

Abstract
Background
The molecular mechanisms through which high‐demand pacing induce myocardial dysfunction remain unclear.
Methods and Results
We created atrioventricular block in pigs using dependent right ventricular septal pacing for 6 months. Echocardiography was performed to evaluate dyssynchrony between pacing (n=6) and sham control (n=6) groups. Microarray and enrichment analyses were used to identify differentially expressed genes (DEGs) in the left ventricular (LV) myocardium between pacing and sham control groups. Histopathological and protein changes were also analyzed and an A cell pacing model was also performed. Pacing significantly increased mechanical dyssynchrony. Enrichment analysis using Ingenuity Pathway Analysis and the activation z‐score analysis method demonstrated that there were 5 DEGs (ABCA1, APOD, CLU, LY96, and SERPINF1) in the LV septum (z‐score=−0.447) and 5 DEGs (APOD, CLU, LY96, MSR1, and SERPINF1) in the LV free wall (z‐score=−1.000) inhibited the liver X receptor/retinoid X receptor (LXR/RXR) pathway, and 4 DEGs (ACTA2, MYL1, PPP2R3A, and SNAI2) activated the integrin‐linked kinase (ILK) pathway in the LV septum (z‐score=1.000). The pacing group had a larger cell size, higher degree of myolysis and fibrosis, and increased expression of intracellular lipid, inflammatory cytokines, and apoptotic markers than the sham control group. The causal relationships between pacing and DEGs related to LXR/RXR and ILK pathways, apoptosis, fibrosis, and lipid expression after pacing were confirmed in the cell pacing model. Luciferase reporter assay in the cell pacing model also supported inhibition of the LXR pathway by pacing.
Conclusions
Right ventricular septal‐dependent pacing was associated with persistent LV dyssynchrony–induced cardiomyopathy through inhibition of the LXR/RXR pathway.
Keywords: atrioventricular block, cardiomyopathy, liver X receptor/retinoid X receptor pathway, pacing
Subject Categories: Basic Science Research, Mechanisms, Pathophysiology, Cardiomyopathy
Clinical Perspective
What Is New?
Lipotoxicity was induced by right ventricular septal‐dependent pacing associated with persistent left ventricular dyssynchrony and the inhibition of the liver X receptor/retinoid X receptor pathway was the key mechanism.
What Are the Clinical Implications?
How to reduce lipotoxicity in cardiomyocytes might be a potential strategy to prevent pacemaker‐induced cardiomyopathy, and further studies should be conducted to validate this hypothesis.
Introduction
Cardiac pacing is a common treatment for patients with atrioventricular block, and high‐demand pacing is always noted in such patients. However, results from several large randomized clinical trials have suggested that long‐term right ventricular (RV) pacing may elicit left ventricular (LV) systolic dysfunction, manifesting in an iatrogenic form of heart failure and increasing the risks of hospitalization and death. The DAVID (Dual Chamber and VVI Implantable Defibrillator)1 and MADIT(Multicenter Automatic Defibrillator Implantation) II2 trials have indicated that frequent RV pacing (>40% of the pacing time) resulted in significantly worse outcomes in implantable cardioverter defibrillation recipients with poor LV systolic function. In patients with preserved LV systolic function, the MOST (The mode selection trial)3 trial suggested that frequent RV pacing can lead to heart failure hospitalization and atrial fibrillation. Accordingly, pacemaker‐induced heart failure has generated increased attention in relation to patients who are pacemaker dependent.
Pacemaker‐induced cardiomyopathy (PICM) is a recently created term for LV systolic dysfunction induced by cardiac pacing and is defined as at least 10% decrease in the LV systolic function or <50% of LV ejection fraction after excluding other causes of cardiomyopathy.4 According to some observation studies, the prevalence of PICM was 10% to 20% in patients who received a pacemaker implantation because of an atrioventricular block.5 Therefore, several clinical and animal studies have been conducted to clarify the mechanisms of PICM and thus prevent its development in patients who are pacemaker dependent. LV mechanical dyssynchrony caused by RV pacing is generally considered to be a precursor of PICM. Several studies have reported an increase in mechanical dyssynchrony and reduced LV ejection fraction following acute or chronic RV apical pacing using real‐time 3‐dimensional echocardiography6, 7 or strain‐rate echocardiography.8 Some studies have reported that RV septal pacing is superior to RV apical pacing in terms of mechanical dyssynchrony9 and LV systolic function, but other studies have suggested no difference in the outcome.8, 10, 11 In recent clinical practice, RV septal pacing has been the most favorable pacing site. Animal studies have demonstrated that altered electrical activation, redistribution of blood flow, myocardial work and oxygen uptake, collagen degradation, cardiomyocyte apoptosis, and fibrosis induced by RV apical pacing contribute to PICM.12, 13 However, the molecular pathway and genetic analyses in these animal studies only examined inflammatory‐ and structure‐related proteins and genes.14 A systemic elucidation of the molecular regulatory mechanisms of myocardial remodeling associated with mechanical dyssynchrony induced by RV pacing has never been attempted. For this reason, we conducted this porcine RV septal‐dependent pacing study using high‐density oligonucleotide microarrays and functional network enrichment analysis to identify the molecular regulatory mechanisms of LV myocardial remodeling associated with persistent mechanical dyssynchrony induced by RV septal pacing.
Methods
The authors declare that all supporting data are available within the article and its online supplementary files.
Study Animals and the Creation of a RV Septal‐Dependent Pacing Model
This study enrolled 12 female Lanyu miniature pigs, which were divided into 2 groups: a pacing group (n=6) and a sham control group (n=6). All pigs received appropriate care as defined by the Guide for the Care and Use of Laboratory Animals, published by Taiwan's National Institutes of Health (NIH publication No. 85‐23, revised in 1996). All procedures and echocardiography measurements were performed with animals under anesthesia and after food restriction for 12 hours and water restriction for 4 hours. All pigs received a surgical procedure for the creation of a pacemaker‐dependent model at an average age of 6 to 7 months and were euthanized at the follow‐up 6 months after the surgery. The procedures are briefly described, and a flowchart of the study procedure is shown in Figure 1.
Figure 1.

Flowchart of the creation of a right ventricular septal‐dependent pig pacing model, echocardiographic examination, microarray analysis, functional network enrichment analysis and genes, proteins, and histochemical studies. Echocardiograms were performed twice, first within 1 wk after pacemaker implantation and then within 1 wk before all pigs were euthanized at the 6‐mo follow‐up in the pacing and sham control groups. Atrioventricular (AV) nodal ablation and generator placement for pacing (in ventricular pacing and dual chamber sensing [VDD] mode) were only performed in the pacing group. In both groups, the atrial (A) lead was placed in the right atrial appendage (RAA) and the ventricular (V) lead was placed in the right ventricular (RV) septum. Myocardial tissues of the septum and free wall of the left ventricle (LV) at the level of the papillary muscle were dissected and collected after euthanasia at the 6‐mo follow‐up in the pacing and sham control groups and some tissues were stored in optimum cutting temperature compound (OCT) for histochemical studies. The midmyocardial layer of the myocardial tissues was sent for histochemical and microarray analysis, and Western blotting/polymerase chain reaction (PCR) studies. BW indicates body weight.
Placement of Pacemaker and Leads and Creation of an Atrioventricular Nodal Block
All pigs received the surgical procedure under general anesthesia with 2.0% to 4.0% isoflurane through endotracheal intubation and premedication with atropine (1 mg/kg), ketamine (12 mg/kg), and xylazine (1.2 mg/kg) by intramuscular injection. After general anesthesia was performed, an ear vein was punctured for the placement of a catheter for an intravenous infusion with 500 mg of cefamezine, and then the pacemaker and lead placement procedure followed. A pacemaker pocket was created over the left paratracheal area and was ≈4 cm in length. Pacing leads were inserted into an external jugular vein using the cutdown method. A RV screw‐in bipolar lead (St Jude) was first inserted into the RV under fluoroscopic guidance and was fixed over the RV septum (Figure S1) after 2 criteria were met—one was that the threshold was <1 V under the pulse width of 0.4 ms and the R‐wave amplitude was higher than 4 mV; the other was that the electric axes of leads I, II, and III were positive. A right atrial screw‐in bipolar lead (St Jude) was then inserted into the right atrium and fixed over the right appendage under fluoroscopic guidance (Figure S1) after confirming that the threshold was <1 V at the pulse width of 0.4 ms and the P‐wave amplitude was higher than 1 mV. After the extravenous portions of the leads were fixed over the paratracheal muscle and the generator was connected to the leads, 1 g of vancomycin powder was scattered over the pacemaker pocket. A 2‐layer method was used to close the surgical wound. The generator was set to the VDD (ventricular pacing and dual sensing) mode and synchronized to the pig's physiological sinus rate; the output setting was 2 times the threshold; and the sensitivity setting was 0.5 times the P or R wave.
After RV lead placement, atrioventricular nodal ablation was performed using a thermocontrol ablation catheter under fluoroscopic and electrophysiological guidance (the setting was 50 W and 60°C) until a complete atrioventricular block was achieved.
Transthoracic Echocardiography
All echocardiographic procedures were performed using a commercially available echocardiography system (Vivid 7; GE‐Vingmed, Horten, Norway). Before transthoracic echocardiography measurements were performed, each animal was anesthetized with atropine (1 mg/kg), ketamine (12 mg/kg), and xylazine (1.2 mg/kg) by intramuscular injection. Two‐dimensional strain and M‐mode echocardiography was performed in the right parasternal area at a left lateral decubitus position. The LV end‐diastolic diameter, LV end‐systolic diameter, and end‐diastolic and end‐systolic thickness of the LV posterior wall were measured using the M‐mode under the right parasternal long‐axis view, as is recommended by the American Society for Echocardiography.15 The LV mass index and LV ejection fraction were computed.16 Intraventricular dyssynchrony was assessed and analyzed using septal‐to‐posterior wall motion delay (SPWMD) in the M‐mode, and 2‐dimensional strain echocardiographic analysis was performed using grayscale images in the midventricular (papillary muscle layer) short‐axis view (60–90 frames per second). The reference point was placed at the beginning of the QRS wave of the ECG. Radial strain dyssynchrony was assessed as the difference between the septal and free wall time‐to‐peak radial strain at midventricular segments.
Specimen Storage
The LV free wall and septal tissues at the papillary muscle level of the pigs were obtained after euthanasia under general anesthesia. Some ventricular tissues were immediately frozen in liquid nitrogen at −80°C for ribonucleic acid (RNA) analyses. Additionally, some ventricular tissues were placed into a tissue Tek container, which was then filled with tissue Tek optimum cutting temperature compound (Sakura Finetek, California); these samples were frozen in liquid nitrogen for later histochemical analysis, and some were immediately fixed in 3.7% buffered formalin and then embedded in paraffin for histological study.
RNA Isolation
RNAs were extracted from the myocardial tissue using a RiboPure kit (Ambion, Grand Island, New York) according to the manufacturer's protocol. The quality of RNA was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies Inc, Santa Clara, California), and samples with an optical density ratio 260/280 >1.8 and RNA integrity number >7.0 were selected for microarray processing and real‐time reverse transcriptase–polymerase chain reaction.
Microarray Analysis and Enrichment Analysis
The Sus scrofa gene (mRNA) was used for microarray analysis on a Porcine Gene 1.0 ST Array (Affymetrix, Santa Clara, California). The arrays were scanned using a GeneChipR Scanner 3000 7G (Affymetrix). Microarray quality control and normalization was performed using the affyQCReport R package and rma function of the affy R package.17 Differentially expressed genes (DEGs) with a fold change of ≥1.5 (Log2FC >0.58) or <0.67 (Log2FC < −0.58) were selected, and functional classes were assigned to all known genes using information from the Gene Ontology database available at the Gene Ontology consortium website (http://amigo.geneontology.org/amigo). Ingenuity Pathway Analysis was used for functional enrichment analysis. The activation states (increased or decreased) of the pathways affected by DEGs were assessed using the z‐score analysis method.18 We adopted a statistical approach by defining a quantity (z‐score) that determined whether a biological function exhibited significantly more “increased” predictions than “decreased” predictions (z>0) or vice versa (z<0).
Quantitative Determination of RNAs Using Real‐Time Polymerase Chain Reaction
The RNA samples were quantified using a spectrophotometer. First‐strand cDNAs were synthesized using reverse transcriptase and oligo (dT) primers. Real‐time quantitative polymerase chain reaction was performed using the ABI Prism 7500 FAST sequence detection system (Applied Biosystems, California) and SYBR Green PCR Master Mix (Applied Biosystems). The RNA results were normalized against glyceraldehyde 3‐phosphate dehydrogenase gene expressions (the endogenous control). We selected 10 genes that were differentially upregulated or downregulated by RV pacing in the microarray analysis and obtained gene expression levels for all specimens using real‐time quantitative polymerase chain reaction. Selected genes and primer sequences are presented in Table S1.
Western Blotting
Protein extracts of ventricular tissue were prepared using a PRO‐PREP protein extraction solution (Intron Biotechnology, Gyeonggi‐do, Korea). Homogenates were centrifuged at 20 817 g for 30 minutes at 4°C to yield supernatants. The concentrations of sample proteins were determined using the Bradford method (Bio‐Rad Inc, Hercules, California) according to the supplier's instructions. Recombinant human tumor necrosis factor‐α protein (Abcam, Cambridge, Massachusetts), HeLa whole cell lysate (Santa Cruz, Texas), Ramos cell lysate (Santa Cruz), rat liver tissue lysate (Santa Cruz), HT‐1080 whole cell lysate (Santa Cruz), HeLa staurosporine‐treated cell lysate (Abcam), and Jurkat apoptosis cell lysate (Cell Signaling Technology, Danvers, Massachusetts) served as positive controls. Protein extracts (30 μg) were electrophoresed on 10% to 15% acrylamide SDS‐PAGE gel at room temperature for 1 hour and electrotransferred onto Polyvinylidene difluoride (PVDF) membranes for 1.5 hours on ice. The membranes were blocked at room temperature for 1 hour in Tris‐buffered saline containing 0.1% Tween‐20 and 5% (w/v) nonfat dry milk or 2% (w/v) bovine serum albumin. The primary antibodies—including anti‐tumor necrosis factor‐α (1:1000 dilution; Abcam), interleukin‐6 (1:1000 dilution; Santa Cruz), interleukin‐10 (1:500 dilution; Biorbyt, Carrickfergus, UK), C‐reactive protein (1:500 dilution; Biorbyt), fibronectin (1:500 dilution; Santa Cruz), cleaved caspase 3 (pig tissues: 1:1000 dilution; rat ventricular cardiomyocytes [RV‐40 strain]: 1:500 dilution; Abcam), cleaved caspase 8 (1:1000 dilution; Santa Cruz), and cleaved caspase 9 (1:1000 dilution; Biovision, Milpitas, CA)—were used to react with the blots at 4°C overnight in 5% nonfat dry milk or 2% bovine serum albumin. The blots were washed 3 times in Tris‐buffered saline containing 0.1% Tween‐20 and incubated at room temperature for 1 hour with horseradish peroxidase–labeled secondary antibody at dilutions of 1:5000 in Tris‐buffered saline containing 0.1% Tween‐20 containing 5% nonfat dry milk or 2% bovine serum albumin. Following 3 washes, blots were incubated with Immobilon Western chemiluminescent HRP substrate (Millipore, Burlington, Massachusetts). All specific values of evaluated proteins were standardized to anti‐α‐sarcomeric actin antibody (1:10 000 dilution; Sigma Aldrich, St Louis, Missouri). Chemiluminescence was quantified using a BioSpectrum 810 imaging system (UVP) (Analytik Jena, Germany).
Histological Analysis
LV tissues obtained from the free wall and septum were deparaffinized in xylene and rehydrated in decreasing concentrations of alcohol. Slides were then stained with hematoxylin and eosin. Tissue sections were observed under an Olympus BX51 microscope, with the analyses including at least 100 randomly selected cells under ×400 magnification. All specimen images were captured using an Olympus DP70 camera, and cardiomyocytes were subsequently analyzed (UTHSCSA, Image tool, version 3.0).
Masson's Trichrome Staining
LV tissue sections were tested using a modified Masson's trichrome stain kit (ScyTek Laboratories, Inc, Logan, Utah) according to the manufacturer's directions. Briefly, 5‐μm sections were deparaffinized and fixed with Bouin's solution, stained with Weigert's iron hematoxylin solution, incubated with Biebrich scarlet/acid fuchsin solution in a phosphomolybdic/phosphotungstic acid solution, and then incubated with aniline blue and acetic acid. After dehydration, sections were mounted and visualized using an Olympus DP70 microscope. The percentage of the positive‐stained area of fibrosis was determined using Image Pro Plus 6.0 software (Media Cybernetics, Silver Spring, Maryland).
Oil Red O Staining
Pig LV tissue sections and rat ventricular cardiomyocytes (RV‐40 strain) were subjected to an Oil red O stain kit (ScyTek Laboratories, Inc) according to the manufacturer's directions. Sections were mounted and visualized using an Olympus DP70 microscope (for pig LV tissues) and a Leica Dmi3000 microscope (for rat ventricular cardiomyocytes). Lipid (neural fat stained by Oil red O) was quantified by analyzing the magnified (×40) images using Cellsens Dimension software (Olympus, Tokyo, Japan) and counting the number of red stain pixels, using porcine fat for the positive control.
Cell Culture and Pacing Model
Rat ventricular cardiomyocytes (RV‐40 strain) were cultured in Prigrow III Medium (ABM Inc, Canada). Culture medium was supplemented with 10% (vol/vol) fetal bovine serum and 1% penicillin/streptomycin. Cells were incubated at 37°C in a humidified atmosphere of 5% CO2 and 95% air. After cells reached 80% confluence, cells received nonpacing or pacing at rates of 0.5, 1.5, and 3 Hz at the output of 1 V and pulse width of 0.4 ms by using a C‐PACE EP culture pacer (IonOptix Corporation, Massachusetts) for 24 hours. Each pacing condition per experiment was tested in quadruple and each experiment was repeated 3 times.
Measurement of Lipids Using Flow Cytometry
Nile red (Sigma Aldrich) was used to stain lipids. Rat ventricular cardiomyocytes were plated in each well of 4‐well plates and allowed to attach for 24 hours. After being paced for 24 hours, the cells were harvested through trypsinization, washed in PBS, and suspended in 5 μg/mL Nile red. After incubation for 15 minutes at 40°C, the cells were washed 3 times and suspended in PBS. To determine the intracellular Nile red content using flow cytometry, 10 000 cells per sample were analyzed using the Cytomic FC500 (Beckman Coulter, Indiana). The fraction of RV‐40 ventricular myocytes expressing Nile red was determined in 10 000 sorted myocytes. Nile red fluorescence was collected through a 530±30 nm wavelength.
Luciferase Reporter Assay
Rat ventricular cardiomyocyte (RV‐40 strain) were transfected with a fixed amount of plasmid DNA using Lipofectamine 2000 (Invitrogen, California) according to the manufacturer's instructions. The plasmid was pGreenFire1‐LXRE in ABCA1 (System Biosciences, California). The pGreenFire1‐LXRE‐in‐ABCA1 Lentivector co‐expresses a destabilized copepod GFP and luciferase from the LXR response elements and neighboring regions in the ABCA1 promoter paired with a minimal CMV promoter. Pacing protocol in the pacing groups followed the cell pacing model. After 24 hours, harvested ventricular cardiomyocytes were washed with PBS and lysed in 200 μL of 1 Glo Lysis Buffer (Promega, Wisconsin) using a Dual‐Glo Luciferase Reporter Assay System kit (Promega). Briefly, 75 μL of the same aliquot of ventricular cardiomyocytes lysate was transferred to a 96‐well white microtiter plate, and 75 μL of each luciferase substrate was added simultaneously with a multichannel pipette. The firefly luminescence was measured in EnSpire Multi mode reader (PerkinElmer, Massachusetts) after cell lysis was completed. In addition, pGF‐mCMV (vector only)/pRL‐TK vector (renilla luciferase) served as a negative and internal control and the ventricular cardiomyocytes treated with LXR‐agonist (1 μmol/L, T090131; Sigma, MO) but without pacing served as a positive control. The LXR activity (firefly/renilla) at the ABCA1 promoter of ventricular cardiomyocytes of each experiment was standardized to pGF‐mCMV. The LXR activity of ventricular cardiomyocytes in each pacing group was normalized against the nonpacing group. Each experiment was tested in triplicate and each experiment was repeated 5 times.
Statistical Analysis
Data are presented as the mean±SD or SEM. Mann–Whitney U test was used to compare the pacing group and the sham control group. Wilcoxon signed rank test was used to compare 2 repeated measurements within the pacing group or the sham control group. Friedman test was used to detect differences in 3 or more repeated measurements within each group. Statistical analysis was performed using commercial statistical software (SPSS for Windows, version 13; SPSS Inc, Chicago, Illinois). A P value (2‐tailed) of <0.05 was considered statistically significant.
Results
Study Animals
This study enrolled 6 female pigs in the pacing group, with an average age of 7.2±0.7 months, and 6 female pigs in the sham control group, with an average age of 6.0±0.6 months. During the operation for pacing model creation, the mean body weight was 31.8±1.60 kg in the sham control group and 30.7±0.92 kg in the pacing group (P=0.873). All pigs were euthanized at follow‐up 6 months after the creation of the pacing model and during these 6 months their feeding was unlimited. On the day of euthanizing, there was no significant difference in body weight between the 2 groups (mean body weight was 42.2±2.13 kg in the sham control and 41.5±2.06 kg in the pacing group; P=0.522).
Echocardiographic Parameters
Echocardiograms were performed twice, first within 1 week after pacemaker implantation and then within 1 week before all pigs were euthanized. During the perioperative period, the heart rate was 97.2±4.6 bpm in the sham control and 100.0±5.9 bpm in the pacing group (P=0.872). In the pre‐euthanizing period, the heart rate was 93.50±7.55 bpm in the sham control and 91.8±2.74 bpm in the pacing group (P=0.261). The LV end‐diastolic diameter of the pacing group at 6‐month follow‐up was significantly larger than that at the baseline (P=0.028), and this was not observed in the sham control group (Figure 2A). No significant change in LV mass index was identified after the 6‐month observation period in the sham control group, whereas LV mass index had increased in the pacing group (Figure 2B). No difference in LV systolic function was measured after the 6‐month observation period in either group (Figure 2C). Dyssynchrony parameters, including SPWMD and septal to free wall radial strain over the papillary muscle level, were measured to determine dyssynchrony induced by pacing. The pacing group had significantly increased dyssynchrony parameters compared with the sham control group in the immediate postoperative period (SPWMD: −73.67±14.98 versus 5.50±5.869 ms, P=0.004; radial strain: −50.33±9.77 versus −2.00±6.56 ms, P=0.010). Likewise, the pacing group exhibited significantly higher dyssynchrony parameters compared with the sham control group at the 6‐month follow‐up before euthanizing (SPWMD: −76.00±6.23 versus −10.83±5.52 ms, P=0.004; radial strain: −64.67±16.64 versus 16.17±14.15 ms, P=0.005) (Figure 2D and 2E). However, no significant change in dyssynchrony parameters, including SPWMD and septal‐to‐free‐wall radial strain, was identified after the 6‐month observation period in the pacing group.
Figure 2.

Echocardiography showed increased left ventricular (LV) size, mass, and dyssynchrony induced by 6 mo of right ventricular septal‐dependent pacing. A, The left ventricular end‐diastolic diameter (LVEDD) of the pacing group at 6‐mo follow‐up was significantly larger than that at the baseline (P=0.028). B, LV mass index increased in the pacing group at 6‐mo follow‐up compared with baseline. C, No difference in LV ejection fraction (LVEF) was measured after the 6‐mo observation period in the pacing and sham control groups. D, The pacing group had significantly increased septal‐to‐posterior wall motion delay (SPWMD) compared with the sham control group in the immediate postoperative period (P=0.004). Likewise, the pacing group exhibited significantly increased SPWMD compared with the sham control group at the 6‐mo follow‐up before euthanasia (P=0.004). However, no significant change in SPWMD was identified after the 6‐mo observation period in the pacing group. E, The pacing group had significantly increased septal‐to‐free‐wall radial strain compared with the sham control group in the immediate postoperative period (P=0.010). Likewise, the pacing group exhibited significantly increased septal‐to‐free‐wall radial strain compared with the sham control group at the 6‐mo follow‐up before euthanasia (P=0.005). However, no significant change in septal‐to‐free‐wall radial strain was identified after the 6‐mo observation period in the pacing group. Gray bar: sham control group; black bar: pacing group.
Microarray Analysis and Functional Enrichment Analysis: Inhibition of LXR/RXR Pathway and Activation of ILK Signaling Pathway in the LV Myocardium Following 6 Months of Pacing
The midmyocardial layer of the septum and free wall of the LV at the level of the papillary muscle were dissected after euthanasia at 6‐month follow‐up in the pacing and sham control groups. A total of 19 212 Sus scrofa genes were used to identify the gene expression profile in the LV tissues of the sham control and pacing groups. To identify the DEGs in the LV myocardium after pacing, the gene expression profile in the LV tissues of septum and free wall derived from 3 sham control pigs were compared with LV tissues from comparable locations derived from 3 pacing pigs through microarray analysis. A total of 301 genes were identified as exhibiting more than 1.5‐fold changes in LV septal tissues; 148 of these genes were differentially upregulated (Log2FC [pacing/sham control] >0.58) (Table S2), and 153 were differentially downregulated (Log2FC < −0.58) (Table S3). A total of 293 genes were identified as exhibiting 1.5‐fold changes in LV free‐wall tissues; 119 genes were differentially upregulated (Log2FC >0.58) (Table S4), and 174 genes were differentially downregulated (Log2FC < −0.58) (Table S5). The heat map graphs are depicted in Figure 3A and 3B.
Figure 3.

Inhibition of the LXR/RXR pathway and activation of ILK signaling pathway induced by 6 mo of right ventricular septal‐dependent pacing. A, Unsupervised hierarchical clustering of RNA microarray expression sequence tags in the left ventricular septum of the pacing group and sham control group. A total of 301 genes were identified, of which 148 were differentially upregulated and 153 were differentially downregulated. B, Unsupervised hierarchical clustering of RNA microarray expression sequence tags in the left ventricular free wall of the pacing group and sham control group. A total of 293 genes were identified, of which 119 were differentially upregulated and 174 genes were differentially downregulated. Bar color indicates mRNA expression level. Red indicates upregulation; black, no change; green, downregulation. C, Activation z‐score analysis method was used to measure activation states of the canonical pathways affected by differentially expressed genes in the left ventricular septum. Only 2 pathways were identified to present z‐scores: the LXR/RXR and ILK signaling pathways (z‐score=−0.447 and 1.000, respectively); that is, the LXR/RXR pathway was inhibited and the ILK signaling was activated. D, Only the LXR/RXR pathway was identified to exhibit a z‐score in the left ventricular free wall; that is, the LXR/RXR pathway was inhibited (z‐score=−1.000). ILK indicates integrin‐linked kinase; LXR/RXR, liver X receptor/retinoid X receptor.
To elucidate the molecular mechanisms of RV septal pacing on LV gene expression, we used Ingenuity Pathway Analysis (Qiagen, Hilden, Germany) for functional enrichment analysis. Networks with the highest scores (P‐score=42, ie, P<10−42) were generated from 301 DEGs from the LV septum and 293 DEGs from the LV free wall using the Ingenuity Pathway Analysis Global Molecular Network algorithm, as depicted in Figure S2.
The most commonly involved canonical pathways, diseases, and functions of DEGs in LV sputum included liver X receptor/retinoid X receptor (LXR/RXR) activation, lipid metabolism, integrin‐linked kinase (ILK) signaling, and apoptosis, whereas the most commonly involved canonical pathways, diseases, and functions of DEGs in LV free wall included LXR/RXR activation, metabolism, cardiac hypertrophy, and calcium signaling (Table 1). These results demonstrated that the molecular mechanisms in both the LV free wall and LV septum following 6 months of RV septal pacing were significantly associated with lipid metabolism, whereas others related to ventricular structural remodeling. Furthermore, we used activation z‐score analysis to measure the activation states (increased or decreased) of the pathways affected by the DEGs, and only 2 pathways in the LV septum presented z‐scores: inhibition of the LXR/RXR pathway, and activation of ILK signaling (z‐score=−0.447 and 1.000, respectively) (Figure 3C). The canonical pathways of LXR/RXR and ILK signaling are depicted in Figure S3. As shown in Table 2, 5 DEGs (ABCA1, APOD, CLU, LY96, and SERPINF1) significantly involved in the LXR/RXR pathway in the LV septum had an activation z‐score=−0.447, which suggested that these 5 genes mildly inhibited the LXR/RXR activation pathway. Four DEGs (ACTA2, MYL1, PPP2R3A, and SNAI2) significantly involved in the ILK signaling pathway in the LV septum had an activation z‐score=1.000, which suggested that these 4 genes moderately activated the ILK signaling pathway. We similarly used the activation z‐score analysis method to determine the activation states of the pathways affected by the DEGs in the LV free wall and discovered that only the LXR/RXR pathway had a z‐score=−1.000 (Figure 3D), which suggested that APOD, CLU, LY96, MSR1, and SERPINF1 genes moderately inhibited the LXR/RXR pathway in the LV free wall (Table 2). The canonical LXR/RXR pathway is depicted in Figure S4.
Table 1.
Top Involved Canonical Pathways and Top Diseases and Functions in the Network Between Pacing and Sham Control Groups Using Ingenuity Pathway Analysis Global Molecular Network Algorithm
| Location | Top Canonical Pathways and Diseases and Functions | Involved Genes | P Value |
|---|---|---|---|
| Septum* | LXR/RXR activation | LY96, SERPINF1, CLU, ABCA1, APOD | 4.47E‐04 |
| Clathrin‐mediated endocytosis signaling | OR1A1, ACTA2, FGF7, CLU, APOD | 2.95E‐03 | |
| ILK signaling | SNAI2, ACTA2, PPP2R3A, MYL1 | 1.70E‐02 | |
| Atherosclerosis signaling | VCAM1, CLU, APOD | 2.75E‐02 | |
| Lipid metabolism, metabolic disease, and adipose tissue | ABCA1, ARNTL, CES1, CLU, EPHX2, GNMT, INSR, POMC, VCAM1, MSTN, PLIN2, PRLR, RDH16, APOD, FGF7, PTGFR, XDH, SLCO1A2, ACTA2, CCK, HK2, IL15, KCNA5, MME, P2RY1, ROBO2, SERPINF1, TGFBI, TSPO, ZBTB12, ZNRD1 | 2.53E‐06 to 1.00E‐04 | |
| Cell death and survival (apoptosis) | ABCA1, ALDOC, APOD,CCK, CES1, CLU, CXCL2, DOCK8, EPHX1, EPHX2, EYA4, FAP, FBXO32, FGF7, FOXF2, FRZB, GNMT, HK2, IFNA1/IFNA13, IL15, INSR, KCNA5, KLF8, LUM, MME, MSTN, NDEL1, NTRK3, OR10A3, PLK2, POMC, PPP2R3A, PRLR, PTGFR, RASGRP2, RPS6KL1, SERPINF1, SLC22A3, SNAI2, STC1, TDP2, TSPO, VCAM1, XDH | 9.54E‐05 to 1.22E‐03 | |
| Cellular movement | ACTA2, CCK, CCL21, CLU, CXCL2, DOCK8, FAP, FGF7, IL15, INSR, KLF8, LUM, LY96, MME, NDEL1, NTRK3, P2RY1, PHACTR1, POMC, RASGRP2, ROBO2, SERPINF1, SNAI2, STC1, TDP2, TGFBI, TSPO, VCAM1 | 1.63E‐04 to 2.25E‐04 | |
| Cardiovascular disease (atherosclerosis) | ABCA1, ALDH5A1, CES1, CLU, CXCL2, EPHX2, P2RY1, PLIN2, VCAM1, XDH | 7.40E‐04 | |
| Free wall† | LXR/RXR Activation | LY96, MSR1, SERPINF1, CLU, APOD | 1.82E‐04 |
| Clathrin‐mediated endocytosis signaling | OR1A1, TFRC, FGF7, CLU, APOD | 1.26E‐03 | |
| Calcium signaling | RCAN1,TRDN, MYL1 | ||
| Cardiac hypertrophy | ARNTL, CSRP3, EPOR, LUM, MSTN, RCAN1, EPOR, mir‐30, RCAN1, NOX4, CLU, EPHX2, LOX, NTRK3, SDC4, TFRC, UTF1,TRDN | 2.05E02 to 4.20E‐05 | |
| Cell‐to‐cell signaling and interaction | F5, GYLTL1B, HRH4, LOX, LY96, MSR1, SDC4, SERPINF1, TFRC | 5.96E‐04 to 4.34E‐03 | |
| Metabolic disease | APOD, CLU, MSR1, PTGFR, ROBO2, SERPINF1, XDH, ARNTL, CCDC12, CTH, CYP4A11, EPOR, mir‐30, MS4A6A, MSR1, NOX4, TRIM31, ZNRD1 | 4.45E‐05 to 7.01E‐03 |
ILK indicates integrin‐linked kinase; LXR/RXR, liver X receptor/retinoid X receptor.
The canonical pathways/diseases/functions in the network derived from 301 differentially expressed genes of left ventricular septum.
The canonical pathways/diseases/functions in the network derived from 293 differentially expressed genes of left ventricular free wall.
Table 2.
Log2 Fold Change Values and Predictive Activity of the Differentially Expressed Genes Significantly Involved in LXR/RXR Activation and ILK Signaling Pathways
| Tissue | Pathways | Symbol | Entrez Gene Name | Z Score | Log2FC Value | Predictive Activity to Pathway (IPA Knowledge Base and Reference Base*) |
|---|---|---|---|---|---|---|
| Septum | LXR/RXR pathway | ABCA1 | ATP‐binding cassette, subfamily A, member 1 | −0.447 | −0.708 | Inhibition* |
| APOD | Apolipoprotein D | −1.460 | Inhibition | |||
| CLU | Clusterin | −1.147 | Inhibition | |||
| LY96 | Lymphocyte antigen 96 | −0.668 | Activation | |||
| SERPINF1 | Serpin peptidase inhibitor, clade F (alpha‐2 antiplasmin, pigment epithelium derived factor), member 1 | −0.628 | Inhibition | |||
| ILK signaling | ACTA2 | Actin, alpha 2, smooth muscle, aorta | 1.000 | 0.679 | Activation | |
| MYL1 | Myosin, light chain 1 | 1.98 | Activation | |||
| PPP2R3A | Protein phosphatase 2 regulatory subunit B, alpha | 0.586 | Inhibition | |||
| SNAI2 | Snail homolog 2 | 0.581 | Activation | |||
| Free wall | LXR/RXR activation | APOD | Apolipoprotein D | −1.000 | −1.139 | Inhibition |
| CLU | Clusterin | −1.298 | Inhibition | |||
| LY96 | lymphocyte antigen 96 | −0.656 | Activation | |||
| MSR1 | Macrophage scavenger receptor 1 | −0.888 | Unknown | |||
| SERPINF1 | Serpin peptidase inhibitor, clade F (alpha‐2 antiplasmin, pigment epithelium derived factor), member 1 | −0.640 | Inhibition |
To validate the changes in the expression of these 10 genes, RNAs from comparable locations of LV tissues obtained for microarray analysis in the 6 sham control pigs and the 6 pacing pigs were sampled and quantitated using real‐time reverse transcriptase–polymerase chain reaction. Among the LXR/RXR pathway–related genes in the LV septal tissue, ABCA1, APOD, CLU, and LY96 were downregulated but SERPINF1 was upregulated by pacing (Figure 4A). All genes related to the ILK signaling pathway in the LV septal tissues (ACTA2, MYL1, PPP2R3A, and SNAI2) were upregulated by pacing (Figure 4B). Among the LXR/RXR pathway–related genes in the LV free wall tissue, APOD, CLU, LY96, and MSR1 were downregulated but SERPINF1 was upregulated by pacing (Figure 4C). Therefore, a discrepancy between the microarray analysis and real‐time reverse transcriptase–polymerase chain reaction analysis was identified only for SERPINF1 expression (Table S6).
Figure 4.

Differentially expressed LXR/RXR pathway‐related and ILK signaling pathway‐related genes induced by 6 mo of right ventricular septal‐dependent pacing. A, LXR/RXR pathway–related genes in the left ventricular septal tissue, ABCA1, APOD, CLU, and LY96 were downregulated but SERPINF1 was upregulated by pacing. B, All genes related to the ILK signaling pathway in the left ventricular septal tissues (MYL1, PPP2R3A, SNAI2 and ACTA2) were upregulated by pacing. C, LXR/RXR pathway–related genes in the left ventricular free wall tissue, APOD, CLU, LY96, and MSR1 were downregulated but SERPINF1 was upregulated by pacing. Gray bar: sham control group; black bar: pacing group. ILK indicates integrin‐linked kinase; LXR/RXR, liver X receptor/retinoid X receptor.
According to these findings on the inhibition of the LXR/RXR pathway and activation of the ILK signaling pathway in LV tissue following 6 months of RV septal pacing, we focused on examining the lipid expression, cardiac hypertrophy, inflammatory‐related cytokines, apoptotic markers, degree of myolysis, and fibrosis status related to the LXR/RXR pathway and ILK signaling pathway in the LV tissue following RV septal‐dependent pacing for 6 months.
Hypertrophy, Myolysis, Fibrosis, and Lipid Accumulation Developed in LV Myocardium Following 6 Months of Pacing
In terms of hypertrophy of cardiomyocytes, cell size in the pacing group was significantly larger than that in the sham control group at both the LV septum and free wall (Figure 5A). The degree of myolysis was also significantly higher in the pacing group than in the sham control group at both the LV septum and free wall (Figure 5B). The area of extracellular fibrosis was significantly greater in the pacing group than the sham control group at both the LV septum and free wall, and the intergroup difference was more prominent in the LV septum than in the LV free wall (Figure 5C). With regard to lipid metabolism disorder, Oil red O staining was used to evaluate the lipid expression in cardiomyocytes. The lipid expression in cardiomyocytes was significantly greater in the pacing group than the sham control group in the LV septum and free wall; additionally, the intergroup difference in lipid expression was more prominent in the LV septum than in the LV free wall (Figure 5D).
Figure 5.
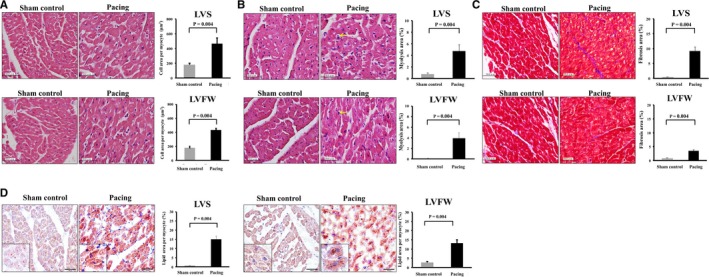
Hypertrophy, myolysis, fibrosis, and expression of lipid developed in left ventricular myocardium following 6 mo of right ventricular septal‐dependent pacing. A, Histochemical study with hematoxylin and eosin showed that cell size in the pacing group was significantly larger than that in the sham control group at both the left ventricular septum (LVS) and LV free wall (LVFW). B, Histochemical study with hematoxylin and eosin showed that the degree of myolysis was significantly higher in the pacing group than in the sham control group at both the LVS and LVFW. C, Histochemical study with Masson's trichrome staining showed that the area of extracellular fibrosis was significantly greater in the pacing group than the sham control group at both the LVS and LVFW, and the intergroup difference was more prominent in the LVS than in the LVFW. D, Histochemical study with Oil red O showed that the lipid expression in cardiomyocytes was significantly greater in the pacing group than the sham control group in the LVS (left figure) and LVFW (right figure). The intergroup difference in lipid expression was more prominent in the LVS than in the LVFW. Insets: distribution of intracellular lipid. Bar=50 μm. Gray bar: sham control group; black bar: pacing group.
Increased Expression of Inflammatory and Apoptosis Markers in LV Myocardium Following 6 Months of Pacing
The expression of inflammatory cytokines (tumor necrosis factor‐α, interleukin‐6, C‐reactive protein) and an anti‐inflammatory cytokine (interleukin‐10) was significantly increased following RV septal pacing for 6 months compared with the sham control, and the intergroup difference in the inflammatory‐related cytokines was again more prominent in the LV septum than in the LV free wall (Figure 6A through 6D). In terms of apoptosis, the expression of cleaved caspase 3, 8, and 9 was significantly increased following RV septal pacing for 6 months compared with the sham control, and the intergroup difference in the expression of cleaved caspase 3, 8, and 9 was more prominent in the LV septum than in the LV free wall (Figure 6E through 6G).
Figure 6.
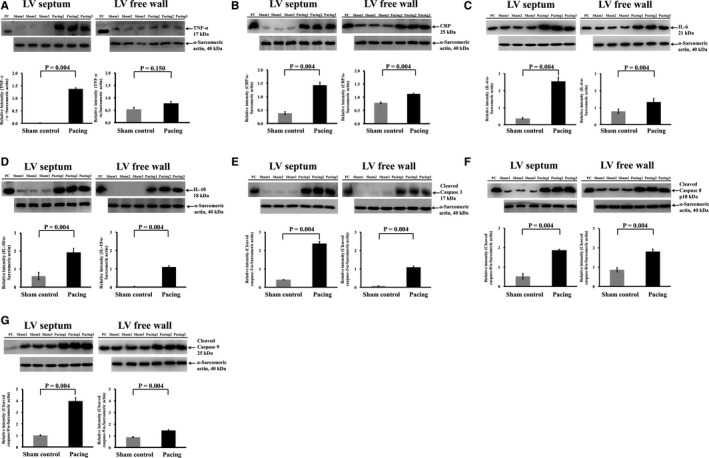
Increased expression of inflammatory and apoptosis markers in left ventricular myocardium following 6 mo of right ventricular septal‐dependent pacing. Immunoblotting study showed that the expression of inflammatory markers of (A) tumor necrosis factor alpha (TNF‐α), (B) C‐reactive protein (CRP), (C) interleukin‐6 (IL‐6), and (D) interleukin‐10 (IL‐10) in left ventricular (LV) septum and LV free wall was significantly increased following right ventricular septal pacing for 6 mo compared with the sham control, and the intergroup difference in the inflammatory‐related cytokines was more prominent in the LV septum than in the LV free wall. Immunoblotting study showed that the expression of apoptosis markers of (E) cleaved caspase 3, (F) cleaved caspase 8, and (G) cleaved caspase 9 was significantly increased following right ventricular septal pacing for 6 mo compared with the sham control, and the intergroup difference in the expression of cleaved caspase 3, 8, and 9 was more prominent in the LV septum than in the LV free wall. Gray bar: sham control group; black bar: pacing group. PC indicates positive control.
Cell Pacing Model to Validate the Changes in DEGs Related to the LXR/RXR Pathway and ILK Signaling Pathway in the Pig Pacing Model
To determine the causal relationship between pacing and the DEGs related to the LXR/RXR pathway and the ILK signaling pathway and lipid expression after pacing, we created a cell pacing model. We also used this model to test the hypothesis that pacing with different rates rather than a fixed current accrued different changes in DEGs and lipid expression following pacing. In this cell pacing model, rat ventricular cardiomyocytes (RV‐40 strain) received nonpacing or pacing for 24 hours at 0.5, 1.5, or 3 Hz. Notably, consistent with the gene expression profile of the pig pacing model, the expression of ABCA1, APOD, CLU, LY96, and MSR1 was significantly downregulated but the expression of SERPINF1, ACTA2, MYL1, PPP2R3A, and SNAI2 was significantly upregulated for higher pacing rates, especially at 1.5 and 3 Hz compared with nonpacing and 0.5‐Hz pacing (Figure 7A and 7B). There was no significant difference in the expression of ABCA1, APOD, CLU, LY96, SERPINF1, MSR1, MYL1, and SNAI2 between the nonpacing and 0.5‐Hz pacing groups.
Figure 7.
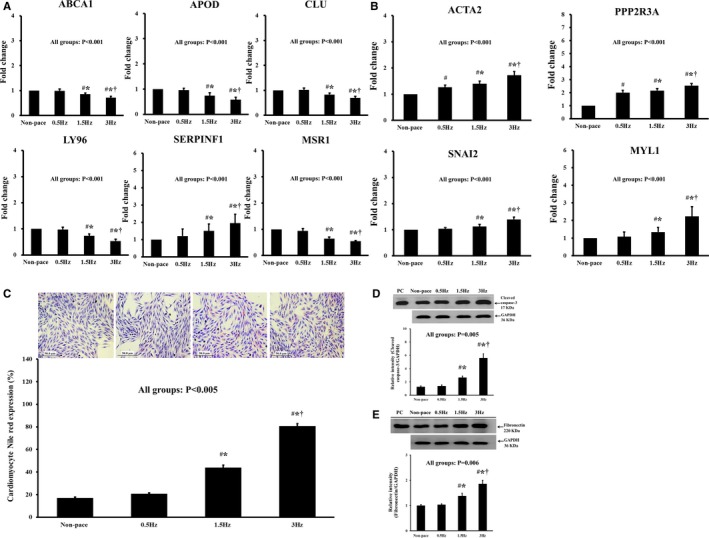
Cell pacing model validated the changes in differentially expressed genes related to the LXR/RXR pathway and ILK signaling pathway and downstream expressions (lipid expression, apoptosis, and fibrosis) observed in the pig pacing model. In this cell pacing model, rat ventricular cardiomyocytes (RV‐40 strain) received nonpacing or pacing for 24 h at 0.5, 1.5, or 3 Hz. A, Among the LXR/RXR pathway–related genes in the rat ventricular cardiomyocytes, the expression of ABCA1, APOD, CLU, LY96, and MSR1 was significantly downregulated but the expression of SERPINF1 was significantly upregulated for higher pacing rates, especially at 1.5 and 3 Hz compared with nonpacing and 0.5‐Hz pacing. There was no significant difference in the expression of ABCA1, APOD, CLU, LY96, SERPINF1, and MSR1 between the nonpacing and 0.5‐Hz pacing groups. B, Among the ILK signaling pathway–related genes in the rat ventricular cardiomyocytes, the expression of ACTA2, PPP2R3A, SNAI2, and MYL1 was significantly upregulated for higher pacing rates, especially at 1.5 and 3 Hz compared with nonpacing and 0.5‐Hz pacing. There was no significant difference in the expression of SNAI2 and MYL1 between the nonpacing and 0.5‐Hz pacing groups. C, The lipid expression was significantly higher with pacing at 1.5 and 3 Hz compared with nonpacing and pacing at 0.5 Hz, and there was no difference between the nonpacing and 0.5‐Hz pacing groups. D, The expression of cleaved caspase 3 was significantly higher with pacing at 1.5 and 3 Hz compared with nonpacing and pacing at 0.5 Hz, and there was no difference in cleaved caspase 3 expression between the nonpacing and 0.5‐Hz pacing groups. E, The expression of fibronectin was significantly higher with pacing at 1.5 and 3 Hz compared with nonpacing and pacing at 0.5 Hz, and there was no difference between the nonpacing and 0.5‐Hz pacing groups. PC indicates positive control. # P value was <0.05 when pacing group was compared with nonpacing group; *P value was <0.05 when 1.5 and 3 Hz compared with 0.5 Hz group; † P value was <0.05 when 3 Hz compared with 1.5 Hz. ILK indicates integrin‐linked kinase; LXR/RXR, liver X receptor/retinoid X receptor.
Cell Pacing Model to Validate the Changes in the Expression of Lipid, Apoptosis, and Fibrosis in the Pig Pacing Model
Lipid expression in rat ventricular cardiomyocytes was evaluated by histochemical study with Oil red O and by flow cytometry analysis with Nile red. Nile red expression was significantly higher with pacing at 1.5 and 3 Hz compared with nonpacing and pacing at 0.5 Hz (Figure 7C). The expression of cleaved caspase 3 was significantly higher with pacing at 1.5 and 3 Hz compared with nonpacing and pacing at 0.5 Hz (Figure 7D). The expression of fibronectin was significantly higher with pacing at 1.5 and 3 Hz compared with nonpacing and pacing at 0.5 Hz (Figure 7E). Notably, there was no difference in the expression of lipid, caspase 3, and fibronectin between the nonpacing and 0.5‐Hz pacing groups. Therefore, it was the pacing rate rather than the pacing current (1 V at 0.4 ms) that inhibited the LXR/RXR pathway and activated the ILK signaling pathway, resulting in lipid accumulation, apoptosis, and fibrosis.
Luciferase Reporter Assay to Evaluate the LXR Activity at the ABCA1 Promoter After Pacing
According to the result of microarray analysis/IPA, ABCA1 was a downstream gene of the LXR/RXR pathway. Hence, we evaluated the LXR activity at the ABCA1 promoter in the rat ventricular cardiomyocytes (RV‐40 strain) after pacing. The LXR activity at the ABCA1 promoter of ventricular cardiomyocytes was significantly suppressed by pacing (1.5 and 3 Hz pacing) and was significantly enhanced by treatment with LXR agonist (Figure 8). These results provided additional support of inhibition of the LXR pathway by pacing.
Figure 8.

Luciferase reporter assay. Rat ventricular cardiomyocytes (RV‐40 strain) received nonpacing, pacing for 24 h at 1.5 or 3 Hz or treatment with LXR agonist (1 μmol/L, T0901317; Sigma, Missouri) but without pacing. The LXR activity at the ABCA1 promoter of ventricular cardiomyocytes in each group was evaluated by luciferase reporter assay and measured by fluorescence and luciferase activity. The luciferase activity of ventricular cardiomyocytes was significantly suppressed by pacing at 1.5 and 3 Hz and was significantly enhanced by treatment with LXR agonist. Therefore, the LXR pathway was inhibited by pacing. mCMV indicates minimal CMV promoter; LXRE, Liver X receptor response element.
Discussion
Our animal study indicated that RV septal‐dependent pacing for 6 months led to persistent LV mechanical dyssynchrony induced by pacing and increased the LV end‐diastolic diameter and LV mass. The genetic and protein pathways analyzed in prior studies were limited,14, 22 whereas our study used high‐density oligonucleotide microarrays and conducted enrichment analysis to systemically explore the molecular regulatory mechanisms of LV myocardial remodeling associated with persistent mechanical dyssynchrony induced by RV pacing. A RNA microarray and functional network enrichment analysis showed that the LXR/RXR pathway was inhibited in both the septum and free wall of the LV, and the ILK signaling pathway was activated in the LV septum following 6 months of RV septal pacing. The biological changes related to inhibition of the LXR/RXR pathway and activation of the ILK signaling pathway—including cardiomyocyte hypertrophy, activation of apoptosis, increased fibrosis, myolysis, and intracellular lipid accumulation—were confirmed using histochemical studies. Notably, the causal relationships between pacing and the DEGs related to the LXR/RXR pathway, ILK signaling pathway, and lipid expression, apoptosis, and fibrosis were confirmed using a cell pacing model. Moreover, the LXR activity at the ABCA1 promoter of rat ventricular cardiomyocytes (RV‐40 strain) evaluated by luciferase reporter assay was found to be significantly suppressed by pacing.
LV mechanical dyssynchrony caused by RV pacing is generally considered to be a precursor of PICM. Our study and prior clinical and animal studies7, 22 have demonstrated pacing‐induced LV dyssynchrony through M‐mode imaging and strain echocardiography. In addition, our study demonstrated that the LV end‐diastolic diameter was enlarged and an LV mass was increased after 6 months of RV septal pacing even though no significant change in the degree of LV dyssynchrony existed after 6 months of pacing. Although systolic function should be theoretically reduced by pacing, the difference in LV systolic function between the pacing and sham control groups did not reach statistical significance. This might be related to the small number of experimental animals. For the first time, our genetic and molecular results demonstrated, after 6 months of RV septal‐dependent pacing associated with persistent pacing‐induced LV dyssynchrony was performed, the genetic inhibition of the LXR/RXR pathway and biological changes related to inhibition of the LXR/RXR pathway such as cardiomyocyte hypertrophy, activation of apoptosis, increased fibrosis, myolysis, and intracellular lipid accumulation.
The LXR/RXR pathway is the principal pathway related to lipid metabolism, and inhibition of the LXR/RXR pathway leads to intracellular lipid accumulation.23, 24 An excess accumulation of intracellular lipid increases endoplasmic reticulum stress, mitochondrial dysfunction, oxidative stress, and defective intracellular signaling, eventually leading to apoptosis and inflammation. This pathophysiological phenomenon has been described as “lipotoxicity.”25 In terms of cardiomyocytes, this pathophysiological phenomenon has been identified as “lipotoxic cardiomyopathy,”26 which also produces excessive levels of cardiac hypertrophy.27 Several in vivo and in vitro studies of lipotoxic cardiomyopathy have been conducted, mainly focusing on aging, diabetes mellitus, a high lipid diet, and ischemic heart disease.28 Using animal models, LXR/RXR pathways and some associated genetic changes were evaluated, including changes in expression of ABCA1, CD36, and scavenger receptors. In our study, significant intracellular lipid accumulation, histologic changes such as prominent fibrosis, myolysis, and the activation of apoptosis, and elevated inflammation markers were discovered in the LV myocardium after 6 months of RV septal pacing. Moreover, a RNA microarray and enrichment analysis indicated that the LXR/RXR pathway was inhibited in both the septum and free wall of the LV following 6 months of RV septal pacing. With the combined results of these analyses, our study showed that RV septal pacing induced lipotoxic cardiomyopathy through inhibition of the LXR/RXR pathway and that this is a novel regulatory mechanism of LV myocardial remodeling associated with persistent mechanical dyssynchrony.
Among the LXR/RXR pathway–related genes identified in the Ingenuity Pathway Analysis, ABCA1, APOD, CLU, LY96, and MSR1 were discovered to be downregulated but SERPINF1 was upregulated in the LV tissue following 6 months of RV septal pacing; these genetic changes were also confirmed using the cell pacing model. Notably, the cell pacing model demonstrated that pacing rate rather than pacing current inhibited the LXR/RXR pathway and consequently resulted in lipid accumulation. ABCA1 is an ATP‐binding cassette transporter and is mainly involved in mediating the cellular efflux of phospholipids and cholesterol in the LXR/RXR pathway. The suppression of ABCA1 expression leads to intracellular lipid accumulation.19, 20, 21 APOD is an atypical apolipoprotein, a component of HDL, a multiligand multifunctional transporter, and associated with antioxidation and antistress activities in fruit flies. Increased APOD deposition was found in atherosclerotic lesions of humans with cardiovascular disease and was established to be a compensatory response.29 CLU is also a component of HDL and plays a protective role in vascular disease progression. CLU reduces the tumor necrosis factor‐α–stimulated expression of chemokines, cell adhesion, and MMP‐9 expression.30, 31 Therefore, downregulation of APOD and CLU should increase oxidative stress and inflammation and activate cell apoptosis. Our findings are consistent with those of other reports. SERPINF1, also called pigment epithelium derived factor, interacts with adipose triglyceride lipase to increase lipolysis and reduce fatty acid oxidation, contributing to insulin resistance in cardiomyocytes.32 Studies have demonstrated that upregulation of SERPINF1 leads to lipid accumulation and lipotoxicity and induces apoptosis and fibrosis.33 SERPINF1 is associated with several cardiovascular diseases.34 In our study, although SERPINF1 was found to be downregulated in the microarray results, SERPINF1 was discovered to be upregulated using quantitative polymerase chain reaction not only in the animal model, but also in the cell pacing model. MSR1 has multifunctional polyanionic ligands, including lipoproteins, lipopolysaccharides, and amyloid, and acts as a ligand for acetylated low‐density lipoprotein in macrophage.35 One article reported an increased hepatocyte cytoplasmic cholesterol accumulation in MSR1−/− mice.36 LY96, also known as myeloid differentiation 2, is a coreceptor of toll‐like receptor 4 and is required for bacterial lipopolysaccharide binding. The expression of toll‐like receptor 4 induces the inflammation process and lipid accumulation, and 1 study discovered that oxidized cholesterol esters binding LY96 activated the expression of toll‐like receptor 4.37 In our study, downregulation instead of upregulation of LY96 was observed, as a result of pacing. We speculate that the downregulation of LY96 by pacing may be a compensatory response to increased inflammation by other LXR/RXR pathway–related genes.
Our study showed that among the ILK signaling pathway–related genes identified by the Ingenuity Pathway Analysis, ACTA2, MYL1, PPP2R3A, and SNAI2 were upregulated in the LV septal tissue following 6 months of RV septal pacing. These genetic changes were also confirmed using the cell pacing model. Similar to our histologic results in a pig pacing model, a canine model indicated that endothelial–mesenchymal transition can be triggered and induce cardiac fibrosis under dyssynchronous pacing.38 However, unlike our study, which examined physiological pacing rate and conducted an enrichment analysis with high‐density oligonucleotide microarrays, the canine‐model study used high‐frequency pacing and evaluated selected and limited molecular mechanisms. ILK is an intracellular serine/threonine kinase and is mainly involved in cell–matrix interaction to regulate cytoskeletal remodeling, cardiac growth, physiological hypertrophy, proliferation, contractility, repair, and endothelial–mesenchymal transition. ILK is most abundantly expressed in the heart.39 Articles have reported an overexpression of the ILK pathway in failing human hearts and cardiac hypertrophy animal models, possibly as a compensatory stress response.40 SNAI2, a snail family member, is the key mediator in the transforming growth factor‐β–induced endothelial–mesenchymal transition, and its upregulation was also discovered in a dyssynchrony pacing animal model.38 ACTA2 and MYL1 are structure‐related genes,41, 42 and the expression of PPP2R3A was sufficiently demonstrated to be a downstream reaction of the inflammation process and related to structural remodeling in a heart failure model.43
Conclusion
RV septal‐dependent pacing associated with persistent LV dyssynchrony induced cardiomyopathy through inhibition of the LXR/RXR pathway as a novel regulatory mechanism, leading to pacing‐induced cardiomyopathy. Our results provide an avenue for future studies targeting the LXR/RXR pathway to prevent structural remodeling associated with pacemaker‐induced cardiomyopathy.
Accession Codes
The data discussed in this manuscript have been deposited in NCBI's Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number GSE112065 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE112065).
Sources of Funding
This work was supported by grants from Chang Gung Memorial Hospital, Taiwan (grant CMRPG6C0381, CMRPG6C0382, CMRPG6F0361) and the Ministry of Science and Technology, Taiwan (grants MOST 104‐2314‐B‐182A‐130, MOST 105‐2314‐B‐182A‐101‐MY3).
Disclosures
None.
Supporting information
Table S1. Primer Sequences for Real‐Time Polymerase Chain Reaction
Table S2. Selected Signature Upregulated Gene Expression in Left Ventricular Septum of Pacing Group Versus Sham Control Group
Table S3. Selected Signature Downregulated Gene Expression in Left Ventricular Septum of Pacing Group Versus Sham Control Group
Table S4. Selected Signature Upregulated Gene Expression in Left Ventricular Free Wall of Pacing Group Versus Sham Control Group
Table S5. Selected Signature Downregulated Gene Expression in Left Ventricular Free Wall of Pacing Group Versus Sham Control Group
Table S6. Analysis of mRNA Levels Via Quantitative Real‐Time PCR and RNA Microarray
Figure S1. Locations of pacing leads at fluoroscopy and at autopsy. Upper panel: Tip of atrial lead was fixed over the right atrial appendage (RAA) and tip of ventricular lead was fixed over septum of the right ventricle under fluoroscopic guidance (posterior‐anterior [PA] view, right anterior oblique [RAO] view, and left anterior oblique [LAO] view). Lower panel: Tip (white arrow) of atrial lead was fixed over the right atrial appendage (RAA) and tip (white arrow) of ventricular lead was fixed over septum of the right ventricle at autopsy.
Figure S2. Regulatory network of differentially expressed genes (DEGs) associated with LXR/RXR pathway and ILK signaling pathway derived by the Ingenuity Pathway Analysis Global Molecular Network algorithm between the sham control and pacing groups. A, The regulatory network constructed from DEGs associated with ILK signaling in the left ventricular sputum. B, The regulatory network constructed from DEGs associated with LXR/RXR activation in the left ventricular sputum. C, The regulatory network constructed from DEGs associated with LXR/RXR activation in the left ventricular free wall. The networks were derived from DEGs in the left ventricular sputum and free wall using the Ingenuity Pathway Analysis Global Molecular Network algorithm, and the network score (P‐score; ie P<10−p‐score) of networks (A, B, and C) were 42, 32, and 51, respectively. The genes with a blue color have functions related to the respective pathway in cardiovascular system development.
Figure S3. Differentially expressed genes (DEGs) involved in the LXR/RXR and ILK signaling pathways in the left ventricular septum between the sham control and pacing groups. A, Functional enrichment analysis showed that 5 DEGs (APOD, CLU, ABCA1, LY96, and SERPINF1) were identified as significantly involved in the LXR/RXR activation pathway, with P=0.00016 and activation z‐score=−0.447. B, Functional enrichment analysis showed that 4 DEGs (PPP2R3A, ACTA2, MYL1, and SNAI2) were identified as significantly involved in the ILK signaling pathway, with P=0.02 and activation z‐score=1.000.
Figure S4. Differentially expressed genes (DEGs) involved in the left ventricular free wall between the sham control and pacing groups. Functional enrichment analysis showed that 5 DEGs (APOD, CLU, MSR1, LY96, and SERPINF1) were identified as significantly involved in the LXR/RXR pathway, with P=0.00036 and activation z‐score=−1.000.
Acknowledgments
We warmly thank the Chang Gung Medical Foundation, Kaohsiung Chang Gung Memorial Hospital, Tissue Bank Core Lab (CLRPG8B0033 and CLRPG8E0161) for their excellent technical support.
(J Am Heart Assoc. 2019;8:e009146 DOI: 10.1161/JAHA.118.009146)
References
- 1. Wilkoff BL, Cook JR, Epstein AE, Greene HL, Hallstrom AP, Hsia H, Kutalek SP, Sharma A. Dual‐chamber pacing or ventricular backup pacing in patients with an implantable defibrillator: the Dual Chamber and VVI Implantable Defibrillator (DAVID) Trial. JAMA. 2002;288:3115–3123. [DOI] [PubMed] [Google Scholar]
- 2. Moss AJ, Zareba W, Hall WJ, Klein H, Wilber DJ, Cannom DS, Daubert JP, Higgins SL, Brown MW, Andrews ML. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med. 2002;346:877–883. [DOI] [PubMed] [Google Scholar]
- 3. Sweeney MO, Hellkamp AS, Ellenbogen KA, Greenspon AJ, Freedman RA, Lee KL, Lamas GA. Adverse effect of ventricular pacing on heart failure and atrial fibrillation among patients with normal baseline QRS duration in a clinical trial of pacemaker therapy for sinus node dysfunction. Circulation. 2003;107:2932–2937. [DOI] [PubMed] [Google Scholar]
- 4. Khurshid S, Epstein AE, Verdino RJ, Lin D, Goldberg LR, Marchlinski FE, Frankel DS. Incidence and predictors of right ventricular pacing‐induced cardiomyopathy. Heart Rhythm. 2014;11:1619–1625. [DOI] [PubMed] [Google Scholar]
- 5. Kiehl EL, Makki T, Kumar R, Gumber D, Kwon DH, Rickard JW, Kanj M, Wazni OM, Saliba WI, Varma N, Wilkoff BL, Cantillon DJ. Incidence and predictors of right ventricular pacing‐induced cardiomyopathy in patients with complete atrioventricular block and preserved left ventricular systolic function. Heart Rhythm. 2016;13:2272–2278. [DOI] [PubMed] [Google Scholar]
- 6. Wolber T, Haegeli L, Huerlimann D, Brunckhorst C, Luscher TF, Duru F. Altered left ventricular contraction pattern during right ventricular pacing: assessment using real‐time three‐dimensional echocardiography. Pacing Clin Electrophysiol. 2011;34:76–81. [DOI] [PubMed] [Google Scholar]
- 7. Liu WH, Chen MC, Chen YL, Guo BF, Pan KL, Yang CH, Chang HW. Right ventricular apical pacing acutely impairs left ventricular function and induces mechanical dyssynchrony in patients with sick sinus syndrome: a real‐time three‐dimensional echocardiographic study. J Am Soc Echocardiogr. 2008;21:224–229. [DOI] [PubMed] [Google Scholar]
- 8. ten Cate TJ, Scheffer MG, Sutherland GR, Verzijlbergen JF, van Hemel NM. Right ventricular outflow and apical pacing comparably worsen the echocardiographic normal left ventricle. Eur J Echocardiogr. 2008;9:672–677. [DOI] [PubMed] [Google Scholar]
- 9. Tse HF, Yu C, Wong KK, Tsang V, Leung YL, Ho WY, Lau CP. Functional abnormalities in patients with permanent right ventricular pacing: the effect of sites of electrical stimulation. J Am Coll Cardiol. 2002;40:1451–1458. [DOI] [PubMed] [Google Scholar]
- 10. Cho GY, Kim MJ, Park JH, Kim HS, Youn HJ, Kim KH, Song JK. Comparison of ventricular dyssynchrony according to the position of right ventricular pacing electrode: a multi‐center prospective echocardiographic study. J Cardiovasc Ultrasound. 2011;19:15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alhous MH, Small GR, Hannah A, Hillis GS, Broadhurst P. Impact of temporary right ventricular pacing from different sites on echocardiographic indices of cardiac function. Europace. 2011;13:1738–1746. [DOI] [PubMed] [Google Scholar]
- 12. Delhaas T, Arts T, Prinzen FW, Reneman RS. Regional fibre stress‐fibre strain area as an estimate of regional blood flow and oxygen demand in the canine heart. J Physiol. 1994;477:481–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Prinzen FW, Peschar M. Relation between the pacing induced sequence of activation and left ventricular pump function in animals. Pacing Clin Electrophysiol. 2002;25:484–498. [DOI] [PubMed] [Google Scholar]
- 14. Yamazaki KG, Ihm SH, Thomas RL, Roth D, Villarreal F. Cell adhesion molecule mediation of myocardial inflammatory responses associated with ventricular pacing. Am J Physiol Heart Circ Physiol. 2012;302:H1387–H1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lang RM, Bierig M, Devereux RB, Flachskampf FA, Foster E, Pellikka PA, Picard MH, Roman MJ, Seward J, Shanewise JS, Solomon SD, Spencer KT, Sutton MS, Stewart WJ. Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr. 2005;18:1440–1463. [DOI] [PubMed] [Google Scholar]
- 16. Teichholz LE, Kreulen T, Herman MV, Gorlin R. Problems in echocardiographic volume determinations: echocardiographic‐angiographic correlations in the presence of absence of asynergy. Am J Cardiol. 1976;37:7–11. [DOI] [PubMed] [Google Scholar]
- 17. Gautier L, Cope L, Bolstad BM, Irizarry RA. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. [DOI] [PubMed] [Google Scholar]
- 18. Kramer A, Green J, Pollard J Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30:523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schmitz G, Langmann T. Transcriptional regulatory networks in lipid metabolism control ABCA1 expression. Biochim Biophys Acta. 2005;1735:1–19. [DOI] [PubMed] [Google Scholar]
- 20. Tarling EJ, de Aguiar Vallim TQ, Edwards PA. Role of ABC transporters in lipid transport and human disease. Trends Endocrinol Metab. 2013;24:342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu Z, Dong A, Feng Z, Li J. Interleukin‐32 promotes lipid accumulation through inhibition of cholesterol efflux. Exp Ther Med. 2017;14:947–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Paslawska U, Gajek J, Kiczak L, Noszczyk‐Nowak A, Skrzypczak P, Bania J, Tomaszek A, Zacharski M, Sambor I, Dziegiel P, Zysko D, Banasiak W, Jankowska EA, Ponikowski P. Development of porcine model of chronic tachycardia‐induced cardiomyopathy. Int J Cardiol. 2011;153:36–41. [DOI] [PubMed] [Google Scholar]
- 23. Zhang Y, Breevoort SR, Angdisen J, Fu M, Schmidt DR, Holmstrom SR, Kliewer SA, Mangelsdorf DJ, Schulman IG. Liver LXRalpha expression is crucial for whole body cholesterol homeostasis and reverse cholesterol transport in mice. J Clin Invest. 2012;122:1688–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Padovani AM, Molina MF, Mann KK. Inhibition of liver x receptor/retinoid X receptor‐mediated transcription contributes to the proatherogenic effects of arsenic in macrophages in vitro. Arterioscler Thromb Vasc Biol. 2010;30:1228–1236. [DOI] [PubMed] [Google Scholar]
- 25. Drosatos K, Schulze PC. Cardiac lipotoxicity: molecular pathways and therapeutic implications. Curr Heart Fail Rep. 2013;10:109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. D'Souza K, Nzirorera C, Kienesberger PC. Lipid metabolism and signaling in cardiac lipotoxicity. Biochim Biophys Acta. 2016;1861:1513–1524. [DOI] [PubMed] [Google Scholar]
- 27. Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. 2001;107:813–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goldberg IJ, Trent CM, Schulze PC. Lipid metabolism and toxicity in the heart. Cell Metab. 2012;15:805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rassart E, Bedirian A, Do Carmo S, Guinard O, Sirois J, Terrisse L, Milne R. Apolipoprotein D. Biochim Biophys Acta. 2000;1482:185–198. [DOI] [PubMed] [Google Scholar]
- 30. Park S, Mathis KW, Lee IK. The physiological roles of apolipoprotein J/clusterin in metabolic and cardiovascular diseases. Rev Endocr Metab Disord. 2014;15:45–53. [DOI] [PubMed] [Google Scholar]
- 31. Kim HJ, Yoo EK, Kim JY, Choi YK, Lee HJ, Kim JK, Jeoung NH, Lee KU, Park IS, Min BH, Park KG, Lee CH, Aronow BJ, Sata M, Lee IK. Protective role of clusterin/apolipoprotein J against neointimal hyperplasia via antiproliferative effect on vascular smooth muscle cells and cytoprotective effect on endothelial cells. Arterioscler Thromb Vasc Biol. 2009;29:1558–1564. [DOI] [PubMed] [Google Scholar]
- 32. Borg ML, Andrews ZB, Duh EJ, Zechner R, Meikle PJ, Watt MJ. Pigment epithelium‐derived factor regulates lipid metabolism via adipose triglyceride lipase. Diabetes. 2011;60:1458–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Crowe S, Wu LE, Economou C, Turpin SM, Matzaris M, Hoehn KL, Hevener AL, James DE, Duh EJ, Watt MJ. Pigment epithelium‐derived factor contributes to insulin resistance in obesity. Cell Metab. 2009;10:40–47. [DOI] [PubMed] [Google Scholar]
- 34. Yamagishi S, Matsui T. Pigment epithelium‐derived factor (PEDF) and cardiometabolic disorders. Curr Pharm Des. 2014;20:2377–2386. [DOI] [PubMed] [Google Scholar]
- 35. Tsujita K, Kaikita K, Hayasaki T, Honda T, Kobayashi H, Sakashita N, Suzuki H, Kodama T, Ogawa H, Takeya M. Targeted deletion of class A macrophage scavenger receptor increases the risk of cardiac rupture after experimental myocardial infarction. Circulation. 2007;115:1904–1911. [DOI] [PubMed] [Google Scholar]
- 36. Bieghs V, Verheyen F, van Gorp PJ, Hendrikx T, Wouters K, Lutjohann D, Gijbels MJ, Febbraio M, Binder CJ, Hofker MH, Shiri‐Sverdlov R. Internalization of modified lipids by CD36 and SR‐A leads to hepatic inflammation and lysosomal cholesterol storage in Kupffer cells. PLoS One. 2012;7:e34378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Han J, Zou C, Mei L, Zhang Y, Qian Y, You S, Pan Y, Xu Z, Bai B, Huang W, Liang G. MD2 mediates angiotensin II‐induced cardiac inflammation and remodeling via directly binding to Ang II and activating TLR4/NF‐kappaB signaling pathway. Basic Res Cardiol. 2017;112:9. [DOI] [PubMed] [Google Scholar]
- 38. Mai J, Hu Q, Xie Y, Su S, Qiu Q, Yuan W, Yang Y, Song E, Chen Y, Wang J. Dyssynchronous pacing triggers endothelial‐mesenchymal transition through heterogeneity of mechanical stretch in a canine model. Circ J. 2015;79:201–209. [DOI] [PubMed] [Google Scholar]
- 39. Hannigan GE, Leung‐Hagesteijn C, Fitz‐Gibbon L, Coppolino MG, Radeva G, Filmus J, Bell JC, Dedhar S. Regulation of cell adhesion and anchorage‐dependent growth by a new beta 1‐integrin‐linked protein kinase. Nature. 1996;379:91–96. [DOI] [PubMed] [Google Scholar]
- 40. Hannigan GE, Coles JG, Dedhar S. Integrin‐linked kinase at the heart of cardiac contractility, repair, and disease. Circ Res. 2007;100:1408–1414. [DOI] [PubMed] [Google Scholar]
- 41. Yuan SM. Alpha‐smooth muscle actin and ACTA2 gene expressions in vasculopathies. Braz J Cardiovasc Surg. 2015;30:644–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Burguiere AC, Nord H, von Hofsten J. Alkali‐like myosin light chain‐1 (myl1) is an early marker for differentiating fast muscle cells in zebrafish. Dev Dyn. 2011;240:1856–1863. [DOI] [PubMed] [Google Scholar]
- 43. DeGrande ST, Little SC, Nixon DJ, Wright P, Snyder J, Dun W, Murphy N, Kilic A, Higgins R, Binkley PF, Boyden PA, Carnes CA, Anderson ME, Hund TJ, Mohler PJ. Molecular mechanisms underlying cardiac protein phosphatase 2A regulation in heart. J Biol Chem. 2013;288:1032–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer Sequences for Real‐Time Polymerase Chain Reaction
Table S2. Selected Signature Upregulated Gene Expression in Left Ventricular Septum of Pacing Group Versus Sham Control Group
Table S3. Selected Signature Downregulated Gene Expression in Left Ventricular Septum of Pacing Group Versus Sham Control Group
Table S4. Selected Signature Upregulated Gene Expression in Left Ventricular Free Wall of Pacing Group Versus Sham Control Group
Table S5. Selected Signature Downregulated Gene Expression in Left Ventricular Free Wall of Pacing Group Versus Sham Control Group
Table S6. Analysis of mRNA Levels Via Quantitative Real‐Time PCR and RNA Microarray
Figure S1. Locations of pacing leads at fluoroscopy and at autopsy. Upper panel: Tip of atrial lead was fixed over the right atrial appendage (RAA) and tip of ventricular lead was fixed over septum of the right ventricle under fluoroscopic guidance (posterior‐anterior [PA] view, right anterior oblique [RAO] view, and left anterior oblique [LAO] view). Lower panel: Tip (white arrow) of atrial lead was fixed over the right atrial appendage (RAA) and tip (white arrow) of ventricular lead was fixed over septum of the right ventricle at autopsy.
Figure S2. Regulatory network of differentially expressed genes (DEGs) associated with LXR/RXR pathway and ILK signaling pathway derived by the Ingenuity Pathway Analysis Global Molecular Network algorithm between the sham control and pacing groups. A, The regulatory network constructed from DEGs associated with ILK signaling in the left ventricular sputum. B, The regulatory network constructed from DEGs associated with LXR/RXR activation in the left ventricular sputum. C, The regulatory network constructed from DEGs associated with LXR/RXR activation in the left ventricular free wall. The networks were derived from DEGs in the left ventricular sputum and free wall using the Ingenuity Pathway Analysis Global Molecular Network algorithm, and the network score (P‐score; ie P<10−p‐score) of networks (A, B, and C) were 42, 32, and 51, respectively. The genes with a blue color have functions related to the respective pathway in cardiovascular system development.
Figure S3. Differentially expressed genes (DEGs) involved in the LXR/RXR and ILK signaling pathways in the left ventricular septum between the sham control and pacing groups. A, Functional enrichment analysis showed that 5 DEGs (APOD, CLU, ABCA1, LY96, and SERPINF1) were identified as significantly involved in the LXR/RXR activation pathway, with P=0.00016 and activation z‐score=−0.447. B, Functional enrichment analysis showed that 4 DEGs (PPP2R3A, ACTA2, MYL1, and SNAI2) were identified as significantly involved in the ILK signaling pathway, with P=0.02 and activation z‐score=1.000.
Figure S4. Differentially expressed genes (DEGs) involved in the left ventricular free wall between the sham control and pacing groups. Functional enrichment analysis showed that 5 DEGs (APOD, CLU, MSR1, LY96, and SERPINF1) were identified as significantly involved in the LXR/RXR pathway, with P=0.00036 and activation z‐score=−1.000.