

Abstract
Background
Sustained pressure overload leads to changes in cardiac metabolism, function, and structure. Both time course and causal relationships between these changes are not fully understood. Therefore, we studied spontaneously hypertensive rats (SHR) during early hypertension development and compared them to control Wistar Kyoto rats.
Methods and Results
We serially evaluated myocardial glucose uptake rates (Ki) with dynamic 2‐[18F] fluoro‐2‐deoxy‐D‐glucose positron emission tomography, and ejection fraction and left ventricular mass to body weight ratios with cardiac magnetic resonance imaging in vivo, determined glucose uptake and oxidation rates in isolated perfused hearts, and analyzed metabolites, mammalian target of rapamycin activity and endoplasmic reticulum stress in dissected hearts. When compared with Wistar Kyoto rats, SHR demonstrated increased glucose uptake rates (Ki) in vivo, and reduced ejection fraction as early as 2 months of age when hypertension was established. Isolated perfused SHR hearts showed increased glucose uptake and oxidation rates starting at 1 month. Cardiac metabolite analysis at 2 months of age revealed elevated pyruvate, fatty acyl‐ and branched chain amino acid‐derived carnitines, oxidative stress, and inflammation. Mammalian target of rapamycin activity increased in SHR beginning at 2 months. Left ventricular mass to body weight ratios and endoplasmic reticulum stress were elevated in 5 month‐old SHR.
Conclusions
Thus, in a genetic hypertension model, chronic cardiac pressure overload promptly leads to increased myocardial glucose uptake and oxidation, and to metabolite abnormalities. These coincide with, or precede, cardiac dysfunction while left ventricular hypertrophy develops only later. Myocardial metabolic changes may thus serve as early diagnostic markers for hypertension‐induced left ventricular hypertrophy.
Keywords: hypertension, hypertrophy/remodeling, metabolic imaging, myocardial metabolism, rats
Subject Categories: Hypertension, Hypertrophy, Nuclear Cardiology and PET, Magnetic Resonance Imaging (MRI)
Clinical Perspective
What Is New?
Serial in vivo imaging and ex vivo analyses establish sequential metabolic, functional and structural cardiac changes during early hypertension in a rat model of hypertensive heart disease.
Metabolic abnormalities precede and coincide with impaired cardiac function. Left ventricular hypertrophy only develops later.
What Are the Clinical Implications?
Left ventricular hypertrophy is a secondary manifestation of hypertension, independently predicts future cardiovascular disease events, and leads to heart failure.
The current studies establish a model in which to investigate relationships between metabolic, functional and structural cardiac abnormalities, and thereby identify specific molecular targets for prevention of left ventricular hypertrophy and consequently heart failure in hypertensive patients.
Introduction
Chronic hypertension leads to left ventricular hypertrophy (LVH) and often, in turn, to heart failure (HF). HF is associated with significant morbidity and mortality, with 1‐year mortality rates as high as 22%.1 LVH plays a significant role in the pathogenesis of HF. Early detection and prevention of LVH is thus important. It is well established that fully developed LVH and HF are associated with metabolic abnormalities.2 However, the temporal and causal relationships between gradually increasing chronic pressure overload, metabolic changes, impaired cardiac function and LVH remain unknown.
Accurate non‐invasive imaging of myocardial substrate metabolism in animals and humans offers a unique opportunity to identify changes in myocardial metabolism before and during the development of LVH, and to establish treatment strategies to prevent LVH.3, 4 We recently developed an improved method for non‐invasive quantification of rates of myocardial glucose uptake and use in vivo in mouse hearts using dynamic 2‐[18F] fluoro‐2‐deoxy‐D‐glucose positron emission tomography (FDG PET).5 Using this method, together with cardiac magnetic resonance (CMR) imaging for evaluating cardiac structure and function in vivo, we previously described that increased myocardial FDG uptake precedes diminished cardiac function and LVH in the transverse aortic constriction induced acute pressure overload mouse model.6, 7, 8 Ex vivo studies in perfused rat hearts under high workload further revealed that increased glucose uptake not matching reduced glucose oxidation resulted in glucose 6‐phosphate (G6P) accumulation in the myocardial wall, activation of mammalian target of rapamycin (mTOR), endoplasmic reticulum stress, and loss in left ventricular function.7
The acute pressure overload mouse model9 used by our laboratory6 and others10 lacks a key feature of the human disease; the slow progressive increase in pressure overload. Therefore, in the current study we used a rat model with slowly developing hypertension, the spontaneously hypertensive rat (SHR). SHR are widely used to study the transition from compensated LVH to systolic HF.11 Prior studies longitudinally imaged SHR using small animal PET/CT (Computed Tomography) from 8 to 22 months of age.12, 13 They observed increased glucose and fatty acid use at 8 months of age in SHR that was sustained up to 20 months, but found decreased cardiac function and increased cardiac volume only at 20 months. Diffusion tensor CMR imaging at 2 years (≈24 months) of age revealed significantly altered myocardial fiber and laminar sheet structure in SHR.14, 15 Another study measured local strain in the left ventricle (LV) directly from gated microPET image data sets of SHR. Preliminary regional analysis in SHR indicated greatest deformation in the lateral free wall when compared with other wall segments during the second year of life (12 to 17 months of age).16 However, systematic analyses of metabolic, functional, and structural changes have not been reported in SHR during the early development of hypertension. In this study we used serial dynamic FDG PET imaging and CMR in vivo, measured myocardial glucose uptake and oxidation rates in perfused SHR hearts ex vivo, and performed a comprehensive cardiac metabolite analysis of SHR and WKY rats between 1 and 5 months of age. Our data suggest that slowly progressive hypertension in SHR leads to early myocardial metabolic changes that coincide with, or even precede, reduced ejection fraction (EF). LVH only develops later.
Methods
The authors declare that all supporting data are available within the article and supplemental material.
Rat Model
Male 3‐week old SHR and WKY rats, purchased from Charles River (Kingston, NY), were housed under controlled conditions (temperature 21±1°C, humidity 60±10%, 12 hours light/12 hours dark cycle, and free access to standard rat chow and water). Measurements described below were performed at baseline (1 month of age), and at 2, 3, and 5 months of age as SHR develop hypertension at ≈6 to 8 weeks of age.17 Animal experiments were approved by the Institutional Animal Care and Use Committees of the University of Virginia and the University of Texas Health Science Center in Houston and performed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Blood Pressure Measurements
SHR and WKY rats were subjected to invasive carotid artery catheterization to measure mean arterial pressure (MAP). Rats were anesthetized with Inactin Hydrate (Sigma; 100 mg/kg body weight) via intraperitoneal injection. A polyethylene (PE)‐50 catheter was inserted into the right carotid artery and MAP recorded over 30 minutes using a Blood Pressure Analyzer (Micromed Inc.). At completion of measurements hearts were harvested, washed with PBS, snap frozen in liquid nitrogen, and stored at −80°C.
FDG PET Imaging In Vivo
Myocardial rates of FDG uptake (Ki) were determined by dynamic 2‐[18F] fluoro‐2‐deoxy‐D‐glucose (18F‐FDG) PET imaging of SHR and WKY rats using the Siemens Focus F 120 microPET scanner as we described for mice.5, 6 Briefly, rats were fasted for 6 hours with free access to water before FDG imaging. They were then subjected to a 60‐minute dynamic PET scan under 2% isoflurane anesthesia. Data acquisition was initiated a few seconds before the slow (over 20 seconds) administration of 1 to 1.5 millicurie (mCi) FDG via a tail‐vein catheter. A Small Animal Gating Instrument (model 1025L for PET) was used to continuously monitor heart rate, respiration, and core body temperature. For attenuation correction a transmission scan using a Co57 point source was done before FDG administration. The list mode data was sorted into 23 time bins (frames×time in seconds (11×8, 1×12, 2×60, 1×180, 8×400 seconds) and the sinograms reconstructed with attenuation correction using filtered back projection algorithms (ramp filter cutoff at the Nyquist frequency).5, 6
Region of Interest Analysis and Computation of Myocardial FDG Uptake Rate, Ki
Briefly, data representing FDG concentrations in the myocardium and left ventricle were taken from 2 regions of interest in a single frame of the 4‐D dynamic PET image. The slices from the last time frame of the image were manually parsed until the left ventricle was clearly visible. The latest time frame most clearly showed the myocardium, since most FDG had been taken up from the blood by that time point. Once a slice in the midsection of the heart was identified, the 2 regions of interest were manually drawn as continuous lines using a series of connected points. Time activity curves (TAC) for each of the 2 regions of interest were generated by averaging the image data of a given region of interest at each time frame. To more accurately represent the uptake in the entire heart and prevent anomalies in a single slice from representing activity in the entire heart, regions of interest were first tested in several slices in the midsection of the heart. Slices were manually parsed from the tail‐most slice towards the head. The first slice in which the ventricle could be resolved from the myocardium (first slice showing the donut shape) was designated as the bottom slice of the midsection of the heart. The last slice, where the ventricle was visible, was designated as the top slice of the midsection. The average slice of the top and bottom slices was designated as the mid‐slice of the heart. The mid‐slice, as well as the slices directly above and below, were analyzed to create 3 TACs. If the TAC from the mid‐slice was consistent with the other 2 TACs based on visual comparison, it was used for modeling. If the mid‐slice contained some heterogeneity relative to the other slices, which was a rare occurrence, then the next slice towards the head was used for modeling. Using the formalism developed in our laboratory for mouse hearts,5, 6 a 3‐compartment kinetic model that can simultaneously correct for spill over and partial volume effects for both the blood pool and myocardium, generate a model corrected blood input function (MCIF), and compute kinetic rate parameters (K1‐k3), was used to compute rates of myocardial FDG uptake, Ki. A blood sample was also collected at around 56 minutes after FDG administration from the tail vein to validate model corrected blood input function (MCIF) at the same time point. The above analysis was performed using the MATLAB_r2018a (Mathworks Inc., Natick MA) computing environment.
Because of the limited spatial resolution of the small animal PET scanner, and small size of rat heart during early stages of hypertension, partial volume averaging can become a significant problem resulting in spill over contamination from the blood to the tissue at early and vice‐versa at late time points. Our model developed in mouse heart5 accounted for partial volume and spill over contamination effects and generated MCIF as described above. In addition, implementation of the convolution function used to represent the myocardium first found poor fits using MATLAB's convolution (conv) function, which may have been attributable to the fact that the MATLAB conv function calculates a convolution on a set of discrete data points. To improve the approximation of the tissue function, the equation was coded as a triangular approximation of the continuous convolution integral, rather than computed using a discrete convolution algorithm. Because of the high number of parameters involved in optimizing the model,5 the model is sensitive to the initial guesses for some of the variables. Because FDG is a non‐reversible tracer, the rate constant k4 was assumed to be small relative to the rate constants K1‐k3. To achieve the best fit, it was necessary to place the initial guess for k4 much lower than the other rate constants. Similarly, the value for A1,5 which helps to determine the height of the concentration peak in the 7 parameter blood concentration equation, required a large enough initial guess to support adequate spill over contamination correction from blood to tissue.
CMR Imaging In Vivo
Myocardial mass and left ventricular (LV) function were determined using CMR 2 days after FDG PET scans on the same rats using an ECG‐triggered cine black blood pulse sequence on a 7T Bruker‐Siemens scanner (ClinScan) as described.18
Briefly, ECG leads were attached to the rats’ chests and rats, anesthetized with 2% isoflurane, were placed in a prone position inside the radiofrequency coil. The LV ejection fraction (EF) and LV mass measurements were performed using an ECG‐triggered cine black blood pulse sequence18 with a slice thickness of 1.5 mm and an in‐plane resolution of 100×100 μm2. During each session, the entire LV was imaged with 9 to 10 contiguous short‐axis slices. The black blood cine images were imported to a workstation and analyzed with SEGMENT (Medviso, AB, Sweden, open source) software. Specifically, end‐diastolic (ED) and end‐systolic (ES) frames were identified and thereafter endocardial and epicardial contours were drawn on the frames for all the slices. Using the SEGMENT software, ED volume (EDV), ES volume, ejection fraction (EF), and LV mass were computed. The LV mass (LVM) was then normalized to body weight (BW) to obtain LVM to BW ratios.
Measurements of Glucose Uptake and Oxidation in Perfused Rat Hearts Ex Vivo
SHR and WKY rat hearts were perfused ex vivo and glucose uptake and oxidation determined as described.7, 19, 20 Briefly, after excision and cannulation, hearts were perfused at 37°C with non‐recirculating Krebs‐Henseleit buffer equilibrated with 95% O2 to 5% CO2 supplemented with 5 mM glucose and 0.4 mM sodium oleate bound to 1% bovine serum albumin. Preload was set at 15 cm H2O. After equilibration, hearts were perfused at normal workload (afterload set to 100 cm H2O) for 30 minutes. All hearts were then subjected for 30 minutes to increased workload (with epinephrine bitartrate 1 μmol/L, plus afterload of 120 cm H2O for 1‐ and 2‐month old rats, or 140 cm H2O for 3‐ and 5‐month old rats). Glucose transport and phosphorylation were determined by [3H]2O production from [2‐3H] glucose (0.05 μCi/mL). Rates of glucose oxidation were determined by quantifying [14C]O2 released into the coronary effluent from [U‐14C] glucose (0.08 μCi/mL).19
Evaluation of Protein Expression and Phosphorylation by Immunoblotting
Frozen hearts, obtained after blood pressure measurements, were weighed (total heart weight), powdered and homogenized in Mammalian Protein Extraction Reagent (MPER; Thermo Scientific) containing Halt protease and phosphatase inhibitor cocktail (Thermo Scientific). Protein concentrations were determined using a bicinchoninic acid assay (Pierce, Thermo Scientific). SDS samples (40 μg total protein per lane) were separated by SDS‐PAGE (10% Tris‐HCl polyacrylamide gels), transferred onto nitrocellulose membranes by electroblotting, and immunoblotted as described.7 Specifically, membranes were blocked in 5% milk/Tris‐buffered saline (TBS)/0.1% Tween‐20 for 2 hours at 4°C and incubated overnight at 4°C, with phospho‐p70S6 kinase (Thr389) antibodies (Cell Signaling; Cat# 9205; 1:1000) in 5% bovine serum albumin/Tris‐buffered saline (TBS)/0.1% Tween‐20, or with p70S6 kinase (Cell Signaling; Cat# 2708; 1:1000), GRP78 (Enzo; Cat# ADI‐SPA‐826‐F; 1:1000), collagen α1 type I (COL1A1) (Santa Cruz; Cat # sc‐293182; 1:200), or collagen α1 type III (COL3A1) (Santa Cruz; Cat # sc‐271249; 1:200) antibodies in 5% milk/Tris‐buffered saline (TBS)/0.1% Tween‐20. Membranes were subsequently incubated with Horseradish Peroxidase (HRP)‐conjugated secondary antibodies (GE Healthcare; Cat# NA934V or NA931V; 1:2500) in 5% milk/Tris‐buffered saline (TBS)/0.1% Tween‐20 for 2 hours at room temperature. Signals were detected using chemiluminescence and band densities determined with ImageJ software (National Institutes of Health). Blots were rinsed in TBS, incubated with GAPDH antibodies (Ambion Life Technologies; Cat# AM4300; 1:2500) overnight at 4°C and then with infrared secondary antibodies (LI‐COR; anti‐mouse IRDye 680; Cat# 925‐68072; 1:15000) in LI‐COR blocking buffer for 2 hours at room temperature. Immunoreactivity was determined using the Odyssey Infrared Imaging System (LI‐COR), and images were analyzed with ImageJ software. All signal intensities were normalized to GAPDH expression.
Metabolite Analyses
Blood glucose was measured in tail vein blood of SHR and WKY rats after a 6 hour fast using a glucometer (ACCU‐CHEK Nano, Bayer). Rats were then anesthetized with Inactin as described above and blood samples collected through a PE‐50 catheter inserted into the right carotid artery. Hearts were harvested and processed for metabolite analysis by Metabolon Inc. (see Data S1). Serum was stored at −80°C for subsequent measurements of free fatty acids (FFA), insulin, and branched chain amino acids (BCAA) using the HR Series NEFA‐HR(2) kit (Wako), insulin ELISA kit (80‐INSHU‐E01.1, ALPCO) and Branched Chain Amino Acid Assay kit (ab83374, ABCAM), respectively.
Statistical Analyses
Due to the invasive and terminal method used for MAP measurements, MAP was measured repetitively at only a single time point (age) for each rat, and not repetitively over time. The MAP data were thus analyzed by 2‐way ANOVA with replicate measurements for each rat considered as subsampling units. In terms of the 2‐way ANOVA model, the dependent variable was MAP, and the independent variables were the rat type (SHR and WKY) and assessment age (1–5 months). For hypothesis testing, a set of linear contrasts of the ANOVA least‐squared means was used to conduct between rat type comparisons of MAP distribution means at 1, 2, 3, and 5 months. A Bonferroni corrected P<0.05 decision rule was used as the null hypothesis rejection rule.
The PET and CMR serial imaging data (Ki, EF, and LVM/BW) were analyzed by way of linear mixed models for time point by time point between rat type comparisons. Hypothesis testing was with respect to the mean response, and a comparison wise P<0.05 decision rule was used for the null hypothesis rejection criterion for between rat type comparisons. With‐in rat type trends in the serial PET and CMR in vivo imaging data were examined by Piecewise Random Coefficient Regression (PRCR). PRCR change points of 2 (for EF and LVM/BW ratios) or 3 (for Ki) months of age were selected for modeling differential time trends based on visual inspection of the rat type empirical mean time profiles. PRCR regression model parameters were estimated via restricted maximum likelihood, and null hypothesis tests related to the regression slope parameters were conducted by way of F‐tests. A P<0.05 decision rule was used as the null hypothesis rejection criterion for testing for non‐zero slope, and for testing with‐in rat type differences in the PRCR slope parameter values.
Repeated glucose uptake and oxidation measurements in perfused rat hearts were analyzed by way of linear mixed models. Hypothesis testing was with respect to the mean response, and a Bonferroni corrected P<0.05 decision rule was used as the null hypothesis rejection rule for between rat type comparisons.
Ex vivo data including immunoblots, heart weight/BW ratios, circulating glucose, insulin, FFA, BCAA levels, and collagen I/collagen III ratios were analyzed using paired Student t‐tests. Ex vivo metabolomics data were analyzed using Welch 2‐sample t‐tests. A P<0.05 decision rule was used as the null hypothesis rejection criterion.
The statistical software package SAS version 9.4 (SAS Institute Inc., Cary, NC) and GraphPad Prism 7 (GraphPad Inc., La Jolla, CA) were used to conduct all the analyses.
Results
Blood Pressure as a Function of Age in SHR
Mean arterial pressure (MAP) was higher in SHR than WKY at 1 month of age (P=0.002), but was still within the normotensive range (<120 mm Hg). SHR were hypertensive at 2 months of age (>160 mm Hg) while WKY remained normotensive (P<0.001). At 3 and 5 months of age, MAP increased further in SHR, while in WKY rats it remained stable and within the normal range (SHR versus WKY; P<0.001 and P<0.001, respectively) (Figure 1A).
Figure 1.
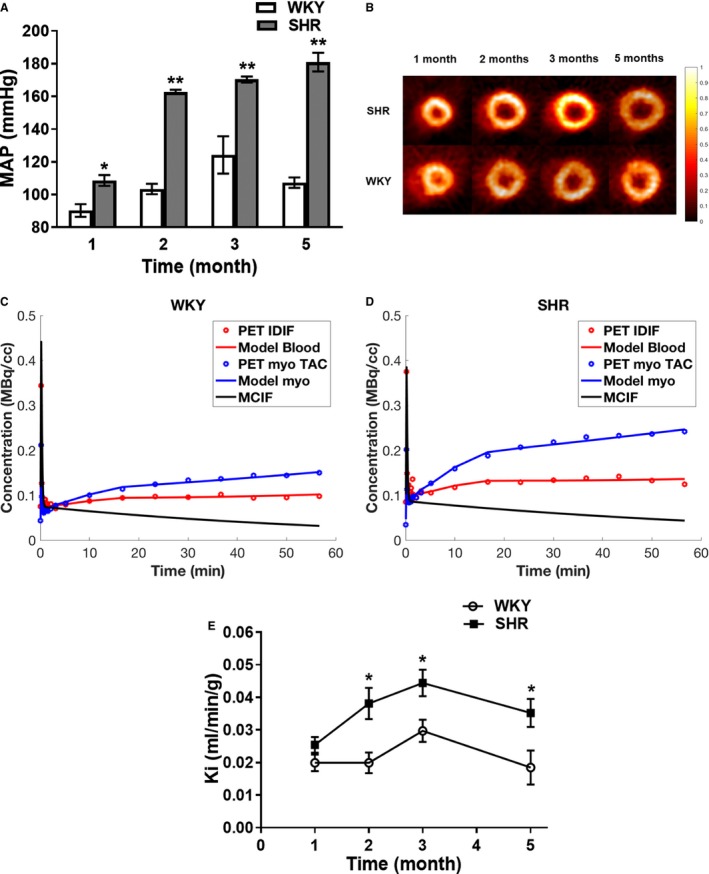
Increased glucose uptake during hypertension development in SHR. A, Blood pressure. MAP was measured in SHR (n=6) and WKY (n=6) at 1, 2, 3, and 5 months of age. MAP were recorded every minute and values averaged to obtain the reported data with standard errors. *P=0.020 SHR vs WKY. B, Transverse in vivo FDG PET images. SHR (n=8) and WKY (n=5) rats were serially imaged at 1, 2, 3, and 5 months of age using FDG PET imaging. Representative transverse last time frame intensity images of SHR and WKY hearts normalized to the injected dose and animal weight are shown. C and D, Time activity curves for WKY (C) and SHR (D) at 2 months of age. Representative time activity curves for LV blood pools and myocardium, model fits, and model corrected blood input function (MCIF). E, myocardial glucose uptake rates in vivo in SHR. Glucose uptake rates were computed from serial FDG PET images of SHR (n=8) and WKY (n=5) hearts. All data are shown as mean±SE. MAP indicates mean arterial pressure; PET, positron emission tomography; SHR, spontaneously hypertensive rats; TAC, time activity curve; IDIF, image‐derived input function; WKY, Wistar Kyoto rats. *P<0.05 SHR vs WKY.
Myocardial Glucose Uptake In Vivo
Transverse in vivo FDG PET images of SHR and WKY hearts demonstrated that, when compared with WKY, FDG uptake was significantly elevated in SHR hearts starting at 2 months of age while LV dilatation was only present at 5 months (Figure 1B).
Representative time activity curves with computed model corrected blood input function (MCIF) for WKY and SHR at 2 months of age are shown in Figure 1C and 1D. As determined by paired t‐tests, computed MCIF radioactivity concentration values, for example in control WKY rats at 2 months of age, were not significantly different from sampled blood values (P=0.201). Computed rates of FDG uptake (Ki) for SHR and WKY showed age‐dependent changes in myocardial glucose uptake for both SHR and WKY (Figure 1E). However, in SHR hearts glucose uptake significantly increased from 1 to 3 months of age (slope=0.0095 Ki units/month, P<0.001), concurrently with blood pressures increasing into the hypertensive range. Meanwhile myocardial glucose uptake remained stable in normotensive WKY (slope=0.0049 Ki units/month, P=0.117). This resulted in a marked difference in Ki between SHR and WKY hearts at 2 months (P=0.009). From 2 to 5 months of age changes in glucose uptake in SHR and WKY hearts were similar but Ki in SHR remained significantly higher than in WKY (P=0.018 and P=0.035 at 3 and 5 months, respectively). A significant decline in Ki was observed in SHR from 3 to 5 months of age (slope=−0.0051 Ki units/month, P=0.047), whereas WKY exhibited no change in Ki from 3 to 5 months of age (slope=−0.0048 Ki units/month, P=0.138). Comparison of Ki in WKY over the 5 months (1–3 versus 3–5 months of age) revealed no significant difference (slope=0.0097 Ki units/month, P=0.068). Thus, for WKY Ki did not significantly change from 1 to 5 months. The reason for the decrease in Ki in SHR at 5 months is not clear, but it could represent an adaptation to increased left ventricular mass (LVM) and/or improved EF that we observe at this age (see below).
Ejection Fraction and Left Ventricular Mass
Cardiac ejection fraction (EF), and left ventricular mass (LVM) were measured in SHR and WKY using in vivo CMR. EF significantly decreased in SHR (slope=−5.0%/month, P= 0.031) but did not change in WKY rats from 1 to 2 months of age (slope=−0.8%/month, P=0.750) (Figure 2A). This resulted in a marked difference in EF between SHR and WKY at 2 months of age (P<0.001) paralleling increased glucose uptake rates (Ki) at the same age (Figure 1E). Between 2 and 5 months of age, EF did not significantly change in SHR (slope=0.4%/month, P=0.498) or WKY (slope=−0.9%/month, P=0.263). However, EF for WKY remained higher than for SHR (P<0.02). For LVM/BW ratios, both SHR and WKY exhibited a decline from 1 to 3 months of age (slope=−0.54 units/month, P=0.001 and slope=−0.64 units/month, P<0.001 respectively) and no change from 3 to 5 months (slope=0.164 units/month, P=0.145 and slope=0.175 units/month, P=0.215 respectively) (Figure 2B). When compared with WKY, SHR showed a significantly higher LVM/BW ratio at 5 months (P=0.022). Note, the decline in LVM/BW ratios from 1 to 2 months was because of a more pronounced increase in BW than LVM in both SHR and WKY. Consistent with the increased LVM/BW in SHR detected by CMR, we also observed a significant increase in weights of harvested SHR hearts when normalized to body weights at 5 months (P<0.05) (Figure 2C). Thus, early LVH is present in SHR at 5 months of age. However, we did not detect fibrosis at this age in SHR based on collagen I/III ratios (WKY 0.31 ±0.03, SHR 0.27±0.01, n=5–6, P=0.21).
Figure 2.
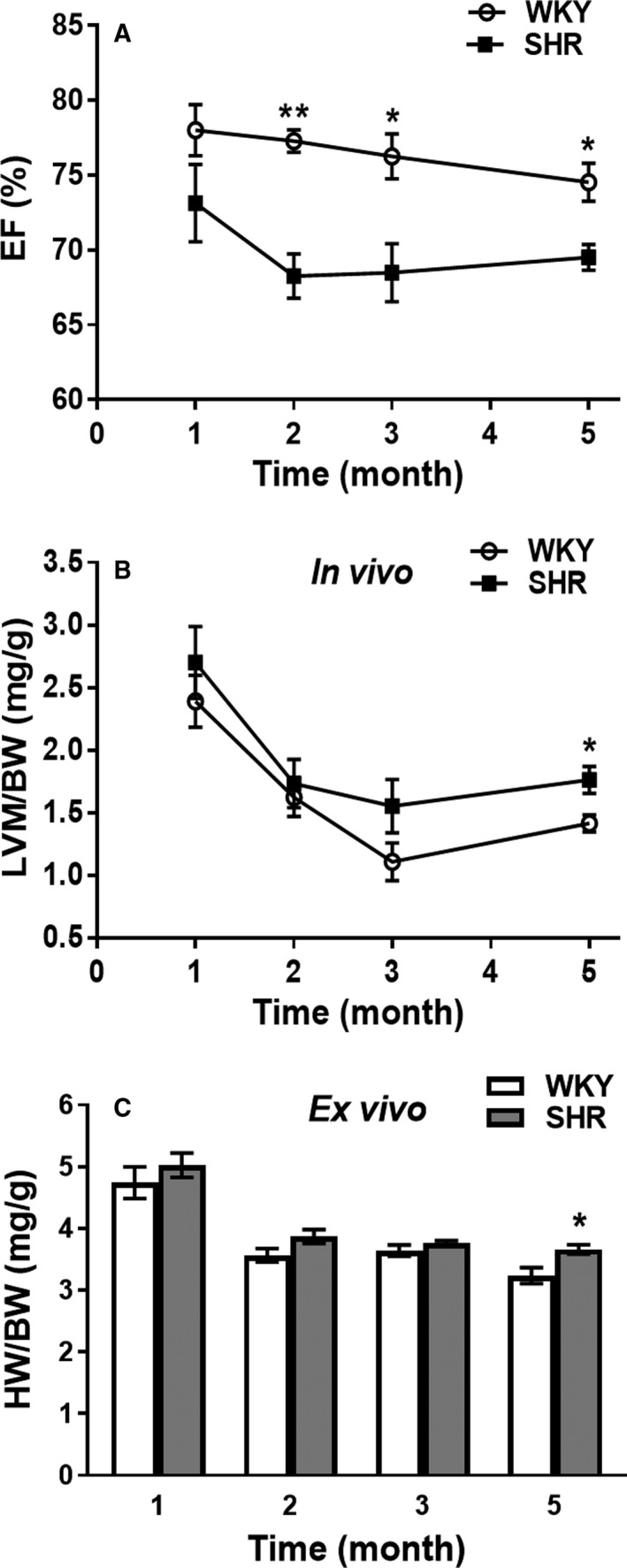
Early deteriorating cardiac function, and development of left ventricular hypertrophy (LVH) in SHR in vivo. A, Ejection fractions (EF) and B, left ventricular mass to body weight ratios (LVM/BW), were determined in SHR (n=8) and WKY (n=5) at 1, 2, 3, and 5 months of age using CMR. C, Heart weight to body weight ratios ex vivo. Entire heart weights were determined after dissection from SHR (n=6) and WKY (n=6) and normalized to body weight (HW/BW). All data are shown as mean±SE. BW indicates body weight; CMR, cardiac magnetic resonance; EF, ejection fraction; HW, heart weight; LVH, left ventricular hypertrophy; LVM, left ventricular mass; SHR, spontaneously hypertensive rats; WKY, Wistar Kyoto rats. *P<0.05 and † P<0.001 SHR vs WKY.
Glucose Uptake and Oxidation in Hearts Ex Vivo
To corroborate the in vivo FDG PET imaging results, rates of myocardial glucose uptake and oxidation were measured in isolated perfused working SHR and WKY hearts. Baseline rates of glucose uptake were significantly increased in SHR hearts at all ages with the largest difference (2‐fold increase versus WKY, P<0.001) occurring at 2 months (Figure 3A). Glucose uptake with increased workload was greater in SHR only at 2 months (P<0.001) (Figure 3B). Glucose oxidation rates were markedly elevated in SHR at all ages (P<0.001) at normal and increased workload (except at 3 months afterload; P=0.359) (Figure 3C and 3D).
Figure 3.

Increased glucose uptake and oxidation in isolated perfused SHR hearts. Rates of myocardial glucose uptake (A and B) and oxidation (C and D) were measured in isolated perfused working hearts of SHR (n=6) and WKY (n=6) rats at 1, 2, 3, and 5 months of age at normal workload (Baseline) (A and C) and at increased workload (Afterload) (B and D). All data are shown as mean±SE. SHR indicates spontaneously hypertensive rats; WKY, Wistar Kyoto rats. *P<0.001 SHR vs WKY.
mTOR and GRP78 Activation
We observed increased mTOR activation (measured by determining levels of p70S6 kinase [p70S6K] phosphorylation) concomitant with increased myocardial glucose‐6‐phosphate (G6P) accumulation in the transverse aortic constriction mouse model.7 In here, we observed significant increases in p70S6K phosphorylation, indicating increased mTOR activity, in SHR hearts at 2, 3, and 5 months of age (Figure 4A), with the highest fold difference between SHR and WKY at 2 months (P<0.001) (Figure 4B). However, G6P levels were normal in SHR hearts at 1, 2, 3, and 5 months (data not shown). Total p70S6K expression in SHR and WKY hearts was similar at all ages. As a marker of endoplasmic reticulum stress we evaluated GRP78 expression. GRP78 was only increased in 5‐month‐old SHR (Figure 4C and 4D) when LVH was apparent (Figure 2B and 2C).
Figure 4.
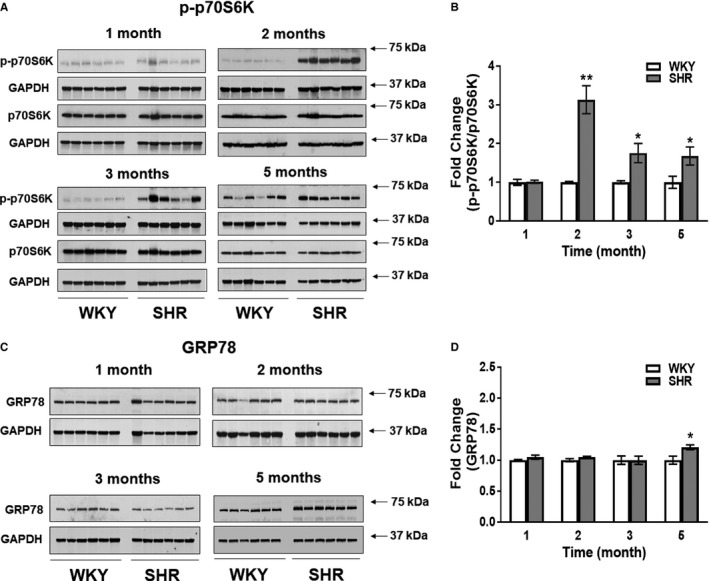
mTOR and ER stress activation in SHR. A, SHR (n=6) and WKY (n=6) hearts were immunoblotted for phospho‐p70S6K (p‐p70S6K), a marker for mTOR activity, total p70S6K, and GAPDH at 1, 2, 3, and 5 months of age. B, Signal intensities for p‐p70S6K and p70S6K were normalized to GAPDH, and p‐p70S6K/p70S6K ratios determined. Results were normalized within age groups (for which samples were analyzed on the same immunoblots) and fold changes reported. C, SHR (n=6) and WKY (n=6) hearts were immunoblotted for GRP78, a marker for endoplasmic reticulum stress, and GAPDH. D, Signal intensities for GRP78 normalized to GAPDH are presented. Results were normalized within age groups (for which samples were analyzed on the same immunoblots) and fold changes reported. All data are shown as mean±SE. *P<0.05 and † P<0.001 SHR vs WKY. SHR indicates spontaneously hypertensive rats; WKY, Wistar Kyoto rats.
Metabolite Profiling
To further assess metabolic changes in SHR hearts, we performed a detailed metabolite analysis (metabolomics) at 2 months of age. As above studies demonstrate, at this age hypertension was established, in vivo myocardial glucose uptake first increased, and EF reduced. The metabolomics data revealed significant changes not only in the 2 predominant energy‐providing metabolic pathways, fatty acid and glucose metabolism, but also in branched chain amino acid (BCAA) metabolism that can contribute to energy provision in the heart. Most remarkably, short‐, medium‐, and long‐chain fatty acyl‐carnitines and BCAA‐derived carnitines were increased (Figure 5A and Table S1). Concomitantly, all BCAAs (leucine, isoleucine, and valine) were modestly elevated and a few predominantly long‐chain free fatty acids were increased (Figure 5A and Table S1). Changes in glucose metabolism were reflected in increased pyruvate and lactate and decreased dihydroxyacetone phosphate levels (Figure 5A and Table S1). Metabolites within the glycogen breakdown pathway were decreased while glycogen synthesis intermediates were unchanged (Table S1). Products of the aldose reductase/polyol pathway were increased (Table S1). Levels of Krebs cycle intermediates were similar between SHR and WKY with the exception of a small decrease in α‐ketoglutarate (SHR/WKY=0.78, P<0.05) that was paired with a similar decrease in glutamate (SHR/WKY=0.82, P<0.05) but increased glutamine (SHR/WKY=1.26, P<0.05). Several nucleotide (purine and pyrimidine) metabolites were substantially decreased in SHR hearts, but Adenosine Monophosphate (AMP), Adenosine Diphosphate (ADP), and dihydroorotate levels were elevated (Table S1). In the nicotinate/nicotinamide and riboflavin metabolic pathways that generate the electron carriers nicotinamide adenine dinucleotide (NAD) and flavin adenine dinucleotide (FAD), metabolites were increased (Table S1). Within the metabolic pathway responsible for synthesis of Coenzyme A (CoA), a necessary co‐factor for fatty acid and glucose oxidation, cysteine (SHR/WKY=0.6, P<0.05) and phosphopantetheine (SHR/WKY=0.13, P<0.05) were reduced.
Figure 5.
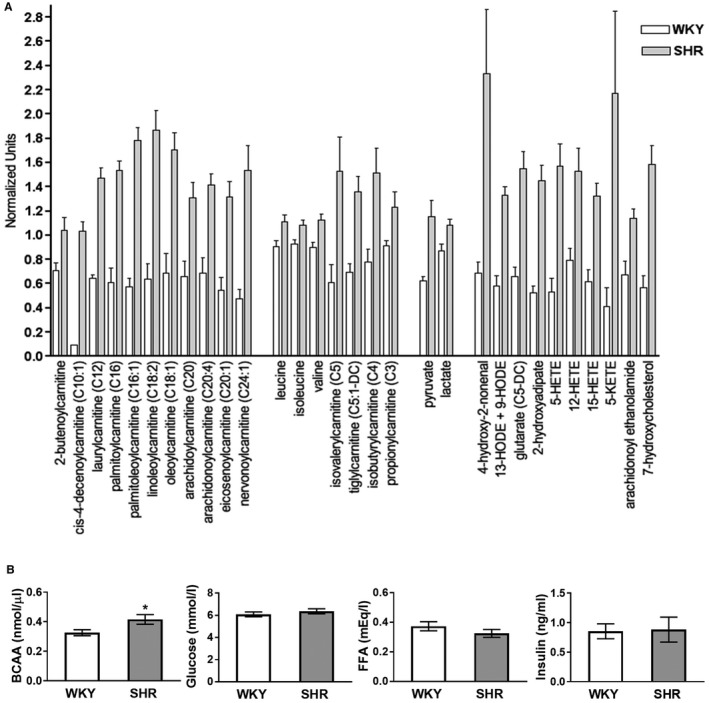
A, Changes in cardiac fatty acid, BCAA, and glucose metabolites and markers of oxidative stress and inflammation in SHR. Metabolites were analyzed in hearts of SHR (n=9) and WKY (n=10) at 2 months of age as described in S1. Data for biochemicals in raw area counts were rescaled to set medians equal to 1 and plotted. Data are shown as mean±SE. P<0.05 for all metabolites comparing SHR with WKY. B, Circulating BCAA, glucose, FFA, and insulin levels in SHR. Total circulating metabolite levels were measured in SHR (n=8) and WKY (n=8) at 2 months of age. BCAA were significantly higher in SHR (*P<0.05). There were no significant differences in circulating glucose, FFA and insulin concentrations between SHR and WKY. BCAA indicates branched chain amino acids; FFA; free fatty acids; SHR, spontaneously hypertensive rats; WKY, Wistar Kyoto rats. *P<0.05.
Oxidative stress in SHR hearts was indicated by striking increases in lipid oxidation and peroxidation products (oxidized fatty acid 4‐hydroxy‐2‐noneal, a major end product of lipid peroxidation; monohydroxy fatty acids; fatty acid dicarboxylates; eicosanoids; 7‐hydroxycholesterol; and long‐chain fatty acid‐derived endocannabinoids) (Figure 5A and Table S1). Although oxidized glutathione (GSSH) was unchanged, reduced glutathione (GSH) was decreased (SHR/WKY=0.55, 0.05< P<0.1) and concomitantly the GSH/GSSH ratio. At the same time the 3 amino acids necessary for glutathione synthesis were decreased; glutamate (SHR/WKY=0.82, P<0.05), glycine (SHR/WKY=0.89, P<0.05) and cysteine (SHR/WKY=0.6, P<0.05). Other notable lipid abnormalities in SHR hearts were changes in various diacylglycerols, membrane phospholipids, sphingolipids, and ceramide levels (Table S1).
Lastly, when circulating levels of the main energy‐providing substrates, FFA, glucose and BCAA, were measured, we found that total BCAA levels were significantly increased in SHR (SHR/WKY=1.3), but glucose and free fatty acid levels were not different from WKY (Figure 5B). Serum insulin levels (after 6 hours of fasting) were also similar between SHR and WKY (Figure 5B).
Discussion
In this study we evaluated time‐dependent changes in cardiac metabolism, function, and structure during the development of hypertension in male SHR. We observed increased glucose uptake and oxidation, and profound changes in levels of metabolites associated with energy provision, oxidative stress, and inflammation, at early time points during the development of hypertension (1 and 2 months of age), before or concomitant with impaired cardiac function (at 2 months of age). These changes occurred before LVH developed at 5 months of age (this study) and long before HF occurs in SHR at 24 months.11
Several studies have reported cardiac metabolic abnormalities, including increased cardiac glucose uptake and myocardial use of glucose for energy provision, in hypertension‐induced LVH and HF in animal models as well as humans.2, 21, 22 However, none have evaluated metabolic changes simultaneously with functional and structural abnormalities during the early stages of slowly progressive hypertension in SHR. A recent study used longitudinal CMR to quantify LV mass, volume, and cardiac work in SHR and control WKY hearts at 3, 14 and 21 months of age in vivo.23 Similar to our findings, lower EF was observed in SHR at all ages. One other study evaluated cardiac structure and function invasively in SHR over a period of 90 weeks (starting at 13 weeks of age).24 Decreased cardiac function was present from 52 to 90 weeks of age in SHR along with significant thickening of the free left ventricular wall indicative of LVH. Metabolic changes were not evaluated in these studies with SHR. However, Kato et al25 analyzed gene and protein expression and metabolite levels in parallel with measurements of cardiac function, substrate uptake, and cardiac energy reserve in salt‐treated Dahl salt‐sensitive rats at 11 and 17 weeks of age when LVH and congestive HF, respectively, had developed. They observed increased glucose uptake at both ages, and normal and decreased fatty acid uptake at 11 and 17 weeks of age, respectively. Interestingly, when these rats were treated with a compound known to enhance glucose oxidation, cardiac function and animal survival improved, suggesting a causal relationship between metabolic and functional changes.
The comprehensive metabolite analysis of SHR hearts at 2 months of age offers insights into changes in cardiac energy metabolism early in hypertension development. Although glucose use is augmented in SHR hearts, as indicated by increased glucose uptake and oxidation, increased pyruvate levels suggest that glycolysis produces more pyruvate than is oxidized. Surplus pyruvate is then used for anaerobic glycolysis to produce lactate. Increases in long‐, medium‐, and short‐chain fatty acyl carnitines are consistent with fatty acyl‐CoA supply to mitochondria exceeding β‐oxidation and/or Krebs cycle flux. Substantially increased BCAA‐carnitines in the presence of only marginally increased BCAAs in the heart and the circulation suggests increased metabolism of BCAAs, exceeding the capacity of the Krebs cycle. The changes in glucose, fatty acid, and BCAA metabolites thus suggest that in response to chronic pressure overload a surfeit of substrate is available to cardiomyocytes, but mitochondria cannot keep up with supply and are not able to produce enough Adenosine Triphosphate (ATP) to meet increased demand and maintain normal function (as reduced EF suggests). That increased substrate supply may trigger cardiac malfunction and hypertrophy is supported by a calorie restriction study in SHR.26 Consistent with decreased oxidative phosphorylation, enzymatic activities of mitochondrial complex I and II are significantly reduced in 24‐week‐old SHR.27 This may be because of oxidative stress and inflammatory processes.28 Our metabolomics data support this hypothesis as we observe increases in proinflammatory lipid oxidation and peroxidation products that are made when oxidative stress is present. A limitation of the metabolomics studies is that they only provide single time point steady state metabolite measurements. In the future, to clearly identify the defect(s) responsible for changes in specific metabolites in SHR hearts, metabolic flux studies will need to be performed.
Metabolomics analyses have been performed on hearts obtained from acute (transverse aortic constriction mice) and chronic pressure overload (Dahl salt‐sensitive rats) animal models at different time points during the development of LVH and HF.29, 30 Although all studies report differences in cardiac metabolite levels, commonalities between the previous analyses and our data are limited. Similar to the present study, increased BCAAs leucine and isoleucine were observed in mouse hearts with LVH 1 week after transverse aortic constriction, markedly increased fatty acyl‐carnitines, hydroxylated and dicarboxylated fatty acids, and lactate in failing hearts after transverse aortic constriction, and increased leucine, isoleucine, and pyruvate in Dahl salt‐sensitive rats with congestive HF.25, 30 Discrepancies between studies may be due to differences in mouse and rat models used and stages in LVH and HF development analyzed.
A recent study of plasma metabolites in HF patients reported increased long‐chain acyl carnitine levels31 that inversely correlated with left ventricular ejection fraction (LVEF). Impaired or dysregulated fatty acid oxidation and spillover of fatty acyl carnitines into the circulation was considered the likely cause for the elevated long‐chain acyl carnitine levels. This interpretation agrees with our observation of increased long‐chain fatty acyl carnitines in SHR hearts. Whether circulating long‐chain fatty acyl carnitines are increased in SHR will need to be determined. If increased, they may serve as markers for impaired cardiac metabolism during early stages of hypertension.
Changes in BCAA metabolism have only recently been described in animal models of HF. Increases in BCAA metabolites were consistently observed,32 but the specific BCAA metabolites that were changed varied. Sun et al33 found increased branched chain ketoacids but normal levels of BCAAs together with decreased activities of BCAT (branched chain amino acid aminotransferase) and BCKDH (branched chain alpha‐keto acid dehydrogenase). We observed significantly increased levels of BCAA‐derived carnitines and mildly elevated BCAAs suggesting increased activities of both BCAT and BCKDH. Whether observed changes reflect different stages in LVH development and are responsible for cardiac dysfunction and promotion of LVH remains to be determined. BCAAs, especially leucine, increase mTOR activity and thereby promote protein synthesis, cellular metabolism, and cell growth.21 In addition, BCAAs and their metabolites may also have a direct impact on mitochondrial function and cellular viability as BCAA abnormalities are associated with defects in the mitochondrial respiratory chain.21 Our study shows upregulated mTOR activity that may be due to elevated leucine, and possibly other significantly upregulated BCAA metabolites. Also, glutamine, increased in SHR hearts, could activate mTOR as shown in neonatal cardiomyocytes.34 A previous study evaluated the role of mTOR in the development of cardiac hypertrophy in SHR.35 Consistent with our observation, mTOR activity was increased by 10 weeks of age. However, in the earlier study it returned to normal by 17 weeks while we still observe mTOR activation at 5 months. Similar to our study, cardiac hypertrophy was present by 17, but not 10, weeks of age. Interestingly, treatment of rats with rapamycin from 13 to 16 weeks reduced cardiac hypertrophy despite increased blood pressure suggesting a role for increased mTOR activity in the development of LVH in SHR.35
Typically, decreased glucose oxidation in the presence of increased glucose uptake characterizes the end stage of disease development,32 as we observed in our earlier acute mouse model of transverse aortic constriction induced LVH and HF.6, 7 However, in the current study we observed that glucose oxidation together with glucose uptake was increased in young SHR hearts (Figure 3). This indicates that during early stages of hypertension and development of LVH glucose uptake and oxidation are coupled, and no G6P accrues in the myocardium, unlike in the transverse aortic constriction model.7 Thus, mTOR may be activated through different mechanisms depending on the model used (SHR versus transverse aortic constriction), the time over which LVH develops (chronic versus acute), or the stage analyzed during LVH and HF progression.
Although SHR represent a good model to assess metabolic changes during the early development of hypertension it needs to be considered that genetic mutations/deletions in the long chain fatty acid transporter CD36 have been reported in some strains of SHR.36 In the Charles River/North American SHR strain (SHR/NCrl) different mRNA transcripts of CD36 were detected, but FAT/CD36 protein expression was reduced by only 26% and palmitate uptake was normal in SHR hearts.37 The Charles River SHR strain we used lacks CD36 (data not shown) which could affect cardiac metabolism by limiting long chain fatty acid uptake as observed in CD36 knockout mice.38 However, earlier studies observing normal fatty acid uptake in hearts of 9 month‐old fed SHR and increased fasting fatty acid uptake rates in hearts of SHR at 8 and 13 months of age12, 13 suggest that CD36 is not the only determinant of fatty acid uptake in SHR hearts. Furthermore, our metabolomics analysis does not support a limited supply of long chain fatty acids to SHR hearts.
Ischemia, endothelial dysfunction, or cardiovascular disease may significantly compromise blood flow in SHR hearts and thus affect cardiac metabolism. However, when Hernandez et al12 evaluated myocardial blood flow in SHR from 8 to 21 months of age, the authors observed significantly increased myocardial blood flow in SHR compared with WKY independent of age. They concluded that elevated myocardial blood flow in SHR may reflect increased myocardial oxygen consumption and decreased capillary density in the hypertrophied heart. In a mouse model of transverse aortic constriction, previously evaluated in our laboratory, we found that changes in myocardial metabolism may have preceded and triggered impaired blood flow in transverse aortic constriction hearts.39 The temporal and causal relationship between myocardial metabolism and impaired blood flow in SHR during the early stages of hypertension will need to be evaluated in the future.
We used only male SHR in our study. Sex has a major impact on the development of LVH in response to pressure overload40 and maladaptive cardiac remodeling occurs more likely in men than in women.41 One study using PET and echocardiography in hypertension‐induced LVH in humans established abnormalities in myocardial fatty acid metabolism as an independent predictor of increased left ventricular mass and dysfunction, a relationship that was stronger in men even after correcting for larger body mass.42 A recent multimodality imaging study using CMR and echocardiography observed that men with aortic stenosis developed LVH with more detrimental abnormalities in left ventricular structure and function than women.43 The mechanisms responsible for the sex differences remain unclear. In the future, we will also evaluate female SHR to establish sex‐specific changes in cardiac metabolism, structure and function.
Conclusion
Our study shows that increased blood pressure in young male SHR is accompanied by profound myocardial metabolic abnormalities that are present before or concomitant with decreased cardiac function, and before LVH develops. The data suggest that excess metabolites not used in ATP production and increased oxidative stress may trigger inflammation and promote cardiac dysfunction early in response to chronic hypertension. mTOR activation in SHR, possibly because of increased BCAA or other metabolites, may drive increased heart mass. The sequence of events corroborates the concept previously formulated by our6, 7 and other groups44, 45 that early metabolic abnormalities could serve as antecedent indicators for hypertension‐induced LVH, and early targeting of metabolic remodeling may offer a novel approach to prevent contractile dysfunction and structural remodeling in patients with hypertension‐induced LVH. Further studies will be required to establish the sequence and causality of observed metabolic, functional, and structural abnormalities at the earliest stages of hypertension. This will include metabolomics analysis at 1 month of age when isolated perfused SHR hearts already show increased glucose uptake and oxidation but cardiac function is relatively normal. We propose SHR as a model to further investigate these issues, and to test metabolic interventions to prevent LVH and HF.
Sources of Funding
This work was supported by grants from the National Institutes of Health, R01 HL123627 (to Kundu), R01 EB 001763 (to Epstein), R01 HL128189 (to Carey) and R01 HL61483 (to Taegtmeyer) as well as start‐up funds from the Department of Radiology and Medical Imaging at the University of Virginia (to Kundu).
Disclosures
None.
Supporting information
Data S1. Supplemental Methods.
Table S1. Changes in Cardiac Metabolites in SHR
Acknowledgments
We thank Jeremy Gatesman in the Center for Comparative Medicine for tail‐vein catheterizations during the FDG PET imaging studies and Kevin Aylor in the Division of Endocrinology at the University of Virginia for technical assistance with BCAA, Insulin and FFA assays and Bruce Gaylinn in the Division of Endocrinology at the University of Virginia for G6P data analysis.
(J Am Heart Assoc. 2019;8:e010926 DOI: 10.1161/JAHA.118.010926.)
Contributor Information
Susanna R. Keller, Email: srk4b@virginia.edu.
Bijoy K. Kundu, Email: bkk5a@virginia.edu.
References
- 1. Bhatia RS, Tu JV, Lee DS, Austin PC, Fang J, Haouzi A, Gong Y, Liu PP. Outcome of heart failure with preserved ejection fraction in a population‐based study. N Engl J Med. 2006;355:260–269. [DOI] [PubMed] [Google Scholar]
- 2. De Jong KA, Lopaschuk GD. Complex energy metabolic changes in heart failure with preserved ejection fraction and heart failure with reduced ejection fraction. Can J Cardiol. 2017;33:860–871. [DOI] [PubMed] [Google Scholar]
- 3. Gropler RJ, Beanlands RS, Dilsizian V, Lewandowski ED, Villanueva FS, Ziadi MC. Imaging myocardial metabolic remodeling. J Nucl Med. 2010;51:88S–101S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Osterholt M, Sen S, Dilsizian V, Taegtmeyer H. Targeted metabolic imaging to improve the management of heart disease. JACC Cardiovasc Imaging. 2012;5:214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhong M, Kundu BK. Optimization of a model corrected blood input function from dynamic FDG‐PET images of small animal heart in vivo. IEEE Trans Nucl Sci. 2013;60:3417–3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhong M, Alonso CE, Taegtmeyer H, Kundu BK. Quantitative PET imaging detects early metabolic remodeling in a mouse model of pressure‐overload left ventricular hypertrophy in vivo. J Nucl Med. 2013;54:609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sen S, Kundu BK, Wu HC, Hashmi SS, Guthrie P, Locke LW, Roy RJ, Matherne GP, Berr SS, Terwelp M, Scott B, Carranza S, Frazier OH, Glover DK, Dillmann WH, Gambello MJ, Entman ML, Taegtmeyer H. Glucose regulation of load‐induced mTOR signaling and ER stress in mammalian heart. J Am Heart Assoc. 2013;2:e004796 DOI: 10.1161/JAHA.113.004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kundu BK, Zhong M, Sen S, Davogustto G, Keller SR, Taegtmeyer H. Remodeling of glucose metabolism precedes pressure overload‐induced left ventricular hypertrophy: review of a hypothesis. Cardiology. 2015;130:211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rockman HA, Ross RS, Harris AN, Knowlton KU, Steinhelper ME, Field LJ, Ross J Jr, Chien KR. Segregation of atrial‐specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc Natl Acad Sci USA. 1991;88:8277–8281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM, Kim JB, Schmitt JP, Molkentin JD, Norris RA, Tager AM, Hoffman SR, Markwald RR, Seidman CE, Seidman JG. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non‐myocyte proliferation and requires Tgf‐beta. J Clin Invest. 2010;120:3520–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brooks WW, Shen SS, Conrad CH, Goldstein RH, Bing OH. Transition from compensated hypertrophy to systolic heart failure in the spontaneously hypertensive rat: structure, function, and transcript analysis. Genomics. 2010;95:84–92. [DOI] [PubMed] [Google Scholar]
- 12. Hernandez AM, Huber JS, Murphy ST, Janabi M, Zeng GL, Brennan KM, O'Neil JP, Seo Y, Gullberg GT. Longitudinal evaluation of left ventricular substrate metabolism, perfusion, and dysfunction in the spontaneously hypertensive rat model of hypertrophy using small‐animal PET/CT imaging. J Nucl Med. 2013;54:1938–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huber JS, Hernandez AM, Janabi M, O'Neil JP, Brennan KM, Murphy ST, Seo Y, Gullberg GT. Longitudinal evaluation of myocardial fatty acid and glucose metabolism in fasted and nonfasted spontaneously hypertensive rats using MicroPET/CT. Mol Imaging. 2017;16:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Giannakidis A, Rohmer D, Veress AI, Gullberg GT. Chapter 53: diffusion tensor MRI‐derived myocardial fiber disarray in hypertensive left ventricular hypertrophy: visualization, quantification and the effect on mechanical function In: Shenasa M, Hindricks G, Borggrefe M, Breithardt G, Josephson ME. and Zipe DP, eds. Cardiac Mapping, 4th Edition, Wiley. 2012:574–588. [Google Scholar]
- 15. Tran N, Giannakidis A, Gullberg GT, Seo Y. Quantitative analysis of hypertrophic myocardium using diffusion tensor magnetic resonance imaging. J Med Imaging (Bellingham). 2016;3:046001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Veress AI, Weiss JA, Huesman RH, Reutter BW, Taylor SE, Sitek A, Feng B, Yang Y, Gullberg GT. Measuring regional changes in the diastolic deformation of the left ventricle of SHR rats using microPET technology and hyperelastic warping. Ann Biomed Eng. 2008;36:1104–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Adams MA, Bobik A, Korner PI. Differential development of vascular and cardiac hypertrophy in genetic hypertension. Relation to sympathetic function. Hypertension. 1989;14:191–202. [DOI] [PubMed] [Google Scholar]
- 18. Berr SS, Roy RJ, French BA, Yang Z, Gilson W, Kramer CM, Epstein FH. Black blood gradient echo cine magnetic resonance imaging of the mouse heart. Magn Reson Med. 2005;53:1074–1079. [DOI] [PubMed] [Google Scholar]
- 19. Goodwin GW, Cohen DM, Taegtmeyer H. [5‐3H]glucose overestimates glycolytic flux in isolated working rat heart: role of the pentose phosphate pathway. Am J Physiol Endocrinol Metab. 2001;280:E502–E508. [DOI] [PubMed] [Google Scholar]
- 20. Taegtmeyer H, Hems R, Krebs HA. Utilization of energy‐providing substrates in the isolated working rat heart. Biochem J. 1980;186:701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang Y, Zhou M, Sun H, Wang Y. Branched‐chain amino acid metabolism in heart disease: an epiphenomenon or a real culprit? Cardiovasc Res. 2011;90:220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Bilsen M, van Nieuwenhoven FA, van der Vusse GJ. Metabolic remodelling of the failing heart: beneficial or detrimental? Cardiovasc Res. 2009;81:420–428. [DOI] [PubMed] [Google Scholar]
- 23. Wilson AJ, Wang VY, Sands GB, Young AA, Nash MP, LeGrice IJ. Increased cardiac work provides a link between systemic hypertension and heart failure. Physiol Rep. 2017;5:e13104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pfeffer JM, Pfeffer MA, Fishbein MC, Frohlich ED. Cardiac function and morphology with aging in the spontaneously hypertensive rat. Am J Physiol. 1979;237:H461–H468. [DOI] [PubMed] [Google Scholar]
- 25. Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y, Iwanaga Y, Narazaki M, Matsuda T, Soga T, Kita T, Kimura T, Shioi T. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ Heart Fail. 2010;3:420–430. [DOI] [PubMed] [Google Scholar]
- 26. Dolinsky VW, Morton JS, Oka T, Robillard‐Frayne I, Bagdan M, Lopaschuk GD, Des RC, Walsh K, Davidge ST, Dyck JR. Calorie restriction prevents hypertension and cardiac hypertrophy in the spontaneously hypertensive rat. Hypertension. 2010;56:412–421. [DOI] [PubMed] [Google Scholar]
- 27. Tang Y, Mi C, Liu J, Gao F, Long J. Compromised mitochondrial remodeling in compensatory hypertrophied myocardium of spontaneously hypertensive rat. Cardiovasc Pathol. 2014;23:101–106. [DOI] [PubMed] [Google Scholar]
- 28. Yu YS, Zheng H. Chronic hydrogen‐rich saline treatment reduces oxidative stress and attenuates left ventricular hypertrophy in spontaneous hypertensive rats. Mol Cell Biochem. 2012;365:233–242. [DOI] [PubMed] [Google Scholar]
- 29. Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J, Kapoor K, Koves TR, Stevens R, Ilkayeva OR, Vega RB, Attie AD, Muoio DM, Kelly DP. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ Heart Fail. 2014;7:1022–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sansbury BE, DeMartino AM, Xie Z, Brooks AC, Brainard RE, Watson LJ, DeFilippis AP, Cummins TD, Harbeson MA, Brittian KR, Prabhu SD, Bhatnagar A, Jones SP, Hill BG. Metabolomic analysis of pressure‐overloaded and infarcted mouse hearts. Circ Heart Fail. 2014;7:634–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hunter WG, Kelly JP, McGarrah RW III, Khouri MG, Craig D, Haynes C, Ilkayeva O, Stevens RD, Bain JR, Muehlbauer MJ, Newgard CB, Felker GM, Hernandez AF, Velazquez EJ, Kraus WE, Shah SH. Metabolomic profiling identifies novel circulating biomarkers of mitochondrial dysfunction differentially elevated in heart failure with preserved versus reduced ejection fraction: evidence for shared metabolic impairments in clinical heart failure. J Am Heart Assoc. 2016;5:e003190 DOI: 10.1161/JAHA.115.003190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ikegami R, Shimizu I, Yoshida Y, Minamino T. Metabolomic analysis in heart failure. Circ J. 2017;82:10–16. [DOI] [PubMed] [Google Scholar]
- 33. Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, Jeyaraj D, Youn JY, Ren S, Liu Y, Rau CD, Shah S, Ilkayeva O, Gui WJ, William NS, Wynn RM, Newgard CB, Cai H, Xiao X, Chuang DT, Schulze PC, Lynch C, Jain MK, Wang Y. Catabolic defect of branched‐chain amino acids promotes heart failure. Circulation. 2016;133:2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pascual F, Coleman RA. Fuel availability and fate in cardiac metabolism: a tale of two substrates. Biochim Biophys Acta. 2016;1861:1425–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Soesanto W, Lin HY, Hu E, Lefler S, Litwin SE, Sena S, Abel ED, Symons JD, Jalili T. Mammalian target of rapamycin is a critical regulator of cardiac hypertrophy in spontaneously hypertensive rats. Hypertension. 2009;54:1321–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gotoda T, Iizuka Y, Kato N, Osuga J, Bihoreau MT, Murakami T, Yamori Y, Shimano H, Ishibashi S, Yamada N. Absence of Cd36 mutation in the original spontaneously hypertensive rats with insulin resistance. Nat Genet. 1999;22:226–228. [DOI] [PubMed] [Google Scholar]
- 37. Bonen A, Han XX, Tandon NN, Glatz JF, Lally J, Snook LA, Luiken JJ. FAT/CD36 expression is not ablated in spontaneously hypertensive rats. J Lipid Res. 2009;50:740–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Coburn CT, Knapp FF Jr, Febbraio M, Beets AL, Silverstein RL, Abumrad NA. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J Biol Chem. 2000;275:32523–32529. [DOI] [PubMed] [Google Scholar]
- 39. Zhong M, Mistry M, Dimastromatteo J, Taegtmeyer H, Glover D.K, Kundu BK. PET imaging of myocardial blood flow in the stressed mouse heart in vivo. J Nucl Med. 54:1635. [Google Scholar]
- 40. de Simone G, Devereux RB, Daniels SR, Meyer RA. Gender differences in left ventricular growth. Hypertension. 1995;26:979–983. [DOI] [PubMed] [Google Scholar]
- 41. Petrov G, Dworatzek E, Schulze TM, Dandel M, Kararigas G, Mahmoodzadeh S, Knosalla C, Hetzer R, Regitz‐Zagrosek V. Maladaptive remodeling is associated with impaired survival in women but not in men after aortic valve replacement. JACC Cardiovasc Imaging. 2014;7:1073–1080. [DOI] [PubMed] [Google Scholar]
- 42. de las Fuentes L, Herrero P, Peterson LR, Kelly DP, Gropler RJ, Davila‐Roman VG. Myocardial fatty acid metabolism: independent predictor of left ventricular mass in hypertensive heart disease. Hypertension. 2003;41:83–87. [DOI] [PubMed] [Google Scholar]
- 43. Treibel TA, Kozor R, Fontana M, Torlasco C, Reant P, Badiani S, Espinoza M, Yap J, Diez J, Hughes AD, Lloyd G, Moon JC. Sex dimorphism in the myocardial response to aortic stenosis. JACC Cardiovasc Imaging. 2018;11:962–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Horowitz JD, Chirkov YY, Kennedy JA, Sverdlov AL. Modulation of myocardial metabolism: an emerging therapeutic principle. Curr Opin Cardiol. 2010;25:329–334. [DOI] [PubMed] [Google Scholar]
- 45. Revenco D, Morgan JP. Metabolic modulation and cellular therapy of cardiac dysfunction and failure. J Cell Mol Med. 2009;13:811–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental Methods.
Table S1. Changes in Cardiac Metabolites in SHR