognized as a central, defining feature of a prosenescent cellular phenotype.47 Furthermore, concordant with suppressed sirtuin 1, miR‐34a expression was significantly higher in the HIV‐1 microparticle‐treated cells. miR‐34a targets the 3′ untranslated region of sirtuin 1 mRNA, resulting in translational repression and lower sirtuin 1 bioavailability.46 In line with this, we observed a robust inverse correlation between miR‐34a expression and sirtuin 1 levels (r=−0.75; P<0.05). The accumulation of endothelial senescence has emerged as a key factor underlying the pathogenesis of vascular aging, atheroma formation, and atherosclerotic CVD.44, 48 Prosenescent endothelial effects of HIV‐1–related circulating microparticles suggest a novel mechanism contributing to accelerated vascular disease associated with HIV‐1.
With regard to apoptosis, HUVECs treated with microparticles from the HIV‐1–seropositive men exhibited markedly higher total caspase‐3 and active caspse‐3 protein expression than cells treated with microparticles from healthy adults. Intracellular concentration of caspase‐3, particularly the active form of caspase‐3, provides specific biological insight into the apoptotic tendency of a cell.49 HIV‐1 infection is known to escalate cellular apoptosis through various mechanisms that ultimately heighten caspase‐3 activity.50, 51 For example, we have shown that HIV‐1 proteins gp120 and Tat induce a proapoptotic endothelial phenotype by increasing intracellular caspase‐3 activity.51 Circulating microparticles may represent another mechanism facilitating the cellular apoptotic effects of HIV‐1 infection. Consistent with this notion is the finding that the expression of miR‐Let‐7a was significantly lower in cells treated with microparticles from the HIV‐1–seropositive adults compared with microparticles from the healthy adults. miR‐Let‐7a directly targets caspase‐3 mRNA. The 3′ untranslated region of the caspase‐3 gene perfectly matches the seed sequence of miR‐Let‐7a, triggering mRNA translational repression and/or degradation.52 In fact, miR‐Let‐7a has been shown to suppress drug‐induced apoptosis in cells directly via its effect on caspase‐3 expression.52 In the present study, we observed a significant, inverse relation between cellular caspase‐3 and miR‐Let‐7a expression, supporting miR‐Let‐7a involvement in the HIV‐1–related microparticle‐induced increase in cellular caspase‐3. Increased active caspase‐3 heightens the rate of cellular apoptosis and is associated with atherosclerosis and thrombosis.53, 54 Circulating microparticles may play an important role in HIV‐1–related atherosclerosis through a proapoptotic influence.6
Changes in the inflammatory and oxidative state of the cell can affect caspase‐3 activity,39 and it is therefore plausible that the increase in caspase‐3 activity in cells treated with microparticles from the HIV‐1–seropositive men may, at least in part, be a consequence of increased inflammatory and oxidative burden. Regardless of the mechanisms involved, the proapoptotic endothelial effects of HIV‐1–related circulating microparticles provide additional insight about the detrimental vascular effects associated with HIV‐1 infection.
eNOS activity and NO generation are critical to endothelial cell homeostasis.55, 56, 57 Herein, we demonstrate that microparticles from HIV‐1–seropositive adults blunt eNOS activation. Reduced eNOS activation is, arguably, the most prominent factor in endothelial dysfunction and, in turn, atherogenesis.58 Given the importance of eNOS activation to endothelial vasodilator function, it is reasonable to posit that circulating microparticles contribute to impaired endothelial vasodilation observed in HIV‐1–infected individuals.6 Interestingly, eNOS activation, and not eNOS protein expression, was affected by the HIV‐1–related microparticles. The cause for the specificity of the microparticle effect on eNOS is not clear. However, the observed changes in the cellular expression of miR‐21, miR‐126, and miR‐155, miRs that directly influence eNOS expression and activation, provide some insight. In line with suppressed eNOS activation, expression of miR‐21 and miR‐126 was significantly lower in the cells treated with microparticles from the HIV‐1–seropositive men compared with microparticles from the healthy men. miR‐21 suppresses phosphatase and tensin homolog (PTEN), a negative regulator of eNOS activation,55 whereas miR‐126 protects the activity of the phosphatidylinositol 3‐kinase/protein kinase B/eNOS signaling pathway and, in turn, eNOS activation.56 Thus, taken together, reduction in the expression of both miR‐21 and miR‐126 affects 2 independent regulatory pathways involved in eNOS activation; disruption in each pathway likely diminished the activation potential of the enzyme. Of note, miR‐155 was not altered by the HIV‐1–related microparticles. This is noteworthy because miR‐155 directly targets eNOS mRNA, reducing its expression,59 and no change in miR‐155 is fully concordant with our finding of no reduction in eNOS protein expression in the cells treated with microparticles from the HIV‐1–infected men.
There are a few experimental considerations about the present study that deserve mention. First, inherent with all cross‐sectional human studies, we cannot dismiss the possibility that genetic and/or lifestyle behaviors may have influenced the results of our group comparisons. However, to minimize the effects of other lifestyle behaviors, all subjects were sedentary, nonobese, and nonsmokers. Moreover, all subjects were carefully screened to eliminate the confounding effects of clinically overt metabolic disease and/or CVD. Indeed, smoking, diabetes mellitus, and hypertension have been shown to have deleterious effects on circulating microparticles.26, 60, 61 Now that we have identified significant differences between our HIV‐1‐seropositive and seronegative study populations, the next question to address is whether these differences are found in more heterogeneous populations. Second, we did not inhibit or counteract the observed changes in miR expression within the cells; therefore, we are unable to precisely discern the exact contribution of each miR on either its specific cellular target or functional outcome. Third, the in vitro nature of our study precludes us from making definitive statements about clinical risk. However, it is important to emphasize that our rationale for selecting these endothelial cell makers of function was because of their established association and causative link with clinical risk and adverse cardiovascular outcomes. Fourth, although all of the HIV‐1–seropositive men were on their first ART regimen, the regimens were not uniform across the subjects. Our sample size is not sufficient to determine whether differences exist between ART regimens and microparticle number and function. Fifth, our study involved only men. We cannot, and should not, assume similar numerical patterns and/or functional microparticle phenotypes in HIV‐1–seropositive women. Considering that HIV‐1–infected women have increased rates of acute myocardial infarctions and ischemic strokes compared with HIV‐1–infected men,62 future studies are essential to characterize the microparticle phenotype in seropositive women. Finally, the in vitro results of the present study involve the use of HUVECs and not an arterial‐derived cell line, raising a potential concern that the expression of proteins and the function of venous endothelial cells in culture may not be representative of arterial endothelial cells and, in turn, may have limited implications for arterial atherosclerotic disease. However, much has been learned about endothelial cell function and atherogenesis using cultured HUVECs.63, 64 Moreover, it has been demonstrated that the expression of key proteins involved in the regulation of vascular endothelial function and disease risk (eg, NF‐κB and eNOS) is similar in endothelial cells acquired from an artery and vein in humans.65, 66 Indeed, venous and arterial cells have been shown to demonstrate similar, reproducible responses to various stimuli (physiologic and pathologic) both in vitro and in vivo.63, 67, 68, 69 Thus, cultured HUVECs are a reliable and useful in vitro cell line for studying endothelial cell function and atherogenic processes.69
In conclusion, the results of this study demonstrate that circulating concentrations of cell‐derived microparticles (EMPs, PMPs, MMPs, and LMPs) are elevated in ART‐treated HIV‐1–seropositive men. In addition, microparticles from ART‐treated HIV‐1–seropositive men adversely affect endothelial cell function, promoting endothelial cell inflammation, oxidative stress, senescence, and apoptosis as well as reducing eNOS activation. It is well established that these changes in endothelial cell phenotype are highly proatherogenic and prothombotic.5, 39, 44 Circulating microparticles represent novel mechanistic mediators of the atherogenic vascular environment associated with HIV‐1 infection and viable targets for therapeutic intervention.
Sources of Funding
This study was supported by National Institutes of Health (NIH) awards HL131458 and HL135598 and NIH/National Center for Advancing Translational Sciences UL1 TR001082.
Disclosures
These disclosures are correct.
Acknowledgments
We thank all the subjects who participated in the study; the clinical staff at the Clinical and Translational Research Center, University of Colorado Boulder, for their assistance; and the staff at the University of Colorado Anschutz Medical Campus Allergy & Clinical Immunology, Infectious Disease Flow Core for their technical assistance.
(J Am Heart Assoc. 2019;8:e011134 DOI: 10.1161/JAHA.118.011134.)
References
- 1. Freiberg MS, Chang C‐CH, Kuller LH, Skanderson M, Lowy E, Kraemer KL, Butt AA, Bidwell Goetz M, Leaf D, Oursler KA, Rimland D, Rodriguez Barradas M, Brown S, Gibert C, McGinnis K, Crothers K, Sico J, Crane H, Warner A, Gottlieb S, Gottdiener J, Tracy RP, Budoff M, Watson C, Armah KA, Doebler D, Bryant K, Justice AC. HIV infection and the risk of acute myocardial infarction. JAMA Intern Med. 2013;173:614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beckman JA, Duncan MS, Alcorn CW, So‐Armah K, Butt AA, Goetz MB, Tindle HA, Sico J, Tracy RP, Justice AC, Freiberg MS. Association of HIV infection and risk of peripheral artery disease. Circulation. 2018;138:255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Islam FM, Wu J, Jansson J, Wilson DP. Relative risk of cardiovascular disease among people living with HIV: a systematic review and meta‐analysis. HIV Med. 2012;13:453–468. [DOI] [PubMed] [Google Scholar]
- 4. Gibellini D, Borderi M, Clò A, Morini S, Miserocchi A, Bon I, Ponti C, Re MC. HIV‐related mechanisms in atherosclerosis and cardiovascular diseases. J Cardiovasc Med (Hagerstown). 2013;14:780–790. [DOI] [PubMed] [Google Scholar]
- 5. Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–3488. [DOI] [PubMed] [Google Scholar]
- 6. Kline ER, Sutliff RL. The roles of HIV‐1 proteins and antiretroviral drug therapy in HIV‐1‐associated endothelial dysfunction. J Investig Med. 2008;56:752–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lovren F, Verma S. Evolving role of microparticles in the pathophysiology of endothelial dysfunction. Clin Chem. 2013;59:1166–1174. [DOI] [PubMed] [Google Scholar]
- 8. Trzepizur W, Martinez MC, Priou P, Andriantsitohaina R, Gagnadoux F. Microparticles and vascular dysfunction in obstructive sleep apnoea. Eur Respir J. 2014;44:207–216. [DOI] [PubMed] [Google Scholar]
- 9. Koganti S, Eleftheriou D, Brogan PA, Kotecha T, Hong Y, Rakhit RD. Microparticles and their role in coronary artery disease. Int J Cardiol. 2017;230:339–345. [DOI] [PubMed] [Google Scholar]
- 10. Rautou P‐E, Vion A‐C, Amabile N, Chironi G, Simon A, Tedgui A, Boulanger CM. Microparticles, vascular function, and atherothrombosis. Circ Res. 2011;109:593–606. [DOI] [PubMed] [Google Scholar]
- 11. Badimon L, Suades R, Arderiu G, Peña E, Chiva‐Blanch G, Padró T. Microvesicles in atherosclerosis and angiogenesis: from bench to bedside and reverse. Front Cardiovasc Med. 2017;4:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Diehl P, Fricke A, Sander L, Stamm J, Bassler N, Htun N, Ziemann M, Helbing T, El‐Osta A, Jowett JBM, Peter K. Microparticles: major transport vehicles for distinct microRNAs in circulation. Cardiovasc Res. 2012;93:633–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jansen F, Yang X, Proebsting S, Hoelscher M, Przybilla D, Baumann K, Schmitz T, Dolf A, Endl E, Franklin BS, Sinning J, Vasa‐Nicotera M, Nickenig G, Werner N. MicroRNA expression in circulating microvesicles predicts cardiovascular events in patients with coronary artery disease. J Am Heart Assoc. 2014;3:e001249 DOI: 10.1161/JAHA.114.001249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hulsmans M, Holvoet P. MicroRNA‐containing microvesicles regulating inflammation in association with atherosclerotic disease. Cardiovasc Res. 2013;100:7–18. [DOI] [PubMed] [Google Scholar]
- 15. Nomura S. Microparticle and atherothrombotic diseases. J Atheroscler Thromb. 2016;23:1–9. [DOI] [PubMed] [Google Scholar]
- 16. Hergenreider E, Heydt S, Treguer K, Boettger T, Horrevoets AJG, Zeiher AM, Scheffer MP, Frangakis AS, Yin X, Mayr M, Braun T, Urbich C, Boon RA, Dimmeler S. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol. 2012;14:249–256. [DOI] [PubMed] [Google Scholar]
- 17. Chironi GN, Boulanger CM, Simon A, Dignat‐George F, Freyssinet J‐M, Tedgui A. Endothelial microparticles in diseases. Cell Tissue Res. 2009;335:143–151. [DOI] [PubMed] [Google Scholar]
- 18. Jansen F, Li Q, Pfeifer A, Werner N. Endothelial‐ and immune cell‐derived extracellular vesicles in the regulation of cardiovascular health and disease. JACC Basic Transl Sci. 2017;2:790–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lohman TG, Roche AF, Martorell R. Anthropometric Standardization Reference Manual. Champaign, IL: Human Kinetics Books; 1988. [Google Scholar]
- 20. Bammert TD, Hijmans JG, Kavlich PJ, Lincenberg GM, Reiakvam WR, Fay RT, Greiner JJ, Stauffer BL, DeSouza CA. Influence of sex on the number of circulating endothelial microparticles and microRNA expression in middle‐aged adults. Exp Physiol. 2017;102:894–900. [DOI] [PubMed] [Google Scholar]
- 21. Jansen F, Yang X, Hoelscher M, Cattelan A, Schmitz T, Proebsting S, Wenzel D, Vosen S, Franklin BS, Fleischmann BK, Nickenig G, Werner N. Endothelial microparticle‐mediated transfer of microRNA‐126 promotes vascular endothelial cell repair via SPRED1 and is abrogated in glucose‐damaged endothelial microparticles. Circulation. 2013;128:2026–2038. [DOI] [PubMed] [Google Scholar]
- 22. Kirby ED, Kuwahara AA, Messer RL, Wyss‐Coray T. Adult hippocampal neural stem and progenitor cells regulate the neurogenic niche by secreting VEGF. Proc Natl Acad Sci USA. 2015;112:4128–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu S, He Y, Vokurkova M, Touyz RM. Endothelial cells negatively modulate reactive oxygen species generation in vascular smooth muscle cells: role of thioredoxin. Hypertension. 2009;54:427–433. [DOI] [PubMed] [Google Scholar]
- 24. Hijmans JG, Stockleman K, Reiakvam W, Levy MV, Brewster LM, Bammert TD, Greiner JJ, Connick E, DeSouza CA. Effects of HIV‐1 gp120 and tat on endothelial cell senescence and senescence‐associated microRNAs. Physiol Rep. 2018;6:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1α‐mediated NF‐κB activation and down‐regulation of TRAF2 expression. Mol Cell Biol. 2006;26:3071–3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Berezin A, Zulli A, Kerrigan S, Petrovic D, Kruzliak P. Predictive role of circulating endothelial‐derived microparticles in cardiovascular diseases. Clin Biochem. 2015;48:562–568. [DOI] [PubMed] [Google Scholar]
- 27. Boulanger CM, Loyer X, Rautou P‐E, Amabile N. Extracellular vesicles in coronary artery disease. Nat Rev Cardiol. 2017;14:259–272. [DOI] [PubMed] [Google Scholar]
- 28. Kearns A, Gordon J, Burdo TH, Qin X. HIV‐1–associated atherosclerosis: unraveling the missing link. J Am Coll Cardiol. 2017;69:3084–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mayne E, Funderburg NT, Sieg SF, Asaad R, Kalinowska M, Rodriguez B, Schmaier AH, Stevens W, Lederman MM. Increased platelet and microparticle activation in HIV infection: upregulation of Pselectin and tissue factor expression. J Acquir Immune Defic Syndr. 2012;59:340–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Halim ATA, Ariffin NAFM, Azlan M. Review: the multiple roles of monocytic microparticles. Inflammation. 2016;39:1277–1284. [DOI] [PubMed] [Google Scholar]
- 31. Angelillo‐Scherrer A. Leukocyte‐derived microparticles in vascular homeostasis. Circ Res. 2012;110:356–369. [DOI] [PubMed] [Google Scholar]
- 32. Mudaliar H, Pollock C, Ma J, Wu H, Chadban S, Panchapakesan U. The role of TLR2 and 4‐mediated inflammatory pathways in endothelial cells exposed to high glucose. PLoS One. 2014;9:e108844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ma S, Tian XY, Zhang Y, Mu C, Shen H, Bismuth J, Pownall HJ, Huang Y, Wong WT. E‐selectin‐targeting delivery of microRNAs by microparticles ameliorates endothelial inflammation and atherosclerosis. Sci Rep. 2016;6:22910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sun X, Icli B, Wara AK, Belkin N, He S, Kobzik L, Hunninghake GM, Vera MP, Blackwell TS, Baron RM, Feinberg MW. MicroRNA‐181b regulates NF‐κB–mediated vascular inflammation. J Clin Invest. 2012;122:1973–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Collins T, Cybulsky MI. NF‐κB: pivotal mediator or innocent bystander in atherogenesis? J Clin Invest. 2001;107:255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liang Y, Li X, Zhang X, Li Z, Wang L, Sun Y, Liu Z, Ma X. Elevated levels of plasma TNF‐α are associated with microvascular endothelial dysfunction in patients with sepsis through activating the NF‐κB and p38 mitogen‐activated protein kinase in endothelial cells. Shock. 2014;41:275–281. [DOI] [PubMed] [Google Scholar]
- 37. Hedayat M, Mahmoudi MJ, Rose NR, Rezaei N. Proinflammatory cytokines in heart failure: double‐edged swords. Heart Fail Rev. 2010;15:543–562. [DOI] [PubMed] [Google Scholar]
- 38. Ramkaran P, Khan S, Phulukdaree A, Moodley D, Chuturgoon AA. miR‐146a polymorphism influences levels of miR‐146a, IRAK‐1, and TRAF‐6 in young patients with coronary artery disease. Cell Biochem Biophys. 2014;68:259–266. [DOI] [PubMed] [Google Scholar]
- 39. Incalza MA, D'Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol. 2018;100:1–19. [DOI] [PubMed] [Google Scholar]
- 40. Lee YJ, Corry PM. Metabolic oxidative stress‐induced HSP70 gene expression is mediated through SAPK pathway: role of Bcl‐2 and c‐Jun NH2‐terminal kinase. J Biol Chem. 1998;273:29857–29863. [DOI] [PubMed] [Google Scholar]
- 41. Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol. 2003;91:7–11. [DOI] [PubMed] [Google Scholar]
- 42. Vassalle C, Bianchi S, Battaglia D, Landi P, Bianchi F, Carpeggiani C. Elevated levels of oxidative stress as a prognostic predictor of major adverse cardiovascular events in patients with coronary artery disease. J Atheroscler Thromb. 2012;18:712–717. [PubMed] [Google Scholar]
- 43. Laufs U, Wassmann S, Czech T, Münzel T, Eisenhauer M, Böhm M, Nickenig G. Physical inactivity increases oxidative stress, endothelial dysfunction, and atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:809–814. [DOI] [PubMed] [Google Scholar]
- 44. Bhayadia R, Schmidt BMW, Melk A, Hömme M. Senescence‐induced oxidative stress causes endothelial dysfunction. J Gerontol A Biol Sci Med Sci. 2016;71:161–169. [DOI] [PubMed] [Google Scholar]
- 45. Tabuchi T, Satoh M, Itoh T, Nakamura M. MicroRNA‐34a regulates the longevity‐associated protein SIRT1 in coronary artery disease: effect of statins on SIRT1 and microRNA‐34a expression. Clin Sci. 2012;123:161–171. [DOI] [PubMed] [Google Scholar]
- 46. Ito T, Yagi S, Yamakuchi M. MicroRNA‐34a regulation of endothelial senescence. Biochem Biophys Res Commun. 2010;398:735–740. [DOI] [PubMed] [Google Scholar]
- 47. Ota H, Eto M, Ogawa S, Iijima K, Akishita M, Ouchi Y. SIRT1/eNOS axis as a potential target against vascular senescence, dysfunction and atherosclerosis. J Atheroscler Thromb. 2010;17:431–435. [DOI] [PubMed] [Google Scholar]
- 48. Minamino T, Komuro I. Vascular cell senescence. Circ Res. 2007;100:15–26. [DOI] [PubMed] [Google Scholar]
- 49. Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ullrich CK, Groopman JE, Ganju RK. HIV‐1 gp120‐ and gp160‐induced apoptosis in cultured endothelial cells is mediated by caspases. Blood. 2000;96:1438–1442. [PubMed] [Google Scholar]
- 51. MacEneaney OJ, Connick E, DeSouza CA. Effects of HIV‐1 gp120 and protease inhibitors on apoptotic susceptibility of CD34+ hematopoietic progenitor cells. J Acquir Immune Defic Syndr. 2011;56:49–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tsang WP, Kwok TT. Let‐7a microRNA suppresses therapeutics‐induced cancer cell death by targeting caspase‐3. Apoptosis. 2008;13:1215–1222. [DOI] [PubMed] [Google Scholar]
- 53. Choy JC, Granville DJ, Hunt DWC, McManus BM. Endothelial cell apoptosis: biochemical characteristics and potential implications for atherosclerosis. J Mol Cell Cardiol. 2001;33:1673–1690. [DOI] [PubMed] [Google Scholar]
- 54. Durand E, Scoazec A, Lafont A, Boddaert J, Hajzen AA, Addad F, Mirshahi M, Desnos M, Tedgui A, Mallat Z. In vivo induction of endothelial apoptosis leads to vessel thrombosis and endothelial denudation. Circulation. 2004;109:2503–2506. [DOI] [PubMed] [Google Scholar]
- 55. Weber M, Baker MB, Moore JP, Searles CD. MiR‐21 is induced in endothelial cells by shear stress and modulates apoptosis and eNOS activity. Biochem Biophys Res Commun. 2010;393:643–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang H‐H, Chen Y, Gao C‐Y, Cui Z‐T, Yao J‐M. Protective effects of microRNA‐126 on human cardiac microvascular endothelial cells against hypoxia/reoxygenation‐induced injury and inflammatory response by activating PI3K/Akt/eNOS signaling pathway. Cell Physiol Biochem. 2017;42:506–518. [DOI] [PubMed] [Google Scholar]
- 57. Balakumar P, Kathuria S, Taneja G, Kalra S, Mahadevan N. Is targeting eNOS a key mechanistic insight of cardiovascular defensive potentials of statins? J Mol Cell Cardiol. 2012;52:83–92. [DOI] [PubMed] [Google Scholar]
- 58. Balligand J‐L, Feron O, Dessy C. eNOS activation by physical forces: from short‐term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol Rev. 2009;89:481–534. [DOI] [PubMed] [Google Scholar]
- 59. Sun H‐X, Zeng D‐Y, Li R‐T, Pang R‐P, Yang H, Hu Y‐L, Zhang Q, Jiang Y, Huang L‐Y, Tang Y‐B, Yan G‐J, Zhou J‐G. Essential role of microRNA‐155 in regulating endothelium‐dependent vasorelaxation by targeting endothelial nitric oxide synthase. Hypertension. 2012;60:1407–1414. [DOI] [PubMed] [Google Scholar]
- 60. Jansen F, Nickenig G, Werner N. Extracellular vesicles in cardiovascular disease. Circ Res. 2017;120:1649–1657. [DOI] [PubMed] [Google Scholar]
- 61. Badrnya S, Baumgartner R, Assinger A. Smoking alters circulating plasma microvesicle pattern and microRNA signatures. Thromb Haemost. 2014;112:128–136. [DOI] [PubMed] [Google Scholar]
- 62. Adekunle R. Review of cardiovascular disease in HIV‐infected women. J AIDS Clin Res. 2016;7:2–12. [Google Scholar]
- 63. Cao Y, Gong Y, Liu L, Zhou Y, Fang X, Zhang C, Li Y, Li J. The use of human umbilical vein endothelial cells (HUVECs) as an in vitro model to assess the toxicity of nanoparticles to endothelium: a review. J Appl Toxicol. 2017;37:1359–1369. [DOI] [PubMed] [Google Scholar]
- 64. Baudin B, Bruneel A, Bosselut N, Vaubourdolle M. A protocol for isolation and culture of human umbilical vein endothelial cells. Nat Protoc. 2007;2:481–485. [DOI] [PubMed] [Google Scholar]
- 65. Silver AE, Christou DD, Donato AJ, Beske SD, Moreau KL, Magerko KA, Seals DR. Protein expression in vascular endothelial cells obtained from human peripheral arteries and veins. J Vasc Res. 2010;47:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Colombo PC, Ashton AW, Celaj S, Talreja A, Banchs JE, Dubois NB, Marinaccio M, Malla S, Lachmann J, Ware JA, Le Jemtel TH. Biopsy coupled to quantitative immunofluorescence: a new method to study the human vascular endothelium. J Appl Physiol. 2002;92:1331–1338. [DOI] [PubMed] [Google Scholar]
- 67. Rossman MJ, Kaplon RE, Hill SD, McNamara MN, Santos‐Parker JR, Pierce GL, Seals DR, Donato AJ. Endothelial cell senescence with aging in healthy humans: prevention by habitual exercise and relation to vascular endothelial function. Am J Physiol Heart Circ Physiol. 2017;313:890–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Donato AJ, Eskurza I, Silver AE, Levy AS, Pierce GL, Gates PE, Seals DR. Direct evidence of endothelial oxidative stress with aging in humans. Circ Res. 2007;100:1659–1666. [DOI] [PubMed] [Google Scholar]
- 69. Onat D, Brillon D, Colombo PC, Schmidt AM. Human vascular endothelial cells: a model system for studying vascular inflammation in diabetes and atherosclerosis. Curr Diab Rep. 2011;11:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]