

Abstract
Background
Canonical studies indicate that cytochrome P450 2E1 (CYP2E1) plays a critical role in the metabolism of xenobiotics and ultimately participates in tissue damage. CYP2E1 upregulates in the pathophysiological development of multiple diseases; however, the mechanism of CYP2E1 upregulation, particularly in heart disease, remains elusive.
Methods and Results
We found that the level of CYP2E1 increased in heart tissues from patients with hypertrophic cardiomyopathy; multiple mouse models of heart diseases, including dilated cardiomyopathy, hypertrophic cardiomyopathy, and myocardial ischemia; and HL‐1 myocytes under stress. We determined that Myc bound to the CYP2E1 promoter and activated its transcription by bioinformatics analysis, luciferase activity, and chromatin immunoprecipitation, and Myc expression was modulated by extracellular signal–regulated kinases 1/2 and phosphatidylinositol 3 kinase/protein kinase B pathways under stress or injury in myocardium by signal transduction analysis. In addition, the level of oxidative stress and apoptosis gradually worsened with age in transgenic mice overexpressing CYP2E1, which was significantly inhibited with CYP2E1 knockdown.
Conclusions
Our results demonstrated that CYP2E1 is likely a sensor of diverse pathophysiological factors and states in the myocardium. Upregulated CYP2E1 has multiple pathophysiological roles in the heart, including increased oxidative stress and apoptosis as well as energy supply to meet the energy demand of the heart in certain disease states. Our discovery thus provides a basis for a therapeutic strategy for heart diseases targeting Myc and CYP2E1.
Keywords: apoptosis, cytochrome P450 2E1, gene expression/regulation, heart disease, Myc, oxidative stress
Subject Categories: Cardiomyopathy, Heart Failure, Hypertrophy, Gene Expression & Regulation
Clinical Perspective
What Is New?
Cytochrome P450 2E1 is likely a sensor of diverse pathophysiological factors and states in the myocardium.
Upregulated cytochrome P450 2E1 has multiple pathophysiological roles in the heart, including increase oxidative stress and apoptosis as well as energy supply to meet the energy demand of the heart in certain disease states.
What Are the Clinical Implications?
Our findings provide a basis for a therapeutic strategy for heart diseases targeting Myc and cytochrome P450 2E1, as cytochrome P450 2E1 upregulation is related to multiple heart diseases.
Introduction
Cytochrome P450 2E1 (CYP2E1) is a member of the cytochrome P450 family of enzymes. Cytochrome P450 participates in the metabolism of a large number of exogenous and endogenous substrates.1 CYP2E1 is expressed in the liver, brain, and heart in humans and animals.2, 3, 4 CYP2E1 locates in different cell compartments, including the plasma membrane, endoplasmic reticulum, Golgi apparatus, and mitochondria.5
CYP2E1 plays a critical role in the metabolism and activation of many important substrates, including small organic molecules (such as ethanol and acetone), toxic chemicals from diet and environmental contamination (such as carbon tetrachloride, chloroform, nitrosamines, benzene, and acrylamide), and endogenous compounds such as fatty acids, ketone bodies, and glycerol.2, 6 The CYP2E1 gene is inducible in response to endogenous and exogenous substrates. In the presence of oxygen and nicotinamide adenine dinucleotide phosphate hydrogen, CYP2E1 can produce reactive oxygen species (ROS) in the presence or absence of substrate, and the nicotinamide adenine dinucleotide phosphate hydrogen oxidase activity of CYP2E1 is remarkably higher than those of other members of the cytochrome P450 family.7 CYP2E1 is a major source of cellular and mitochondrial ROS/reactive nitrogen species, which induces tissue damages mainly through damage to cellular and mitochondrial macromolecules, including mitochondrial DNA.8
CYP2E1 has been implicated in the pathophysiological development of a wide variety of diseases, including obesity, diabetes mellitus, alcoholic liver disease, inflammation, Parkinson disease, and cancer.8, 9, 10, 11, 12 However, upregulated CYP2E1 levels are also observed under normal physiological conditions, such as in overfed rat, in high‐fat fed rat, and in fasted rat.13, 14, 15 In addition, several studies have demonstrated that hormones can mediate the regulation of CYP2E1 expression, including insulin, leptin, thyroid, and growth hormones.16, 17, 18
Accumulating evidence suggests that numerous genes associated with mechanotransduction, mitochondrial energetics, oxidative stress, and extracellular matrix are regulated in the development of heart diseases, including CYP2E1.19, 20 We previously demonstrated that the phenotype of transgenic mice overexpressing CYP2E1 in the myocardium is similar to that of dilated cardiomyopathy (DCM) and that knockdown of endogenous CYP2E1 expression markedly prevents the development of DCM in mouse models.3 Thus, upregulation of CYP2E1 may be a key factor in the pathogenesis of heart failure (HF), and CYP2E1 is likely a sensor of diverse pathophysiological factors and states in the myocardium.
Furthermore, we have previously established that CYP2E1 is upregulated under multiple pathological states of the heart, including familiar DCM, as studies in cTnTR141W and LMNAE82K transgenic mouse models and in drug‐induced DCM/HF mouse models, as to adriamycin‐treated mice.3, 21 Moreover, CYP2E1 levels are increased in several cardiovascular diseases, including ischemia, DCM, and spontaneously hypertensive, and in several species, including human, mice, rats, and dogs.9, 21, 22, 23, 24, 25, 26
In the present study, we determined that CYP2E1 is also upregulated in the myocardium from mouse models of ischemia induced by ligation of left anterior descending (LAD) coronary artery, mouse models of hypertrophic cardiomyopathy (HCM)/HF induced by thoracic aorta constriction (TAC), and patients with HCM.
Systematic examinations of the mechanisms that CYP2E1 is upregulated under multiple pathophysiological states, especially in heart disease, are lacking. Thus, in the present study, we determined for the first time the molecular mechanism involved in the CYP2E1 upregulation in the myocardium based on promoter interaction and signal pathway analysis in mice and myocyte models under stress and/or injury. In addition, the characteristics of the pathological role of CYP2E1 in oxidative stress and apoptosis were also determined.
Methods
The data, analytic methods, and study materials will be made available upon request to other researchers for purposes of reproducing results or replicating procedures. Material will be available from Dr Lu at the Institute of Laboratory Animal Science. All procedures were performed in accordance with the Animal Care and Use Committees of the Institute of Laboratory Animal Science of Peking Union Medical College.
Animals
Cardiac‐specific CYP2E1‐overexpression transgenic mice (referred to as CYP2E1‐ov) and the cardiac‐specific CYP2E1‐knockdown transgenic mice (referred to as CYP2E1‐kd) were previously generated in our laboratory, and genotyping of the transgenic mice were performed by polymerase chain reaction (PCR) as previously described (see Table S1 for primer sequences).3, 21
Mice Model of Myocardial Ischemia
C57BL6 mice at 8 to 12 weeks of age were assigned to groups receiving either myocardial ischemia via LAD or sham surgery as previously described.27 Briefly, the mice were anesthetized with tribromoethanol and then intubated to allow artificial ventilation with room air. The heart was exposed through left thoracotomy and the LAD coronary artery was ligated with an 8‐0 nylon suture. The mice were euthanized at 1 week after the operation. All surgeries and subsequent analyses were performed in a blinded fashion.
TAC Treatment
The TAC operation was performed in 8‐week‐old C57BL6 mice as previously described.28 Briefly, wild‐type (WT) mice were anesthetized with intraperitoneally administered tribromoethanol at 216 mg/kg body weight. The surgery was performed using a ventilator to acquire passive respiration. The aorta was constricted using a 27‐gauge blunted needle as a calibrator. The innominate artery and left common carotid artery blood flow was detected with Doppler analysis at 1 week after the surgery. The innominate artery/left common carotid artery ratio from 5.9 to 10.7 marked a comparable pressure gradient and was selected in this study. The sham operation was the same except that the aorta was not constricted. Mice that survived were chosen for the following studies at 2, 4, or 8 weeks after the surgery.
Adriamycin Treatment
Adriamycin treatment was administered to 8‐week‐old C57BL6 mice as previously described.3 Adriamycin (Shenzhen Wanle Pharmaceutical Co, Ltd) was administered intraperitoneally with a constant volume of saline at 4 mg/kg every other day for a total of 14 days. Echocardiographic analysis was performed on mice at day 0 (before Adriamycin treatment), and at 2 weeks after cessation of adriamycin treatment.
Human Cardiac Samples
All procedures using human samples followed the principles summarized in the Declaration of Helsinki and were approved by the ethics committee of Fuwai Hospital. The interventricular septum of the left ventricle was acquired from 14 patients with obstructive HCM who had Morrow septal myectomy. To minimize any potential confounding effects, we excluded patients with congenital heart disease, myocardial infarction, and valvular heart diseases. Control cardiac tissues were acquired from the same region of 8 healthy donors who died because of an accident and with no history of heart disease. Written informed consent was acquired from all participants or their relatives.
Protein Extraction and Immunoblotting
Total protein lysates were prepared as previously described.3, 29, 30 Proteins were separated by SDS‐PAGE and transferred to the nitrocellulose membranes. Then the membranes were incubated with antibodies against CYP2E1 (Abcam; ab28146, 1:5000), phosphor‐p44/42 MAPK (Erk1/2) (Thr201/Tyr204) (Cell Signaling; 4370, 1:2000), p44/42 MAPK (Erk1/2) (Cell Signaling; 9102, 1:1000), phosphor‐PI3 Kinase p85 (Tyr458)/p55 (Tyr199) (Cell Signaling; 4228, 1:1000), PI3 Kinase p85 (Cell Signaling; 11889, 1:1000), phosphor‐Akt (Ser473) (Cell Signaling; 4060, 1:2000), Akt (Cell Signaling; 8596, 1:1000), Myc (Abcam; ab17355, 1:500), CEBP Alpha (Abcam, ab40764), CEBP Beta (Abcam, ab32358), or C/EBP zdta (ThermoFisher, PA5‐37925). Antibody binding was detected with horseradish peroxidase–conjugated immunoglobulin G using a chemiluminescence detection system after incubation with the appropriate secondary antibodies. GAPDH was used as normalization (Abcam; ab9482, 1:5000). Bands were quantitatively analyzed with ImageJ software.
Constructs
The overexpression plasmid of Myc (pMyc) contained Mus muculus Myc cDNA and was purchased from OriGene (MC216121). A series of truncated CYP2E1 promoter fragments containing genomic sequence of the mouse CYP2E1 gene at different 5′ end positions and the same 3′ end position, +37, was amplified by PCR to introduce the KpnI and XhoI restriction sites, respectively. The functional consequences were analyzed with the mutated vectors that with the Myc binding site (−846 to −835, CAAGCACATGGA) or the CCAAT enhancer binding protein (C/EBP) binding site (−756 to −743, CATTTTTACAAATA) deleted out in the p0.9Cyp2e1‐luc plasmid, individually or synchronously. The PCR products were cloned into the pGL3‐Basic vector with the firefly luciferase gene as reporter. The pRL‐CMV vector containing Renilla luciferase gene as the reporter was used as normalization (Promega, E2261). The constructs were all confirmed by DNA sequencing (see Table S1 for primer sequences and Table S2 for the sequencing results of the mutated vectors in this study).
Cell Culture and Treatment
Cells of HL‐1 adult mouse cardiac muscle were grown in Claycomb Medium supplemented with 10% FCS, 4 mmol/L l‐glutamine, and 50 μmol/L noradrenaline (MedChem Express, HY‐13715B), in a humidified 5% CO2 incubator as previously described.31 The HL‐1 cells were changed to a medium without noradrenaline 5 days before the treatment. The stress treatments were starvation in a medium without serum for 8 hours or treatment with isoprenaline (Sigma; I6504, 10 μmol/L) for 12 hours. The inhibitor treatment was performed by PD098059 (mitogen‐activated protein kinase kinase [MEK]/extracellular signal–regulated kinase [ERK] inhibitor [Cell Signaling; 9900, 50 μmol/L]) or LY294002 (PI3K inhibitor [Cell Signaling; 9901, 25 μmol/L]) for 12 hours. After the treatment, cells were harvested for protein extraction.
Transient Transfections and Luciferase Assays
HL‐1 cells were cultured as above. Upon reaching ≈35% confluence, the cells were transfected with DNA using ViaFect transfection reagent according to the instructions (Promega, E4982). Firefly reporter plasmid (0.5 μg) was cotransfected with pMyc in each treatment. The entire amount of transfected DNA was equalized using pcDNA3 between each experiment. pRL‐CMV vector (0.02 mg) was always cotransfected as normalization. The cells were harvested at 40 hours after transfection and ≈100% confluence. Luciferase activity was analyzed using the Dual‐Luciferase system (Promega, E1910). The cells were treated with 20 μmol/L 10058‐F4 (Sigma, F3680), an Myc inhibitor, for 6 hours in the specific regulation analysis experiment.
Chromatin Immunoprecipitation Assay
HL‐1 cells were cultured and transfected with either 3 μg of pMyc or 3 μg of pcDNA3 as a control using ViaFect transfection reagent, and 1.5 μg of the −855/+37 luciferase reporter plasmid (p0.9Cyp2e1‐luc) would be cotransfected when indicated. To monitor the transfection efficiency, 0.3 μg of the pGFP plasmid was added. At 42 hours after transfection and ≈100% confluence, cells were cross‐linked with 1% formaldehyde for 10 minutes and then treated with lysis buffer containing a protease inhibitor cocktail (Thermo Scientific, 87785). Chromatin was fragmented by sonication, then precleared with a salmon sperm DNA‐protein A‐agarose 50% slurry. Immunoprecipitation was operated by incubation with mouse monoclonal Myc antibody (Abcam; ab17355, 1 μg) at 4°C overnight. After the immune complexes were washed and reversed from the cross‐linking, the DNA fragments were extracted and a fragment of the CYP2E1 promoter was amplified by PCR (see Table S1 for primer sequences).
Survival Analysis
The cumulative percent mortality was calculated every 2 weeks for TAC‐treated mice or every 1 week for adriamycin‐treated mice. Upon death, mice underwent autopsy, and the pathological changes in the heart were recorded. Kaplan–Meier curves were assayed using log‐rank test (SPSS version 16.0 software [SPSS Inc]).
Echocardiography
Echocardiographic inspection was performed with a small animal echocardiography system as previously described (Vevo770).3, 29, 30
Histological Analysis
Heart tissues were fixed in 4% formaldehyde, and then mounted in paraffin. The sections were stained with hematoxylin and eosin or Masson trichrome as previously described.3 Myocardial fibrosis were measured using Aperio Image Scope version 8.2.5 software (Leica Biosystems Imaging).
H2O2, Malondialdehyde, Glutathione, and Total Antioxidant Capacity Measurements
Heart tissues were homogenized rapidly in buffer with 0.15 mol/L KCl and 1.0 mmol/L EDTA, followed by centrifugation at 13 000g at 4°C for 30 minutes, then the supernatant were collected for the following assays. H2O2 (Abcam, ab102500), malondialdehyde (Abcam, ab118970), glutathione (Promega, V6612), and total antioxidant capacity (T‐AOC; BioVision, K274‐100) were measured following the manufacturer's instructions.3
Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling Assay
The terminal deoxynucleotidyl transferase dUTP nick end labeling assay was performed using an In Situ Apoptosis Kit (Millipore, S7101) as previously described.3 The results are expressed as the percentage of apoptotic cells among the total cell population.
Measurement of CYP2E1 Activity
CYP2E1 activity was determined by the rate of oxidation of p‐nitrophenol to p‐nitrocatechol in the presence of nicotinamide adenine dinucleotide phosphate hydrogen and oxygen as previously described.16 The activity was determined using the equation: p‐nitrophenol activity (nmol/min per mg protein)=D546/9.53/0.2/60/7.1×103.3, 21
Coimmunoprecipitation and Liquid Chromatography Tandem Mass Spectrometry
Immunocomplexes samples run with SDS‐PAGE and Western blot analysis. After in‐gel digestion, the peptides were extracted from the gel and then analyzed by nano–coimmunoprecipitation and liquid chromatography tandem mass spectrometry (MS) on a Q Exactive mass spectrometer. Chromatography was performed with solvent A (Milli‐Q [Millipore] water with 2% acetonitrile and 0.1% formic acid) and solvent B (90% acetonitrile with 0.1% formic acid). Q Exactive was operated in information‐dependent data acquisition mode to switch automatically between MS and MS/MS acquisition. MS spectra were acquired across the mass range of 350 to 2000 m/z. Precursor ions were then analyzed using Xcalibur software (Thermo Scientific). All MS/MS data were analyzed using Mascot (Matrix Science, version 2.3.0). Only those proteins with P<0.05 were accepted.
Statistical Analysis
Normal distribution of the sample sets was determined by Shapiro–Wilk normality test. For sample sets with gaussian distribution, Student 2‐tailed t test or 2‐way ANOVA was used. For the sample sets with a nongaussian distribution, Mann–Whitney test or Kruskal–Wallis test was used. All analyses were considered statistically significant at P<0.05. Statistical analysis was performed using Prism software (GraphPad).
Results
CYP2E1 Upregulated in Heart Disease in Patients and Mouse Models
CYP2E1 is a major source of cellular and mitochondrial ROS/reactive nitrogen species and therefore induces tissue damage in a variety of diseases. The expression of CYP2E1 is altered under multiple conditions, including fasting, nutrition intake, and various pathophysiological statuses.
We analyzed the expression of CYP2E1 in heart tissues from patients with HCM, an ischemia mouse model, an HCM mouse model, and a DCM mouse model. CYP2E1 expression was significantly increased in heart tissues from patients with HCM compared with normal controls (Figure 1A and 1B; n=8 in the normal group, n=14 in the HCM group [P<0.001]). CYP2E1 levels were also significantly increased in myocardium in the LAD ligation‐induced ischemia mouse model (Figure 1C and 1D; n=4 mice in the WT‐LAD group, n=5 mice in the WT‐sham group [P<0.05]), the TAC‐induced cardiomyopathy/HF mouse model (Figure 1C and 1D; n=4 mice in the WT‐TAC group, n=5 mice in the WT‐sham group [P<0.01]), and the adriamycin‐induced DCM mouse model (Figure 1C and 1D; n=4 mice in the WT‐adriamycin group, n=5 mice in the WT‐saline group [P<0.05]), consistent with the results from patients with HCM.
Figure 1.
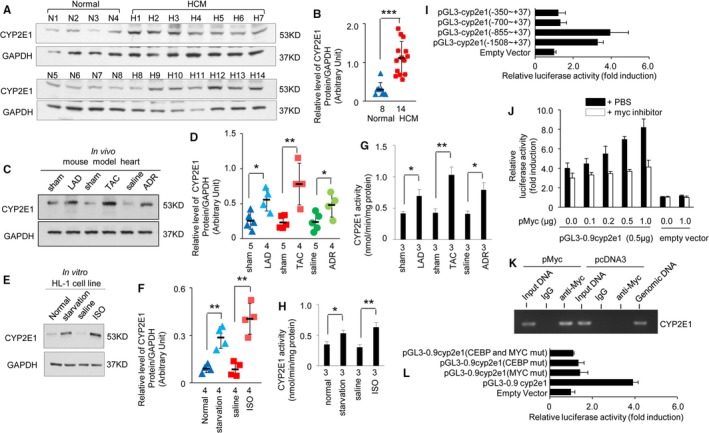
Upregulation of cytochrome P450 2E1 (CYP2E1) in multiple heart diseases and Myc activated CYP2E1 gene promoter. CYP2E1 expression (A) in heart tissues from patients with hypertrophic cardiomyopathy (HCM) (n=14) and normal controls (n=8), and quantitative analysis (B) using GAPDH for normalization (***P<0.001 vs normal). CYP2E1 expression (C and D) in heart tissues of the left anterior descending (LAD) coronary artery–, thoracic aorta constriction (TAC)–, and adriamycin‐induced mouse models, and quantitative analysis using GAPDH for normalization (*P<0.05 or **P<0.01 vs sham or saline). CYP2E1 expression (E and F) in HL‐1 cells under starvation or isoprenaline stress, and quantitative analysis using GAPDH for normalization (**P<0.01 vs normal or saline). CYP2E1 activity in vivo models (G, *P<0.05 or **P<0.01 vs sham or saline) and in vitro models (H, *P<0.05 or **P<0.01 vs normal or saline). Transcriptional activity of a series of truncated CYP2E1 promoter constructs that transfected to HL‐1 cells (I, n=4 independent experiments; see Table S1 for polymerase chain reaction primer sequences). Promoter activities of the p0.9cyp2e1‐luc and pGL3‐Basic construct alone or cotransfected with the indicated amounts of Myc expression plasmid with or without Myc inhibitor 10058‐F4 (J, n=3 independent experiments). Chromatin immunoprecipitation assay on extracts from HL‐1 cells transfected with the Myc expression plasmid or with pcDNA3 plamid as a control (K, n=4 independent experiments). Transcriptional activity of a series of mutated CYP2E1 promoter constructs that transfected to HL‐1 cells (L, n=4 independent experiments).
CYP2E1 was also strongly upregulated in cultured HL‐1 myocytes under stress induced by starvation (Figure 1E and 1F; n=4 independent experiment, cultured without serum for 8 hours [P<0.01 for starvation versus normal]) or isoprenaline treatment (Figure 1E and 1F; n=4 independent experiment, treated with isoprenaline [10 μmol/L] for 12 hours [P<0.01 for isoprenaline versus saline]).
In addition, we analyzed CYP2E1 activity and found that it was in accordance with the level of CYP2E1 protein in vivo mouse models (Figure 1G; n=3, WT‐LAD versus WT‐sham, [P<0.05]; WT‐TAC versus WT‐sham [P<0.01]; WT‐adriamycin versus WT‐saline [P<0.05]) and in vitro HL‐1 cells under stress (Figure 1H; starvation treatment, n=3 independent experiment, cells cultured without serum for 8 hours [P<0.05 for starvation versus normal]; isoprenaline treatment, n=3 independent experiment, cells treated with isoprenaline [10 μmol/L] for 12 hours [P<0.01 for isoprenaline versus saline]).
Myc‐Activated CYP2E1 Gene Promoter
CYP2E1 is upregulated in multiple heart diseases; therefore, clarification of the regulation of the CYP2E1 promotor may provide insights on the pathological development of heart disease.
The promoter region of mouse CYP2E1 was determined by searching for putative transcription factor and binding sites (http://www.cbrc.jp/research/db/TFSEARCH.html). A series of CYP2E1 5′‐flanking region‐luciferase reporter plasmids was then constructed in the back bone of the pGL3 basic vector and transfected into HL‐1 mouse cardiac muscle cells.
The highest transcriptional activation was produced by the −855/+37 construct. The 5′ deletion to −700 resulted in an 88.9% decrease in luciferase activity compared with the −855/+37 construct. However, 5′ deletion to −350 only mildly reduced the luciferase activity compared with the −700/+37 construct (Figure 1I, n=4 independent experiment). These results suggest that the −855 to −700 region of the CYP2E1 promoter is crucial for opening the CYP2E1 promoter.
In silico analysis using bioinformatics provides additional information to identify direct targets of the transcription factor studied, and the functionality of these response elements can be analyzed using promoter gene reporter assays. The proximal promoter sequence of the promoter of interest, the CYP2E1, can be downloaded from the reference mice genome by uploading the promoter sequence and using the promoter search at http://www.cbrc.jp/research/db/TFSEARCH.html. The identified response elements are compared with the consensus sequence. Then, mutagenesis analysis and cell transfection can be performed for visual analysis.
The analysis showed that the −855 to −700 region of the CYP2E1 promoter contains binding sites for Myc and C/EBP, which have been implicated in heart diseases.27, 28, 29, 30 In addition, in astrocytes, lipopolysaccharide induces the activation of MAPK kinase, which subsequently provokes a binding element of C/EBP‐β and ‐δ and mediates the transcriptional activation of CYP2E1.27
Furthermore, we performed coimmunoprecipitation, followed by liquid chromatography coupled with tandem MS to screen the upstream and interacted proteins with CYP2E1, and Myc and C/EBP were both the upstreams with CYP2E1 (Table S3).
To explore the function of Myc in opening the CYP2E1 promoter, pMyc was cotransfected with p0.9Cyp2e1‐luc into HL‐1 cells. The CYP2E1 promoter was activated by the expression of Myc in a dose‐dependent manner, whereas promoter activation was completely inhibited by the Myc inhibitor 10058‐F4 (20 μmol/L for 6 hours; Figure 1J, n=3 independent experiment). Furthermore, pMyc and p0.9Cyp2e1‐luc were cotransfected, and PCR was performed with specific primers after immunoprecipitation of the cross‐linked material with an anti‐Myc antibody. We obtained a band corresponding to sequences of the Myc binding site in CYP2E1 promoter, indicating that Myc binds to the mouse CYP2E1 promoter (Figure 1K, n=3 independent experiments).
To further assess the functional consequences of the loss of the Myc and/or C/EBP binding site in CYP2E1 promoter, the Myc binding site (−846 to −835, CAAGCACATGGA) and the C/EBP binding site (−756 to −743, CATTTTTACAAATA) were mutated in the p0.9Cyp2e1‐luc plasmid. Then, the plasmids carrying the mutated binding sites were individually or simultaneously transfected into the HL‐1 cells. Interestingly, transfection of the p0.9Cyp2e1‐luc plasmid carrying a mutated Myc or C/EBP binding site reduced the transcriptional activity of CYP2E1 by 85.4% and 88.1%, respectively, and simultaneous transfection of the 2 binding site mutants reduced the transcriptional activity of CYP2E1 by 96.3% (Figure 1L, n=4 independent experiment). These results suggest that both C/EBP and Myc are necessary to open the CYP2E1 promoter in myocytes.
Myc Expression is Upregulated in Response to Stress in the Myocardium
To assess the response of Myc and C/EBP expression facing stress in the myocardium, we employed TAC‐ and adriamycin‐treated mouse models, which are used widely to evaluate experiments under stress and injury in the myocardium.31, 32
The TAC‐treated mice displayed typical HCM/HF phenotypes, with smaller chambers, thick‐walled ventricles, cardiac dysfunction, myocytes disarray, and fibrosis (Figure 2A through 2F). The adriamycin‐treated mice displayed typical DCM/HF phenotypes with dilated chambers, thin walls, cardiac dysfunction, myocytes disarray, and fibrosis (Figure 2A, 2F through 2J). The echocardiographic parameters of left ventricular diameter, left ventricular posterior wall thickness, left ventricular fractional shortening, and stroke volume of the TAC‐treated mouse model at 2, 4, and 8 weeks after TAC treatment and the adriamycin‐treated mouse model at 2 weeks after cessation of adriamycin treatment are shown in Tables S4 through S7. The survival analysis of these 2 models is shown in Figure S1.
Figure 2.
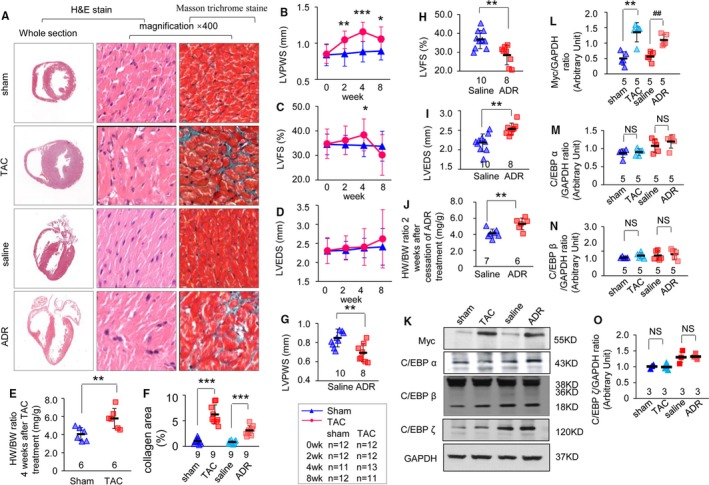
Myc expression is upregulated in response to stress in the myocardium. A, Hematoxylin and eosin (H&E) staining of the whole‐heart transverse sections after thoracic aorta constriction (TAC) or adriamycin treatment (left line). Magnification of H&E‐stained sections of the left ventricle (middle line; magnification ×400, scan bar is 20 μm). Magnification of Masson trichrome–stained left ventricle section (right line; magnification ×400, scan bar is 20 μm). Myocytes stained red and collagen stained green. Echocardiographic parameters of left ventricular posterior wall thickness in systole (LVPWS) (B), left ventricular fractional shortening (LVFS) (C), and left ventricular end‐diastolic diameter (LVEDS) (D) were analyzed in TAC‐treated mice at 0, 2, 4, and 8 weeks after TAC treatment (* P<0.05, **P<0.01, or ***P<0.001 vs sham group). E, Heart to body weight (HW/BW) ratio in TAC‐treated mice (n=6 mice per group, **P<0.001 vs sham group). F, Quantitative analysis of collagen area for mouse models (n=3 mice per group, n=3 microscopic fields per mice; ***P<0.001 vs control group). Echocardiographic parameters of LVPWS (G), LVFS (H), and LVEDS (I) were analyzed in adriamycin‐treated mice at 2 weeks after cessation of adriamycin treatment (n=8 mice in the adriamycin group, n=10 mice in the saline group; **P<0.01 vs saline group). J, HW/BW ratio in adriamycin‐treated mice (n=6 mice in the adriamycin group, n=7 mice in the saline group; **P<0.01 vs saline group). K, Myc, CCAAT enhancer binding protein (C/EBP)‐α, C/EBP‐β, and C/EBP‐ζ expression in heart tissues of mouse models. Quantitative analysis of Myc (L), C/EBP‐α (M), C/EBP‐β (N), and C/EBP‐ζ (O) proteins expression using GAPDH for normalization (n=6 mice per group, **P<0.01 TAC vs sham; ## P<0.01 adriamycin vs saline. NS indicates nonsignificant.
We observed that Myc (Figure 2K and 2L; n=5 mice per group [P<0.01], model versus control) but not C/EBP‐α, C/EBP‐β, or C/EBP‐ζ (Figure 2K, 2M through 2O; n=3 or 5 mice per group) was obviously upregulated in heart tissues from these 2 mice models.
The results presented in Figures 1 and 2 suggest that Myc is the main transcriptional factor that responded to stress and opened the CYP2E1 promoter in the presence of C/EBP in the myocardium.
CYP2E1 Expression is Associated With Myc Expression and ERK1/2 Pathway and PI3K/Akt Pathway Activities Under Stress in the Myocardium
The ERK1/2 and phosphatidylinositol 3 kinase (PI3K)/protein kinase B (Akt) pathway was activated under stress and injury in the myocardium, along with an increase in Myc levels.33, 34
Furthermore, we performed coimmunoprecipitation followed by liquid chromatography coupled with tandem MS to screen the canonical pathways, and ERK/mitogen‐activated protein kinase (MAPK) and PI3K signaling were both interacted with the CYP2E1 (Table S8). All MS/MS data were analyzed using Mascot (Matrix Science; version 2.3.0). Mascot was set up to search the UniProt human database containing 20 199 protein sequences. Mascot was searched with a fragment ion mass tolerance of 0.050 Da and a parent ion tolerance of 10.0 PPM. Carbamidomethyl of cysteine was specified as fixed modifications. Oxidation of methionine was specified in Mascot as variable modifications. Only those proteins with P<0.05 were accepted.
We observed that ERK1/2, PI3K, and Akt phosphorylation were all significantly increased in associated with increased Myc and CYP2E1 levels in heart tissues from both TAC‐ and adriamycin‐treated mice (Figure 3A through 3F; n=5 mice per group, WT‐TAC versus WT‐sham [P<0.01]; WT‐adriamycin versus WT‐saline [P<0.05]).
Figure 3.
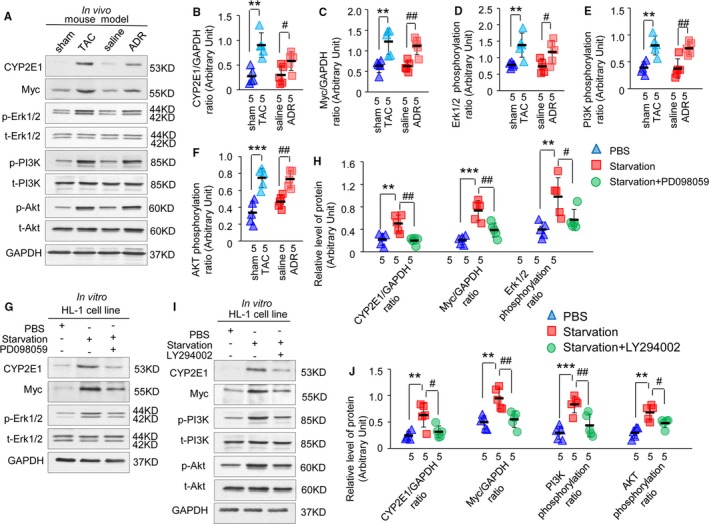
Activation of the extracellular signal–regulated kinase (ERK) 1/2 and phosphatidylinositol 3 kinase (PI3K)/protein kinase B (Akt) pathway is pivotal for the expression of cytochrome P450 2E1 (CYP2E1) and Myc in HL‐1 cells under stress. Expression of CYP2E1, Myc, phosphorylated, and total level of ERK1/2, PI3K, and Akt in heart tissues of thoracic aorta constriction (TAC)– and adriamycin‐induced mouse models (A). Quantitative analysis of protein expression using GAPDH for normalization (B through F, **P<0.01 or ***P<0.001 TAC vs sham; # P<0.05 or ## P<0.01 adriamycin vs saline). Expression of CYP2E1, Myc, phosphorylated, and total level of ERK1/2 in HL‐1 cells under starvation with or without mitogen‐activated protein kinase kinase/ERK inhibitor (G). Quantitative analysis of protein expression using GAPDH for normalization (H, **P<0.01 or ***P<0.001 starvation vs PBS; # P<0.05 or ## P<0.01 starvation+PD098059 vs starvation). Expression of CYP2E1, Myc, phosphorylated, and total level of PI3K and Akt in HL‐1 cells under starvation with or without a PI3K inhibitor (I). Quantitative analysis of protein expression using GAPDH for normalization (J, **P<0.01 or ***P<0.001 starvation vs PBS; # P<0.05 or ## P<0.01 starvation+LY294002 vs starvation).
This result suggested that CYP2E1 expression is associated with activation of the ERK1/2 and PI3K/Akt pathway, which is involved in the regulation of Myc expression in the heart under stress.
Activation of the ERK1/2 and PI3K/Akt Pathway is Pivotal for the Expression of CYP2E1 and Myc in HL‐1 Cells Under Stress
To confirm the relationship between the activities of ERK1/2 and PI3K/Akt signal pathways and the expression of CYP2E1 and Myc, we analyzed the expression of CYP2E1 and Myc in HL‐1 cardiomyocytes treated with an MEK/ERK inhibitor and a PI3K inhibitor.
First, the activities of the ERK1/2 and PI3K/Akt signal pathway and the expression of CYP2E1 and Myc were all upregulated by starvation in HL‐1 cardiomyocytes (Figure 3G through 3J; n=5 independent experiment, P<0.01 or P<0.001 for starvation versus normal), in accordance with the results obtained in vivo (Figure 3A through 3F).
Second, when MEK/ERK phosphorylation was inhibited by PD098059 (MEK/ERK inhibitor, 50 μmol/L, treatment for 1 hour) under starvation, the expression of CYP2E1 and Myc decreased by 60.3% (Figure 3G and 3H; n=5 independent experiment, starvation+PD098059 versus starvation [P<0.01]) and 47.2% (Figure 3G and 3H; n=5 independent experiment, starvation+PD098059 versus starvation [P<0.01]), respectively, compared with starving cells treated without PD098059.
Third, when PI3K phosphorylation was inhibited by LY294002 (PI3K inhibitor, 25 μmol/L, treatment for 1 hour) under starvation, the expression of CYP2E1 and Myc decreased by 50.2% (Figure 3I and 3J; n=5 independent experiment, starvation+LY294002 versus starvation [P<0.05]) and 42.2% (Figure 3I and 3J; n=5 independent experiment, starvation+LY294002 versus starvation [P<0.01]), respectively, compared with starving cells treated without LY294002.
These results suggest that activation of the MEK/ERK and PI3K signal pathway is pivotal in the regulation of CYP2E1 and Myc expression in myocardium under stress and/or injury.
Inhibiting Myc Blunted CYP2E1 and Consequently Reduced Oxidative Stress and Apoptosis
The molecular mechanism involved in CYP2E1 upregulation in the myocardium based on promoter interaction and signal pathway analysis in mice and myocyte models under stress and/or injury were determined above. Then, we further verified whether inhibiting Myc would result in blunting of CYP2E1 and consequently reduced indices of oxidative stress and release of cytochrome c and activation of caspase 3 and 9. We measured H2O2, malondialdehyde, antioxidant glutathione, and T‐AOC, which reflect the level of oxidative stress. H2O2 and malondialdehyde were all increased, while glutathione and T‐AOC were all decreased after the starvation treatment (in a medium without serum for 8 hours). All 4 indexes reversed after the treatment of Myc inhibitor 10058‐F4 (20 μmol/L for 8 hours, Figure 4A through 4D; n=3 independent experiment, starvation versus PBS and starvation+10058‐F4 versus starvation [P<0.05]).
Figure 4.
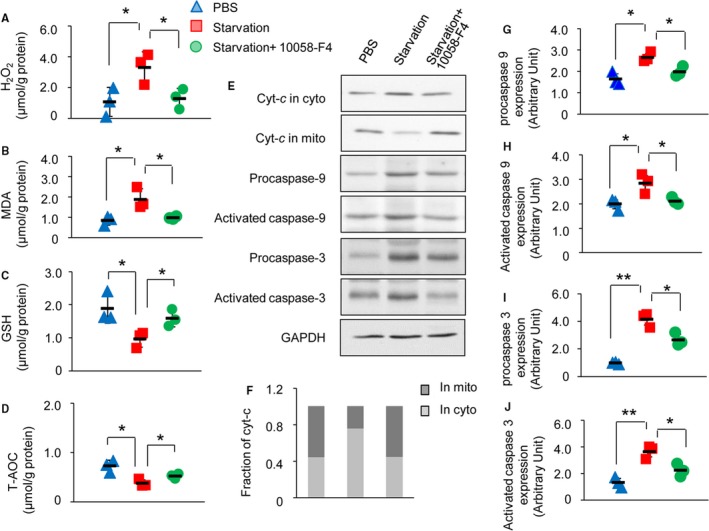
Inhibiting Myc blunted cytochrome P450 2E1 (CYP2E1) and consequently reduced Oxidative stress and apoptosis. Level of H2O2 (A), malondialdehyde (B), glutathione (C), and total antioxidant capacity (T‐AOC) (D) in HL‐1 cells under starvation with or without Myc inhibitor 10058‐F4 (*P<0.05 starvation vs PBS or starvation+100058‐F4 vs starvation). Mitochondrial cytochrome c (Cyt‐c) release and the activation of caspase 9 and caspase 3 were measured by Western blot in lysate from HL‐1 cells under starvation with or without Myc inhibitor 10058‐F4 (E). Quantitative analysis of Cyt‐c in cytoplasm and mitochondria (F). Quantitative analysis of activation of caspase 9 and caspase 3 using GAPDH for normalization (G through J, *P<0.05 or **P<0.01 starvation vs PBS or starvation+100058‐F4 vs starvation).
We then tested the apoptosis relative proteins, release of cytochrome c, and activation of caspase 9 and 3 and they were all increased after the starvation treatment (in a medium without serum for 8 hours). All of these indexes reversed after the treatment of Myc inhibitor 10058‐F4 (20 μmol/L for 8 hours, Figure 4E through 4J; n=3 independent experiment, starvation versus PBS and starvation+10058‐F4 versus starvation [P<0.05 or P<0.01]).
In addition, the characteristics of the pathological role of CYP2E1 in oxidative stress and apoptosis were also determined. We comparatively analyzed ROS production and myocytes apoptosis in CYP2E1‐ov and CYP2E1‐kd mice at 1, 5, and 8 months of age.
CYP2E1 overexpression significantly increased the levels of the oxidants of H2O2 (Figure S2A; n=3 mice per group at each age [P<0.05]) and malondialdehyde (Figure S2B; n=3 mice per group at each age [P<0.05 or P<0.01]), and decreased the levels of the antioxidant glutathione (Figure S2C; n=3 mice per group at each age [P<0.05 or P<0.001]) and T‐AOC (Figure S2D; n=3 mice per group at each age [P<0.05]) in heart tissues, compared with the non‐transgenic group.
By contrast, knockdown of the expression of CYP2E1 resulted in an oxidation‐resistance tendency, especially with respect to glutathione and T‐AOC levels in heart tissues at a young age, compared with the non‐transgenic group (Figure S2A through S2D; n=3 mice per group at each age [P<0.05]).
CYP2E1 overexpression increased ROS production and, in turn, caused progressive myocyte apoptosis. The apoptotic index reached up to 14.60% in CYP2E1‐ov transgenic mice at 8 months of age (Figure S2E and S2F; n=3 mice per group, n=3 slides per mouse, n=9 microscopic fields per slide [P<0.01]), a 7‐fold increase compared with the non‐transgenic group.
By contrast, apoptotic myocytes were rare in both non‐transgenic and CYP2E1‐kd mice, with apoptotic index of 1.74% and 1.13%, respectively, at 8 months of age (Figure S2E and S2F; n=3 mice per group, n=3 slides per mouse, n=9 microscopic fields per slide), Moreover, CYP2E1 knockdown tended to inhibit apoptosis.
Discussion
Our results demonstrated that ERK1/2 and PI3K/Akt pathways and multiple environmental stimulus modulate Myc expression, at least in part. Increased Myc is the key transcriptional factor for opening CYP2E1 promoter especially under stress or injury. Furthermore, CYP2E1 is likely a sensor of diverse pathophysiological factors and states in the myocardium, and upregulated CYP2E1 has multiple pathophysiological roles in the heart, including increased oxidative stress and apoptosis as well as energy supply to meet the energy demand of the heart in certain disease states (Figure 5). Our discovery thus provides a basis for a therapeutic strategy for heart diseases targeting Myc and CYP2E1.
Figure 5.
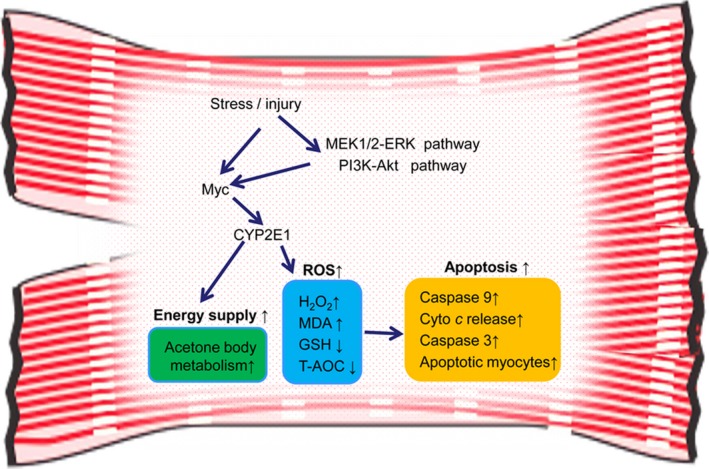
Schematic diagram showing the mechanism of cytochrome P450 2E1 (CYP2E1) expression and its pathophysiological roles in myocardium. Akt indicates protein kinase B; Cyt‐c, cytochrome c; ERK, extracellular signal–regulated kinase; MEK mitogen‐activated protein kinase kinase, PI3K, phosphatidylinositol 3 kinase; T‐AOC, total antioxidant capacity.
Major Findings
CYP2E1 upregulation has been associated with multiple conditions, such as fasting, nutrition intake, and various pathophysiological states, including liver disease, cardiovascular injury, tumorigenesis, neuronal dysfunction, and diabetes mellitus.4, 9, 11, 21 Upregulated expression of CYP2E1 is involved not only in pathways related to energy metabolism but also oxidative damage of heart tissues, with direct importance for heart disease.2, 9, 21, 22, 23, 24, 25, 26, 35
We previously found that CYP2E1 upregulated under multiple pathophysiological status in the myocardium, including cTnTR141W DCM transgenic mice, LmnaE82K DCM transgenic mice, and the adriamycin‐induced DCM/myocardial injury mouse model.3, 21 Consistent with these findings, in the present study, we demonstrated that CYP2E1 levels were also increased significantly in the myocardium of a LAD‐induced ischemia mouse model, TAC‐induced cardiomyopathy/HF mouse model, and HL‐1 myocyte under stress due to starvation or isoprenaline treatment. In addition, we also observed a significant increase in CYP2E1 levels in heart tissues from patients with HCM.
Taken together, these findings suggest that the upregulation of CYP2E1 in the myocardium is a common event in multiple heart diseases and states, including hypertension, cardiomyopathy, myocardial ischemia, starvation, and stress. CYP2E1 is likely a sensor of diverse pathophysiological factors and status in the myocardium.
Another important new perspective provided by this work is that Myc interacted with the CYP2E1 promoter and activated its transcription. Myc and C/EBP were both important transcription factors for opening the CYP2E1 promoter in the myocardium. However, the expression of C/EBP is stable, and Myc exhibited a significant increase under stress or injury. Therefore, Myc was the key transcription factor for opening the CYP2E1 promoter especially under stress or injury.
We then determined that the characteristic of the pathological role that CYP2E1 involved in oxidative stress and apoptosis with age. We found that the extent of oxidative stress worse with age in CYP2E1‐overexpressing transgenic mice, indicated by levels of H2O2, malondialdehyde, antioxidant glutathione, and T‐AOC. By contrast, oxidation resistance was extremely high in CYP2E1 knockdown transgenic mice at a young age, and this resistance decreased with age. The trends in the levels of H2O2, malondialdehyde, glutathione, and T‐AOC in CYP2E1 knockdown mice were opposite of those observed in CYP2E1‐overexpressing mice. Our and others’ findings demonstrate that CYP2E1 overexpression could increase oxidative stress and apoptosis in heart tissues, and energy supply to meet the energy requirement in disease status and the injury degree increases gradually with age. By contrast, CYP2E1 knockdown inhibits this process.
Strengths and Limitations
The activation of both PI3K/Akt and ERK1/2 has been implicated in the association with ischemic preconditioning, cardioprotection, and hypertrophy.36 Both of these signaling pathways and multiple stresses are indicated in the involvement in the upregulation of Myc.33, 37, 38 In recent years, it has been demonstrated that Myc is rapidly upregulated in response to virtually all forms of pathologic stress, and Myc plays an important role in the development of cardiac hypertrophy and HF and energy metabolism in the myocardium.29, 39, 40 However, the mechanism involved in the interaction between Myc and CYP2E1 has not been reported until now. Taken together, our results suggest that Myc participates in apoptosis and energy metabolism, at least in part, through CYP2E1.
The PI3K/Akt pathway and the MAPK pathway are usually known as protective pathways; however, it has been determined that ROS induction is often accompanied by activation of PI3K, and this has been proven by using inhibitors of PI3K and PI3K knockout mice. Therefore, PI3K seems to be commonly involved in the ROS accumulation induced by cytokines and growth factors. ROS, such as H2O2 and oxygen, have emerged as key mediators of intracellular signaling, which is elevated by various types of extracellular stimuli, including growth factors, cytokines, and environmental stresses. It is widely accepted that ROS can modulate cell functions by activating MAPKs, phospholipase C, protein kinase C, and various other types of signaling components. In addition to the role of PI3K in ROS induction, PI3K in various cell types was activated in response to the exogenous application of H2O2. H2O2 stimulation leads to the initiation of downstream signaling events, such as stimulation of phospholipase Cγ2 and MAPK and activation of PI3K. In addition, hydrogen peroxide treatment caused an increase in the amount of p85 PI3K associated with the particulate fraction. Furthermore, upregulated CYP2E1 participated in oxidative stress and apoptosis via bioactivation, leading to pronounced formation of toxic secondary metabolites and ROS, which ultimately induces tissue and cellular damage.7, 8
In addition, our recent research has shown that CYP2E1 is also involved in energy supply in myocardium, and this may be one of the reasons for the activation of ERK1/2 and PI3K/Akt pathways, which usually play a role in the organism. CYP2E1 is involved in the metabolism of endogenous ketones and also participates in the metabolism of gluconeogenesis through ketone bodies. Ketone bodies can be efficient fuel in special disease statuses.41 Thus, CYP2E1 has multiple pathophysiological roles in the heart, including increased oxidative stress and apoptosis as well as energy supply to meet the energy demand of the heart in certain disease states.
Conclusions
ERK1/2 and PI3K/Akt pathways and multiple environmental stimuli modulate Myc expression, at least in part. Increased Myc is the key transcriptional factor for opening the CYP2E1 promoter especially under stress or injury. CYP2E1 has multiple pathophysiological roles in the heart, including increased oxidative stress and apoptosis as well as energy supply to meet the energy demand of the heart in certain disease states. Thus, these findings suggest that targeting CYP2E1 could potentially be a therapeutic strategy for pathological cardiac hypertrophy and HF.
Sources of Funding
This research was supported by the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (CAMS‐I2M, 2016‐I2M‐1‐015), the Beijing Natural Science Foundation (5172027), and the National Natural Science Foundation (31872314).
Disclosures
None.
Supporting information
Table S1. Oligonucleotide/Primer Sequences in this Study
Table S2. Sequencing Results of the Mutated Vectors in this Study
Table S3. Upstream Regulators of CYP2E1 Screened by Co‐IP‐MS
Table S4. Echocardiographic Characteristics of WT Mice 2 Weeks After TAC Treatment
Table S5. Echocardiographic Characteristics of WT Mice 4 Weeks After TAC Treatment
Table S6. Echocardiographic Characteristics of WT Mice 8 Weeks After TAC Treatment
Table S7. Echocardiographic Characteristics of WT Mice 2 Weeks After Cessation of Adriamycin Treatment
Table S8. Canonical Pathways Interacted With CYP2E1 Screened by Co‐IP‐MS/
Figure S1. Cumulative percent mortality for mouse models.
Figure S2. Cytochrome P450 2E1 (CYP2E1) increased oxidative stress and apoptosis in mice.
(J Am Heart Assoc. 2019;8:e009871 DOI: 10.1161/JAHA.118.009871.)
References
- 1. Neve EP, Ingelman‐Sundberg M. Molecular basis for the transport of cytochrome P450 2E1 to the plasma membrane. J Biol Chem. 2000;275:17130–17135. [DOI] [PubMed] [Google Scholar]
- 2. Cederbaum AI. Alcohol metabolism. Clin Liver Dis. 2012;16:667–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lu D, Ma Y, Zhang W, Bao D, Dong W, Lian H, Huang L, Zhang L. Knockdown of cytochrome P450 2E1 inhibits oxidative stress and apoptosis in the cTnT(R141W) dilated cardiomyopathy transgenic mice. Hypertension. 2012;60:81–89. [DOI] [PubMed] [Google Scholar]
- 4. Boussadia B, Ghosh C, Plaud C, Pascussi JM, de Bock F, Rousset MC, Janigro D, Marchi N. Effect of status epilepticus and antiepileptic drugs on CYP2E1 brain expression. Neuroscience. 2014;281:124–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guengerich FP. Cytochrome p450 and chemical toxicology. Chem Res Toxicol. 2008;21:70–83. [DOI] [PubMed] [Google Scholar]
- 6. Tanaka E, Terada M, Misawa S. Cytochrome P450 2E1: its clinical and toxicological role. J Clin Pharm Ther. 2000;25:165–175. [DOI] [PubMed] [Google Scholar]
- 7. Leung TM, Nieto N. CYP2E1 and oxidant stress in alcoholic and non‐alcoholic fatty liver disease. J Hepatol. 2013;58:395–398. [DOI] [PubMed] [Google Scholar]
- 8. Caro AA, Cederbaum AI. Oxidative stress, toxicology, and pharmacology of CYP2E1. Annu Rev Pharmacol Toxicol. 2004;44:27–42. [DOI] [PubMed] [Google Scholar]
- 9. Hunter AL, Cruz RP, Cheyne BM, McManus BM, Granville DJ. Cytochrome p450 enzymes and cardiovascular disease. Can J Physiol Pharmacol. 2004;82:1053–1060. [DOI] [PubMed] [Google Scholar]
- 10. Gonzalez FJ. Role of cytochromes P450 in chemical toxicity and oxidative stress: studies with CYP2E1. Mutat Res. 2005;569:101–110. [DOI] [PubMed] [Google Scholar]
- 11. Shahabi HN, Andersson DR, Nissbrandt H. Cytochrome P450 2E1 in the substantia nigra: relevance for dopaminergic neurotransmission and free radical production. Synapse. 2008;62:379–388. [DOI] [PubMed] [Google Scholar]
- 12. Linhart K, Bartsch H, Seitz HK. The role of reactive oxygen species (ROS) and cytochrome P‐450 2E1 in the generation of carcinogenic etheno‐DNA adducts. Redox Biol. 2014;3:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hong JY, Pan JM, Gonzalez FJ, Gelboin HV, Yang CS. The induction of a specific form of cytochrome P‐450 (P‐450j) by fasting. Biochem Biophys Res Commun. 1987;142:1077–1083. [DOI] [PubMed] [Google Scholar]
- 14. Raucy JL, Lasker JM, Kraner JC, Salazar DE, Lieber CS, Corcoran GB. Induction of cytochrome P450IIE1 in the obese overfed rat. Mol Pharmacol. 1991;39:275–280. [PubMed] [Google Scholar]
- 15. Yoo JS, Ning SM, Pantuck CB, Pantuck EJ, Yang CS. Regulation of hepatic microsomal cytochrome P450IIE1 level by dietary lipids and carbohydrates in rats. J Nutr. 1991;121:959–965. [DOI] [PubMed] [Google Scholar]
- 16. Peng HM, Coon MJ. Regulation of rabbit cytochrome P450 2E1 expression in HepG2 cells by insulin and thyroid hormone. Mol Pharmacol. 1998;54:740–747. [PubMed] [Google Scholar]
- 17. Chen GF, Ronis MJ, Ingelman‐Sundberg M, Badger TM. Hormonal regulation of microsomal cytochrome P4502E1 and P450 reductase in rat liver and kidney. Xenobiotica. 1999;29:437–451. [DOI] [PubMed] [Google Scholar]
- 18. Lee GH, Oh KJ, Kim HR, Han HS, Lee HY, Park KG, Nam KH, Koo SH, Chae HJ. Effect of BI‐1 on insulin resistance through regulation of CYP2E1. Sci Rep. 2016;6:32229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Raghow R. An ‘omics’ perspective on cardiomyopathies and heart failure. Trends Mol Med. 2016;22:813–827. [DOI] [PubMed] [Google Scholar]
- 20. Tayal U, Prasad S, Cook SA. Genetics and genomics of dilated cardiomyopathy and systolic heart failure. Genome Med. 2017;9:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang W, Lu D, Dong W, Zhang L, Zhang X, Quan X, Ma C, Lian H, Zhang L. Exprssion of CYP2E1 increases oxidative stress and induces apoptosis of cardiomyocytes in transgenic mice. FEBS J. 2011;278:1484–1492. [DOI] [PubMed] [Google Scholar]
- 22. Thum T, Borlak J. Cytochrome P450 mono‐oxygenase gene expression and protein activity in cultures of adult cardiomyocytes of the rat. Br J Pharmacol. 2000;130:1745–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thum T, Borlak J. Testosterone, cytochrome P450, and cardiac hypertrophy. FASEB J. 2002;16:1537–1549. [DOI] [PubMed] [Google Scholar]
- 24. Aberle NS II, Ren J. Short‐term acetaldehyde exposure depresses ventricular myocyte contraction: role of cytochrome P450 oxidase, xanthine oxidase, and lipid peroxidation. Alcohol Clin Exp Res. 2003;27:577–583. [DOI] [PubMed] [Google Scholar]
- 25. Sidorik L, Kyyamova R, Bobyk V, Kapustian L, Rozhko O, Vigontina O, Ryabenko D, Danko I, Maksymchuk O, Kovalenko VN, Filonenko VV, Chaschin NA. Molecular chaperone, HSP60, and cytochrome P450 2E1 co‐expression in dilated cardiomyopathy. Cell Biol Int. 2005;29:51–55. [DOI] [PubMed] [Google Scholar]
- 26. Jing L, Jin CM, Li SS, Zhang FM, Yuan L, Li WM, Sang Y, Li S, Zhou LJ. Chronic alcohol intake‐induced oxidative stress and apoptosis: role of CYP2E1 and calpain‐1 in alcoholic cardiomyopathy. Mol Cell Biochem. 2012;359:283–292. [DOI] [PubMed] [Google Scholar]
- 27. Fuchs O, Kostecka A, Provazníková D, Krásná B, Kotlín R, Stanková M, Kobylka P, Dostálová G, Zeman M, Chochola M. CCAAT/enhancer binding protein alpha (CEBPA) polymorphisms and mutations in healthyindividuals and in patients with peripheral artery disease, ischaemic heart disease and hyperlipidaemia. Folia Biol (Praha). 2010;56:51–57. [PubMed] [Google Scholar]
- 28. Olson AK, Ledee D, Iwamoto K, Kajimoto M, O'Kelly Priddy C, Isern N, Portman MA. C‐Myc induced compensated cardiac hypertrophy increases free fatty acid utilization for the citric acid cycle. J Mol Cell Cardiol. 2013;55:156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Redondo‐Angulo I, Mas‐Stachurska A, Sitges M, Giralt M, Villarroya F, Planavila A. C/EBPβ is required in pregnancy‐induced cardiac hypertrophy. Int J Cardiol. 2016;202:819–828. [DOI] [PubMed] [Google Scholar]
- 30. Carvalho FS, Burgeiro A, Garcia R, Moreno AJ, Carvalho RA, Oliveira PJ. Doxorubicin‐induced cardiotoxicity: from bioenergetic failure and cell death to cardiomyopathy. Med Res Rev. 2014;34:106–135. [DOI] [PubMed] [Google Scholar]
- 31. Varga ZV, Ferdinandy P, Liaudet L, Pacher P. Drug‐induced mitochondrial dysfunction and cardiotoxicity. Am J Physiol Heart Circ Physiol. 2015;309:H1453–H1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Appari M, Breitbart A, Brandes F, Szaroszyk M, Froese N, Korf‐Klingebiel M, Mohammadi MM, Grund A, Scharf GM, Wang H, Zwadlo C, Fraccarollo D, Schrameck U, Nemer M, Wong GW, Katus HA, Wollert KC, Müller OJ, Bauersachs J, Heineke J. C1q‐TNF‐related protein‐9 promotes cardiac hypertrophy and failure. Circ Res. 2017;120:66–77. [DOI] [PubMed] [Google Scholar]
- 33. Reuss DE, Mucha J, Hagenlocher C, Ehemann V, Kluwe L, Mautner V, von Deimling A. Sensitivity of malignant peripheral nerve sheath tumor cells to TRAIL is augmented by loss of NF1 through modulation of MYC/MAD and is potentiated by curcumin through induction of ROS. PLoS One. 2013;8:e57152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tan J, Li Z, Lee PL, Guan P, Aau MY, Lee ST, Feng M, Lim CZ, Lee EY, Wee ZN, Lim YC, Karuturi RK, Yu Q. PDK1 signaling toward PLK1‐MYC activation confers oncogenic transformation, tumor‐initiating cell activation, and resistance to mTOR‐targeted therapy. Cancer Discov. 2013;3:1156–1171. [DOI] [PubMed] [Google Scholar]
- 35. Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ Res. 2004;95:135–145. [DOI] [PubMed] [Google Scholar]
- 36. Hausenloy DJ, Iliodromitis EK, Andreadou I, Papalois A, Gritsopoulos G, Anastasiou‐Nana M, Kremastinos DT, Yellon DM. Investigating the signal transduction pathways underlying remote ischemic conditioning in the porcine heart. Cardiovasc Drugs Ther. 2012;26:87–93. [DOI] [PubMed] [Google Scholar]
- 37. Wang Z, Ge L, Wang M, Carr BI. Phosphorylation regulates Myc expression via prolonged activation of the mitogen‐activated protein kinase pathway. J Cell Physiol. 2006;208:133–140. [DOI] [PubMed] [Google Scholar]
- 38. Kalkat M, De Melo J, Hickman KA, Lourenco C, Redel C, Resetca D, Tamachi A, Tu WB, Penn LZ. MYC deregulation in primary human cancers. Genes (Basel). 2017;8:E151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ahuja P, Zhao P, Angelis E, Ruan H, Korge P, Olson A, Wang Y, Jin ES, Jeffrey FM, Portman M, Maclellan WR. Myc controls transcriptional regulation of cardiac metabolism and mitochondrial biogenesis in response to pathological stress in mice. J Clin Invest. 2010;120:1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Le A, Dang CV. Studying Myc's role in metabolism regulation. Methods Mol Biol. 2013;1012:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lommi J, Kupari M, Koskinen P, Näveri H, Leinonen H, Pulkki K, Härkönen M. Blood ketone bodies in congestive heart failure. J Am Coll Cardiol. 1996;28:665–672. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Oligonucleotide/Primer Sequences in this Study
Table S2. Sequencing Results of the Mutated Vectors in this Study
Table S3. Upstream Regulators of CYP2E1 Screened by Co‐IP‐MS
Table S4. Echocardiographic Characteristics of WT Mice 2 Weeks After TAC Treatment
Table S5. Echocardiographic Characteristics of WT Mice 4 Weeks After TAC Treatment
Table S6. Echocardiographic Characteristics of WT Mice 8 Weeks After TAC Treatment
Table S7. Echocardiographic Characteristics of WT Mice 2 Weeks After Cessation of Adriamycin Treatment
Table S8. Canonical Pathways Interacted With CYP2E1 Screened by Co‐IP‐MS/
Figure S1. Cumulative percent mortality for mouse models.
Figure S2. Cytochrome P450 2E1 (CYP2E1) increased oxidative stress and apoptosis in mice.