

Abstract
Background
Heterogeneity in the underlying processes that contribute to heart failure with preserved ejection fraction (HFpEF) is increasingly recognized. Diabetes mellitus is a frequent comorbidity in HFpEF, but its impact on left ventricular and arterial structure and function in HFpEF is unknown.
Methods and Results
We assessed the impact of diabetes mellitus on left ventricular cellular and interstitial hypertrophy (assessed with cardiac magnetic resonance imaging, including T1 mapping pregadolinium and postgadolinium administration), arterial stiffness (assessed with arterial tonometry), and pulsatile arterial hemodynamics (assessed with in‐office pressure‐flow analyses and 24‐hour ambulatory monitoring) among 53 subjects with HFpEF (32 diabetic and 21 nondiabetic subjects). Despite few differences in clinical characteristics, diabetic subjects with HFpEF exhibited a markedly greater left ventricular mass index (78.1 [95% CI, 70.4–85.9] g versus 63.6 [95% CI, 55.8–71.3] g; P=0.0093) and indexed extracellular volume (23.6 [95% CI, 21.2–26.1] mL/m2 versus 16.2 [95% CI, 13.1–19.4] mL/m2; P=0.0008). Pronounced aortic stiffening was also observed in the diabetic group (carotid‐femoral pulse wave velocity, 11.86 [95% CI, 10.4–13.1] m/s versus 8.8 [95% CI, 7.5–10.1] m/s; P=0.0027), with an adverse pulsatile hemodynamic profile characterized by increased oscillatory power (315 [95% CI, 258–373] mW versus 190 [95% CI, 144–236] mW; P=0.0007), aortic characteristic impedance (0.154 [95% CI, 0.124–0.183] mm Hg/mL per second versus 0.096 [95% CI, 0.072–0.121] mm Hg/mL per second; P=0.0024), and forward (59.5 [95% CI, 52.8–66.1] mm Hg versus 40.1 [95% CI, 31.6–48.6] mm Hg; P=0.0010) and backward (19.6 [95% CI, 16.2–22.9] mm Hg versus 14.1 [95% CI, 10.9–17.3] mm Hg; P=0.0169) wave amplitude. Abnormal pulsatile hemodynamics were also evident in 24‐hour ambulatory monitoring, despite the absence of significant differences in 24‐hour systolic blood pressure between the groups.
Conclusions
Diabetes mellitus is a key determinant of left ventricular remodeling, arterial stiffness, adverse pulsatile hemodynamics, and ventricular‐arterial interactions in HFpEF.
Clinical Trial Registration
URL: https://www.clinicaltrials.gov. Unique identifier: NCT01516346.
Keywords: arterial stiffness, diabetes mellitus, hemodynamics, heart failure with preserved ejection fraction, left ventricular hypertrophy, magnetic resonance imaging, myocardial fibrosis
Subject Categories: Heart Failure, Remodeling, Magnetic Resonance Imaging (MRI)
Clinical Perspective
What Is New?
Compared with nondiabetic subjects with heart failure with preserved ejection fraction, diabetic subjects with heart failure with preserved ejection fraction exhibit more left ventricular hypertrophy, myocardial extracellular volume expansion, pronounced aortic stiffening, and an adverse pulsatile hemodynamic profile, characterized by increased oscillatory power, aortic characteristic impedance, and forward and backward (reflected) wave amplitude.
Abnormal pulsatile hemodynamics in the diabetic subgroup with heart failure with preserved ejection fraction are also evident in 24‐hour ambulatory monitoring, despite the absence of significant differences in 24‐hour systolic blood pressure between the groups.
What Are the Clinical Implications?
Heterogeneity in the underlying processes that contribute to heart failure with preserved ejection fraction is increasingly recognized.
Diabetes mellitus is responsible for important differences in relevant underlying phenotypes in this patient population.
Introduction
The burden of heart failure (HF) has markedly increased over the past several years. Approximately half of HF is secondary to HF with preserved ejection fraction (HFpEF), which is anticipated to represent an even larger proportion of the total burden of HF as the population ages.1 Multiple therapies that provide clinical benefit in HF with reduced EF are available. However, proven pharmacologic interventions to improve outcomes in HFpEF are currently lacking. Patients with HFpEF have a substantial burden of comorbidities that may contribute to its pathophysiological characteristics and may impact its prognosis and response to therapy.
Heterogeneity in the underlying processes that contribute to HFpEF is increasingly recognized. Diabetes mellitus is a key risk factor for HF, and a frequent comorbidity in patients with HFpEF, which is associated with poor outcomes in this patient population.2 Increased aortic stiffness, arterial dysfunction, and cardiac remodeling have been reported in diabetic patients in the general population, in patients with chronic kidney disease, and in patients with left ventricular (LV) systolic dysfunction.3, 4, 5, 6, 7, 8, 9, 10 However, no data are available about the influence of diabetes mellitus on LV remodeling/fibrosis, arterial stiffness, pulsatile arterial hemodynamics, and ventricular‐vascular interactions in HFpEF. HFpEF is often accompanied by diabetes mellitus, obesity, and various other comorbidities that impact LV remodeling and arterial function,3, 11, 12 but is associated with structural cardiovascular abnormalities in its own right.13 Furthermore, HFpEF differs from HF with reduced EF in terms of ventricular remodeling and ventricular‐arterial coupling patterns.14, 15, 16, 17, 18 Detailed hemodynamic and cardiac phenotyping can provide important insights into the pathophysiological characteristics of LV remodeling, fibrosis, and abnormal ventricular‐arterial interactions, which contribute to the pathophysiological characteristics of HFpEF.19, 20
In this study, we aimed to assess the impact of diabetes mellitus on LV hypertrophy, diffuse myocardial fibrosis, large artery stiffness, ventricular‐vascular interactions, and in‐office and 24‐hour pulsatile arterial hemodynamics in HFpEF.
Methods
We analyzed data from a previous phase 2 trial that was designed to assess the effect of isosorbide dinitrate, isosorbide dinitrate plus hydralazine, or placebo on pulsatile hemodynamics and LV remodeling in HFpEF.21 The current study used only baseline (preintervention) phenotypic data.
The data, analytic methods, and study materials are not publicly available for purposes of reproducing the results or replicating the procedures. Such data may be made available to other researchers for collaborative research, through the establishment of appropriate data sharing agreements and regulatory approvals.
Study Population
Inclusion criteria for the trial included symptomatic HFpEF (LVEF >50%), in addition to at least one of the following: (1) prior hospitalization for decompensated HF; (2) short‐term treatment for HF requiring intravenous diuretics or hemofiltration; (3) echocardiographic evidence for elevated filling pressures22; (4) long‐term treatment with a loop diuretic for control of symptoms; (5) or an elevated NT‐proBNP (N‐terminal pro‐B‐type natriuretic peptide). Subjects needed to be on stable medical therapy for the past month. Exclusion criteria included any rhythm other than sinus with native conduction; noncardiac conditions that significantly limit exercise (orthopedic or neuromuscular); known hypertrophic, infiltrative, or inflammatory cardiomyopathy; pericardial disease; significant pulmonary disease; primary pulmonary arteriopathy; acute coronary syndrome or coronary revascularization within the past 60 days; clinically significant perfusion defects on stress imaging without subsequent revascularization; significant valvular disease (eg, moderate or greater mitral regurgitation or aortic stenosis); uncontrolled hypertension (systolic blood pressure [SBP] >180 mm Hg or diastolic blood pressure [DBP] >100 mm Hg); prior reduced LVEF <50% (ie, recovered EF); hemoglobin <10 g/dL; current therapy with organic nitrates or hydralazine; and elevations in liver function test results. The presence of HFpEF was adjudicated by 2 cardiologists with expertise in HFpEF (J.A.C. and P.Z.), and subjects with an alternative explanation for symptoms were also excluded. The protocol was approved by the institutional review boards of the Philadelphia Veterans Affairs Hospital and the Hospital of the University of Pennsylvania. All subjects provided written informed consent. Diabetes mellitus was defined as the use of antidiabetic medications, a fasting plasma glucose level ≥126 mg/dL, and/or a hemoglobin A1c level >6.5%. Enrollment was not performed in regards to diabetic status and, therefore, there was no matching procedure to enroll diabetic versus nondiabetic subjects in the parent trial.
Echocardiography and Arterial Tonometry
Echocardiography was performed using a Vivid e9 or Vivid I machine (General Electric, Fairfield, CT). Echocardiographic parameters of diastolic function were quantified according to American Society of Echocardiography guidelines by a trained cardiologist (P.Z.)22; each metric was quantified in triplicate, with average values presented. Volumetric flow was quantified using pulse‐wave Doppler measurements from the LV outflow tract in the 5‐chamber view (Figure 1) and the LV outflow tract cross‐sectional area measured in the parasternal long‐axis view, providing a time‐resolved aortic flow waveform.
Figure 1.
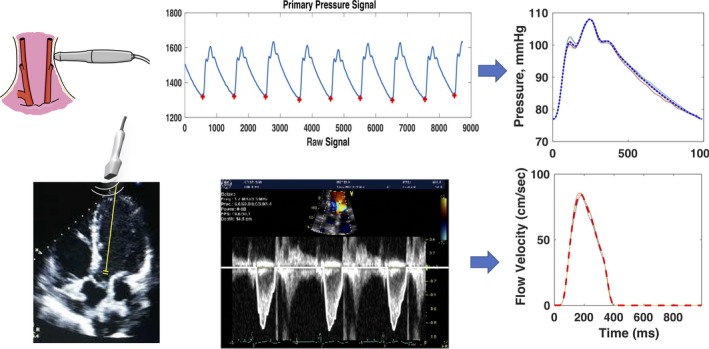
Measurement and signal averaging of central pressure and flow using arterial tonometry and Doppler echocardiography.
Applanation tonometry was performed at the carotid, radial, and femoral arteries using a high‐fidelity tonometer (Millar Instruments, Houston, TX), with a single‐lead ECG recording. The ECG complex of the ECG was used as a fiducial point. Surface measurements were obtained from the sternal notch to the site of interrogation at the carotid and femoral arteries to compute carotid‐femoral pulse wave velocity (CF‐PWV), a measure of large artery stiffness. Specifically, the path length was computed as the distance from the sternal notch to the femoral interrogation site minus the distance from the sternal notch to the carotid interrogation site.
Radial tonometric waveforms were calibrated using the brachial SBP and DBP, obtained using a validated oscillometric device (Omron HEM‐705CP [Omron Corp, Kyoto, Japan] or Accutorr Plus [Datascope Corp, Paramus, NJ]), assuming no brachial‐to‐radial amplification. Mean arterial pressure was computed as the mean pressure from the radial pressure waveform. Carotid tonometry, calibrated using mean arterial pressure and DBP, was used to obtain a central pressure waveform (Figure 1). Tonometric signals were processed using Sphygmocor software (AtCor Medical, Australia).
Central Pulsatile Arterial Hemodynamics
The pressure‐flow pair was used to perform pulsatile hemodynamic analyses (Figure 2). Pressure‐flow analyses were performed by a technician and overread by a cardiologist with expertise in pressure‐flow analyses (J.A.C.). Custom‐designed software programmed in Matlab (R2014b; MathWorks, Natick, MA) was used to assess aortic pressure‐flow relations, as described previously in detail.19, 23 In brief, after alignment of signal‐averaged central pressure and flow waveforms, we computed aortic input impedance as the ratio of central pressure/flow in the frequency domain. Aortic root characteristic impedance was quantified as the average of impedance modulus at higher frequencies (Figure 2). Wave separation analysis was performed to obtain the amplitude of the forward and backward pressure waves. Reflection magnitude was defined as the ratio of backward/forward pressure.
Figure 2.
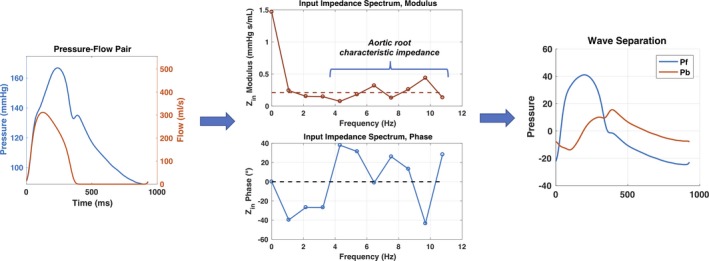
Quantification of the aortic input impedance spectrum, aortic characteristic impedance, and wave separation analysis. Pb indicates backward pressure; Pf, forward pressure; Zin, input impedance.
Ambulatory Central BP Monitoring and Pulse Wave Analysis
Ambulatory BP monitoring over 24 hours was performed using the Mobilograph device (IEM, Stolberg, Germany), which measures brachial and central (aortic) BP, as well as time‐resolved waveforms, allowing for ambulatory pulse wave analysis (including wave separation into forward and backward waves, as shown in Figure 3).24, 25, 26 We analyzed data over 24 hours, as well as daytime and nighttime indexes separately.
Figure 3.
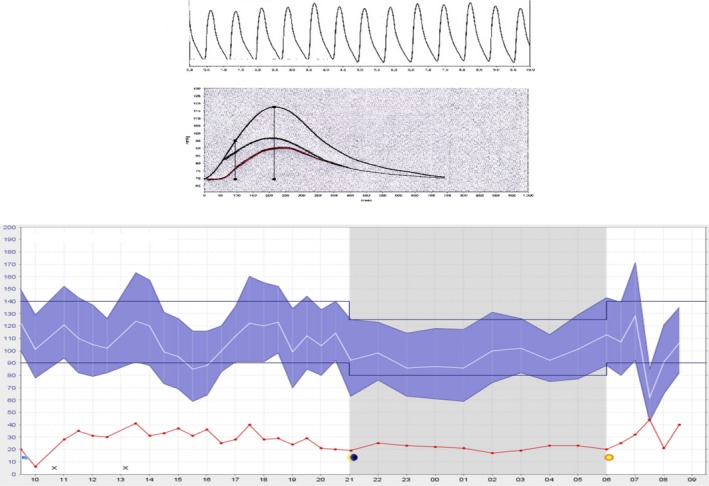
Assessment of ambulatory central blood pressure and pulsatile hemodynamics.
Cardiac Magnetic Resonance Imaging: LV Structure and Function
Cardiac magnetic resonance imaging (CMR) was performed in 44 of the studied subjects. Participants underwent a CMR examination to assess LV structure and function using a 1.5‐T whole body magnetic resonance imaging scanner (Avanto or Espree; Siemens Healthcare, Malvern, PA) equipped with a phased‐array cardiac coil. LV volumes and EF were determined using balanced steady‐state free‐precession cine imaging. Typical parameters were as follows: repetition time=2.6 milliseconds; echo time=1.3 milliseconds; phases=30; slice thickness=8 mm; bandwidth=898 Hz/pixel; flip angle=70°; field of view=300 to 340 mm2; matrix size=192×192; and parallel imaging factor=2. LV short‐axis stack cine images were manually traced at end diastole and end systole using CMR42 software (Circle CVI, Calgary, AB, Canada). All magnetic resonance imaging measurements were performed by a trained technician and overread by a cardiologist with expertise in CMR (J.A.C.). LV mass (LVM) was computed as the difference between epicardial and endocardial volumes, multiplied by myocardial density. LVM was normalized for height in meters raised to the power of 1.7.27
We used a modified look‐locker inversion recovery sequence to assess T1 times before and after the IV administration of gadolinium contrast (gadopentetate dimeglumine, 0.15 mmol/kg or equivalent) in a midventricular short‐axis slice.28, 29 Scan parameters for modified look‐locker inversion recovery were as follows: field of view=340 mm2; matrix size=144×192; slice thickness=6 mm; repetition time=2.4 milliseconds; echo time=1.18 milliseconds; flip angle=30°; bandwidth=1000 Hz/pixel; and parallel imaging factor=2. Myocardial T1 measurements were performed before and at several time points (≈5, 10, 15, and 20–40 minutes) after gadolinium administration. Modified look‐locker inversion recovery was performed with a 5‐3‐3 schema with 2 inversions (5 TIs (inversion times) after inversion 1, 3 T1 recovery heartbeats, and 3 TIs after inversion 2). All available blood and myocardial T1 measurements were used to compute λ (the myocardium‐blood partition coefficient) as the slope of the myocardial 1/T1 over the blood 1/T1 change, via linear regression.29 The fraction of myocardial tissue comprised by the extracellular space (extracellular volume [ECV] fraction) equals λ×(1−hematocrit). Extracellular LV volume was computed as LV wall volume multiplied by ECV. Cellular LV volume was computed as LV wall volume multiplied by (1−ECV).30
Subjects enrolled in the trial who demonstrated impaired renal function precluding the administration of gadolinium (estimated glomerular filtration rate <30 mL/min per 1.73 m2) or significant claustrophobia did not undergo CMR measurements.
Statistical Analyses
Patient characteristics were summarized using standard descriptive statistics. We compared general characteristics of study subjects using the t test of the Mann‐Whitney U test, as appropriate. Proportions were compared with the χ2 or the Fisher exact test, as appropriate.
For comparisons of cardiac phenotypes, we performed adjusted and unadjusted analyses using t tests and ANCOVA, respectively. For all comparisons of continuous variables, normality was assessed with the Anderson‐Darling test and log transformations were applied as needed to improve normality. In all cases, means and 95% CIs are expressed in the native (linear) scale. Statistical significance was defined as a 2‐tailed P<0.05. All probability values presented are 2 tailed. Statistical analyses were performed using the Matlab statistics and machine learning toolbox (Matlab 2016b; Mathworks, Natwick, MA) and SPSS for Mac v22 (SPSS Inc, Chicago, IL).
Results
General characteristics of study subjects are shown in Table 1. Our sample included 32 diabetic and 21 nondiabetic subjects with HFpEF. Compared with nondiabetic subjects, diabetic subjects exhibited greater office SBP (143 versus 129 mm Hg; P=0.0241) and a greater frequency of statin use (75% versus 47.62%; P=0.0420). There were no significant differences between the groups in age, sex, race/ethnicity, body mass index, DBP, history of coronary disease, history of hypertension, vasoactive medication use, renal function, hematocrit, NT‐proBNP levels, or echocardiographic parameters of diastolic function. Mean hemoglobin A1c in the diabetic group was 7.9% (interquartile range, 6.9%–9.28%), and mean fasting glucose was 154 mg/dL (interquartile range, 99–212 mg/dL). Insulin, metformin, sulfonylurea, and Gulcan‐like peptide‐1 receptor agonists use were present in 56.25%, 31.25%, 16.13%, and 3.23% of diabetic subjects, respectively. No subjects were receiving α‐glucosidase inhibitors, thiazolidinediones, dipeptidyl peptidase‐4 inhibitors, or sodium‐glucose cotransporter‐2 inhibitors.
Table 1.
Baseline Characteristics of the Study Population[Link]
| Characteristics | Nondiabetic Subjects (n=21) | Diabetic Subjects (n=32) | P Value |
|---|---|---|---|
| Age, y | 64 (59.3–69.5) | 63 (59–69) | 0.92 |
| Male sex | 13 (61.90) | 27 (84.38) | 0.06 |
| Race/ethnicity | |||
| White | 8 (38.10) | 13 (40.62) | 0.69 |
| Black | 13 (61.90) | 18 (56.25) | |
| Other | 0 (0.00) | 1 (3.12) | |
| Body mass index, kg/m2 | 34.5±7.8 | 37.7±6.2 | 0.11 |
| Systolic BP, mm Hg | 129±20 | 143±22 | 0.0241 |
| Diastolic BP, mm Hg | 77.5±11.6 | 76.2±15.8 | 0.77 |
| Medical history | |||
| Myocardial infarction | 1 (4.76) | 6 (18.75) | 0.23 |
| Dyslipidemia | 15 (71.43) | 29 (90.62) | 0.13 |
| Coronary artery disease | 7 (33.33) | 13 (40.62) | 0.59 |
| Hypertension | 16 (76.19) | 30 (93.75) | 0.10 |
| Current smoker | 5 (23.81) | 4 (12.50) | 0.46 |
| Diabetic neuropathy | ··· | 14 (45.16) | ··· |
| Diabetic retinopathy | ··· | 12 (38.71) | ··· |
| Medication use | |||
| β Blockers | 10 (47.62) | 21 (65.62) | 0.19 |
| Aspirin | 13 (61.90) | 25 (78.12) | 0.20 |
| Clopidogrel | 3 (14.29) | 2 (6.25) | 0.379 |
| ACE inhibitors | 9 (42.86) | 15 (46.88) | 0.77 |
| ARBs | 2 (9.52) | 9 (28.12) | 0.17 |
| Loop diuretics | 8 (38.10) | 19 (59.38) | 0.13 |
| MRAs | 1 (4.76) | 1 (3.12) | 1.00 |
| Statins | 10 (47.62) | 24 (75.00) | 0.04 |
| Calcium‐channel blocker | 5 (23.81) | 14 (43.75) | 0.14 |
| Thiazide diuretics | 7 (33.33) | 11 (34.38) | 0.94 |
| Warfarin | 0 (0.00) | 1 (3.12) | 1.00 |
| Insulin | ··· | 18 (56.25) | ··· |
| Metformin | ··· | 10 (31.25) | ··· |
| Sulfonylureas | ··· | 5 (16.13) | ··· |
| Gulcan‐like peptide‐1 receptor agonists | ··· | 1 (3.23) | ··· |
| Laboratory tests | |||
| NT‐proBNP, pg/mL | 166 (65–317) | 317 (134–960) | 0.10 |
| eGFR, mL/min per 1.73 m2 | 77 (60–99.8) | 66 (50.3–97.8) | 0.39 |
| Hematocrit, % | 40.1±3.5 | 38.2±5.1 | 0.16 |
| Hemoglobin A1c, % | 6 (5.68–6.43) | 7.9 (6.9–9.28) | ··· |
| Fasting glucose, mg/dL | 96 (89–103) | 154 (99–212) | ··· |
| Echocardiographic diastolic function parameters | |||
| E wave velocity, cm/s | 77.9±24 | 82.7±23.9 | 0.51 |
| A wave velocity, cm/s | 78.7±21.1 | 74.2±24.8 | 0.54 |
| Mitral deceleration time, ms | 223±56 | 225±42 | 0.89 |
| Mitral annular septal e′, cm/s | 62.2 (53.7–75) | 59 (44.2–81.1) | 0.62 |
| Mitral annular lateral e′, cm/s | 97.8 (74–111.4) | 84.3 (53.2–106.5) | 0.11 |
| E/e′ ratio | 10.5 (8.4–12.8) | 11.1 (9.4–14.2) | 0.29 |
| LV ejection fraction, % | 61.8±6.6 | 61.5±8.1 | 0.88 |
Numbers represent the mean±SD, median (interquartile range), or count (percentage). ACE indicates angiotensin‐converting enzyme; ARB, angiotensin II receptor blocker; BP, blood pressure; eGFR, estimated glomerular filtration rate; LV, left ventricular; MRA, mineralocorticoid receptor antagonist; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide; E, mitral peak velocity of early filling; e′, early dialostic mitral annular velocity.
LV Structure and Function
Table 2 shows a comparison of subjects who underwent versus those who did not undergo a CMR. Table 3 shows a comparison of key parameters of LV remodeling and cellular and interstitial expansion between diabetic and nondiabetic subjects. Diabetic subjects exhibited a markedly greater LVM (191 versus 148 g; P=0.0095; Figure 4), which was also true when LVM was adjusted for either body height (P=0.0079) or body surface area (BSA; 78.1 versus 63.6 g; P=0.0093). The ECV fraction tended to be greater in diabetic subjects (30.4% versus 27.1%), but this difference was not significant (P=0.1096). The extracellular LV wall volume was markedly increased in the diabetic group (52.7 versus 35.4 mL; P=0.0003; Figure 4). The extracellular wall volume indexed for BSA was markedly increased in the diabetic group (23.6 versus 16.2 mL/m2; P=0.0008). There were also between‐group differences in cellular wall volume (123 versus 97 mL; P=0.0307), which, however, did not reach statistical significance when indexed for BSA (P=0.0526).
Table 2.
Comparison of General Characteristics Between Subjects Who Underwent Versus Those Who Did Not Undergo CMR
| Characteristics | Subjects Who Underwent CMR (n=44) | Subject Who Did Not Undergo CMR (n=9) | P Value |
|---|---|---|---|
| Age, y | 62 (59–67) | 68 (60.8–80.3) | 0.17 |
| Male sex | 31 (70.45) | 9 (100.00) | 0.09 |
| Race/ethnicity | |||
| White | 17 (38.64) | 4 (44.44) | 0.87 |
| Black | 26 (59.09) | 5 (55.56) | |
| Other | 1 (2.27) | 0 (0.00) | |
| Body mass index, kg/m2 | 36.6±6.8 | 35.6±8.2 | 0.70 |
| Systolic BP, mm Hg | 150±20 | 150±18 | 0.99 |
| Diastolic BP, mm Hg | 85.9±12 | 84±17.4 | 0.72 |
| Medical history | |||
| Myocardial infarction | 6 (13.64) | 0 (0.00) | 0.57 |
| Dyslipidemia | 38 (86.36) | 6 (66.67) | 0.17 |
| Coronary artery disease | 17 (38.64) | 3 (33.33) | 1.00 |
| Hypertension | 40 (90.91) | 6 (66.67) | 0.09 |
| Current smoker | 8 (18.60) | 1 (11.11) | 1.00 |
| Diabetic neuropathy | 12 (27.27) | 2 (25.00) | 1.00 |
| Diabetic retinopathy | 11 (25.00) | 1 (12.50) | 0.66 |
| Medication use | |||
| β Blockers | 26 (59.09) | 5 (55.56) | 1.00 |
| Aspirin | 30 (68.18) | 8 (88.89) | 0.42 |
| Clopidogrel | 4 (9.09) | 1 (11.11) | 1.00 |
| ACE inhibitors | 21 (47.73) | 3 (33.33) | 0.49 |
| ARBs | 10 (22.73) | 1 (11.11) | 0.67 |
| Loop diuretics | 25 (56.82) | 2 (22.22) | 0.08 |
| MRAs | 2 (4.55) | 0 (0.00) | 1.00 |
| Statins | 29 (65.91) | 5 (55.56) | 0.71 |
| Calcium‐channel blocker | 17 (38.64) | 2 (22.22) | 0.47 |
| Thiazide diuretics | 3 (33.33) | 14 (31.82) | 1.00 |
| Warfarin | 1 (2.27) | 0 (0.00) | 1.00 |
| Insulin | 15 (34.09) | 3 (33.33) | 1.00 |
| Metformin | 10 (22.73) | 1 (12.50) | 0.67 |
| Sulfonylureas | 4 (9.09) | 1 (12.50) | 0.58 |
| Gulcan‐like peptide‐1 receptor agonists | 1 (2.27) | 0 (0.00) | 1.00 |
| Laboratory tests | |||
| NT‐proBNP, pg/mL | 228 (85–466) | 415 (147–891) | 0.38 |
| eGFR, mL/min per 1.73 m2 | 66 (56–97.8) | 80 (65.8–100.5) | 0.41 |
| Hematocrit, % | 38.9±4.4 | 39±5.8 | 0.97 |
| Hemoglobin A1c, % | 7.2 (6.2–8.33) | 6.5 (5.93–9.88) | 0.96 |
| Fasting glucose, mg/dL | 103 (90–159) | 104 (90–222) | 0.91 |
| Echocardiographic diastolic function parameters | |||
| E wave velocity, cm/s | 80.8±24.1 | 82.1±23.8 | 0.91 |
| A wave velocity, cm/s | 75.1±23.3 | 82.3±25.7 | 0.52 |
| Mitral deceleration time, ms | 213 (190–251) | 229 (226–257) | 0.18 |
| Mitral annular septal e′, cm/s | 64.8±22.2 | 61.5±12.3 | 0.74 |
| Mitral annular lateral e′, cm/s | 94.3 (56.1–107.8) | 82.3 (57–98.7) | 0.63 |
| E/e′ ratio | 10.7 (8.8–12.9) | 12.9 (9.7–14) | 0.45 |
Numbers represent the mean±SD, median (interquartile range), or count (percentage). ACE indicates angiotensin‐converting enzyme; ARB, angiotensin II receptor blocker; BP, blood pressure; CMR, cardiac magnetic resonance imaging; eGFR, estimated glomerular filtration rate; MRA, mineralocorticoid receptor antagonist; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide; E, mitral peak velocity of early filling; e′, early dialostic mitral annular velocity.
Table 3.
Comparison of Key Cardiovascular Parameters of LV Hypertrophy and Cellular and Interstitial Expansion Between Diabetic and Nondiabetic Subjects
| Parameters | Mean (95% CI) | P Value | |
|---|---|---|---|
| Nondiabetic Subjects | Diabetic Subjects | ||
| Nonadjusted | |||
| LV mass, g | 148 (124–172) | 191 (171–211) | 0.0095 |
| LV mass indexed for BSA, g/m2 | 63.6 (55.8–71.3) | 78.1 (70.4–85.9) | 0.0093 |
| LV mass indexed for height, g/m1.7 | 57 (49.5–64.5) | 71.6 (63.9–79.3) | 0.0079 |
| Extracellular volume fraction, % | 27.1 (24–30.2) | 30.4 (28–32.8) | 0.11 |
| Cellular LV wall volume, mL | 97 (80–114) | 123 (106–139) | 0.0307 |
| Extracellular LV wall volume, mL | 35.4 (29.4–41.4) | 52.7 (45.8–59.6) | 0.0003 |
| Cellular LV wall volume index, mL/m2 | 43.5 (36.8–50.1) | 52.3 (46.1–58.5) | 0.053 |
| Extracellular LV wall volume index, mL/m2 | 16.2 (13.1–19.4) | 23.6 (21.2–26.1) | 0.0008 |
| Adjusted for systolic BP and statin use | |||
| LV mass, g | 149 (124–174) | 190 (169–211) | 0.0220 |
| LV mass indexed for BSA, g/m2 | 64.4 (56.2–72.7) | 77.4 (69.5–85.4) | 0.0299 |
| LV mass indexed for height, g/m1.7 | 58.2 (50.1–66.2) | 70.6 (62.8–78.4) | 0.0336 |
| Extracellular volume fraction, % | 26.9 (23.4–30.4) | 30.6 (27.9–33.2) | 0.13 |
| Cellular LV wall volume, mL | 99 (80–119) | 121 (103–139) | 0.12 |
| Extracellular LV wall volume, mL | 35.8 (28.9–42.8) | 52.3 (44.8–59.8) | 0.0036 |
| Cellular LV wall volume index, mL/m2 | 44.4 (36.6–52.1) | 51.6 (45–58.3) | 0.18 |
| Extracellular LV wall volume index, mL/m2 | 16.4 (12.9–20) | 23.5 (20.8–26.2) | 0.0065 |
BP indicates blood pressure; BSA, body surface area; LV, left ventricular.
Figure 4.
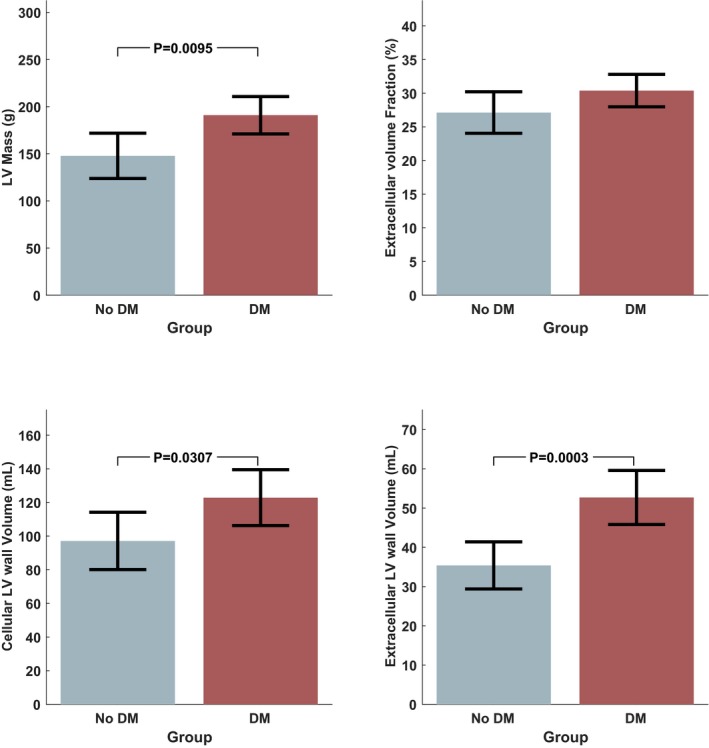
Comparison of key cardiovascular parameters of left ventricular (LV) hypertrophy and cellular and interstitial expansion between diabetic and nondiabetic subjects. DM indicates diabetes mellitus.
The lower panel of Table 3 shows comparisons adjusted for statin use and SBP. In these adjusted comparisons, LVM was significantly greater in diabetic subjects (190 versus 149 g; P=0.0299). Diabetic subjects also exhibited greater LVM indexed for BSA or body height. The extracellular wall volume was markedly greater in the diabetic subgroup (35.8 versus 52.3 mL; P=0.0036), whereas the cellular wall volume was not significantly different between the groups with or without indexing for BSA (P=0.1796 and P=0.1176), respectively.
Differences in Arterial Stiffness and Aortic Pulsatile Hemodynamics
Figure 5 and Table 4 show differences in large artery stiffness and aortic pulsatile hemodynamics between the groups. Diabetic subjects demonstrated a marked increase in large artery stiffness (CF‐PWV, 11.8 versus 8.8 m/s; P=0.0027). Although aortic (central) SBP or DBP was not significantly different between the groups (Table 4), marked differences in pulsatile hemodynamics were present. Aortic (central) pulse pressure was greater in the diabetic group (59.2 versus 44.5 mm Hg; P=0.0118). There was a marked increase in proximal aortic characteristic impedance (0.154 versus 0.096 mm Hg/mL per second; P=0.0024). There was a proportional increase in forward wave amplitude (59.5 versus 40.1 mm Hg) and backward wave amplitude (19.6 versus 14.1 mm Hg; Figure 5), without significant differences in reflection magnitude (backward/forward pressure). Diabetic subjects also exhibited a markedly greater oscillatory power (315 versus 190 mW; P=0.0007), without significant differences in steady power. In analyses adjusted for statin use, these trends did not substantially change (Table 4, bottom part).
Figure 5.
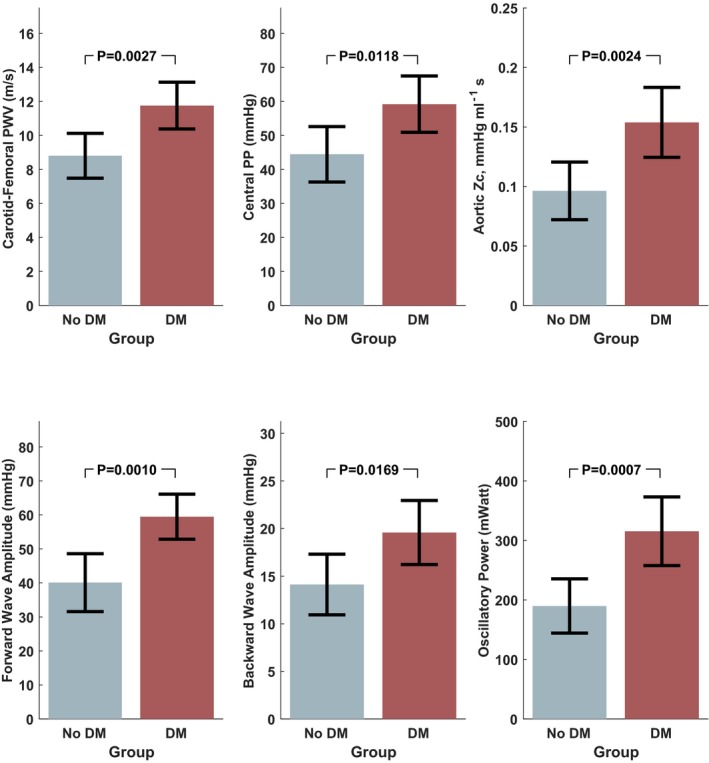
Differences in aortic pulsatile hemodynamics assessed via pressure‐flow analyses, between diabetic and nondiabetic subjects. DM indicates diabetes mellitus; PP, pulse pressure; PWV, pulse wave velocity; Zc, characteristic impedance.
Table 4.
Differences in Aortic Pulsatile Hemodynamics Assessed Via Pressure‐Flow Analyses, Between Diabetic and Nondiabetic Subjects
| Parameters | Mean (95% CI) | P Value | |
|---|---|---|---|
| Nondiabetic Subjects | Diabetic Subjects | ||
| Nonadjusted | |||
| Carotid‐femoral PWV, m/s | 8.8 (7.5–10.1) | 11.8 (10.4–13.1) | 0.0027 |
| Central SBP, mm Hg | 121 (110–131) | 133 (124–142) | 0.0776 |
| Central DBP, mm Hg | 74.5 (67.4–81.6) | 73.1 (67.5–78.6) | 0.76 |
| Central MAP, mm Hg | 93.4 (85.8–101) | 95.4 (89.4–101.4) | 0.67 |
| Central PP, mm Hg | 44.5 (36.3–52.6) | 59.2 (50.9–67.5) | 0.0118 |
| Aortic root Zc, mm Hg/mL per second | 0.096 (0.072–0.121) | 0.154 (0.124–0.183) | 0.0024 |
| Reflection magnitude | 0.386 (0.345–0.427) | 0.357 (0.325–0.389) | 0.29 |
| Forward wave amplitude, mm Hg | 40.1 (31.6–48.6) | 59.5 (52.8–66.1) | 0.0010 |
| Backward wave amplitude, mm Hg | 14.1 (10.9–17.3) | 19.6 (16.2–22.9) | 0.0169 |
| Oscillatory power, mW | 190 (144–236) | 315 (258–373) | 0.0007 |
| Steady power, mW | 980 (819–1142) | 1164 (1017–1311) | 0.09 |
| Adjusted for statin use | |||
| Carotid‐femoral PWV, m/s | 9.1 (7.7–10.5) | 11.5 (10.2–12.9) | 0.0165 |
| Central SBP, mm Hg | 122 (111–133) | 132 (123–141) | 0.16 |
| Central DBP, mm Hg | 74.1 (66.8–81.5) | 73.3 (67.6–78.9) | 0.86 |
| Central MAP, mm Hg | 93.6 (85.7–101.5) | 95.3 (89.1–101.5) | 0.74 |
| Central PP, mm Hg | 46 (37.5–54.6) | 58 (49.9–66.1) | 0.0452 |
| Aortic root Zc, mm Hg/mL per second | 0.102 (0.077–0.127) | 0.149 (0.121–0.176) | 0.0137 |
| Reflection magnitude | 0.381 (0.338–0.423) | 0.36 (0.328–0.393) | 0.47 |
| Forward wave amplitude, mm Hg | 41.4 (32.7–50.2) | 58.7 (52–65.4) | 0.0045 |
| Backward wave amplitude, mm Hg | 14.4 (11–17.7) | 19.4 (16–22.8) | 0.0359 |
| Oscillatory power, mW | 194 (145–242) | 311 (253–370) | 0.0024 |
| Steady power, mW | 966 (801–1132) | 1174 (1022–1326) | 0.07 |
DBP indicates diastolic blood pressure; MAP, mean arterial pressure; PP, pulse pressure; PWV, pulse wave velocity; SBP, systolic blood pressure; Zc, characteristic impedance; E (mitral peak velocity of early filling); e' (early dialostic mitral annular velocity). Please remove GLP and use full expansion gulcan‐like peptide‐1 receptor agonists.
There were no significant differences in stroke volume (88 [95% CI, 80.2–95.7] mL versus 80.4 [95% CI, 70.5–90.3] mL) or cardiac output (5.49 [95% CI, 4.9–6.08] L/min versus 4.73 [95% CI, 4.07–5.39] L/min) between the groups. After adjustment for mean arterial pressure and heart rate, CF‐PWV was significantly greater among diabetic subjects (11.6 [95% CI, 10.2–13.0] m/s) compared with nondiabetic subjects (8.9 [95% CI, 7.6–10.2] m/s; P=0.0069).
Ambulatory BP and Pulsatile Hemodynamics
There were no significant differences in daytime, nighttime, or 24‐hour brachial SBP, DBP, or aortic SBP (Table 5). In contrast, highly significant differences were observed in brachial pulse pressure between the groups, with higher daytime (58.5 versus 48.6 mm Hg; P=0.0078), nighttime (55.1 versus 45.3 mm Hg; P=0.0006), and 24‐hour (58.3 versus 47.5 mm Hg; P=0.0020) values in the diabetic group (Table 5). Similarly, greater values of daytime (45.6 versus 36.8 mm Hg; P=0.0070), nighttime (43.5 versus 35.2 mm Hg; P=0.0051), and 24‐hour (46.5 versus 37.3 mm Hg; P=0.0009) central pulse pressure were found in the diabetic group. Diabetic subjects exhibited greater 24‐hour forward (29.5 versus 23.8 mm Hg; P=0.0011) and backward (19.3 versus 15.3 mm Hg; P=0.0016) wave amplitudes. Figure 6 demonstrates key differences in 24‐hour BP and arterial hemodynamic values between the groups.
Table 5.
Differences in Ambulatory BP and Pulsatile Hemodynamics Between Diabetic and Nondiabetic Subjects
| Parameters | Mean (95% CI) | P Value | |
|---|---|---|---|
| Nondiabetic Subjects | Diabetic Subjects | ||
| Unadjusted | |||
| Daytime brachial SBP | 131 (121–141) | 138 (131–146) | 0.27 |
| Daytime central SBP | 120 (111–130) | 128 (120–136) | 0.23 |
| Daytime DBP | 82.7 (76.6–88.8) | 79.7 (75.2–84.3) | 0.45 |
| Daytime brachial PP | 48.6 (43–54.1) | 58.5 (54.4–62.7) | 0.0078 |
| Daytime central PP | 36.8 (32.4–41.3) | 45.6 (41.3–50) | 0.0070 |
| Nighttime brachial SBP | 117 (107–127) | 129 (122–137) | 0.07 |
| Nighttime central SBP | 108 (99–118) | 117 (109–124) | 0.18 |
| Nighttime DBP | 71.5 (64.2–78.8) | 73.2 (67.7–78.8) | 0.72 |
| Nighttime brachial PP | 45.3 (41.5–49.1) | 55.1 (51.6–58.6) | 0.0006 |
| Nighttime central PP | 35.2 (31–39.4) | 43.5 (40.1–46.8) | 0.0051 |
| 24‐h Brachial SBP | 126 (117–135) | 135 (128–142) | 0.13 |
| 24‐h Central SBP | 116 (107–124) | 125 (119–132) | 0.09 |
| 24‐h DBP | 79 (73.2–84.9) | 77.9 (73.5–82.3) | 0.76 |
| 24‐h Brachial PP | 47.5 (42.4–52.6) | 58.3 (54.5–62.1) | 0.0020 |
| 24‐h Central PP | 37.3 (33.3–41.3) | 46.5 (43.5–49.5) | 0.0009 |
| 24‐h Forward wave amplitude | 23.8 (21.3–26.3) | 29.5 (27.6–31.3) | 0.0011 |
| 24‐h Backward wave amplitude | 15.3 (13.5–17.2) | 19.3 (17.9–20.7) | 0.0016 |
| Adjusted for SBP (office) and statin use | |||
| Daytime brachial SBP | 137 (129–145) | 135 (129–141) | 0.72 |
| Daytime central SBP | 126 (117–135) | 125 (117–132) | 0.82 |
| Daytime DBP | 85.7 (79.8–91.5) | 78.1 (73.8–82.3) | 0.0554 |
| Daytime brachial PP | 51.4 (46.4–56.3) | 57 (53.3–60.6) | 0.10 |
| Daytime central PP | 38.8 (34–43.6) | 44.1 (40–48.3) | 0.12 |
| Nighttime brachial SBP | 123 (114–132) | 126 (119–132) | 0.64 |
| Nighttime central SBP | 112 (103–122) | 114 (107–121) | 0.78 |
| Nighttime DBP | 75.5 (68.7–82.3) | 70.9 (65.9–76) | 0.32 |
| Nighttime brachial PP | 46.8 (42.9–50.8) | 54.1 (50.7–57.4) | 0.0113 |
| Nighttime central PP | 36.3 (31.9–40.7) | 42.8 (39.3–46.3) | 0.0359 |
| 24‐h Brachial SBP | 132 (123–140) | 131 (125–137) | 0.97 |
| 24‐h Central SBP | 120 (112–128) | 123 (117–128) | 0.69 |
| 24‐h DBP | 82.3 (76.7–87.8) | 76.1 (72.1–80.1) | 0.10 |
| 24‐h Brachial PP | 50.1 (45.4–54.8) | 56.8 (53.4–60.2) | 0.0363 |
| 24‐h Central PP | 38.5 (34.4–42.6) | 45.8 (42.8–48.8) | 0.0114 |
| 24‐h Forward wave amplitude | 24.8 (22.3–27.3) | 28.9 (27.1–30.7) | 0.0184 |
| 24‐h Backward wave amplitude | 15.7 (13.8–17.7) | 19.1 (17.7–20.5) | 0.0133 |
All units are in mm Hg. BP indicates blood pressure; DBP, diastolic BP; PP, pulse pressure; SBP, systolic BP.
Figure 6.
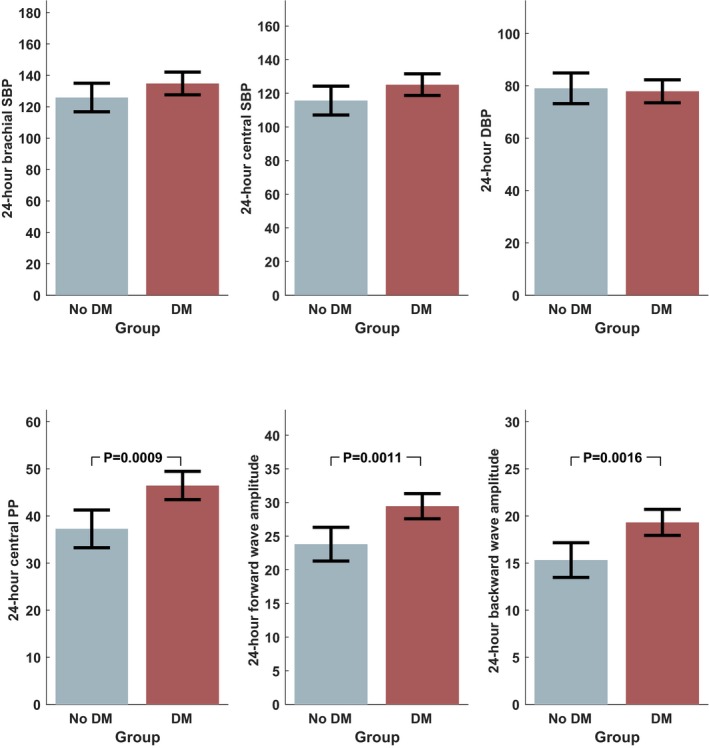
Differences in ambulatory blood pressure (BP) and pulsatile hemodynamics between diabetic and nondiabetic subjects. All units are in mm Hg. DBP indicates diastolic BP; DM, diabetes mellitus; PP, pulse pressure; SBP, systolic BP.
After adjustment for statin use and office SBP, differences persisted in nighttime and 24‐hour central and aortic pulse pressure, as well as in forward and backward wave amplitude (Table 5).
Discussion
Using a combination of echocardiography, arterial tonometry, and CMR, we compared LV remodeling, arterial structure and function, and office and ambulatory arterial pulsatile hemodynamics between diabetic and nondiabetic subjects with HFpEF. Despite similar age and few differences in standard clinical characteristics, diabetic subjects with HFpEF exhibited an increase in LVM, extracellular myocardial volume, and large artery stiffness. Consistent with the hemodynamic effects of large artery stiffening, diabetic subjects demonstrated adverse pulsatile hemodynamics, with increased aortic characteristic impedance, pulsatile power, forward and backward wave amplitude, and aortic pulse pressure, demonstrated with in‐office assessments, as well as 24‐hour ambulatory monitoring. Interestingly, 24‐hour central pulsatile hemodynamics were substantially different between the groups, despite the absence of significant differences in 24‐hour SBP. Our findings underscore the importance of diabetes mellitus as a determinant of LV structure, arterial stiffness, aortic pulsatile hemodynamics, ventricular structure, and ventricular‐arterial interactions in HFpEF.
Aortic stiffness, measured as proximal aortic distensibility with magnetic resonance imaging31 or CF‐PWV,32 has been shown to be increased in HFpEF, and to correlate with aerobic capacity in this population.31 However, the determinants of aortic stiffness in HFpEF are poorly understood. Moreover, the heterogeneity in the underlying processes that contribute to HFpEF is increasingly recognized, and may be responsible for the failure of various candidate therapeutic interventions to improve outcomes in this patient population. Therefore, there is a great interest in a better phenotypic and mechanistic characterization of readily identifiable patient subgroups with HFpEF. Diabetes mellitus is a frequent comorbidity in HFpEF, and is associated with poor outcomes in this patient population, but its impact on arterial stiffness, pulsatile hemodynamics, and ventricular‐arterial interactions in HFpEF is unknown. Our study demonstrates, for the first time, that diabetic patients with HFpEF exhibit a distinct phenotypic profile, with pronounced aortic stiffening compared with their nondiabetic counterparts. The differences in CF‐PWV between diabetic and nondiabetic subjects (≈3 m/s) is striking and equivalent to several decades of aging of the arterial tree. It is also more pronounced than reported in the literature for subjects without HFpEF. In the general population, diabetes mellitus was associated with an ≈0.74‐m/s increase in middle‐aged adults,5 and in general, diabetes mellitus tended to have a relatively small effect on PWV, compared with BP and age33; the effect of diabetes mellitus on CF‐PWV was estimated to be somewhat larger (≈1.67 m/s) among adults with chronic kidney disease.9 In contrast, our study demonstrated pronounced arterial stiffening (3‐m/s greater CF‐PWV values) in diabetic subjects with HFpEF, compared with their nondiabetic HFpEF counterparts, despite the absence of significant differences in age or 24‐hour SBP. Furthermore, large differences in arterial stiffness persisted after adjustment for office SBP, which was slightly higher in the diabetic group.
The mechanisms underlying arterial stiffening in diabetes mellitus remain poorly understood. It has been proposed to result from accelerated advanced glycation end product–mediated collagen cross‐linking,6, 34 aortic wall calcification,35 endothelial dysfunction,36 chronic low‐grade inflammation, increased oxidative stress, and increased sympathetic tone.37 Advanced glycation end product formation results from several reversible and irreversible steps that culminate in the detrimental cross‐linking of collagen molecules within the arterial vessel wall. In this process, sugar moieties (glucose, fructose, and glycolytic adductions) interact with the free amino acid residues of proteins, leading to the formation of Schiff base and Amadori products. In the setting of hyperglycemia, Amadori products are formed and unformed rapidly, and are in equilibrium with the blood glucose concentration.38 These reversible Amadori products undergo slow rearrangements, ultimately leading to the irreversible formation of advanced glycation end products.39 Advanced glycation end product deposition on arterial wall collagen leads to pathologic cross‐linking throughout the collagen molecule, which may contribute to an increased collagen content and stiffness. A relationship between inactive matrix Gla‐protein (MGP; dephospho‐uncarboxylated MGP), an inhibitor of vascular calcification, and CF‐PWV has also been recently demonstrated in diabetic subjects.40 MGP is a small protein secreted by the vascular smooth muscle cells, which undergoes vitamin K–dependent posttranslational activation. Active MGP is a potent inhibitor of vascular calcification. In a mouse model of diabetes mellitus with MGP gene deletion, elevated glucose levels lead to increased BMP (bone morphogenetic protein) signaling, endothelial‐mesenchymal transitioning, and osteoinduction,41 which was limited by transgenic enhancement of MGP expression.
Increased aortic stiffness promotes adverse hemodynamics via several key mechanisms. First, aortic stiffening increases aortic root characteristic impedance, leading to a steeper pressure‐flow relation in early systole, a wider pulse pressure, and greater forward‐wave amplitude. Second, increased aortic PWV from aortic stiffening promotes a premature arrival of reflected waves from the periphery to the aorta, while the LV is still ejecting blood, leading to excessive LV afterload and increased myocardial oxygen consumption.19, 20, 42, 43 A premature arrival of the reflected wave also results in a reduction in diastolic pressure augmentation by the reflected wave, resulting in a reduced coronary perfusion gradient during diastole.42 Finally, an increase in large artery stiffness leads to excessive penetration of pulsatility from large arteries into the microvasculature of organs that require torrential blood flow (such as the kidney and the brain), which may contribute to end‐organ microvascular damage.44, 45, 46
The impact of diabetes mellitus on aortic root stiffness and pulsatile hemodynamics has not been previously examined in HFpEF. Aortic pulsatile hemodynamics are best assessed via analyses of aortic pressure‐flow relations, which allow for a comprehensive characterization of arterial load and ventricular‐arterial interactions.23, 47 We found a pronounced increase in proximal aortic characteristic impedance in the diabetic group. The proximal aorta exerts an important influence on central pulse pressure and is not assessed with usual measurements of CF‐PWV. Aortic root characteristic impedance depends on its size and stiffness, and determines the pulsatile pressure increase for any given flow rate.23, 47 Consistent with the observed increased aortic stiffness, PWV, and characteristic impedance, we found a pronounced increase in oscillatory power (which is the product of pulsatile pressure and flow) and increased forward wave amplitude, backward wave amplitude, and central pulse pressure in the diabetic group. This physiologic pattern was also evident from 24‐hour ambulatory recordings.
Interestingly, despite the markedly abnormal pulsatile hemodynamics found in 24‐hour monitoring, 24‐hour SBP was not significantly different between the groups. SBP is a composite hemodynamic marker, which depends on both mean pressure and pulse pressure. It follows that marked SBP elevations may be absent despite adverse pulsatile hemodynamics when mean pressure is low, which tends to occur at the expense of a lower diastolic pressure. This hemodynamic profile, consistent with elevated arterial stiffness, has been clearly associated with adverse outcomes in non‐HFpEF populations.48, 49 Similarly, in the TOPCAT (Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist Trial), no relationship was observed between baseline SBP quartiles and outcomes,50 whereas a low DBP predicted an increased risk of a composite of all‐cause death, nonfatal myocardial infarction, nonfatal stroke, or hospitalization for HF in this cohort.51 Similarly, a high pulse pressure has been associated with all‐cause death in HFpEF.52
We also found that diabetic subjects exhibited significantly greater office SBP, despite the absence of significant differences in 24‐hour SBP. This may be because of a type II error for ambulatory SBP, although it is possible that diabetic subjects with HFpEF exhibit a greater white coat effect. Future studies should address potential differences in office versus ambulatory BP measurements in diabetic versus nondiabetic subjects with HFpEF and their prognostic implications.
The effects of diabetes mellitus on arterial stiffness are unlikely to be a consequence of the effects of diabetes mellitus on the myocardium. First, our analyses included quantification of pressure‐flow relations, which intrinsically account for the amount of flow generated by the LV. Second, the LVEF and stroke volume were not significantly different between the groups and are unlikely to explain the differences in arterial stiffness and pulsatile load. Rather, our results are consistent with the notion that diabetes mellitus exerts adverse effects on the arterial tree. Interestingly, the relationship between diabetes mellitus and arterial stiffness may be bidirectional, as recent data suggest that arterial stiffening may precede diabetes mellitus and predict the incidence of diabetes mellitus in nondiabetic adults.53 Arterial abnormalities associated with diabetes mellitus can adversely impact the myocardium, given that increased and/or abnormal pulsatile load favors LV hypertrophy, fibrosis, and diastolic dysfunction.3, 15, 17, 18, 43, 54, 55, 56, 57 Diabetes mellitus also has primary effects on the myocardium, and likely provides a “double hit” (ie, hemodynamic and metabolic) that may be important for the development of HFpEF. The interactions between the hemodynamic and the metabolic/neurohormonal effects of diabetes mellitus in the pathogenesis of HFpEF should be the focus of future studies.
Our study should be interpreted in the context of its strengths and limitations. Our study is the first to assess the impact of type 2 diabetes mellitus arterial stiffness on pulsatile hemodynamics and LV remodeling in HFpEF. We assessed CF‐PWV, the gold standard index of large artery stiffness, in addition to comprehensive in‐laboratory assessments of proximal aortic pressure‐flow relations using state‐of‐the‐art techniques. We used measured flow, as opposed to synthetic or triangular flow waveforms. We used carotid high‐fidelity tonometry, which does not rely in the application of transfer functions to peripheral pressure waveforms. In addition, we assessed 24‐hour ambulatory brachial and aortic BP and pulsatile hemodynamics. We enrolled a multiethnic cohort, which may be more generalizable to multiethnic populations. Our assessments of LV remodeling included measurements of LVM, as well as measures of cellular and interstitial expansion using contemporary magnetic resonance imaging techniques. Our study also has several limitations. We applied strict inclusion/exclusion criteria during the parent trial, to increase our confidence in the inclusion of subjects with HFpEF, as opposed to alternative causes of symptoms. In the process, we may have excluded many subjects with HFpEF and comorbidities related to arterial stiffness, such as advanced renal disease and dementia. This may have biased our study toward the null and may have underestimated the effect of diabetes mellitus on arterial stiffness. Our sample size is relatively small, although the marked differences in the measured parameters resulted in highly significant between‐group differences. We did not perform significance adjustments for multiple comparisons, which may increase the α error rate. However, our results on aortic stiffness (CF‐PWV), in‐office pulsatile hemodynamics, and 24‐hour assessments are highly consistent, adding confidence to our results. We did not assess urinary albumin excretion, an established renal and cardiovascular risk marker. All phenotypes were measured in a blinded manner to the randomized arm in the parent trial; strict blinding for diabetic status was not done, as these comparisons were performed retrospectively. Nevertheless, images were analyzed independently of clinical data, and many of the examined phenotypes (such as ECV computations, arterial stiffness measurements, pressure‐flow analyses, and ambulatory BP monitoring–based measurements) are not significantly susceptible to systematic operator bias. An additional limitation is that the potential effect of intraobserver/interobserver variability was not assessed, which may have limited our power to detect some associations. Given the limited sample size, we did not perform comprehensive adjustments for clinical characteristics, and adjusted only for characteristics that differed between the groups. Finally, our sample size was not sufficiently large to explore the relationship between glycemic control and arterial stiffness or LV remodeling/fibrosis among diabetic subjects with HFpEF. This should be the focus of future studies, given that glycemic control has been shown to be an important determinant of LV myocardial interstitial expansion in diabetic subjects without HFpEF.58
In summary, we comprehensively characterized, for the first time, arterial stiffness and in‐office and pulsatile arterial hemodynamic abnormalities in subjects with HFpEF with and without diabetes mellitus. We demonstrate that diabetes mellitus is associated with marked aortic stiffening, and an adverse pulsatile hemodynamic profile, characterized by increased aortic characteristic impedance, pulsatile power, forward and backward wave amplitude, and aortic pulse pressure, despite the absence of significant differences in 24‐hour SBP. Diabetic subjects also demonstrated more pronounced LV hypertrophy, with expansion of the extracellular myocardial compartment. Our findings underscore the importance of diabetes mellitus as a determinant of LV structure, arterial stiffness, aortic pulsatile hemodynamics, and ventricular‐arterial interactions in HFpEF and indicate the need to develop therapeutic approaches to mitigate arterial stiffening and its consequences in diabetic subjects. These approaches may be useful for the prevention and treatment of HFpEF.
Sources of Funding
This study was funded by a grant from the National Institute of Aging (5R21AG043802‐02, Chirinos) and grants from the Veterans Affairs Health Network (VISN‐4 CPPF program, Chirinos), 1R56HL124073‐01A1 (Chirinos) and 1 R01 HL121510‐01A1 (Chirinos). The project was also supported by grant UL1RR024134 from the National Center for Research Resources and by grant UL1TR000003 from the National Center for Advancing Translational Sciences, National Institutes of Health (NIH); the content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Zamani was funded by the Institute for Translational Medicine and Therapeutics of the University of Pennsylvania (grant 5UL1TR000003‐09 from the National Center for Research Resources), 5‐T32‐HL007843‐17, 1‐K23‐HL‐130, and 551‐01.
Disclosures
Chirinos has received consulting honoraria from Bristol‐Myers Squibb, OPKO Healthcare, Fukuda‐Denshi, Microsoft, Ironwood Pharmaceuticals, Sanifit, Pfizer, Akros Pharma, Merck, and Bayer. He received research grants from the National Institutes of Health, the American College of Radiology Network, Fukuda Denshi, Bristol‐Myers Squibb, and Microsoft; and he received device loans from AtCor Medical, Unex, and Uscom. Chirinos is named as inventor in a University of Pennsylvania patent application for the use of inorganic nitrates/nitrites for the treatment of heart failure and preserved ejection fraction (HFpEF) and in a University of Pennsylvania invention disclosure for the use of biomarkers in HFpEF. The remaining authors have no disclosures to report.
(J Am Heart Assoc. 2019;8:e011457 DOI: 10.1161/JAHA.118.011457.)
References
- 1. Lam CS, Donal E, Kraigher‐Krainer E, Vasan RS. Epidemiology and clinical course of heart failure with preserved ejection fraction. Eur J Heart Fail. 2011;13:18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sandesara PB, O'Neal WT, Kelli HM, Samman‐Tahhan A, Hammadah M, Quyyumi AA, Sperling LS. The prognostic significance of diabetes and microvascular complications in patients with heart failure with preserved ejection fraction. Diabetes Care. 2018;41:150–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Townsend RR, Wilkinson IB, Schiffrin EL, Avolio AP, Chirinos JA, Cockcroft JR, Heffernan KS, Lakatta EG, McEniery CM, Mitchell GF, Najjar SS, Nichols WW, Urbina EM, Weber T; American Heart Association Council on Hypertension . Recommendations for improving and standardizing vascular research on arterial stiffness: a scientific statement from the American Heart Association. Hypertension. 2015;66:698–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Loehr LR, Meyer ML, Poon AK, Selvin E, Palta P, Tanaka H, Pankow JS, Wright JD, Griswold ME, Wagenknecht LE, Heiss G. Prediabetes and diabetes are associated with arterial stiffness in older adults: the ARIC study. Am J Hypertens. 2016;29:1038–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chirinos JA, Segers P, Gillebert TC, De Buyzere ML, Van Daele CM, Khan ZA, Khawar U, De Bacquer D, Rietzschel ER, Asklepios I. Central pulse pressure and its hemodynamic determinants in middle‐aged adults with impaired fasting glucose and diabetes: the Asklepios study. Diabetes Care. 2013;36:2359–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Prenner SB, Chirinos JA. Arterial stiffness in diabetes mellitus. Atherosclerosis. 2015;238:370–379. [DOI] [PubMed] [Google Scholar]
- 7. Shah AM, Hung CL, Shin SH, Skali H, Verma A, Ghali JK, Kober L, Velazquez EJ, Rouleau JL, McMurray JJ, Pfeffer MA, Solomon SD. Cardiac structure and function, remodeling, and clinical outcomes among patients with diabetes after myocardial infarction complicated by left ventricular systolic dysfunction, heart failure, or both. Am Heart J. 2011;162:685–691. [DOI] [PubMed] [Google Scholar]
- 8. Townsend RR, Chirinos JA, Parsa A, Weir MA, Sozio SM, Lash JP, Chen J, Steigerwalt SP, Go AS, Hsu CY, Rafey M, Wright JT Jr, Duckworth MJ, Gadegbeku CA, Joffe MP; Chronic Renal Insufficiency Cohort Investigators . Central pulse pressure in chronic kidney disease: a chronic renal insufficiency cohort ancillary study. Hypertension. 2010;56:518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Townsend RR, Wimmer NJ, Chirinos JA, Parsa A, Weir M, Perumal K, Lash JP, Chen J, Steigerwalt SP, Flack J, Go AS, Rafey M, Rahman M, Sheridan A, Gadegbeku CA, Robinson NA, Joffe M. Aortic PWV in chronic kidney disease: a CRIC ancillary study. Am J Hypertens. 2010;23:282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lehrke M, Marx N. Diabetes mellitus and heart failure. Am J Med. 2017;130:S40–S50. [DOI] [PubMed] [Google Scholar]
- 11. Chirinos JA, Rietzschel ER, De Buyzere ML, De Bacquer D, Gillebert TC, Gupta AK, Segers P; Asklepios investigators . Arterial load and ventricular‐arterial coupling: physiologic relations with body size and effect of obesity. Hypertension. 2009;54:558–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Townsend RR, Black HR, Chirinos JA, Feig PU, Ferdinand KC, Germain M, Rosendorff C, Steigerwalt SP, Stepanek JA. Clinical use of pulse wave analysis: proceedings from a symposium sponsored by North American Artery. J Clin Hypertens (Greenwich). 2015;17:503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mohammed SF, Borlaug BA, Roger VL, Mirzoyev SA, Rodeheffer RJ, Chirinos JA, Redfield MM. Comorbidity and ventricular and vascular structure and function in heart failure with preserved ejection fraction: a community‐based study. Circ Heart Fail. 2012;5:710–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chirinos JA, Sweitzer N. Ventricular‐arterial coupling in chronic heart failure. Card Fail Rev. 2017;3:12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weber T, Chirinos JA. Pulsatile arterial haemodynamics in heart failure. Eur Heart J. 2018;39:3847–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chirinos JA, Akers SR, Trieu L, Ischiropoulos H, Doulias PT, Tariq A, Vassim I, Koppula MR, Syed AA, Soto‐Calderon H, Townsend RR, Cappola TP, Margulies KB, Zamani P. Heart failure, left ventricular remodeling, and circulating nitric oxide metabolites. J Am Heart Assoc. 2016;5:e004133 DOI: 10.1161/JAHA.116.004133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chirinos JA. Ventricular‐arterial coupling: invasive and non‐invasive assessment. Artery Res. 2013;7:2–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ky B, French B, May Khan A, Plappert T, Wang A, Chirinos JA, Fang JC, Sweitzer NK, Borlaug BA, Kass DA, St John Sutton M, Cappola TP. Ventricular‐arterial coupling, remodeling, and prognosis in chronic heart failure. J Am Coll Cardiol. 2013;62:1165–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chirinos JA. Deep phenotyping of systemic arterial hemodynamics in HFpEF (part 1): physiologic and technical considerations. J Cardiovasc Transl Res. 2017;10:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chirinos JA. Deep phenotyping of systemic arterial hemodynamics in HFpEF (part 2): clinical and therapeutic considerations. J Cardiovasc Transl Res. 2017;10:261–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zamani P, Akers S, Soto‐Calderon H, Beraun M, Koppula MR, Varakantam S, Rawat D, Shiva‐Kumar P, Haines PG, Chittams J, Townsend RR, Witschey WR, Segers P, Chirinos JA. Isosorbide dinitrate, with or without hydralazine, does not reduce wave reflections, left ventricular hypertrophy, or myocardial fibrosis in patients with heart failure with preserved ejection fraction. J Am Heart Assoc. 2017;6:e004262 DOI: 10.1161/JAHA.116.004262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nagueh SF, Appleton CP, Gillebert TC, Marino PN, Oh JK, Smiseth OA, Waggoner AD, Flachskampf FA, Pellikka PA, Evangelista A. Recommendations for the evaluation of left ventricular diastolic function by echocardiography. J Am Soc Echocardiogr. 2009;22:107–133. [DOI] [PubMed] [Google Scholar]
- 23. Chirinos JA, Segers P. Noninvasive evaluation of left ventricular afterload: part 2: arterial pressure‐flow and pressure‐volume relations in humans. Hypertension. 2010;56:563–570. [DOI] [PubMed] [Google Scholar]
- 24. Hametner B, Wassertheurer S, Kropf J, Mayer C, Holzinger A, Eber B, Weber T. Wave reflection quantification based on pressure waveforms alone—methods, comparison, and clinical covariates. Comput Methods Programs Biomed. 2013;109:250–259. [DOI] [PubMed] [Google Scholar]
- 25. Wassertheurer S, Kropf J, Weber T, van der Giet M, Baulmann J, Ammer M, Hametner B, Mayer CC, Eber B, Magometschnigg D. A new oscillometric method for pulse wave analysis: comparison with a common tonometric method. J Hum Hypertens. 2010;24:498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weber T, Wassertheurer S, Rammer M, Maurer E, Hametner B, Mayer CC, Kropf J, Eber B. Validation of a brachial cuff‐based method for estimating central systolic blood pressure. Hypertension. 2011;58:825–832. [DOI] [PubMed] [Google Scholar]
- 27. Chirinos JA, Segers P, De Buyzere ML, Kronmal RA, Raja MW, De Bacquer D, Claessens T, Gillebert TC, St John‐Sutton M, Rietzschel ER. Left ventricular mass: allometric scaling, normative values, effect of obesity, and prognostic performance. Hypertension. 2010;56:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Messroghli DR, Greiser A, Frohlich M, Dietz R, Schulz‐Menger J. Optimization and validation of a fully‐integrated pulse sequence for modified look‐locker inversion‐recovery (MOLLI) T1 mapping of the heart. J Magn Reson Imaging. 2007;26:1081–1086. [DOI] [PubMed] [Google Scholar]
- 29. Liu CY, Liu YC, Wu C, Armstrong A, Volpe GJ, van der Geest RJ, Liu Y, Hundley WG, Gomes AS, Liu S, Nacif M, Bluemke DA, Lima JA. Evaluation of age‐related interstitial myocardial fibrosis with cardiac magnetic resonance contrast‐enhanced T1 mapping: MESA (Multi‐Ethnic Study of Atherosclerosis). J Am Coll Cardiol. 2013;62:1280–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McDiarmid AK, Swoboda PP, Erhayiem B, Lancaster RE, Lyall GK, Broadbent DA, Dobson LE, Musa TA, Ripley DP, Garg P, Greenwood JP, Ferguson C, Plein S. Athletic cardiac adaptation in males is a consequence of elevated myocyte mass. Circ Cardiovasc Imaging. 2016;9:e003579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hundley WG, Kitzman DW, Morgan TM, Hamilton CA, Darty SN, Stewart KP, Herrington DM, Link KM, Little WC. Cardiac cycle‐dependent changes in aortic area and distensibility are reduced in older patients with isolated diastolic heart failure and correlate with exercise intolerance. J Am Coll Cardiol. 2001;38:796–802. [DOI] [PubMed] [Google Scholar]
- 32. Weber T, Wassertheurer S, O'Rourke MF, Haiden A, Zweiker R, Rammer M, Hametner B, Eber B. Pulsatile hemodynamics in patients with exertional dyspnea: potentially of value in the diagnostic evaluation of suspected heart failure with preserved ejection fraction. J Am Coll Cardiol. 2013;61:1874–1883. [DOI] [PubMed] [Google Scholar]
- 33. Cecelja M, Chowienczyk P. Dissociation of aortic pulse wave velocity with risk factors for cardiovascular disease other than hypertension: a systematic review. Hypertension. 2009;54:1328–1336. [DOI] [PubMed] [Google Scholar]
- 34. Monnier VM, Vishwanath V, Frank KE, Elmets CA, Dauchot P, Kohn RR. Relation between complications of type I diabetes mellitus and collagen‐linked fluorescence. N Engl J Med. 1986;314:403–408. [DOI] [PubMed] [Google Scholar]
- 35. Towler DA. Arteriosclerotic calcification: a serpi(n)ginous path to cardiovascular health? Circ Res. 2015;117:744–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brillante DG, O'Sullivan AJ, Johnstone MT, Howes LG. Arterial stiffness and haemodynamic response to vasoactive medication in subjects with insulin‐resistance syndrome. Clin Sci (Lond). 2008;114:139–147. [DOI] [PubMed] [Google Scholar]
- 37. Weber T. Arterial stiffness, wave reflections, and diabetes: a bidirectional relationship? Am J Hypertens. 2010;23:1047–1048. [DOI] [PubMed] [Google Scholar]
- 38. Brownlee M, Vlassara H, Kooney A, Ulrich P, Cerami A. Aminoguanidine prevents diabetes‐induced arterial wall protein cross‐linking. Science. 1986;232:1629–1632. [DOI] [PubMed] [Google Scholar]
- 39. Sell DR, Monnier VM. Molecular basis of arterial stiffening: role of glycation—a mini‐review. Gerontology. 2012;58:227–237. [DOI] [PubMed] [Google Scholar]
- 40. Sardana M, Vasim I, Varakantam S, Kewan U, Tariq A, Koppula MR, Syed AA, Beraun M, Drummen NE, Vermeer C, Akers SR, Chirinos JA. Inactive matrix Gla‐protein and arterial stiffness in type 2 diabetes mellitus. Am J Hypertens. 2017;30:196–201. [DOI] [PubMed] [Google Scholar]
- 41. Yao Y, Jumabay M, Ly A, Radparvar M, Cubberly MR, Bostrom KI. A role for the endothelium in vascular calcification. Circ Res. 2013;113:495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hoffman JI, Buckberg GD. The myocardial oxygen supply: demand index revisited. J Am Heart Assoc. 2014;3:e000285 DOI: 10.1161/JAHA.113.000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chirinos JA. Deciphering systolic‐diastolic coupling in the intact heart. Hypertension. 2017;69:575–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cardoso CR, Ferreira MT, Leite NC, Barros PN, Conte PH, Salles GF. Microvascular degenerative complications are associated with increased aortic stiffness in type 2 diabetic patients. Atherosclerosis. 2009;205:472–476. [DOI] [PubMed] [Google Scholar]
- 45. Kim WJ, Park CY, Park SE, Rhee EJ, Lee WY, Oh KW, Park SW, Kim SW, Song S. The association between regional arterial stiffness and diabetic retinopathy in type 2 diabetes. Atherosclerosis. 2012;225:237–241. [DOI] [PubMed] [Google Scholar]
- 46. Smith A, Karalliedde J, De Angelis L, Goldsmith D, Viberti G. Aortic pulse wave velocity and albuminuria in patients with type 2 diabetes. J Am Soc Nephrol. 2005;16:1069–1075. [DOI] [PubMed] [Google Scholar]
- 47. Chirinos JA, Segers P. Noninvasive evaluation of left ventricular afterload: part 1: pressure and flow measurements and basic principles of wave conduction and reflection. Hypertension. 2010;56:555–562. [DOI] [PubMed] [Google Scholar]
- 48. Somes GW, Pahor M, Shorr RI, Cushman WC, Applegate WB. The role of diastolic blood pressure when treating isolated systolic hypertension. Arch Intern Med. 1999;159:2004–2009. [DOI] [PubMed] [Google Scholar]
- 49. Chirinos JA, Zambrano JP, Chakko S, Veerani A, Schob A, Perez G, Mendez AJ. Relation between ascending aortic pressures and outcomes in patients with angiographically demonstrated coronary artery disease. Am J Cardiol. 2005;96:645–648. [DOI] [PubMed] [Google Scholar]
- 50. Selvaraj S, Claggett B, Shah SJ, Anand I, Rouleau JL, Desai AS, Lewis EF, Pitt B, Sweitzer NK, Pfeffer MA, Solomon SD. Systolic blood pressure and cardiovascular outcomes in heart failure with preserved ejection fraction: an analysis of the TOPCAT trial. Eur J Heart Fail. 2018;20:483–490. [DOI] [PubMed] [Google Scholar]
- 51. Tsujimoto T, Kajio H. Low diastolic blood pressure and adverse outcomes in heart failure with preserved ejection fraction. Int J Cardiol. 2018;263:69–74. [DOI] [PubMed] [Google Scholar]
- 52. Teng TK, Tay WT, Dahlstrom U, Benson L, Lam CSP, Lund LH. Different relationships between pulse pressure and mortality in heart failure with reduced, mid‐range and preserved ejection fraction. Int J Cardiol. 2018;254:203–209. [DOI] [PubMed] [Google Scholar]
- 53. Muhammad IF, Borne Y, Ostling G, Kennback C, Gottsater M, Persson M, Nilsson PM, Engstrom G. Arterial stiffness and incidence of diabetes: a population‐based cohort study. Diabetes Care. 2017;40:1739–1745. [DOI] [PubMed] [Google Scholar]
- 54. Chirinos JA, Phan TS, Syed AA, Hashmath Z, Oldland HG, Koppula MR, Tariq A, Javaid K, Miller R, Varakantam S, Dunde A, Neetha V, Akers SR. Late systolic myocardial loading is associated with left atrial dysfunction in hypertension. Circ Cardiovasc Imaging. 2017;10:e006023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chirinos JA, Segers P, Gupta AK, Swillens A, Rietzschel ER, De Buyzere ML, Kirkpatrick JN, Gillebert TC, Wang Y, Keane MG, Townsend R, Ferrari VA, Wiegers SE, St John Sutton M. Time‐varying myocardial stress and systolic pressure‐stress relationship: role in myocardial‐arterial coupling in hypertension. Circulation. 2009;119:2798–2807. [DOI] [PubMed] [Google Scholar]
- 56. Chirinos JA, Segers P, Raina A, Saif H, Swillens A, Gupta AK, Townsend R, Emmi AG Jr, Kirkpatrick JN, Keane MG, Ferrari VA, Wiegers SE, St John Sutton MG. Arterial pulsatile hemodynamic load induced by isometric exercise strongly predicts left ventricular mass in hypertension. Am J Physiol Heart Circ Physiol. 2010;298:H320–H330. [DOI] [PubMed] [Google Scholar]
- 57. Chirinos JA, Segers P, Gillebert TC, Gupta AK, De Buyzere ML, De Bacquer D, St John‐Sutton M, Rietzschel ER, Asklepios I. Arterial properties as determinants of time‐varying myocardial stress in humans. Hypertension. 2012;60:64–70. [DOI] [PubMed] [Google Scholar]
- 58. Al‐Badri A, Hashmath Z, Oldland GH, Miller R, Javaid K, Syed AA, Ansari B, Gaddam S, Witschey WR, Akers SR, Chirinos JA. Poor glycemic control is associated with increased extracellular volume fraction in diabetes. Diabetes Care. 2018;41:2019–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]