

Abstract
Background
Reduced miR‐133a was previously found to be associated with thoracic aortic (TA) dilation, as seen in aneurysm disease. Because wall tension increases with vessel diameter (Law of Laplace), this study tested the hypothesis that elevated tension led to the reduction of miR‐133a in the TA.
Methods and Results
Elevated tension (1.5 g; 150 mm Hg) applied to murine TA ex vivo reduced miR‐133a tissue abundance compared with TA held at normotension (0.7 g; 70 mm Hg). Cellular miR‐133a levels were reduced with biaxial stretch of isolated murine TA fibroblasts, whereas smooth muscle cells were not affected. Mechanisms contributing to the loss of miR‐133a abundance were further investigated in TA fibroblasts. Biaxial stretch did not reduce primary miR‐133a transcription and had no effect on the expression/abundance of 3 microRNA‐specific exoribonucleases. Remarkably, biaxial stretch increased exosome secretion, and exosomes isolated from TA fibroblasts contained more miR‐133a. Inhibition of exosome secretion prevented the biaxial stretch‐induced reduction of miR‐133a. Subsequently, 2 in vivo models of hypertension were used to determine the effect of elevated wall tension on miR‐133a abundance in the TA: wild‐type mice with osmotic pump–mediated angiotensin II infusion and angiotensin II–independent spontaneously hypertensive mice. Interestingly, the abundance of miR‐133a was decreased in TA tissue and increased in the plasma in both models of hypertension compared with a normotensive control group. Furthermore, miR‐133a was elevated in the plasma of hypertensive human subjects, compared with normotensive patients.
Conclusions
Taken together, these results identified exosome secretion as a tension‐sensitive mechanism by which miR‐133a abundance was reduced in TA fibroblasts.
Keywords: exosome secretion, fibroblasts, hypertension, microRNA, thoracic aorta, vascular biology, wall tension
Subject Categories: Basic Science Research, Vascular Biology
Clinical Perspective
What Is New?
Tension applied to thoracic aortic rings resulted in the acute reduction in miR‐133a.
Mechanical tension in the form of biaxial cyclic stretch applied to isolated primary aortic fibroblasts and smooth muscle cells revealed that fibroblasts preferentially responded to mechanical tension, resulting in the acute reduction of miR‐133a.
Tension‐dependent loss of miR‐133a in fibroblasts was mediated through exosome secretion.
Elevated blood pressure (increased wall tension) was sufficient to induce the loss of miR‐133a from the descending thoracic aorta (mouse models) and was associated with increased plasma levels of miR‐133a (mouse models and human plasma).
What Are the Clinical Implications?
Increased vascular wall tension (hypertension) is a stimulus driving the loss of miR‐133a in the thoracic aorta.
These results identified a specific tension‐sensitive mechanism by which miR‐133a was reduced in a cell type that plays a key role in adverse vascular remodeling (thoracic aortic fibroblasts).
Introduction
Alterations in microRNA abundance have been associated with multiple cardiovascular diseases, and although much effort has been directed toward understanding their role in modulating key cellular targets, less is known about how microRNA abundance is regulated within the cell. Previously, this laboratory identified that several microRNAs, including miR‐1, miR‐21, miR‐29a, miR‐486, miR‐720, and miR‐133a, were reduced in ascending aortic tissue from patients with thoracic aortic aneurysm (TAA).1 Many of these microRNAs, including miR‐133a, displayed an inverse linear correlation with aortic diameter, such that, as diameter increased, the abundance of miR‐133a was reduced; however, the underlying mechanisms responsible for this observation remained to be determined. On the basis of the fundamentals of the Law of Laplace, we know that vessel wall tension is dependent on the relationship between pressure and diameter (wall tension equals pressure multiplied by diameter). Accordingly, during aneurysm formation, wall tension increases as the aorta dilates, and this may play a role in determining microRNA levels in the thoracic aorta (TA). Therefore, this study sought to determine a mechanism responsible for the reduction of miR‐133a and tested the hypothesis that elevated wall tension induces the loss of miR‐133a from the TA.
The importance of miR‐133a in the regulation of extracellular matrix (ECM) remodeling in cardiovascular tissue is becoming increasingly recognized. Care et al demonstrated, in a murine model of cardiac hypertrophy, that miR‐133a was reduced in the left ventricular myocardium after transverse aortic arch constriction and that in vivo knockdown of miR‐133a (using anti‐miR‐133a) alone was sufficient to induce cardiac hypertrophy.2 Similarly, Torella et al demonstrated miR‐133a was reduced in the carotid artery after balloon distension injury in a rat model.3 More important, they demonstrated that systemic overexpression of miR‐133a by adenovirus attenuated postinjury remodeling, in contrast to miR‐133a knockdown (using an anti‐miR‐133a oligonucleotide), which enhanced postinjury remodeling. Data from these studies emphasize the key role that miR‐133a plays in cardiovascular remodeling and suggest that loss of miR‐133a–mediated translational control likely contributes to the pathologic changes that were observed in our studies during aneurysm formation.
Furthermore, multiple validated targets of miR‐133a have been demonstrated to play key roles in vascular pathologic characteristics. First, miR‐133a targets transforming growth factor (TGF)‐β,4 which is elevated in TAA tissue, induces connective tissue growth factor expression, alters fibroblast phenotype, and causes apoptosis in smooth muscle cells (SMCs). Second, miR‐133a targets TGF‐β receptor II,4 which is elevated in TAA tissue, and with the concomitant decline in TGF‐β receptor‐I, shifts TGF‐β ligand signaling toward the Activin Receptor‐Like Kinase 1 (ALK‐1), pathway activating the sma‐related + Mothers Against Decapentaplegic Homolog 1, 5, or 8 (SMAD 1/5/8) pathway, which induces a profile of gene expression resulting in matrix degradation.5 Third, miR‐133a targets connective tissue growth factor,6 which is elevated in TAA tissue and also contributes to changes in fibroblast phenotype. Fourth, miR‐133a targets collagen 1a1, a major component of the aortic ECM.7 Finally, miR‐133a targets membrane type‐1 matrix metalloproteinase (MMP),8 which is elevated in TAA tissue, degrades components of the ECM, activates other MMPs, such as MMP‐2, and directly releases ECM‐bound cytokines, including TGF‐β. In addition to the above listed direct targets of miR‐133a, it is apparent that miR‐133a may have the capacity to regulate multiple pathways involved in complex pathologic features, increasing the significance of the loss of miR‐133a observed in the development of TAA. Accordingly, understanding the mechanisms that regulate miR‐133a cellular abundance is essential and may provide insight into potential therapeutic targets.
The current report identifies increased wall tension (hypertension) as a stimulus driving the loss of miR‐133a in TA tissue. The effects of tension were examined on the transcription of miR‐133a, the levels of cellular exoribonucleases, and the cellular export of miR‐133a via exosomes. The unique findings of this study propose a novel mechanism by which increased mechanical tension reduces miR‐133a abundance via exosome secretion from aortic fibroblasts, a cell type believed to play a major role in managing pathologic remodeling.9, 10
Methods
The data, analytic methods, and study materials will be made available to other researchers on request for purposes of reproducing the results or replicating the procedures.
Ex Vivo TA Tension Application
Animal care and surgical procedures were approved by the Medical University of South Carolina Institutional Animal Care and Use Committee (AR3380) and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Wild‐type C57BL/6 mice (10–16 weeks of age; Envigo) underwent thoracotomy, and the descending TA was harvested (n=7; 4 males/3 females). Endothelial‐intact aortic tissue segments were cut transversely into rings of ≈3 mm in length, which were suspended on parallel wires, and bathed in an oxygenated physiologic salt solution (Krebs‐Henseleit solution) in a tissue myograph. Aortic segments were maintained at an experimentally derived optimal tension (normotension; 0.7 g, ≈70 mm Hg equivalent) or elevated tension (1.5 g, ≈150 mm Hg equivalent) for 3 hours using methods previously described.11, 12 Aortic segments were also held at 0.7 g and then treated with or without 100 nm angiotensin II (AngII; A9525; Sigma) and allowed to develop tension by contracting against immobilized parallel wires for 3 hours; the peak tension generated was recorded (n=8; 4 males/4 females).
Cell Culture
The descending TA from wild‐type C57BL/6 mice (10–16 weeks of age; Envigo) was extracted, and primary fibroblast or SMC cultures were established as described previously (n=8; 4 males/4 females).13, 14 The isolated fibroblasts were maintained in a fibroblast‐specific growth medium (fibroblast growth media 2 with added supplemental pack containing 2% fetal calf serum; C‐23020; PromoCell, Heidelberg, Germany) with an additional 10% fetal bovine serum (FBS; Gibco; catalog No. 1600) added. The isolated SMCs were maintained in SMC‐specific growth medium (SMC Growth Medium 2 with added supplemental pack containing 5% fetal calf serum; C‐22062; PromoCell). All primary cultures were maintained at 37°C in a humidified 5% CO2/95% air atmosphere. Primary fibroblasts and SMCs were used between passages 2 and 10.
Primary Cell Biaxial Cyclic Tension Application and AngII Stimulation
Fibroblasts or SMCs were seeded at a density of 5000 cells per cm2 onto an amino‐coated Bioflex‐6 well plate (BF‐3001A; Flexcell International Corporation, Burlington, NC), and allowed to adhere overnight. Culture medium was then replaced with fresh complete medium containing exosome‐depleted FBS (10%; System Biosciences) with or without AngII (100 nmol/L; A9525; Sigma). To inhibit exosome secretion, fibroblasts were pretreated with an inhibitor of neutral sphingomyelinase, GW4869 (20 nmol/L, 24 hours; D1692; Sigma‐Aldrich, St Louis, MO). After the 24‐hour treatment, cells were rinsed with PBS and the appropriate culture medium was replaced. Culture plates were then held static (control) or subjected to 12% biaxial cyclic stretch, at a rate of 1 Hz, mimicking a myocardial‐derived amplitude and waveform in a Flexcell culture system (FX5000; Flexcell International Corporation).
Determination of mRNA Expression and microRNA Abundance by Quantitative Polymerase Chain Reaction
Total RNA was extracted using TRIzol Reagent (Thermo Fisher Scientific; catalog No. 15596026) and quantified by NanoDrop 2000 (Thermo Fisher Scientific). For cellular (fibroblasts or SMCs) and tissue (TA) microRNA quantitation, single‐stranded cDNA was synthesized from 100 ng total RNA. Mature miR‐133a levels were standardized to total RNA by methods previously described.15 Exosomal microRNA was quantified from precipitated exosomes (ExoQuick‐TC; System Biosciences, Inc; catalog No. EXOTC50A‐1; from 5 mL of cell medium conditioned for 18 hours). Circulating microRNA quantities were similarly measured from total RNA isolated from 250 μL of serum. MicroRNA cDNA synthesis and polymerase chain reaction quantitation were performed using the miR‐133a TaqMan microRNA Assay (Thermo Fisher; catalog No. 002246‐4427975), according to the manufacturer's instructions. Primary microRNA expression was determined by reverse transcription of 1 μg total RNA using the High Capacity RNA‐to‐cDNA kit (Thermo Fisher Scientific; catalog No. 4392339), followed by quantitation using the ThermoFisher primary‐microRNA analysis kit (primary miR‐133a‐1: Mm03306281_pri, and primary miR‐133a‐2: Mm03307401_pri; catalog No. 4427012). For ribonuclease gene expression analysis, total RNA (1 μg) was reverse transcribed with the iScript cDNA Synthesis Kit (170‐8891; Bio‐Rad, Hercules, CA). Each cDNA was amplified with gene‐specific TaqMan expression assays (5′‐3′ Exoribonuclease (XRN‐1): Mm00496326_m1, 5′‐3′ Exoribonuclease (XRN‐2): Mm00457212_m1, Exosome Component 4; 3′‐5′ Exoribonuclease (EXOSC4): Mm00615045_g1, and GAPDH: Mm99999915_g1; Applied Biosystems). Quantitative polymerase chain reactions were performed on a Bio‐Rad CFX96 Real‐Time System. All samples were tested in duplicate and averaged. The relative expression of each mRNA and primary microRNA was calculated and normalized to GAPDH (used as an internal control), using the comparative cycle threshold (CT) method. Relative expression intensity values were calculated as 2−ΔCT, in which ΔCT are CT values normalized to the reference control.
Immunoblot Analysis
Relative abundance levels of XRN1, XRN2, ExoSC4, and β‐actin were determined by immunoblotting. Briefly, 20 μg of each aortic fibroblast homogenate was fractionated on a 4% to 12% bis‐tris gradient gel. The proteins were transferred to nitrocellulose membranes (0.45 μm; Bio‐Rad) and incubated in 5% nonfat dry milk containing antisera specific for XRN1 (1 μg/mL; Thermo Fisher Scientific; PA5‐41888), XRN2 (1:1000; Thermo Fisher Scientific; PA5‐38789), ExoSC4 (5 μg/mL; Thermo Fisher Scientific; PA5‐41890), or β‐actin (1:10 000; Biovision Inc; 3662‐100). A secondary peroxidase‐conjugated antibody (primary antibody, species specific) was applied (1:5000; 5% nonfat dry milk), and signals were detected with a chemiluminescent substrate (Western Lighting Chemiluminescence Reagent Plus; PerkinElmer, San Jose, CA) and recorded on film. Band intensity was quantified using Gel‐Pro Analyzer, version 3.1.14 (Media Cybernetics, Silver Spring, MD).
Exosome Precipitation and Quantitation
Fibroblasts were maintained in growth media containing exosome‐depleted FBS (10%) and exposed to 12% biaxial cyclic tension, as before. After 18 hours, the culture medium was collected and centrifuged at 3000g for 15 minutes to remove cells and cell debris. Supernatant (5 mL) was transferred to a separate tube and exosomes were precipitated, as described above. Relative exosome abundance was compared using acetylcholinesterase activity as a surrogate for exosome number, as previously described.16 The incubation was performed at 37°C for 30 minutes, and the change in absorbance was measured at 412 nm on a Spectramax M3 (Molecular Devices).
Murine Models of Hypertension
Two distinct murine models of hypertension were used. First, a well‐established model of AngII infusion by osmotic minipump (Alzet; model 2004, providing continuous infusion for 28 days; 1.46 mg/kg per day) was used; and second, a commercially available, spontaneously hypertensive mouse strain (BPH/2J (BPH2); Jackson Laboratories; C57BL/6 background; catalog No. 003005), in which elevated blood pressure can be measured as early as 5 weeks of age, while peaking at 21 weeks, was used. The corresponding normotensive strain (BPN/3J (BPN3); Jackson Laboratories; C57BL/6 background; catalog No. 003004) was used as a control for exosome measurements and blood pressure determination using a noninvasive tail‐cuff system (CODA; Kent Scientific Corporation, Torrington, CT).
Human Subjects
Informed consent was obtained for all patients before blood/plasma collection, and analysis of patient plasma was approved by each respective Institutional Review Board of the collection centers involved. The inclusion/exclusion criteria for all subjects were previously described.17 Control patients fulfilled the inclusion criteria, and they did not have a medical history of hypertension. Hypertensive patients fulfilled the inclusion criteria, they had a documented history of hypertension and left ventricular hypertrophy in their electronic medical record, and all were receiving antihypertensive medications at the time of blood/plasma collection. No patient included in this study had evidence of heart failure, as specified by the criteria defined by the European Society of Cardiology and the Heart Failure Society of America.18, 19
Statistical Analysis
Statistical tests were performed using STATA (Intercooled STATA v8.2, College Station, TX) and SAS Statistics. The sample sizes for all experiments performed in this project were calculated by power analysis using SigmaPlot, version 14. For ex vivo studies, sample sizes were based on initial analysis comparing the primary readouts between experimental groups (aortic tissue miR‐133a levels), and power calculations using a t test model were completed assuming a 69.1% difference in means between groups with a pooled SD (across both groups) of 28.3%. To provide hypothesis testing at a desired power of 0.95 with an α level of 0.05, the sample sizes for tissue analyses were determined to be a minimum of 6 samples per group. For in vitro studies, sample sizes were based on initial analysis comparing the primary readouts between experimental groups (fibroblast miR‐133a levels), and power calculations using a t test model were completed assuming a 78.9% difference in means between groups with a pooled SD of 28.2%. To provide hypothesis testing at a desired power of 0.95 with an α level of 0.05, the sample sizes for cell culture analyses were determined to be a minimum of 5 samples per group. For in vivo studies, sample sizes were based on initial analysis comparing the primary readouts between experimental groups (tissue miR‐133a levels), and power calculations using an ANOVA model were completed assuming a 25.5% minimum detectable difference in means with a pooled SD (across all 3 groups) of 7.4%. To provide hypothesis testing at a desired power of 0.95 with an α level of 0.05, the sample sizes for tissue analyses were determined to be a minimum of 5 samples per group. All data were assessed for normality using the Shapiro‐Wilk test. Data sets in Figures 1, 2, 3, 4 through 5 were subjected to 2‐sample t tests (unpaired, 2 tailed). Data sets in Figure 6 were subjected to a 1‐way ANOVA, followed by pairwise comparison of means by the Ryan/Einot‐Gabriel/Welsch method. Data for Figure 6B and 6C were plotted and analyzed as log of miR‐133a expression values using methods described by Schmittgen and Livak.20 Data sets in Figure 7 were subjected to 2‐sample t tests (unpaired, 2 tailed). Data in Table 1 are presented as mean and SD. In Table 2, a Fisher's exact test was performed on sex and ethnicities. Data were expressed as fold change from control values, unless otherwise stated in the figure legend. Data are represented as mean±SEM in the text. In the figures, data are represented in dot plots with the mean and SEM shown next to each group. P<0.05 was considered to be statistically significant.
Figure 1.
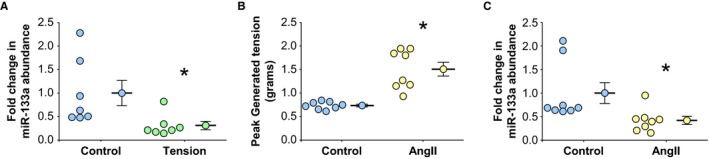
Effects of ex vivo mechanical tension on miR‐133a levels in thoracic aortic rings. Descending aortas from wild‐type C57BL/6 mice were transversely cut into 3‐mm rings and suspended in a tissue myograph on parallel wires in oxygenated Krebs‐Henseleit solution. A, miR‐133a abundance in aortic rings held at 0.7 g (normotension; Control, n=7) or with 1.5 g applied tension (Tension, n=7) for 3 hours. B, Peak‐developed tension (grams) in aortic rings held at normotension (0.7 g) in the absence (Control, n=8) or presence of 100 nmol/L angiotensin II (AngII, n=8) for 3 hours. C, Effect of developed tension on miR‐133a abundance in aortic rings held at normotension (0.7 g) in the absence (Control, n=8) or presence of 100 nmol/L AngII (AngII, n=8). A through C, Data are represented in dot plots with the mean and SEM shown next to each group. Comparisons were made using a 2‐sample t test (unpaired, 2 tailed). *P<0.05 vs control.
Figure 2.

Effects of mechanical tension and angiotensin II (AngII) on miR‐133a levels in isolated thoracic aortic fibroblasts and smooth muscle cells. Aortic fibroblasts and smooth muscle cells were isolated from the descending aortas from C57BL/6 mice. Each data point represents an independently isolated cell line. A, miR‐133a abundance was measured in aortic fibroblast lines after exposure to 3 hours of 12% biaxial cyclic stretch (Tension, n=8) or held static (Control, n=8). B, miR‐133a abundance was measured in aortic smooth muscle cell (SMC) lines after exposure to 3 hours of 12% biaxial cyclic stretch (Tension, n=7) or held static (Control, n=7). C, miR‐133a abundance was measured in aortic fibroblasts lines treated without (Control, n=8) or with 100 nmol/L AngII (AngII, n=8) for 3 hours. D, miR‐133a abundance was measured in aortic SMC lines treated without (Control, n=7) or with 100 nmol/L angiotensin II (AngII, n=7) for 3 hours. A through D, Data are represented in dot plots with the mean and SEM shown next to each group. Comparisons were made using a 2‐sample t test (unpaired, 2 tailed). *P<0.05 vs control.
Figure 3.
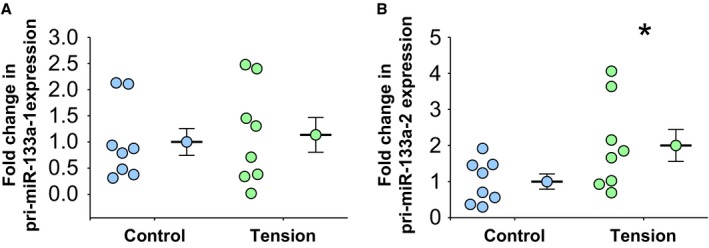
Mechanical tension and transcription of primary miR‐133a‐1 and primary miR‐133a‐2. Total RNA was isolated from each individual cell line, and primary miR‐133a levels were determined by real‐time polymerase chain reaction. A, Primary miR‐133a‐1 expression levels in aortic fibroblasts exposed to 12% biaxial cyclic stretch (Tension, n=8) or held static (Control, n=8) for 3 hours. B, Primary miR‐133a‐2 expression levels in aortic fibroblasts exposed to 12% biaxial cyclic stretch (Tension, n=8) or held static (Control, n=8) for 3 hours. A and B, Data are represented in dot plots with the mean and SEM shown next to each group. Comparisons were made using a 2‐sample t test (unpaired, 2 tailed). *P<0.05 vs control.
Figure 4.

Mechanical tension and microRNA‐specific ribonuclease mRNA expression and protein abundance. Total RNA or protein was isolated from each independent cell line and examined by real‐time polymerase chain reaction or Western blotting, respectively. A, XRN1 mRNA expression levels in aortic fibroblasts exposed to 12% biaxial cyclic stretch (Tension, n=9) or held static (Control, n=9) for 3 hours. B, XRN1 protein abundance and representative immunoblot in aortic fibroblasts exposed to 12% biaxial cyclic stretch (Tension, n=5) or held static (Control, n=5) for 3 hours. C, XRN2 mRNA expression levels in aortic fibroblasts exposed to 12% biaxial cyclic stretch (Tension, n=9) or held static (Control, n=9) for 3 hours. D, XRN2 protein abundance and representative immunoblot in aortic fibroblasts exposed to 12% biaxial cyclic stretch (Tension, n=5) or held static (Control, n=5) for 3 hours. E, ExoSC4 mRNA expression levels in aortic fibroblasts exposed to 12% biaxial cyclic stretch (Tension, n=9) or held static (Control, n=9) for 3 hours. F, ExoSC4 protein abundance and representative immunoblot in aortic fibroblasts exposed to 12% biaxial cyclic stretch (Tension, n=5) or held static (Control, n=5) for 3 hours. A through F, Data are represented in dot plots with the mean and SEM shown next to each group. Comparisons were made using a 2‐sample t test (unpaired, 2 tailed). No significant differences were observed vs control.
Figure 5.

Mechanical tension and exosome secretion of miR‐133a. Exosomes were precipitated from conditioned culture media from each fibroblast and smooth muscle cell (SMC) line. A, Acetylcholinesterase activity was quantitated as a measure of exosome number from each fibroblast line exposed to 12% biaxial cyclic stretch (Tension, n=8) or held static (Control, n=8) for 18 hours. B, miR‐133a abundance in exosomes precipitated from the conditioned media of each fibroblast line exposed to 12% biaxial cyclic stretch (Tension, n=7) or held static (Control, n=7) for 18 hours. C, miR‐133a abundance in each aortic fibroblast line after exposure to 3 hours of 12% biaxial cyclic stretch in the presence of 20 μmol/L GW4869 (Tension+GW4869, n=8) or held static (Control, n=8). D, Acetylcholinesterase activity was quantitated as a measure of exosome number from each SMC line exposed to 12% biaxial cyclic stretch (Tension, n=6) or held static (Control, n=6) for 18 hours. E, miR‐133a abundance in exosomes precipitated from the conditioned media of each SMC line exposed to 12% biaxial cyclic stretch (Tension, n=6) or held static (Control, n=6) for 18 hours. A through E, Data are represented in dot plots with the mean and SEM shown next to each group. Comparisons were made using a 2‐sample t test (unpaired, 2 tailed). *P<0.05 vs control.
Figure 6.

Mean arterial blood pressure and miR‐133a levels in thoracic aortic tissue and plasma from 2 murine models of hypertension. A, Mean arterial blood pressure assessed by noninvasive tail cuff, in control mice (Control, n=12), angiotensin II (AngII)–induced hypertensive mice (AngII, n=12), and spontaneously hypertensive mice (BPH2, n=12). B, miR‐133a abundance in thoracic aortic tissue from control mice (Control, n=12), Ang II‐induced hypertensive mice (AngII, n=12), and spontaneously hypertensive mice (BPH2, n=16). C, Plasma miR‐133a abundance in control mice (Control, n=6), AngII‐induced hypertensive mice (AngII, n=6), and spontaneously hypertensive mice (BPH2, n=8). A through C, Data are represented in dot plots with the mean and SEM shown next to each group. Comparisons were made using a 1‐way ANOVA, followed by pairwise comparison of means by the Ryan/Einot‐Gabriel/Welsch test. *P<0.05 vs control; # P<0.05 vs AngII.
Figure 7.
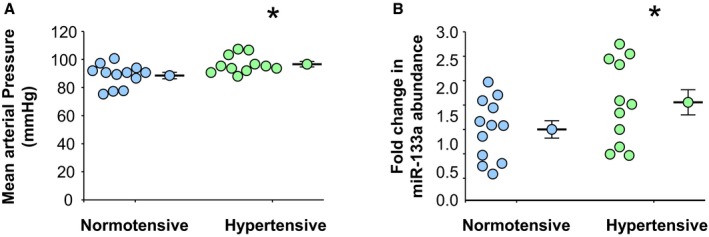
Mean arterial blood pressures and plasma levels of miR‐133a in clinical specimens. A, Patient mean arterial blood pressures assessed at time of plasma collection in normotensive (n=12) and hypertensive (n=11) patients. B, Plasma miR‐133a abundance in normotensive (n=12) and hypertensive (n=11) patients. A and B, Data are represented in dot plots with the mean and SEM shown next to each group. Comparisons were made using a 2‐sample t test (unpaired, 2 tailed). *P<0.05 vs control.
Table 1.
Murine Blood Pressure
| Group | Blood Pressure, mm Hg | MAP, mm Hg | ||||
|---|---|---|---|---|---|---|
| Systolic | Diastolic | |||||
| Mean | SD | Mean | SD | Mean | SD | |
| Control (n=12) | 148 | 9.76 | 108 | 9.08 | 121 | 8.97 |
| AngII (n=12) | 199 | 14.05 | 155 | 11.36 | 170 | 12.11 |
| BPH2 (n=12) | 187 | 9.38 | 146 | 11.06 | 160 | 10.30 |
Murine systolic, diastolic, and calculated MAP blood pressure levels were assessed by noninvasive tail cuff, in 3 groups. Data are presented as mean and SD in the 3 groups of mice used in these studies. AngII indicates angiotensin II–induced hypertensive mice; BPH2, spontaneously hypertensive mice; Control, control mice; MAP, mean arterial pressure.
Table 2.
Patent Demographics
| Demographics | Normotensive (n=12) | Hypertensive (n=11) | P Value |
|---|---|---|---|
| Male sex, % | 25 | 27 | 1.000 |
| Black race, % | 8 | 27 | 1.000 |
| Age, y | 63±4 | 65±3 | 0.193 |
| Heart rate, bpm | 68±9 | 70±12 | 0.654 |
| Body surface area, m2 | 1.76±0.26 | 2.03±0.18 | 0.009 |
| Walk in 6 min, m | 396.8±51.4 | 386.6±45.3 | 0.620 |
| Systolic blood pressure, mm Hg | 122±9 | 131±9 | 0.026 |
| Diastolic blood pressure, mm Hg | 72±8 | 80±6 | 0.014 |
| Mean arterial pressure, mm Hg | 89±8 | 97±6 | 0.014 |
Demograpical information of patients included in these studies. Data are presented as mean±SD, unless otherwise indicated. A Fisher's exact test was performed on sex and ethnicities, and P values were determined using the method of summing small P values. For all other categories, pairwise comparisons were made using 2‐sample t tests. All P values are displayed. Bpm indicates beats per minute.
Results
Increased Mechanical Tension Reduces miR‐133a in TA Tissue
To determine the effects of elevated applied tension on miR‐133a levels, intact murine TA segments (rings) harvested from wild‐type, normotensive mice were hung on parallel wires in an ex vivo tissue myograph. Control TA segments were held at a previously determined level of tension (0.7 g) that approximates in vivo normotension (mean arterial pressure [MAP], ≈70 mm Hg).11, 12 To simulate elevated wall tension (hypertension), an increased amount of tension (1.5 g, MAP, ≈150 mm Hg) was applied directly to the aortic segments for 3 hours. The abundance of miR‐133a was reduced in response to increased applied tension compared with control segments (0.31±0.08‐ versus 1.0±0.25‐fold expression; P<0.05 versus control; n=7) (Figure 1A).
Similarly, to examine the role of developed tension on miR‐133a abundance, TA rings from wild‐type, normotensive mice were exposed to 100 nmol/L AngII for 3 hours in an ex vivo tissue myograph. AngII‐dependent vessel contraction increased developed wall tension compared with rings treated with vehicle alone (1.51±0.15 versus 0.73±0.03 g tension; P<0.05 versus control; n=8) (Figure 1B). In response, the AngII‐induced contraction resulted in a reduction in miR‐133a abundance compared with noncontracting rings treated with sterile PBS (vehicle) alone (0.42±0.09‐ versus 1.0±0.22‐fold expression; P<0.05 versus control; n=8) (Figure 1C).
Increased Mechanical Tension Results in Reduced miR‐133a Abundance in Isolated TA Fibroblasts
When isolated aortic fibroblasts and SMCs were exposed to mechanical tension in the form of biaxial cyclic stretch, miR‐133a abundance was reduced in stretched fibroblasts compared with static fibroblast controls (0.21±0.02‐ versus 1.0±0.27‐fold expression; P<0.05 versus control; n=8) (Figure 2A). Interestingly, no change in miR‐133a abundance was observed in stretched SMCs (0.98±0.43‐ versus 1.0±0.12‐fold expression; P=0.46 versus control; n=7) (Figure 2B). These findings suggested that the reduction in mature miR‐133a abundance observed in aortic tissue segments exposed to elevated tension may have been attributable to a reduction of miR‐133a in the aortic fibroblasts.
To confirm that the loss of miR‐133a was independent of AngII receptor signaling, isolated primary aortic fibroblasts and SMCs were exposed to 100 nmol/L AngII for 3 hours. No change in miR‐133a was observed in fibroblasts (1.20±0.52‐ versus 1.0±0.27‐fold expression; P=0.62 versus control; n=8) (Figure 2C) or SMCs (1.14±0.26‐ versus 1.0±0.11‐fold expression; P=0.69 versus control; n=7) (Figure 2D). These findings confirmed that the alterations in miR‐133a abundance observed in the aortic ring segments was likely attributable to the direct effects of mechanical tension on the fibroblasts within the aortic wall rather than AngII‐mediated receptor signaling.
Mechanical Tension Does Not Reduce Transcription of miR‐133a in TA Fibroblasts
To determine the mechanism of tension‐induced loss of miR‐133a, primary miR‐133a transcript levels (primary miR‐133a‐1 and primary miR‐133a‐2) were measured in isolated aortic fibroblasts after biaxial cyclic stretch. The expression of primary miR‐133a‐1 did not change in response to mechanical stretch (1.14±0.33‐ versus 1.0±0.26‐fold expression; P=0.62 versus control; n=8) (Figure 3A), whereas the expression of primary miR‐133a‐2 increased in response to stretch (2.0±0.44‐ versus 1.0±0.21‐fold expression; P<0.05 versus control; n=8) (Figure 3B). The results suggested that the tension‐induced loss of miR‐133a occurred posttranscriptionally.
Increased Mechanical Tension Does Not Alter the Expression or Abundance of MicroRNA‐Specific Exoribonucleases in TA Fibroblasts
Although mature microRNAs are not likely substrates for most endoribonucleases,21, 22 they do possess unprotected 5′ and 3′ ends, making them susceptible to degradation by specific exoribonucleases.23, 24 Three exoribonucleases that are capable of degrading microRNAs have been identified: XRN‐1,25 XRN‐2,25, 26 and ExoSC4.23 To examine whether mechanical tension induced these exoribonucleases, mRNA expression and protein levels were measured in isolated aortic fibroblasts after application of biaxial cyclic stretch. Neither the mRNA expression nor protein abundance of any of these exoribonucleases was changed compared with static fibroblasts: XRN‐1 mRNA: 1.44±0.29‐ versus 1.0±0.47‐fold expression (n=9, P=0.197) (Figure 4A); XRN‐1 protein: 0.86±0.14‐ versus 1.0±0.17‐fold abundance (n=9, P=0.555) (Figure 4B); XRN‐2 mRNA: 0.82±0.08‐ versus 1.0±0.42‐fold expression (n=9, P=0.286) (Figure 4C); XRN‐2 protein: 0.93±0.29‐ versus 1.0±0.18‐fold abundance (n=5, P=0.851) (Figure 4D); ExoSC4 mRNA: 0.70±0.07‐ versus 1.0±0.20‐fold expression (n=9, P=0.166) (Figure 4E); and ExoSC4 protein: 1.0±0.09‐ versus 1.0±0.10‐fold abundance (n=5, P=0.992) (Figure 4F). These findings suggested that the tension‐induced loss of miR‐133a in fibroblasts was not mediated by the rapid degradation of mature miR‐133a catalyzed by exoribonuclease activity.
Mechanical Tension Induces Exosome Secretion of miR‐133a
It has been well described that many microRNAs are exported/secreted from cells in exosomes.27 Exosomes are 30‐ to 100‐nm‐diameter endosomal vesicles that are packaged into multivesicular bodies and secreted on fusion with the plasma membrane. To determine the effects of stretch on exosome secretion, TA fibroblasts were grown on flexible membranes and either held static or exposed to biaxial cyclic stretch in medium containing exosome‐depleted FBS. At the end of 18 hours, the medium was collected and the newly secreted exosomes were precipitated. Acetylcholinesterase is enriched in the lipid bilayer membrane of exosomes, allowing the measurement of acetylcholinesterase activity to be used as a surrogate for the number of exosomes present.28, 29 Acetylcholinesterase activity was higher in exosomes precipitated from the medium of fibroblasts exposed to stretch compared with the medium from the static control fibroblasts (1.27±0.04‐ versus 1.0±0.08‐fold acetylcholinesterase activity; P<0.05; n=8) (Figure 5A). Furthermore, the abundance of miR‐133a was increased in the exosomes collected from the fibroblasts exposed to stretch compared with static controls (1.65±0.28‐ versus 1.0±0.12‐fold expression; P<0.05; n=7) (Figure 5B). These results demonstrated that biaxial cyclic stretch enhanced TA fibroblast exosome secretion and the loss of cellular miR‐133a.
Previous studies have demonstrated that exosome formation and membrane curvature is dependent on neutral sphingomyelinase 2–mediated hydrolysis of the choline head group, in the conversion of sphingomyelin to ceramide.30, 31, 32 Inhibition of neutral sphingomyelinase 2 with the use of a well‐described noncompetitive inhibitor (GW4869) has been demonstrated to prevent exosome formation and secretion.30, 33, 34, 35 Accordingly, to demonstrate that the stretch‐induced loss of miR‐133a was mediated by exosome formation and secretion, fibroblasts were exposed to biaxial cyclic stretch in the presence of GW4869. The results demonstrated that inhibition of exosome formation prevented the stretch‐induced secretion of miR‐133a; no change in cellular miR‐133a levels were observed with GW4869 compared with static control (1.02±0.35‐ versus 1.0±0.29‐fold expression; P=0.52, n=8) (Figure 5C). Taken together, these studies suggested that one mechanism by which miR‐133a is lost in response to elevated mechanical tension is through increased packaging and secretion of miR‐133a in exosomes.
To determine the effects of stretch on SMC exosome secretion, TA SMCs were grown on flexible membranes and either held static or exposed to biaxial cyclic stretch in medium containing exosome‐depleted FBS. At the end of 18 hours, the medium was collected and the newly secreted exosomes were precipitated. Acetylcholinesterase activity was similar in exosomes precipitated from the medium of SMCs exposed to stretch compared with the medium from the static control SMCs (0.90±0.12‐ versus 1.0±0.11‐fold acetylcholinesterase activity; P=0.21; n=6) (Figure 5D). Furthermore, the abundance of miR‐133a was similar in the exosomes collected from the SMCs exposed to stretch compared with static controls (1.07±0.26‐ versus 1.0±0.14‐fold expression; P=0.86; n=6) (Figure 5E). These results confirmed that TA SMC exosome secretion of miR‐133a was not affected by biaxial cyclic stretch.
Increased Vessel Wall Tension in Vivo Results in a Reduction of TA Tissue miR‐133a Abundance
To further examine the relationship between wall tension and the in vivo loss of miR‐133a, 2 distinct murine models of hypertension were used. Murine blood pressure measurements were obtained, and relative changes in MAP were measured by tail‐cuff using the CODA system. MAP in wild‐type normotensive mice was determined to be 121.16±2.59 mm Hg (normotensive, n=12). Normotensive mice were treated with AngII (1.44 mg/kg per day) delivered by SC osmotic pump for 28 days. This resulted in an increased MAP to 169.94±3.50 mm Hg (AngII, n=12) (P<0.05 versus normotensive and BPH2) (Figure 6A and Table 1). Second, the effects of elevated wall tension on miR‐133a abundance were examined in a spontaneously hypertensive mouse line (BPH2, genetically selected, AngII independent),36 which displayed an elevated MAP of 159.67±2.97 mm Hg (BPH2; P<0.05 versus normotensive and AngII, n=12) (Figure 6A and Table 1). The TA tissue abundance of miR‐133a was reduced in both models of hypertension compared with normotensive control animals (AngII: −7.11±0.04 log expression, n=12, P<0.05 versus control; BPH2: −7.22±0.03 log expression, n=16, P<0.05 versus control; control: −6.97±0.06, n=12) (Figure 6B). Plasma levels of miR‐133a were subsequently assessed in both hypertensive mouse models. Although miR‐133a abundance was elevated in the plasma of the AngII‐induced mice compared with normotensive controls, they failed to reach statistical significance (−5.86±0.18 log expression, n=6, P=0.449). However, miR‐133a levels were significantly increased in the plasma of BPH2 mice (n=8) compared with normotensive control animals (n=6) (BPH2 versus control: −5.49±0.10 versus −6.15±0.20 log expression; P<0.05 versus control) (Figure 6C). Combined, these results demonstrated that miR‐133a abundance is reduced in TA tissue in response to elevated vessel wall tension. Moreover, circulating plasma levels of miR‐133a were increased.
Circulating miR‐133a Abundance Was Increased in Hypertensive Patients
To establish whether there is a relationship between elevated wall tension and circulating levels of miR‐133a in patients, the abundance of miR‐133a was determined in plasma samples collected from patients previously diagnosed with hypertension and compared with normotensive controls (patient demographic information is listed in Table 2). Blood pressure was measured at the time of blood collection. MAP was significantly elevated in the hypertensive patients (n=11) compared with the normotensive patients (n=12) (97±6 versus 89±8 mm Hg; P<0.05) (Figure 7A) (blood pressures are listed in Table 2). Moreover, circulating plasma levels of miR‐133a were increased in the hypertensive group (n=11) compared with the normotensive group (n=12) (1.55±0.26‐ versus 1.0±0.18‐fold expression; P<0.05) (Figure 7B). These results supported the described animal studies and demonstrated that increased MAP was associated with increased plasma levels of miR‐133a.
Discussion
Previous work from this laboratory demonstrated the abundance of miR‐133a was decreased in aortic tissue from patients with TAA and was inversely proportional to aortic diameter.1 We hypothesized, on the basis of the Law of Laplace, that elevated wall tension, as experienced with increased aortic diameter, may play a role in regulating miR‐133a cellular abundance. Accordingly, in this study, we examined mechanisms capable of regulating miR‐133a abundance under conditions of elevated wall tension. The unique findings of this study were 4‐fold. First, it was determined that ex vivo tension applied to intact TA rings resulted in a short‐term reduction in tissue miR‐133a abundance. Second, mechanical tension in the form of biaxial cyclic stretch applied to isolated primary aortic fibroblasts and SMCs revealed that fibroblasts preferentially responded to mechanical tension, resulting in the short‐term reduction of miR‐133a abundance. Third, 3 potential mechanisms capable of regulating the cellular abundance of microRNAs were examined, and results demonstrated that tension‐dependent loss of miR‐133a was primarily mediated through exosome secretion. Finally, using 2 in vivo models of hypertension and human plasma samples from normotensive and hypertensive patients, it was determined that elevated blood pressure (increased wall tension) was sufficient to induce the loss of miR‐133a from the descending TA (mouse models) and was associated with increased plasma levels of miR‐133a (mouse models and human plasma). Taken together, these results identified a specific tension‐sensitive mechanism by which miR‐133a was reduced in a cell type that plays a key role in adverse vascular remodeling.
The association between increased wall tension and vascular remodeling, in aortic aneurysm, has been described.37 In previous results from this laboratory, mechanical tension applied to intact murine TA rings altered gene expression of several key MMPs active in ECM remodeling, specifically MMP‐2 and the membrane type‐1 MMP.11, 12 Combined with previous findings suggesting miR‐133a regulates the abundance of membrane type‐1 MMP,8, 38 these prior studies provided foundational evidence that justified examination of the role of elevated wall tension on the levels of this microRNA. In this report, it was demonstrated that in as little as 3 hours, applied tension (roughly equivalent to a MAP of ≈150 mm Hg) reduced miR‐133a by ≈70% in TA tissue. This is in agreement with a study by Mohamed and colleagues, who performed a genome‐wide analysis on the dysregulation of microRNA abundance in the thoracic diaphragm of mice after ex vivo application of mechanical tension.39 Using microarray analysis, the authors demonstrated that multiple microRNAs were affected by the application of mechanical tension relative to control; applied tension increased the abundance of some microRNAs and decreased the abundance of others, including miR‐133a.39 The present study is in agreement with these findings by demonstrating that the TA is also mechanically sensitive and elevated wall tension results in the short‐term reduction of miR‐133a. Although the application of ex vivo mechanical tension allowed isolation of the effects of tension alone on miR‐133a levels, it is well known that multiple factors influence wall tension in vivo. AngII is one factor that causes increased peripheral vascular resistance through interacting with its receptors and driving vasoconstriction, leading to elevated vascular wall tension. Therefore, using endothelial cell‐intact aortic rings, we demonstrated that developed tension in response to AngII reduces miR‐133a abundance. Interestingly, developed tension had a similar effect to the direct application of mechanical tension. Furthermore, this suggested that physiological changes in wall tension, such as the development of hypertension, may be sufficient to alter miR‐133a abundance in the vasculature.
When isolated aortic fibroblasts and SMCs were examined using a biaxial cyclic stretch paradigm mimicking the cardiac cycle, the short‐term reduction of miR‐133a abundance was observed in fibroblasts, but not in SMCs. Previous studies have suggested that fibroblasts are sensitive to changes in mechanical tension and respond by altering phenotypic characteristics, taking on a “synthetic” or mobile role.40, 41 In pathologic vascular remodeling, these synthetic fibroblasts become activated in the adventitia and migrate inward to the media, remodeling the ECM and enhancing vessel stiffness.42 Previous results from this laboratory defined changes in cellular makeup within the aortic wall during TAA formation, revealing that aortic dilation occurs simultaneously with vessel remodeling and the emergence of a population of active fibroblasts.9 When combined with studies demonstrating that both thoracic and abdominal aortic dilation is accompanied by the apoptotic loss of SMCs,9, 43, 44 it is believed that fibroblasts may be the key cellular mediator of aortic remodeling in TAA development. Because a single microRNA can regulate the translation of multiple targets, the present results may provide support for the idea that the short‐term loss of miR‐133a from aortic fibroblasts contributes to altered fibroblast phenotype and may contribute to the aberrant vascular remodeling that occurs during aneurysm development. This hypothesis will be addressed in future studies. Interestingly, when the isolated fibroblasts and SMCs were exposed to AngII in culture, no change in miR‐133a levels was observed. This suggested that the short‐term loss of miR‐133a abundance was not mediated by direct activation of the classic AngII pathway.
In this study, 3 mechanisms capable of regulating the abundance of miR‐133a were investigated. First, to determine if the reduction of miR‐133a in aortic fibroblasts was attributable to reduced miR‐133a transcription, primary miR‐133a transcript levels were quantitated after the application of mechanical tension. Because miR‐133a is transcribed from 2 separate locations within the genome, both primary miR‐133a‐1 and primary miR‐133a‐2 levels were examined. Although several studies have demonstrated that mechanical tension may alter transcription,11, 12, 45 tension had no effect on primary miR‐133a‐1 levels. Surprisingly, an increase in primary miR‐133a‐2 was induced; however, this was not sufficient to overcome the loss of mature miR‐133a observed. Therefore, it was concluded that tension‐induced reduction of mature miR‐133a was not a result of decreased transcription. Second, ribonuclease‐mediated degradation was examined. Three known exoribonucleases capable of degrading microRNAs are XRN‐1, XRN‐2, and ExoSC4.23, 24 Tension did not alter exoribonuclease mRNA expression or protein abundance, suggesting rapid degradation of miR‐133a was not likely mediated by exoribonucleases. Third, exosome secretion has been identified as an efficient mechanism for rapid reduction of cytoplasmic nucleic acids.46 In the present study, it was determined that acetylcholinesterase activity increased by ≈30% in exosomes precipitated from conditioned culture medium collected from stretched fibroblasts versus their static controls. This suggested that there was an increase in either size or number of exosomes secreted in response to stretch. Nonetheless, when normalizing for the amount of acetylcholinesterase activity, more miR‐133a was detected in the exosomes collected from the culture medium of fibroblasts exposed to stretch, suggesting increased packaging and export. Most important, when exosome secretion was arrested through inhibition of neutral sphingomyelinase 2, the tension‐induced reduction of miR‐133a in fibroblasts was abolished. Therefore, it was concluded that exosome secretion was the major mechanism responsible for the rapid loss of mature miR‐133a from aortic fibroblasts in the presence of elevated tension. Interestingly, cellular neutral sphingomyelinase 2 has been identified to be activated immediately (in <1 minute) in response to elevated vascular pressure.47, 48 This pathway, which is largely initiated within caveolae located on the cell membrane, has been demonstrated to upregulate the secretion of exosomes in response to elevated mechanical stretch.49 Caveolae are primary sites for rapid mechanoinduced tyrosine phosphorylation of proteins and are considered mechanosensing organelles containing signaling molecules, including sphingomyelin and nonreceptor tyrosine kinases.50 Future studies classifying activation of these pathways in TAA may identify novel targets for therapeutic intervention.
The physiological relevance of these findings was determined in vivo through an examination of the effects of increased vessel wall tension on the aortic tissue levels of miR‐133a. For this approach, 2 unique murine models of hypertension were used. In the first model, hypertension was induced in mice by delivering AngII by osmotic pump infusion. The second uses a commercially available, spontaneously hypertensive mouse line (BPH2). In this model, it has been reported that hypertension is angiotensin independent through the observation of low circulating angiotensin I levels.51 In both models, elevated mean arterial blood pressures were confirmed and, as anticipated, miR‐133a was found to be reduced in the tissue of the descending TA. In a similar study performed by Castoldi and colleagues, hypertension was induced in Sprague‐Dawley rats by treatment with AngII via osmotic pump.7 After 4 weeks of hypertension, the levels of miR‐133a were found to be reduced in myocardial tissue.7 Although the loss of miR‐133a was inhibited with an AngII receptor blocker, irbesartan, this study was unable to determine whether this effect was mediated by AngII signaling or elevated tension.7 Interestingly, in the present study, both murine models of hypertension displayed increased plasma levels of miR‐133a. Although statistical significance was reached only in the BPH2 model, this may have been attributable to the time difference in exposure to elevated blood pressures. Although the AngII model of hypertension experienced elevated blood pressures for a total of 4 weeks, the hypertensive BPH2 mice (12–14 weeks of age when examined) had experienced hypertension since ≈5 weeks of age (a total of 7–9 weeks).52 These findings led us to the question of whether this effect could be similarly observed clinically in hypertensive patients. Accordingly, miR‐133a abundance was measured in the plasma of individuals with and without documented hypertension. Plasma levels of miR‐133a were increased in the hypertensive group. This is in agreement with a previous study from this laboratory when investigating circulating microRNA levels in patients with elevated TA wall tension from TAA.53 In this past study, it was demonstrated that plasma miR‐133a levels were elevated in patients with TAA compared with healthy controls. Combined with these past findings, the present results confirmed that in vivo elevated tension was sufficient to induce the loss of miR‐133a in the aortic tissue (mouse) and concomitantly demonstrated an association with increased circulating levels of miR‐133a (mouse and human).
The present study is not without limitations. First, the effects of tension were determined on miR‐133a alone. Although increasing evidence highlights the role miR‐133a plays in maintaining tissue homeostasis, it is anticipated that the abundance of other microRNAs may also be altered with elevated tension. Accordingly, we are unable to conclude whether tension‐induced secretion of miR‐133a in exosomes is limited to miR‐133a or is a characteristic of a specific group of microRNAs. Second, after 3 hours of stretch, the amount of exosomes secreted into the cell culture medium was not sufficient to be detected above that of the exosome‐depleted medium alone, suggesting 3 hours was insufficient for the detection of de novo synthesis of exosomes. Therefore, the length of time was increased to 18 hours, which was sufficient in demonstrating an increase in exosomes with stretch. Third, murine blood pressures were assessed using a noninvasive tail‐cuff system. This system routinely reports systolic and diastolic pressures that contribute to a normotensive MAP of ≈120 mm Hg in untreated wild‐type mice.54 Although a MAP of this value would typically be considered hypertensive, these results are consistent with other investigators using the CODA system for measuring murine blood pressure.54 Therefore, we assessed the relative difference between the groups of mice and were able to determine a significant increase in the blood pressure measurements in both murine models of hypertension. Fourth, although circulating levels of miR‐133a were identified to be significantly elevated in the clinical plasma samples taken from hypertensive patients, the sample size of measurements was limited, preventing advanced modeling of the correlation between pressure and miR‐133a abundance. Last, all hypertensive patients in this study were actively being treated with antihypertensive agents at the time of blood collection. Despite this, blood pressures and plasma levels of miR‐133a were significantly elevated compared with the nonhypertensive control group.
Taken together, the results of this study have identified a specific tension‐sensitive mechanism by which miR‐133a was reduced in aortic fibroblasts through the packaging and secretion of exosomes. These current findings hold significance with regard to the potential advancement in understanding the role of miR‐133a in the regulation of TA remodeling and even suggest the possibility of using circulating levels of this microRNA to detect or monitor the progression of pathologic conditions associated with adverse vascular wall tension.
Author Contributions
Experimental design was conceived by Akerman, Stroud, and Jones. All experimental procedures were performed by Akerman, Blanding, Stroud, and Nadeau. Akerman wrote the manuscript. Clinical samples provided by Zile. Editorial revisions and data analysis and interpretation were performed by Akerman, Blanding, Stroud, Ruddy, Mukherjee, Zile, Ikonomidis, and Jones. All authors reviewed the results and approved the final version of the manuscript.
Sources of Funding
Research reported in this publication was supported by the National Heart, Lung, and Blood Institute (NHLBI) of the National Institutes of Health and the Department of Veterans Affairs under Award Numbers (NHLBI) R01HL102121 (Ikonomidis), 1R01HL123478 (Zile), and (VA‐ORD Merit BLR&D Award) I01BX000904 (Jones). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Department of Veterans Affairs.
Disclosures
Akerman, Mukherjee, Zile, Ikonomidis, and Jones report intellectual property filing related to miR‐133a and hypertension. The remaining authors have no disclosures to report.
(J Am Heart Assoc. 2019;8:e010332 DOI: 10.1161/JAHA.118.010332.)
References
- 1. Jones JA, Stroud RE, O'Quinn EC, Black LE, Barth JL, Elefteriades JA, Bavaria JE, Gorman JH III, Gorman RC, Spinale FG, Ikonomidis JS. Selective microRNA suppression in human thoracic aneurysms: relationship of miR‐29a to aortic size and proteolytic induction. Circ Cardiovasc Genet. 2011;4:605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Hoydal M, Autore C, Russo MA, Dorn GW II, Ellingsen O, Ruiz‐Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G. MicroRNA‐133 controls cardiac hypertrophy. Nat Med. 2007;13:613–618. [DOI] [PubMed] [Google Scholar]
- 3. Torella D, Iaconetti C, Catalucci D, Ellison GM, Leone A, Waring CD, Bochicchio A, Vicinanza C, Aquila I, Curcio A, Condorelli G, Indolfi C. MicroRNA‐133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ Res. 2011;109:880–893. [DOI] [PubMed] [Google Scholar]
- 4. Shan H, Zhang Y, Lu Y, Zhang Y, Pan Z, Cai B, Wang N, Li X, Feng T, Hong Y, Yang B. Downregulation of miR‐133 and miR‐590 contributes to nicotine‐induced atrial remodelling in canines. Cardiovasc Res. 2009;83:465–472. [DOI] [PubMed] [Google Scholar]
- 5. Jones JA, Barbour JR, Stroud RE, Bouges S, Stephens SL, Spinale FG, Ikonomidis JS. Altered transforming growth factor‐beta signaling in a murine model of thoracic aortic aneurysm. J Vasc Res. 2008;45:457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, van der Made I, Herias V, van Leeuwen RE, Schellings MW, Barenbrug P, Maessen JG, Heymans S, Pinto YM, Creemers EE. miR‐133 and miR‐30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res. 2009;104:170–178, 176p following 178. [DOI] [PubMed] [Google Scholar]
- 7. Castoldi G, Di Gioia CR, Bombardi C, Catalucci D, Corradi B, Gualazzi MG, Leopizzi M, Mancini M, Zerbini G, Condorelli G, Stella A. miR‐133a regulates collagen 1A1: potential role of miR‐133a in myocardial fibrosis in angiotensin II‐dependent hypertension. J Cell Physiol. 2012;227:850–856. [DOI] [PubMed] [Google Scholar]
- 8. Xu M, Wang YZ. miR133a suppresses cell proliferation, migration and invasion in human lung cancer by targeting MMP14. Oncol Rep. 2013;30:1398–1404. [DOI] [PubMed] [Google Scholar]
- 9. Jones JA, Beck C, Barbour JR, Zavadzkas JA, Mukherjee R, Spinale FG, Ikonomidis JS. Alterations in aortic cellular constituents during thoracic aortic aneurysm development: myofibroblast‐mediated vascular remodeling. Am J Pathol. 2009;175:1746–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goldsmith EC, Bradshaw AD, Zile MR, Spinale FG. Myocardial fibroblast‐matrix interactions and potential therapeutic targets. J Mol Cell Cardiol. 2014;70:92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ruddy JM, Jones JA, Stroud RE, Mukherjee R, Spinale FG, Ikonomidis JS. Differential effects of mechanical and biological stimuli on matrix metalloproteinase promoter activation in the thoracic aorta. Circulation. 2009;120:S262–S268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ruddy JM, Jones JA, Stroud RE, Mukherjee R, Spinale FG, Ikonomidis JS. Differential effect of wall tension on matrix metalloproteinase promoter activation in the thoracic aorta. J Surg Res. 2010;160:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Flack EC, Lindsey ML, Squires CE, Kaplan BS, Stroud RE, Clark LL, Escobar PG, Yarbrough WM, Spinale FG. Alterations in cultured myocardial fibroblast function following the development of left ventricular failure. J Mol Cell Cardiol. 2006;40:474–483. [DOI] [PubMed] [Google Scholar]
- 14. Ray JL, Leach R, Herbert JM, Benson M. Isolation of vascular smooth muscle cells from a single murine aorta. Methods Cell Sci. 2001;23:185–188. [DOI] [PubMed] [Google Scholar]
- 15. Peltier HJ, Latham GJ. Normalization of microRNA expression levels in quantitative RT‐PCR assays: identification of suitable reference RNA targets in normal and cancerous human solid tissues. RNA. 2008;14:844–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ho IK, Ellman GL. Triton solubilized acetylcholinesterase of brain. J Neurochem. 1969;16:1505–1513. [DOI] [PubMed] [Google Scholar]
- 17. Zile MR, Baicu CF, Ikonomidis JS, Stroud RE, Nietert PJ, Bradshaw AD, Slater R, Palmer BM, Van Buren P, Meyer M, Redfield MM, Bull DA, Granzier HL, LeWinter MM. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation. 2015;131:1247–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Paulus WJ, Tschope C, Sanderson JE, Rusconi C, Flachskampf FA, Rademakers FE, Marino P, Smiseth OA, De Keulenaer G, Leite‐Moreira AF, Borbely A, Edes I, Handoko ML, Heymans S, Pezzali N, Pieske B, Dickstein K, Fraser AG, Brutsaert DL. How to diagnose diastolic heart failure: a consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology. Eur Heart J. 2007;28:2539–2550. [DOI] [PubMed] [Google Scholar]
- 19. Lindenfeld J, Albert NM, Boehmer JP, Collins SP, Ezekowitz JA, Givertz MM, Katz SD, Klapholz M, Moser DK, Rogers JG, Starling RC, Stevenson WG, Tang WH, Teerlink JR, Walsh MN. HFSA 2010 comprehensive heart failure practice guideline. J Card Fail. 2010;16:e1–e194. [DOI] [PubMed] [Google Scholar]
- 20. Schmittgen TD, Livak KJ. Analyzing real‐time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. [DOI] [PubMed] [Google Scholar]
- 21. Ruegger S, Grosshans H. MicroRNA turnover: when, how, and why. Trends Biochem Sci. 2012;37:436–446. [DOI] [PubMed] [Google Scholar]
- 22. Aryani A, Denecke B. In vitro application of ribonucleases: comparison of the effects on mRNA and miRNA stability. BMC Res Notes. 2015;8:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bail S, Swerdel M, Liu H, Jiao X, Goff LA, Hart RP, Kiledjian M. Differential regulation of microRNA stability. RNA. 2010;16:1032–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang Z, Qin YW, Brewer G, Jing Q. MicroRNA degradation and turnover: regulating the regulators. Wiley Interdiscip Rev RNA. 2012;3:593–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chatterjee S, Fasler M, Bussing I, Grosshans H. Target‐mediated protection of endogenous microRNAs in C. elegans . Dev Cell. 2011;20:388–396. [DOI] [PubMed] [Google Scholar]
- 26. Chatterjee S, Grosshans H. Active turnover modulates mature microRNA activity in Caenorhabditis elegans . Nature. 2009;461:546–549. [DOI] [PubMed] [Google Scholar]
- 27. Hunter MP, Ismail N, Zhang X, Aguda BD, Lee EJ, Yu L, Xiao T, Schafer J, Lee ML, Schmittgen TD, Nana‐Sinkam SP, Jarjoura D, Marsh CB. Detection of microRNA expression in human peripheral blood microvesicles. PLoS One. 2008;3:e3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Savina A, Vidal M, Colombo MI. The exosome pathway in K562 cells is regulated by Rab11. J Cell Sci. 2002;115:2505–2515. [DOI] [PubMed] [Google Scholar]
- 29. Gupta S, Knowlton AA. HSP60 trafficking in adult cardiac myocytes: role of the exosomal pathway. Am J Physiol Heart Circ Physiol. 2007;292:H3052–H3056. [DOI] [PubMed] [Google Scholar]
- 30. Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem. 2010;285:17442–17452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brugger B, Simons M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319:1244–1247. [DOI] [PubMed] [Google Scholar]
- 32. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–289. [DOI] [PubMed] [Google Scholar]
- 33. Wang X, Huang W, Liu G, Cai W, Millard RW, Wang Y, Chang J, Peng T, Fan GC. Cardiomyocytes mediate anti‐angiogenesis in type 2 diabetic rats through the exosomal transfer of miR‐320 into endothelial cells. J Mol Cell Cardiol. 2014;74:139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kulshreshtha A, Ahmad T, Agrawal A, Ghosh B. Proinflammatory role of epithelial cell‐derived exosomes in allergic airway inflammation. J Allergy Clin Immunol. 2013;131:1194–1203, 1203.e1–e14. [DOI] [PubMed] [Google Scholar]
- 35. Li J, Liu K, Liu Y, Xu Y, Zhang F, Yang H, Liu J, Pan T, Chen J, Wu M, Zhou X, Yuan Z. Exosomes mediate the cell‐to‐cell transmission of IFN‐alpha‐induced antiviral activity. Nat Immunol. 2013;14:793–803. [DOI] [PubMed] [Google Scholar]
- 36. Elias MF, Sorrentino RN, Pentz CA III, Florini JR. “Spontaneously” hypertensive mice: a potential genetic model for the study of the relationship between heart size and blood pressure. Exp Aging Res. 1975;1:251–265. [DOI] [PubMed] [Google Scholar]
- 37. Coady MA, Rizzo JA, Goldstein LJ, Elefteriades JA. Natural history, pathogenesis, and etiology of thoracic aortic aneurysms and dissections. Cardiol Clin. 1999;17:615–635; vii. [DOI] [PubMed] [Google Scholar]
- 38. Eckhouse SR, Akerman AW, Logdon CB, Oelsen JM, O'Quinn EC, Nadeau EK, Stroud RE, Mukherjee R, Jones JA, Spinale FG. Differential membrane type 1 matrix metalloproteinase substrate processing with ischemia‐reperfusion: relationship to interstitial microRNA dynamics and myocardial function. J Thorac Cardiovasc Surg. 2013;145:267–275, 277.e261–264; discussion 275‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mohamed JS, Hajira A, Lopez MA, Boriek AM. Genome‐wide mechanosensitive microRNA (MechanomiR) screen uncovers dysregulation of their regulatory networks in the mdm mouse model of muscular dystrophy. J Biol Chem. 2015;290:24986–25011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Humphrey JD, Dufresne ER, Schwartz MA. Mechanotransduction and extracellular matrix homeostasis. Nat Rev Mol Cell Biol. 2014;15:802–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jiang M, Qiu J, Zhang L, Lu D, Long M, Chen L, Luo X. Changes in tension regulates proliferation and migration of fibroblasts by remodeling expression of ECM proteins. Exp Ther Med. 2016;12:1542–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sartore S, Chiavegato A, Faggin E, Franch R, Puato M, Ausoni S, Pauletto P. Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: from innocent bystander to active participant. Circ Res. 2001;89:1111–1121. [DOI] [PubMed] [Google Scholar]
- 43. Thompson RW, Liao S, Curci JA. Vascular smooth muscle cell apoptosis in abdominal aortic aneurysms. Coron Artery Dis. 1997;8:623–631. [DOI] [PubMed] [Google Scholar]
- 44. Lopez‐Candales A, Holmes DR, Liao S, Scott MJ, Wickline SA, Thompson RW. Decreased vascular smooth muscle cell density in medial degeneration of human abdominal aortic aneurysms. Am J Pathol. 1997;150:993–1007. [PMC free article] [PubMed] [Google Scholar]
- 45. Mammoto A, Mammoto T, Ingber DE. Mechanosensitive mechanisms in transcriptional regulation. J Cell Sci. 2012;125:3061–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Takahashi A, Okada R, Nagao K, Kawamata Y, Hanyu A, Yoshimoto S, Takasugi M, Watanabe S, Kanemaki MT, Obuse C, Hara E. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat Commun. 2017;8:15287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Czarny M, Liu J, Oh P, Schnitzer JE. Transient mechanoactivation of neutral sphingomyelinase in caveolae to generate ceramide. J Biol Chem. 2003;278:4424–4430. [DOI] [PubMed] [Google Scholar]
- 48. Czarny M, Schnitzer JE. Neutral sphingomyelinase inhibitor scyphostatin prevents and ceramide mimics mechanotransduction in vascular endothelium. Am J Physiol Heart Circ Physiol. 2004;287:H1344–H1352. [DOI] [PubMed] [Google Scholar]
- 49. Pironti G, Strachan RT, Abraham D, Mon‐Wei YuS, Chen M, Chen W, Hanada K, Mao L, Watson LJ, Rockman HA. Circulating exosomes induced by cardiac pressure overload contain functional angiotensin II type 1 receptors. Circulation. 2015;131:2120–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rizzo V, Sung A, Oh P, Schnitzer JE. Rapid mechanotransduction in situ at the luminal cell surface of vascular endothelium and its caveolae. J Biol Chem. 1998;273:26323–26329. [DOI] [PubMed] [Google Scholar]
- 51. Uddin M, Harris‐Nelson N. Renin activity and angiotensin I concentration in genetically selective inbred line of hypertensive mice. Biochem Biophys Res Commun. 2004;316:842–844. [DOI] [PubMed] [Google Scholar]
- 52. Schlager G, Sides J. Characterization of hypertensive and hypotensive inbred strains of mice. Lab Anim Sci. 1997;47:288–292. [PubMed] [Google Scholar]
- 53. Ikonomidis JS, Ivey CR, Wheeler JB, Akerman AW, Rice A, Patel RK, Stroud RE, Shah AA, Hughes CG, Ferrari G, Mukherjee R, Jones JA. Plasma biomarkers for distinguishing etiologic subtypes of thoracic aortic aneurysm disease. J Thorac Cardiovasc Surg. 2013;145:1326–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rateri DL, Davis FM, Balakrishnan A, Howatt DA, Moorleghen JJ, O'Connor WN, Charnigo R, Cassis LA, Daugherty A. Angiotensin II induces region‐specific medial disruption during evolution of ascending aortic aneurysms. Am J Pathol. 2014;184:2586–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]