

Introduction
Pulmonary arterial hypertension (PAH) is a rare but lethal disorder caused by several pathological changes in the pulmonary vasculature. There is endothelial cell dysfunction characterized by exaggerated secretion of vasoconstrictive, pro‐proliferative substances, such as endothelin, and impaired release of vasodilatory, antiproliferative molecules, such as nitric oxide and prostacyclin.1, 2, 3 This imbalance contributes to increases in pulmonary artery (PA) smooth muscle cell (PASMC) tone and proliferation.1, 2, 3 Moreover, endothelial cells exhibit metabolic reprogramming with a switch to anaerobic glycolysis.4 In PASMCs, there is evidence of hypercontractility,5 proliferation and apoptosis resistance due to genetically6, 7 and epigenetically8 controlled mechanisms, calcium mishandling,9, 10, 11 metabolic reprogramming,5, 12 and abnormal mitochondrial dynamics.13, 14 Mitochondrial metabolic reprogramming, creating a Warburg metabolic phenotype that promotes proliferation, is also observed in pulmonary vascular fibroblasts.15 Extracellular matrix (ECM) remodeling promotes PAH by increasing vessel stiffness and thereby altering signaling pathways and inducing metabolic derangements through mechanotransduction.3, 16, 17, 18, 19 Finally, there is evidence of a significant inflammatory response with T cell, B cell, and dendritic cell infiltration into the pulmonary vasculature20, 21 and elevated levels of circulating inflammatory cytokines.20, 22, 23 These molecular, cellular, and histological changes manifest as reduced pulmonary arterial compliance24 and elevated pulmonary vascular resistance (PVR) and pulmonary arterial pressures25 that, in aggregate, augment the workload of the right ventricle.26 As PAH progresses, the heightened demands on the right ventricle lead to right ventricular hypertrophy (RVH), fibrosis, and metabolic derangements that often culminate in RV failure.27 RV dysfunction is the strongest predictor of mortality in PAH28, 29, 30, 31, 32 and is the major reason that the median survival with PAH is only 5 to 7 years, both in registries30, 31, 32 and in population‐based studies.33
Although there have been significant gains in the understanding of the pathophysiology of PAH in recent years, advances in PAH therapeutics have not kept pace, and many promising basic science discoveries have not been tested in patients. Approved PAH medications are predominately pulmonary vasodilators that modulate the endothelin, nitric oxide, and prostacyclin pathways.34 These therapies primarily address the vasoconstrictive phenotype of PAH, which is the predominant feature in only 5% to 10% of patients35 or those patients deemed vasoresponders.36 Although current medications provide benefits to nearly all PAH patients even if they are not vasoresponders, they were approved because they significantly increase exercise capacity (6‐minute walk distance [6MWD]), improve quality of life, and/or reduce morbidity37, 38, 39, 40, 41, 42, 43, 44, 45; however, only epoprostenol confers a clear survival benefit.46 Consequently, there is an urgent need to develop novel, effective therapies that target additional molecular pathways that drive the pathogenesis of PAH in order to supplement our current treatment options with the hopes of accelerating progress toward a cure.
The strategy of expanding the PAH pharmacopeia by repurposing medications that are used as therapy for other medical conditions is attractive because it may accelerate the development of new therapies and reduce the costs associated with new drug discovery for this orphan disease. In PAH, this consideration is important because, worldwide, many PAH patients do not even have access to currently approved therapies,47 so discovery of an inexpensive treatment option would very likely have a significant global impact. Available drugs have established safety profiles, so the considerable time and money required to exclude common toxicities and to demonstrate tolerability and safety can be reduced. Nevertheless, even for available agents, the need to confirm dosing and to establish the disease‐specific adverse effect profiles cannot be circumvented. Experience suggests that PAH patients may be either more or less sensitive to many drugs. For example, PAH patients require higher doses of calcium channel blockers than do patients with angina pectoris due to coronary artery disease.48 Conversely, PAH patients cannot tolerate the equivalent dose of the tyrosine kinase inhibitor sorafenib49 that cancer patients do. Nonetheless, repurposing has been effectively implemented in PAH. Sildenafil, a phophodisesterase type 5 inhibitor used for erectile dysfunction,50 was first tested in an acute treatment protocol in 13 pulmonary hypertension patients and provided a favorable hemodynamic response. Sildenafil administration led to a decrease in mean pulmonary arterial pressure (mPAP) and PVR and an increase in cardiac index.51 Then, a small placebo‐controlled crossover trial conducted in 22 PAH patients showed that sildenafil (dose: 25–100 mg, 3 times/day) increases exercise capacity and cardiac index, calculated using echocardiography, and improves quality of life.52 These findings provided biological plausibility for the SUPER (Sildenafil Use in Pulmonary Arterial Hypertension) trial, which documented significant increases in 6MWD and reductions in mPAP and PVR after 12 weeks of sildenafil treatment.40 These findings ultimately led to US Food and Drug Administration approval of sildenafil for PAH.
Preclinical research has identified many molecular pathways that contribute to pathological pulmonary vascular remodeling and RV dysfunction in PAH that have yet to be exploited therapeutically. mutations in BMPR2 (bone morphogenic protein receptor 2), for example, are associated with hereditable PAH,6, 7 and the BMPR2 pathway is downregulated in diverse rodent models of PAH53, 54; however, no current therapy targets BMPR2. Multiple mechanisms contribute to impaired BMPR2 signaling including inflammation‐mediated dysregulation,55, 56 estrogen‐induced suppression,57 autophagosomal degradation,58 and impaired membrane trafficking of the protein.59 When these mechanisms are targeted in preclinical studies, BMPR2 signaling is partially restored and reductions in pulmonary vascular disease severity are observed.60 Moreover, the cancer‐like,61 autoimmune/inflammatory,20, 62 and Warburg metabolic phenotypes4, 13, 15, 63 that promote vascular obstruction and fibrosis caused by increased proliferation and impaired apoptosis of PASMCs,12, 64 endothelial cells,4 and fibroblasts15 can also be inhibited to halt or even reverse pulmonary hypertension in animal studies, and yet none of these pathways are exploited by approved PAH‐targeted therapies. Another untapped target in PAH is the right ventricle, where ischemia and fibrosis, relating to impaired angiogenesis and a Warburg metabolic phenotype, contribute to RV dysfunction.65 Importantly, the metabolic changes and the related RV dysfunction can be partially reversed via pharmacological intervention.65, 66 Moreover, in the RV cardiomyocyte, microtubule remodeling causes mistrafficking and dysregulation of JPH2 (junctophilin 2) and subsequent pathological t‐tubule remodeling. This pathway can be rectified with colchicine, suggesting a novel therapeutic target to improve RV function.67 These are just a few of the numerous molecular pathways in both the pulmonary vasculature and the right ventricle that could be targeted to expand the PAH pharmacopeia to improve outcomes for PAH patients.
In this review, we discuss and evaluate the rigor of the preclinical data that support the notion that 22 medications could potentially be used to target molecular mechanisms involved in the pathogenesis of pulmonary vascular remodeling and RV dysfunction in PAH. We highlight currently available drugs that have clinical safety profiles with preclinical evidence of physiological changes at the whole‐animal level. We also discuss the available data from completed and ongoing exploratory clinical trials that are attempting to translate the information gleaned from animal models into therapy for PAH patients. Hopefully, this strategy will more rapidly fill the pipeline of drugs for PAH by identifying new agents that can potentially ameliorate or even cure this orphan disease. Repurposing medications may realize benefits for patients by accelerating the flow of ideas from the bench to the bedside.
Aldosterone Antagonists
Aldosterone is a steroid hormone that binds mineralocorticoid receptors, which are present in multiple tissues, including the heart and pulmonary vasculature. Aldosterone alters gene regulation and promotes a wide array of physiological effects including sodium and water retention, cardiac fibrosis, and activation of the sympathetic nervous system.68, 69 The broad distribution of mineralocorticoid receptors and diverse physiological effects underlies the use of aldosterone antagonists for several clinical indications, including left‐sided systolic heart failure,70, 71 systemic hypertension,72 and refractory ascites in cirrhotic patients.73 Aldosterone antagonists are generally well tolerated. The most important adverse effect is hyperkalemia, which is more frequently observed in patients also treated with an angiotensin‐converting enzyme inhibitor74 or in patients with chronic kidney disease.75 Finally, gynecomastia occurs in 6.9% to 10% of patients and can be painful and esthetically problematic in men.68 However, eplerenone, a newer aldosterone antagonist, does not cause gynecomastia and is effective in treating left heart failure.71
Contributing to the biological plausibility of targeting aldosterone in PAH, serum aldosterone levels are elevated in PAH patients and correlate with hemodynamic measures of pulmonary vascular disease. In a study that compared 5 controls with 20 PAH patients, serum levels of aldosterone were significantly higher in PAH patients (control versus PAH: 1200±424 versus 5959±2818 pg/mL, P<0.02).76 Moreover, serum aldosterone levels were positively correlated with PVR (r=0.72, P<0.02) and transpulmonary gradient (r=0.69, P<0.02) and inversely correlated with cardiac output (r=−0.79, P<0.005).76 Likewise, plasma and lung aldosterone levels are elevated in the monocrotaline rat (MCT rat) model of PAH.77
In PAH, increased serum aldosterone levels dampen activation of nitric oxide synthase in endothelial cells,77 promote adverse ECM remodeling in response to hypoxia in endothelial cells,78 and stimulate PASMC proliferation.79 Finally, aldosterone increases expression of the transcription factor Nedd9 (neural precursor cell expressed developmentally downregulated 9) via inhibition of proteolytic degradation in endothelial cells. Nedd9 then transcriptionally activates COL3A1 (collagen type III alpha 1 chain)80 to further promote ECM remodeling.
The preclinical data supporting the use of aldosterone antagonists to counteract mineralocorticoid pathway activation to combat pulmonary vascular disease are robust. Aldosterone negatively regulates endothelin B receptor–mediated nitric oxide production in pulmonary endothelial cells. Aldosterone increases production of reactive oxygen species, which oxidize endothelin receptor B at cysteine 405, an amino acid that lies in the endothelin nitric oxide synthase activating region of the receptor. The oxidation of endothelin receptor B reduces nitric oxide production77 (Figure 1). Treatment of rats with monocrotaline‐PAH with the aldosterone antagonist spironolactone (25 mg/kg per day) beginning at the time of monocrotaline injection increases nitric oxide levels in lung extracts and blunts development of adverse pulmonary vascular remodeling.77 In a reversal study, spironolactone (25 mg/kg per day) given 14 days after monocrotaline injection significantly reduced PA systolic pressure and PVR index.77 Eplerenone also slows the development of pulmonary vascular disease. Eplerenone (0.6 mg/g chow), initiated concurrently with exposure to hypoxia (O2 tension 76 mm Hg for 21 days) in the Sugen‐5416 (SU‐5416) hypoxia rat model, reduces PA systolic pressure.77
Figure 1.
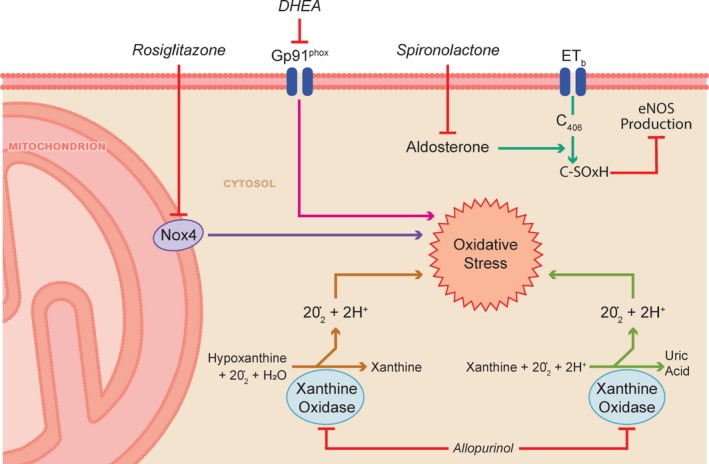
Spironolactone, allopurinol, DHEA, and rosiglitazone combat oxidative stress. DHEA indicates dehydroepiandrosterone; eNOS, endothelial nitric oxide synthase; ET, Endothelin‐b receptor; Nox4, NADPH oxidase 4.
Another pathological mechanism by which excess aldosterone promotes pulmonary vascular disease is ECM remodeling. In human PA endothelial cells, hypoxia enhances c‐Fos/c‐Jun binding to the proximal AP1 (activator protein 1) site of the promoter region of StAR (steroidogenic acute regulatory protein) and increases StAR expression.78 StAR promotes aldosterone synthesis, which in turn induces transcription of CTGF (connective tissue growth factor), collagen III, and MMP2 (matrix metalloprotease 2) and MMP9.78 In a series of in vivo experiments distinct from those described in the previous paragraph, treatment of SU‐5416 hypoxia rats with eplerenone (0.6 mg/g chow for 21 days) starting at the time of SU‐5416 injection reduces CTGF and collagen III levels in the pulmonary vasculature and lessens the severity of experimental PAH.78 In a reversal study, spironolactone (25 mg/kg per day) given 14 days after SU‐5416 for 7 days at hypoxia and continued for 16 to 17 days in normoxia reduces RVH, mPAP, and right atrial pressure.78
Aldosterone also promotes PASMC proliferation. Aldosterone increases the abundance of phosphorylated p70S6K (70‐kDa ribosomal S6 kinase), the active form of the major downstream effector kinase of mTORC1 (mammalian target of rapamycin complex 1), through a mechanism dependent on both Akt79 and the mTORC1 subunit Raptor79 in cultured PASMCs. The activation of mTORC1 promotes proliferation and apoptosis resistance of cultured PASMCs (Figure 2).79 When administered in a preventative manner, spironolactone reduces phosphorylated p70S6K expression in the pulmonary vasculature in MCT rats.79 Furthermore, combining spironolactone and a small interfering RNA targeting Raptor prevents pulmonary vascular remodeling in MCT rats.79 In a regression protocol, spironolactone plus small interfering RNA to Raptor reverses pulmonary hypertension in SU‐5416 hypoxia rats.79 Using a scoring system modified from Provencher et al,81 the scientific rigor score is 4 (Table 1) for the preclinical data supporting the use of aldosterone in PAH.
Figure 2.
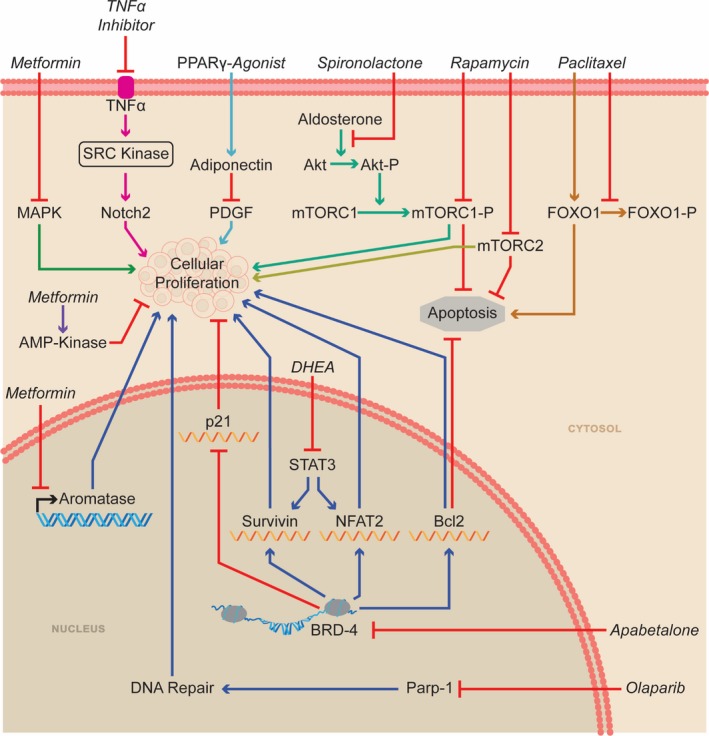
Multiple pathways can be inhibited to alter the proliferation/apoptosis balance of pulmonary artery smooth muscle cells. Bcl‐2 indicates B cell lymphoma 2; BRD‐4, bromodomain‐containing protein 4; FOXO1, forkhead box protein O1; MAPK, mitogen‐activated protein kinases; mTORC, mammalian target of rapamycin complex; NFATC2, nuclear factor of activated T cells 2; P, phosphorylation; Parp‐1, poly(ADP‐ribose) polymerase 1; PDGF, platelet derived growth factor; PPAR‐γ, peroxisome proliferator‐activator‐γ; STAT3, signal transducer and activator of transcription 3; TNF‐α, tumor necrosis factor‐α.
Table 1.
Numerical Score of Preclinical Rigor of Potentially Repurposed Medications
| Drug | Number of PAH Models Used | Regression Evaluateda | Human Tissue/Cells Evaluateda | Randomization Specifieda | Power Calculationa | Multiple Publications Demonstrating Efficacya | Male and Female Sexa | Long‐Term Safety Evaluationa | Total Score |
|---|---|---|---|---|---|---|---|---|---|
| Aldosterone antagonist | 2 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 5 |
| Allopurinol | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 2 |
| Anakinrab | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 |
| Anastrozole | 4 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 7 |
| Apabetalone | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 4 |
| β‐Adrenergic blockers | 2 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 4 |
| Chloroquine | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 2 |
| Colchicine | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 3 |
| DHEA | 3 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 5 |
| Dichloroacetate | 5 | 1 | 1 | 1 | 0 | 1 | 1 | 1 | 11 |
| Metformin | 4 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 8 |
| Nab‐rapamycin | 2 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 6 |
| Olaparib | 2 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 5 |
| Paclitaxel | 2 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 4 |
| Ranolazine | 2 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 4 |
| Rituximabb | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 2 |
| Rosiglitazone/pioglitazone | 4 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 8 |
| Tacrolimus | 3 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 5 |
| Tocilizumab | 2 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 4 |
| Trimetazidine | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 2 |
| TNF‐α inhibitor | 2 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 7 |
| Verteporfin | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 2 |
DHEA indicates dehydroepiandrosterone; PAH, pulmonary arterial hypertension; TNF‐α, tumor necrosis factor α.
1 = yes, 0 = no.
Indicates a molecule with similar mechanism of action was used in preclinical studies.
The impact of spironolactone in PAH is currently being investigated in 2 ongoing clinical trials. The CAPS‐PAH (Combination Ambrisentan Plus Spironolactone in Pulmonary Arterial Hypertension Study) is a single‐center, double‐blind, placebo‐controlled, crossover study that will investigate whether addition of spironolactone to ambrisentan alters exercise capacity in 30 PAH patients (ClinicalTrials.gov identifier NCT02253394). PAH patients on ambrisentan for >90 days who are New York Heart Association (NYHA) functional class II or III will be randomized to 50 mg of spironolactone daily or placebo for 90 days and then will undergo testing. After a 21‐day washout period, patients will cross over to the other arm for another 90 days of treatment, followed by a repeat assessment. The primary end points are change in 6MWD and maximal oxygen consumption. Secondary outcomes will include estimated cardiac output and RV function using echocardiography, biomarkers of RV failure (NT‐pro‐BNP [N‐terminal probrain natriuretic protein], IL6 [interleukin 6], troponin, and collagen III), and quality of life.
Concurrently, a multicenter, double‐blind, randomized, placebo‐controlled trial will also examine whether treatment with spironolactone alters outcomes in 70 PAH patients (ClinicalTrials.gov identifier NCT01712620). Patients in NYHA functional classes I to III who are either on stable PAH‐specific vasodilator therapy for 4 weeks or treatment‐naïve before enrollment will be randomized to placebo or spironolactone (25 mg daily for 7 weeks and, if tolerated, increased to 50 mg daily during week 8). The study will last for 24 weeks with the primary end points being change in 6MWD and clinical worsening. Secondary end points will include change in placebo‐corrected maximal oxygen consumption, RV function (quantified by cardiac magnetic resonance imaging [MRI]), and markers of inflammation. Finally, discontinuation rates due to adverse effects including hyperkalemia and gynecomastia will be recorded.
In summary aldosterone antagonists could combat endothelial dysfunction, prevent ECM remodeling, and slow PASMC proliferation in PAH. The 2 ongoing clinical trials will determine whether aldosterone antagonists are tolerable and effective in PAH.
Allopurinol
Oxidative stress, including an increase in reactive oxygen species formation, is implicated in the pathogenesis of pulmonary vascular remodeling.82 Xanthine oxidase catalyzes the transformation of hypoxanthine to xanthine and then to uric acid with the associated production of 4 superoxide anions.83 Thus, xanthine oxidase is potentially a major regulator of cellular oxidative stress (Figure 1).84 A pathological role for xanthine oxidase in PAH is suggested in several human studies. In a study of 99 PAH patients, the natural logarithmic transformation of serum uric acid is positively correlated with right atrial pressure (r=0.64, P<0.001).85 Higher serum levels of uric acid are associated with lower 6MWD and higher mortality in a study of 29 PAH patients.86 Furthermore, xanthine oxidase activity is elevated in the serum of PAH patients compared with control participants (5201±2836 [n=31] versus 2424±1419 [n=6] arbitrary units, P=0.026).87 In lung extracts of PAH patients, expression of the oxidative stress markers 8‐hydroxyguanosine and nitrotyrosine are increased.88 Mass spectrometry reveals elevated levels of 5‐hydroxyeicosatetraenoic acid, the oxidized product of 5‐oxo‐eicosatetraenoic acid, in lungs of PAH patients who had not been treated with prostacyclin.88 Moreover, expression of the antioxidant enzyme SOD2 (superoxide dismutase), which catalyzes breakdown of superoxide anion to the less toxic H2O2, 89 is reduced in PAH lungs.88 SOD2 downregulation (in PAH patients and experimental PAH) was independently confirmed and shown to result from an epigenetic mechanism mediated by DNMT1 (DNA methyltransferase 1) and DNMT3b. Methylation of the promoter of the SOD2 gene reduces SOD2 protein levels and decreases H2O2, which activates HIF‐1α (hypoxia‐inducible factor 1α), creating a state of pseudohypoxia (normal oxygen tension but activation of hypoxic signaling pathways).8 Interestingly, this pathological process can be reversed by the DNMT inhibitor decitabine, which is used to treat patients with myelodysplastic disorders.90
Allopurinol, a xanthine oxidase inhibitor, is used to prevent gout91 and nephrolithiasis caused by hyperuricosuria.92 Allopurinol is well tolerated for extended periods under these conditions. However, renal dysfunction increases the risk of side effects including gastrointestinal discomfort, lung toxicity, epidermolysis syndrome, and hypersensitivity syndrome.93 Two preclinical studies have examined the utility of allopurinol in pulmonary vascular disease.
Rats exposed to hypoxia (10% oxygen for 7 or 21 days) have elevated levels of PCOOH (phosphatidylcholine hydroperoxide), a marker of oxidative stress that reflects increased xanthine oxidase activity.94 Treating hypoxic rats with allopurinol (50 mg/kg every 12 hours starting the day before hypoxia exposure) decreases PCOOH levels and blunts adverse pulmonary vascular remodeling and reduces RVH.94 Likewise, neonatal rats exposed to hypoxia (13% O2 from birth for 14 days) have increased serum and lung xanthine oxidase activity,95 and allopurinol (50 mg/kg per day starting the first day of hypoxia) normalizes xanthine oxidase activity and reduces RVH and adverse pulmonary vascular remodeling.95
No clinical trials are currently investigating the use of allopurinol in PAH. The ease of administration and favorable side‐effect profile suggests that a trial of allopurinol in PAH is feasible. However, human doses would need to be much lower than those used in rodent studies (Table 2),96 and the rigor of the preclinical studies is low, with a score of 2 (Table 1).
Table 2.
Summary of Preclinical Results of Potentially Repurposed Drugs for PAH
| Drug | Mechanism of Action | Downstream Consequence | In Vivo Effects | Animal Model Used | Animal Model Dose | Equivalent Human Dosea | Maximal Daily Dose in Clinical Practice |
|---|---|---|---|---|---|---|---|
| Aldosterone antagonist | Inhibition of aldosterone signaling |
1. Increased nitric oxide levels in the PV 2. Reduced ECM remodeling in the PV 3. Inhibition of mTORC1 signaling leading to reduced PASMC proliferation |
1.Blunted PV remodeling 2. Reduced RVH |
MCT SU‐5416 hypoxia |
Spironolactone (25 mg/kg/d) Eplerenone (0.6 mg/g chow) |
Spironolactone: 4.0 mg/kg/d Eplerenone: 0.1 mg/g food |
Spironolactone: 200 mg Eplerenone: 100 mg |
| Allopurinol | Xanthine oxidase inhibitor |
1. Reduced PCOOH levels 2. Normalization of xanthine oxidase activity 3. Reduction in overall oxidative stress |
1.Blunted PV remodeling 2. Reduced RVH |
Hypoxic adult and neonatal rats |
50 mg/kg/d 50 mg/kg every 12 h |
8.1 mg/kg/d 8.1 mg/kg every 12 h |
300 mg |
| Anakinra | Block inflammatory cytokine IL1 |
1. Reduced IL1 mRNA in lungs 2. Reduced macrophage infiltration into pulmonary vasculature |
1. Blunted PV remodeling in MCT rats 2. Reduced RVH in MCT rats |
MCT Hypoxia |
Anakinra not used in preclinical study | Anakinra not used in preclinical study | 100 mg |
| Anastrozole | Inhibitor of estrogen signaling |
1. Increased BMPR2 signaling 2. Increased expression of PPAR‐γ 3. Increased expression of CD36 4. Increased insulin sensitivity 5. Reduction in PASMC proliferation |
1. Blunted PV remodeling 2. Reduced RVH |
Hypoxic rats Hypoxic mice SU‐5416 hypoxia BMPR2 R899X mice |
0.03–3 mg/kg/d | 0.005–0.5 mg/kg/d | 1 mg |
| Apabetaloneb | BRD‐4 inhibitor |
1. Reduced levels of oncogenic proteins NFATC2, Bcl‐2, and survivin 2. Increased expression of p21 3. Reduction in PASMC proliferation |
1. Blunted PV remodeling 2. Reduced RVH |
SU‐5416 hypoxia | Apabetalone not used in preclinical study | Apabetalone not used in preclinical study | 300 mg |
| β‐Adrenergic blockers | Counteract excessive sympathetic nervous system activation in right ventricle and pulmonary vasculature |
1. Normalization of β‐adrenergic signaling in the right ventricle 2. Increased SERCA2a mRNA levels |
1. Blunted PV remodeling 2. Decreased RV fibrosis 3. Improved RV function 4. Augmented exercise capacity 5. Improved survival |
MCT, SU‐5416 hypoxia |
Arotinolol (0.25 mg/kg/d) Bisoprolol (10 mg/kg/d) Carvedilol (15 mg/kg/d) |
Arotinolol (0.04 mg/kg/d) Bisoprolol (1.6 mg/kg/d) Carvedilol (2.4 mg/kg/d) |
Arotinolol: NA, Bisoprolol: 10 mg Carvedilol: 100 mg |
| Chloroquine | Inhibitor of lysosomal degradation |
1. Increased BMPR2 signaling via reduction in lysosomal degradation 2. Reduction in PASMC proliferation |
1. Blunted PV remodeling 2. Reduced RVH |
MCT | 50 mg/kg/d | 8.1 mg/kg/d | 2.3 mg/kg |
| Colchicine | Anti‐inflammatory and normalization of JPH2 levels via microtubule depolymerization |
1. Reduction in PASMC proliferation 2. Restoration of structure and function of T‐tubules in RV cardiomyocytes |
1. Reduced PV remodeling 2. Reduced RVH 3. Improved RV function 4. Enhanced exercise capacity |
MCT |
1.0 mg/kg/d for 5 d 0.5 mg/kg 3 times/wk |
0.16 mg/kg for 5 d 0.08 mg/kg 3 times/wk |
2.4 mg |
| DHEA | Inhibits STAT3 which reduces NFATC2 and survivn and increases BMPR2 |
1. Reduction in PASMC proliferation 2. Increased PASMC apoptosis 3. Increased BMPR2 signaling |
1. Reduced PV remodeling 2. Reduced RVH 3. Improved RV function 4. Enhanced exercise capacity |
MCT, SU‐5416 hypoxia |
10 mg/kg/d 30 mg/kg every other day 1% in food |
1.6 mg/kg/d 4.8 mg/kg every other day 0.16% in food |
100 mg |
| Dichloroacetate | Counteract Warburg metabolic effect via PDK inhibition |
1. Improved glucose oxidation 2. Reduced PASMC proliferation 3. Increased PASMC apoptosis 4. Increased potassium channel levels 5. Depolarization of mitochondria |
1. Reduced PV remodeling 2. Improved RV function 3. Enhanced RV contractility 4. Reduced RVH 5. Increased exercise capacity 6. Improved survival |
Hypoxic rats MCT SU‐5416 FHR PAB rats |
70–80 mg/kg/d 0.75 g/L drinking water |
11.3–12.9 mg/kg/d 0.12 g/L of drinking water |
25 mg/kg |
| Metformin | Inhibitor of MAPK activation, inhibitor of aromatase transcription, augments AMP activation |
1. Reduced PASMC proliferation 2. Reduced PASMC contractility 3. Reduced RV lipid deposition |
1. Reduced PV remodeling 2. Reduced RVH |
Hypoxic rats MCT SU‐5416 hypoxia BMPR2 R899X |
100 mg/kg/d 25 g/kg of high‐fat chow |
16.1 mg/kg /d 4.0 g/kg chow |
2550 mg |
| Nab‐rapamycin | Inhibitor of mTORC1 and mTORC2 |
1. Reduced PASMC proliferation 2. Increased PASMC apoptosis |
1. Reduced PV remodeling (dose dependent) 2. Reduced RVH (dose dependent) |
MCT Hypoxic mice |
Nab‐rapamycin not used in preclinical study | Nab‐rapamycin not used in preclinical study | 100 mg/m2 |
| Olaparib | Inhibitor of PARP1 |
1. Reduced PASMC proliferation 2. Increased PASMC apoptosis |
1. Reduced PV remodeling 2. Reduced RVH |
MCT SU‐5416 |
6 mg/kg/d | 0.97 mg/kg/d | 800 mg |
| Paclitaxel | FOXO1 Activator |
1. Reduced PASMC proliferation 2. Increased BMPR2 signaling 3. Increased PASMC apoptosis |
1. Reduced PV remodeling 2. Reduced RVH 3. Improved RV function |
SU‐5416 Hypoxia MCT |
5–7 mg/kg/wk 1 mg/kg/wk aerosolized |
0.8–1.1 mg/kg/wk 0.16 mg/kg/wk aerosolized |
225 mg/m2 every 3 to 4 wks |
| Ranolazine | Reduction of FAO and enhancement of glucose oxidation (by activating Randle cycle) |
1. Reduced Glut1 and HK1 mRNA levels 2. Increased RV glucose oxidation 3. Increased ATP production 4. Decreased FAO |
1. Reduced RVH 2. Improved RV function 3. Decreased RV fibrosis 4. Reduced risk of arrhythmias 5. Increased exercise capacity |
PAB rats MCT |
20 mg/d 0.25–0.5% in chow |
3.2 mg/d 0.04–0.08% in chow |
2000 mg |
| Rituximabb | Anti‐inflammatory via blocking of CD20 |
1. Reduced IL6, HIF‐1α, and VEGF 2. Decreased PASMC proliferation |
1. Reduced PV remodeling 2. Reduced RVH |
Ovalbumin immunization plus SU‐5416 rats | Rituximab not used in preclinical study | Rituximab not used inpreclinical study | 1000 mg every 2 wk |
|
Rosiglitazone/ pioglitazone |
PPAR‐γ activators |
1. Increased adiponectin levels 2. Reduced NOX4 levels 3. Reduced PASMC proliferation 4. Improved mitochondrial organization 5. Induced FAO genes 6. Improved FAO efficacy in cardiomyocytes |
1. Reduced PV remodeling 2. Reduced RVH 3. Improved RV function |
ApoE knockout mice Hypoxic rats Hypoxic mice SU‐5416 rats |
Rosiglitazone (8–10 mg/kg/d) Pioglitazone (20 mg/kg/d) |
Rosiglitazone (1.3–1.6 mg/kg/d) Pioglitazone (3.2 mg/kg/d) |
Rosiglitazone: 8 mg Pioglitazone: 45 mg |
| Tacrolimus | Calcineurin inhibitor |
1. Sequestered FK‐binding protein 2 from BMPR1 receptors 2. Increased BMPR2 signaling 3. Improved endothelial function 4. Reduced PASMC proliferation |
1. Reduced PV remodeling 2. Reduced RVH |
BMRP2 endothelial knockout mice MCT SU‐5416 hypoxia |
0.05 mg/kg/d | 0.008 mg/kg/d | 0.6 mg/kg |
| Tocilizumabb | Inhibit inflammatory cytokine IL6 |
1. Reduced STAT3 activation 2. Induced PASMC apoptosis |
1. Reduced PV remodeling 2. Reduced RVH |
MCT SU‐5416 hypoxia |
Tocilizumab not used in preclinical study | Tocilizumab not used in preclinical study | 800 mg every 4 wk |
| Trimetazidine | Reduce FAO and enhance glucose oxidation (by activating Randle cycle) |
1. Reduced Glut1 and HK1 mRNA levels 2. Increased RV glucose oxidation 3. Increased ATP production 4. Decreased FAO |
1. Reduced RVH 2. Improved RV function 3. Improved exercise capacity |
PAB rats | 0.7 g/L of drinking water | 0.11 g/L of drinking water | 70 mg |
| TNF‐α inhibitor | Anti‐inflammatory via blocking of TNF‐α signaling |
1. Increased BMPR2 signaling 2. Decreased NOTCH2 expression 3. Reduced PASMC proliferation |
1. Reduced PV remodeling 2. Reduced RVH |
MCT SU‐5416 |
Etanercept: 2.5 mg/kg twice weekly | 0.4 mg/kg twice weekly | Etanercept: 100 mg twice weekly |
| Verteporfin | Inhibitor of YAP‐induced glutaminolysis |
1. Decreased lysyl oxidase activity 2. Reduced glutaminase activity 3. Reduced pulmonary arteriolar stiffness 4. Decreased PASMC proliferation |
1. Reduced PV remodeling 2. Reduced RVH |
MCT | 25 mg/kg/d | 4.0 mg/kg/d | 6 mg/m2 every 3 mo |
ApoE indicates apolipoprotein E; Bcl‐2, B cell lymphoma 2; BMPR, bone morphogenic protein receptor; BRD‐4, bromodomain‐containing protein 4; ECM, extracellular matrix; FOXO1, forkhead box protein O1; FHR, Fawn hooded rat; Glut1, glucose transporter 1; HIF‐1α, hypoxia‐inducible factor 1α; HK1, hexokinase 1; JPH2, junctophilin 2; IL, interleukin; MAPK, mitogen‐activated protein kinase; MCT, monocrotaline; mTORC, mammalian target of rapamycin complex; NA, not available; NFATC2, nuclear factor of activated T cells 2; NOTCH2, notch 2; PAB, Pulmonary artery banded; PAH, pulmonary arterial hypertension; Parp‐1, poly(ADP‐ribose) polymerase 1; PASMC, pulmonary artery smooth muscle cell; PCOOH, phosphatidylcholine hydroperoxide; PPAR‐γ, peroxisome proliferator‐activator γ; PV, pulmonary vasculature; RV, right ventricular; RVH, right ventricular hypertrophy; SERCA2a, sarco/endoplasmic reticulum Ca2+‐ATPase; STAT3, signal transducer and activator of transcription 3; SU‐5416, Sugen‐5416; VEGF, vascular endothelial growth factor; YAP, Yes‐associated protein.
Indicates human dose was calculated via differences in body surface area.96
Indicates a molecule with similar mechanism of action was used in preclinical studies.
Anakinra
As discussed, substantial evidence shows that inflammation plays a role in PAH pathogenesis. Serum levels of the inflammatory cytokine IL1, which promotes IL6 synthesis (Figure 3), are increased in PAH patients.22, 23 Moreover, IL1 mRNA levels are elevated in the lungs of MCT rats.97 Furthermore, administration of IL1 to BMPR2 (bone morphogenetic protein receptor type 2) R899X transgenic mice produces a more severe PAH phenotype.98 In aggregate, these findings provide evidence of a direct adverse effect of IL1 and inflammation in the pathogenesis of PAH. Anakinra is a recombinant IL1 receptor antagonist that can be used to treat rheumatoid arthritis99 and recurrent pericarditis.100 Anakinra is safe, but adverse side effects include headache, vomiting, injection‐site irritation, and increased risk of infection due to the immunosuppressive actions.99
Figure 3.
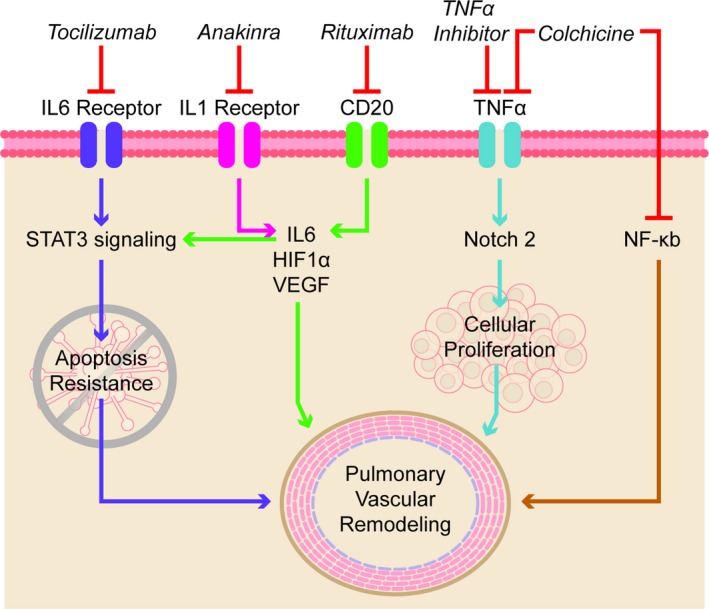
Proinflammatory pathways that can be targeted for treatment of pulmonary arterial hypertension. HIF‐1α indicates hypoxia‐inducible factor‐1α; IL, interleukin; NF‐κB, nuclear factor κB; NOTCH2, notch 2; STAT3, signal transducer and activator of transcription 3; TNF‐α, tumor necrosis factor‐α; VEGF, vascular endothelial growth factor.
IL1 antagonism is beneficial in rats with PAH induced by monocrotaline but not in rats with chronic hypoxic pulmonary hypertension induced by hypobaric hypoxia (simulated altitude of 16 000 ft [4877 m]). MCT rats have increased mRNA levels of IL1 and IL1 receptor in lung extracts, whereas hypoxic rats do not.97 Use of a purified recombinant human IL1 receptor antagonist (2 mg/kg twice a day starting at the time of monocrotaline injection or hypoxia for 2 weeks) reduces mPAP and RVH in MCT rats at the 3‐week time point.97 This approach is associated with a reduction in lung IL1 mRNA levels. In contrast, chronic hypoxic rats experienced no benefit with the IL1 antagonist.97 The differences in phenotype, with much greater lung inflammation in MCT rats, likely explains the divergent effects. Thus, IL1 antagonism appears to be effective in inflammatory, preclinical PAH models.
The safety of anakinra in PAH was recently reported in a single‐arm, open‐label study of 6 PAH patients. Patients with connective tissue–associated, HIV, portal hypertension, or schistosomiasis‐associated PAH were excluded. In this small study, all patients had evidence of RV dysfunction, as defined by RV diastolic diameter >4.3 cm, fractional area change <35%, and/or tricuspid annular plane systolic excursion ≤ 1.5 cm, and NYHA class II or III symptoms despite optimal therapy. Patients received 100 mg of anakinra daily for 14 days by subcutaneous injection.101 After 14 days of treatment, high‐sensitivity C‐reactive protein and symptom burden, as quantified by the Minnesota Living with Heart Failure Questionnaire, were significantly reduced.101 There was no significant change in peak oxygen consumption, minute ventilation over carbon dioxide slope, tricuspid annular plane systolic excursion, or RV fractional area change.101 This study provides evidence that short‐term administration of anakinra is safe with a potential reduction in symptom burden. A larger and longer duration trial will be needed to determine the utility of anakinra for PAH treatment.
Anastrozole
Targeting the estrogen pathway in PAH is rooted in the observation that there is a consistent and substantial (≈3–4:1) female predominance in the incidence of PAH.102, 103, 104, 105 Moreover, estrogen is linked to reduced BMPR2 expression57 and metabolic derangements106 in the pulmonary vasculature. Anastrozole is an antiestrogen compound that inhibits aromatase, an enzyme that catalyzes the formation of estradiol from testosterone.107 Anastrozole is currently used as an adjuvant in postmenopausal women with hormone receptor–positive breast cancer.108 Anastrozole is well tolerated in breast cancer patients, with common side effects including gastrointestinal discomfort, hot flashes, and gynecological disturbances. Long‐term use of anastrozole can reduce bone mineral density.109
The beneficial effects of anastrozole are observed only in female rodents in preclinical PAH models. For example, female mice exposed to chronic hypoxia (10% O2 for 14 days) and then treated with anastrozole (0.3 or 3 mg/kg per day) for 14 additional days at hypoxia have reduced adverse remodeling of pulmonary arteries, lower RV systolic pressure (RVSP), and less RVH.57 In contrast, hypoxic male mice experience no benefit with anastrozole treatment.57 In a regression protocol of SU‐5416 hypoxia rats, anastrozole (0.03, 0.3, or 3 mg/kg per day for 14 days during normoxia) decreases the number of occluded and remodeled pulmonary arterioles but, again, only in female rats.57 The sex‐specific effects may be due to differences in aromatase levels in PASMCs. Specifically, male mice, rats, and humans have less aromatase in PASMCs than their female counterparts.57 Intriguingly, in the hypoxic mice and SU‐5416 hypoxia rat experiments described earlier, anastrozole increases PASMC BMPR2 expression (Figure 4) but only in cells derived from females.57 However, other mechanisms may also exist because the beneficial effects of anastrozole are also observed in inducible BMPR2 R899X transgenic mice. In this study (conducted exclusively in female mice), anastrozole (0.3 mg/kg per day) was used in combination with fulvestrant, a selective estrogen receptor degrader,110 to more fully inhibit estrogen signaling. Anastrozole and fulvestrant increase lung expression of PPAR‐γ (peroxisome proliferator‐activator γ) and CD36 (which regulates fatty acid uptake and insulin sensitivity111) and improve insulin sensitivity (Figure 5).106 Conversely, estrogen reduces insulin‐induced membrane mobilization of GLUT4 (glucose transporter type 4) in pulmonary microvascular endothelial cells, which may underlie the negative effects of estrogen on insulin sensitivity.106 Anastrozole and fulvestrant reduce the percentage of muscularized pulmonary arteries and lower RVSP.106 Thus, anastrozole's sex‐specific efficacy may relate to beneficial effects of estrogen inhibition on BMPR2 signaling and/or altered metabolism.
Figure 4.
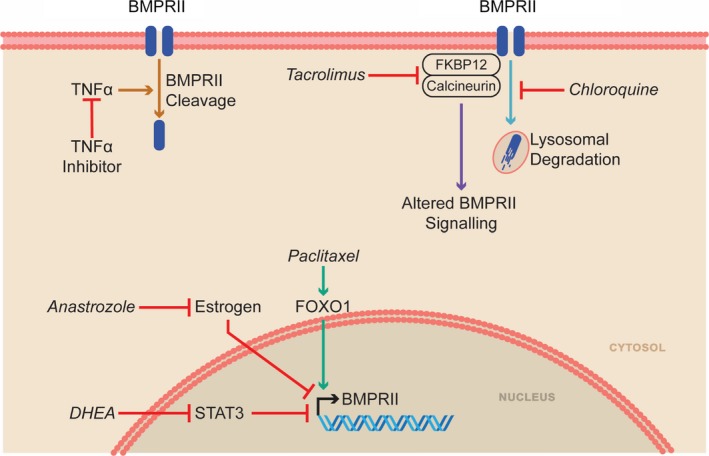
Medications that can augment the BMPR2 pathway as a therapeutic strategy for PAH. BMPR2 indicates bone morphogenic protein receptor 2; DHEA, dehydroepiandrosterone; FKBP12, 12‐kDa FK506‐binding protein; FOXO1, forkhead box protein O1; STAT3, signal transducer and activator of transcription 3; TNF‐α, tumor necrosis factor‐α.
Figure 5.
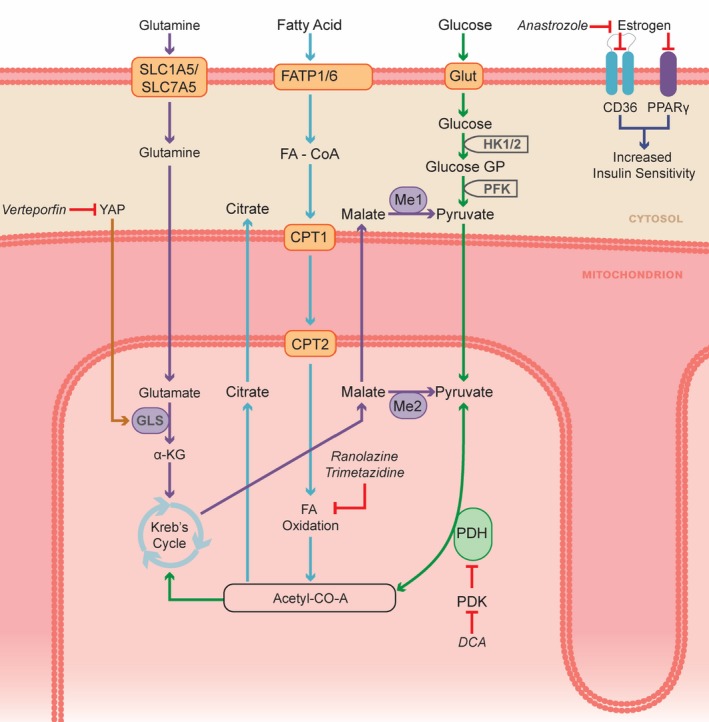
Pathological metabolic changes can be targeted with several available medications. α‐KG, α‐ketoglutarate; CoA, coenzyme A; CPT, carnitine palmitoyltransferase; DCA, dichloroacetate; FA, fatty acid; FATP, fatty acid transport protein; GLS, glutaminase; Glut, glucose transporter; HK, hexokinase; Me, malic enzyme; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; PFK, phosphofructokinase; PPAR‐γ, peroxisome proliferator‐activator‐γ; SLC, solute carrier family; YAP, Yes‐associated protein.
In a clinical trial of 18 male and female PAH patients, randomized in a 2:1 fashion to anastrozole (1 mg/day) or placebo for 3 months, anastrozole significantly decreased 17β‐estradiol levels and increased 6MWD by a median distance of 26 m versus placebo.112 However, there was no improvement in RV function and quality of life, and this trial did not assess invasive hemodynamics. These initial pilot data have led to the PHANTOM (Pulmonary Hypertension and Anastrozole) trial (ClinicalTrials.gov identifier NCT03229499). PHANTOM is a multicenter, double‐blind, randomized, placebo‐controlled trial that will investigate whether anastrozole (1 mg/day) for 1 year alters outcomes in 84 NYHA functional class I to III PAH patients on stable PAH‐specific therapy. Change in 6MWD will be the primary end point, with secondary end points including changes in RV function, NT‐pro‐BNP, biomarkers of anastrozole treatment, symptomatic burden, daily activity, time to clinical worsening, and adverse side effects. The results of PHANTOM will help determine the efficacy of anastrozole in PAH.
The impact of estrogen inhibition with tamoxifen, a selective estrogen receptor blocker,113 is also being investigated in PAH. A single‐center, double‐blind, randomized, placebo‐controlled trial will be conducted at Vanderbilt University. The effects of tamoxifen (20 mg 3 times/day for 24 weeks) will be examined in 24 PAH patients (ClinicalTrials.gov identifier NCT03528902). The inclusion criteria for this trial include patients who have idiopathic, heritable, or drug‐ or toxin‐induced PAH or PAH associated with connective tissue disease and who are classified as World Health Organization (WHO) functional class I to III and able to walk 150 to 550 m during a 6MWD test. Important exclusion criteria include treatment with any therapy that modulates sex hormones, pregnancy, WHO functional class IV, initiation of any PAH therapy within 3 months before visit, and use of either bosentan or selexipag, as these medications may have drug–drug interactions with tamoxifen.
Although estrogen inhibition holds promise for PAH treatment, we need to carefully observe how it will affect RV function. Evidence shows that estrogen augments RV function as 17β‐estradiol supplementation increases exercise capacity in both male and female SU‐5416 hypoxia rats, likely via an anti‐inflammatory effect and inhibition of RV cardiomyocyte apoptosis.114 Furthermore, 17β‐estradiol administration to SU‐5416 hypoxia female rats increases expression of PGC‐1α (peroxisome proliferator–activated receptor γ coactivator 1α) and maintains mitochondrial mass and oxidative capacity in the right ventricle.115
Apabetalone
The balance of PASMC proliferation and apoptosis is an attractive target for PAH therapy because the imbalance of these 2 processes is repeatedly observed in PAH.116 Increased expression of the epigenetic regulator BRD‐4 (bromodomain‐containing protein 4) is observed in lung extracts, distal pulmonary arteries, and isolated PASMCs of PAH patients.117 The upregulation of BRD‐4 depends on the downregulation of miR‐204, a microRNA that represses BRD‐4.117 BRD‐4 regulates transcription of many genes through its interaction with acetylated histones. Increased abundance of BRD‐4 promotes cell survival and inhibits apoptosis.117 Apabetalone is a BRD‐4 inhibitor that is currently being evaluated in patients with coronary artery disease,118 although it is not yet approved for clinical use. In short‐term clinical trials ranging from 3 to 6 months, apabetalone is well tolerated, but evidence suggests it may cause mild transaminase elevation.118
The beneficial effects of BRD‐4 inhibition in PAH is observed in human PASMCs and in SU‐5416 hypoxia rats. In cultured PASMCs, BRD‐4 inhibition with either small interfering RNA or JQ1, a nonspecific BRD protein inhibitor, reverses upregulation of oncogenic proteins, such as NFATC2 (nuclear factor of activated T cells 2), Bcl‐2 (B cell lymphoma 2), and survivin while simultaneously increasing expression of p21, an inhibitory cell‐cycle regulator (Figure 2).117 These molecular changes are accompanied by a reduction in proliferation and heightened apoptosis in cultured PASMCs.117 In SU‐5416 hypoxia rats (3 weeks of 10% O2, then return to normoxia), treatment with nebulized JQ1 (1 μmol every 4 days for 2 weeks at 5 weeks after SU‐5416 injection) prevents proliferation and promotes apoptosis of PASMCs, in turn reducing mPAP and increasing cardiac output.117
Apabetalone is currently being investigated in a single‐arm trial in a 2‐center study. Ten PAH patients who are WHO functional class II or III and on stable PAH therapy for >4 months will receive 100 mg of apabetalone twice a day for 16 weeks (ClinicalTrials.gov identifier NCT03655704). The primary end point in this trial is change in PVR. Secondary end points will include change in mPAP, right atrial pressure, mixed venous saturation, 6MWD, WHO functional class, NT‐proBNP, quality of life, and change in biomarkers of vascular calcification, inflammation, complement, acute phase response, fibrogenesis, and metabolism.
β‐Adrenergic Blockers
Biological plausibility for application of β‐adrenergic blockers in PAH is supported by the observation that there is extreme neurohormonal activation in RV failure secondary to PAH (Figure 6).119 Indeed, the degree of autonomic activation, both systemically and in the RV, is greater in PAH than in left ventricular (LV) failure syndromes.119, 120 β‐Adrenergic blockers antagonize β‐adrenergic receptors and thus could combat the effects of the marked systemic activation of the sympathetic nervous system that characterizes PAH.119, 120
Figure 6.
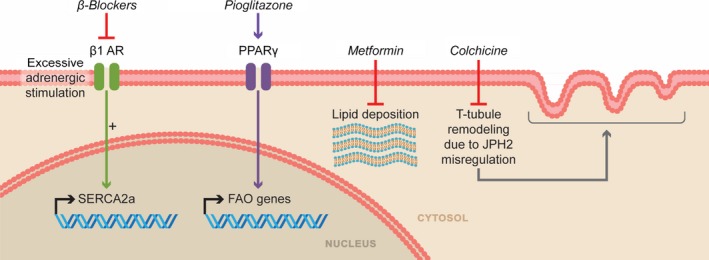
Pathologic pathways in the right ventricle that can be inhibited to improve right ventricular function. β1 AR indicates β1 adrenergic receptor; FAO, fatty acid oxidation; JPH2, junctophilin 2; PPAR‐γ, peroxisome proliferator‐activator‐γ; SERCA2a, sarco/endoplasmic reticulum Ca2+‐ATPase.
β‐Adrenergic blockers are well tolerated if not administered to patients in decompensated heart failure, but common adverse effects include fatigue, bradycardia, and hypotension.121 Clinically, β‐adrenergic blockers have several indications including treatment of systemic hypertension,121 arrhythmias,121 angina pectoris, 121 prophylaxis for variceal bleeding in select cirrhotic patients,122 and left‐sided systolic heart failure.123 In fact, β‐adrenergic blockers are one of the main treatment strategies in LV systolic dysfunction because chronic β‐adrenergic blocker therapy causes beneficial LV reverse remodeling and can increase LV ejection fraction124 while improving survival.123, 125, 126, 127
Preclinical studies demonstrate that antagonism of neurohormonal activation by chronic β‐adrenergic receptor blockade or inhibition of GRK2 (G‐protein‐coupled receptor kinase 2)–mediated β‐adrenergic receptor uncoupling improves RV function, reverses RV remodeling, and restores RV β‐adrenergic receptor signaling pathways.128, 129, 130, 131, 132 Arotinolol (an α‐ and β‐adrenergic receptor blocker) at a dose of 0.25 mg/kg per day starting at the time of monocrotaline injection prevents development of PAH and RVH.130 Bisoprolol (a β1‐cardioselective blocker) at a dose of 10 mg/kg per day starting 10 days after monocrotaline injection decreases RV fibrosis and inflammation, restores RV β‐adrenergic signaling, and improves RV function.129 Carvedilol (a nonselective α1/β1/β2‐adrenergic receptor antagonist) at a dose of 15 mg/kg per day has beneficial effects on the right ventricle in both the SU‐5416 hypoxia and MCT models.128 In SU‐5416 hypoxia rats, carvedilol, initiated at return to normoxia and maintained for 4 weeks, increases SERCA2a (sarco/endoplasmic reticulum Ca2+‐ATPase) mRNA levels (Figure 6), reduces RVH, improves RV function, reverses RV remodeling, and augments exercise capacity.128 In MCT rats, carvedilol given 2 weeks after monocrotaline injection also enhances RV function, blunts RVH, and improves survival.128
Although considerable evidence shows that β‐adrenergic blockers have a favorable effect on the right ventricle, substantial data indicate that β‐adrenergic blockers may also hasten pulmonary vascular remodeling. For instance, arotinolol reduces pulmonary arterial pressures in MCT rats.130 Furthermore, nebivolol increases nitric oxide activity, whereas carvedilol promotes vasodilation through α‐adrenergic receptor antagonism.133 Finally, propranolol blocks protein kinase C activity which, as a result, promotes nitric oxide synthesis.134 These agents may have favorable effects on both the pulmonary vasculature and the right ventricle in PAH.
Several retrospective human studies show chronic β‐adrenergic blocker therapy is safe when given at very low doses and slowly uptitrated in PAH patients who are not in overt heart failure. Under these circumstances, use of multiple β‐adrenergic blockers, including metoprolol, atenolol, carvedilol, propranolol, nadolol, and labetalol, is not associated with increased mortality in PAH patients.135, 136, 137 Based on the preclinical data and retrospective safety data in patients with PAH, several small clinical trials have evaluated the safety and efficacy of β‐adrenergic blockers in PAH. In an open‐label study of 6 PAH patients, treatment with carvedilol (median dose of 18.75 mg twice daily) was well tolerated and significantly improved RV ejection fraction (RVEF), as measured by cardiac MRI.138 Treatment with bisoprolol (up to 10 mg/day for 6 months) in a randomized, double‐blind, placebo‐controlled, cross‐over study of 19 PAH patients was also well tolerated. However, bisoprolol reduced cardiac index and exercise capacity and failed to improve RVEF.139 More recently, in a randomized, double‐blind, placebo‐controlled trial of 20 PAH patients, use of carvedilol improved several surrogate end points.140 Carvedilol treatment reduced glucose uptake in the right ventricle, as quantified by positron emission tomography, and increased β‐adrenergic receptor density in circulating white blood cells. Carvedilol treatment significantly improved RV function (increased fractional area change on echocardiography) at 3 months but not at 6 months of follow‐up.140 However, carvedilol treatment did not increase exercise capacity or cardiac output.140 Carvedilol was well tolerated, with no serious adverse events in participants receiving a fixed low dose (3.125 mg twice a day, n=10) or doses up to 25 mg twice a day (n=4).
Clearly, larger and longer studies are required to definitively examine the safety and efficacy of β‐adrenergic blockers for improving RV function in PAH. Future studies should consider carvedilol over bisoprolol because it offers potentially beneficial vasodilator and antioxidant pleiotropic effects.141, 142 Furthermore, carvedilol, acting as a “biased” ligand, stimulates β‐arrestin signaling and antagonizes G‐protein–mediated signaling.143 This is important because β‐arrestin–dependent signaling is cardioprotective in the presence of chronic catecholamine stimulation,144 which may explain why carvedilol reduces mortality by 50% in animal studies.128 In conclusion, β‐blockers should be tested mainly in PAH patients with reduced RV function because these patients are more likely to have increased neurohormonal activation119 and thus have a higher likelihood of receiving a beneficial effect.
Chloroquine
As discussed, increasing BMPR2 signaling is a promising mechanism for PAH treatment. In cultured PASMCs, chloroquine increases BMPR2 protein levels by modulating autophagy (Figure 4).58 Chloroquine was initially used to treat malaria; however, its uses have expanded to include treatment of rheumatologic disease such as rheumatoid arthritis and systemic lupus erythematosus.145 Chloroquine is well tolerated in long‐term use, although screening is needed for ocular side effects, especially in patients with impaired renal function.145
In cultured PASMCs, chloroquine increases BMPR2 protein levels, slows proliferation, and promotes apoptosis.58 In vivo, chloroquine (20 or 50 mg/kg per day) given to rats at the time of monocrotaline injection attenuates the severity of PAH, as demonstrated by reduction in pulmonary vascular obstruction, RVSP, and RVH.58 When given after PAH is established (3 weeks after monocrotaline injection), chloroquine (50 mg/kg per day) reduces the number of muscularized pulmonary arteries, RVSP, and RVH.58 The scientific rigor score supporting the use of chloroquine is 2 (Table 1), but chloroquine's long and extensive track record of safe use in humans with numerous diseases (including inflammatory conditions relevant to PAH) suggest that it could be considered for PAH patients. However, the doses used in preclinical studies may not be achievable in humans (Table 2), and dose ranging studies will be required. There are currently no trials to examine the use of chloroquine in PAH.
Colchicine
Drugs that target both the pulmonary vasculature and the right ventricle have great potential for PAH. Colchicine, an anti‐inflammatory medication that is used to treat flares of the crystalline arthropathy gout,146 familial Mediterranean fever,147 and chronic or relapsing pericarditis,148 has potential to reduce PASMC proliferation via anti‐inflammatory mechanisms149 and to improve RV function by combating t‐tubule derangements.67 When used clinically, colchicine has several side effects, with the most prominent being dose‐dependent gastrointestinal disturbances (nausea, vomiting, and diarrhea).150 However, at a dose of 0.5 mg twice a day, colchicine was safely tolerated for 6 months in a clinical trial of LV systolic heart failure patients.151
Administration of colchicine (1.0 mg/kg/day) for 5 days beginning 5 days after monocrotaline administration significantly reduces levels of TNF‐α (tumor necrosis factor α), NF‐κb (nuclear factor κB), MMP9, and TGF‐β (transforming growth factor β) in both the lungs and the right ventricle. The improved inflammatory mediator profile achieved by colchicine is associated with diminished severity of pulmonary hypertension (Figure 3).149
In RV cardiomyocytes, colchicine counteracts the pathologically remodeled microtubule cytoskeleton and corrects the detrimental JPH2 downregulation that contributes to RV dysfunction in PAH. JPH2, a protein that is essential for proper t‐tubule structure and function,152 is decreased in monocrotaline RV cardiomyocytes and this leads to disrupted t‐tubule structure.67 The structural changes in the t‐tubules ultimately contribute to RV hypokinesis (Figure 6).67 In MCT rats, colchicine administration (0.5 mg/kg 3 times/week) starting 1 week after monocrotaline injection increases JPH2 protein levels by depolymerizing the hyperstabilized microtubule cytoskeleton, which slows pathological t‐tubule remodeling. These molecular and cellular changes improve RV function, increase cardiac output, and enhance exercise capacity.67 Importantly, although colchicine directly augments RV function, as shown by a disruption of the strong relationship between elevated pulmonary arterial pressures and RV dysfunction, it also regresses pulmonary vascular disease burden.67
Because colchicine has the potential to target pathological processes in both the pulmonary vasculature and the right ventricle, it could be investigated in PAH patients. Barriers leading to a clinical trial include the scientific rigor score of only 3 (Table 1), and the human doses equivalent to those used in animal studies may be difficult to achieve (Table 2).
Dehydroepiandrosterone
Dehydroepiandrosterone, or DHEA, is a naturally occurring, cholesterol‐derived steroid hormone that is a precursor for both estrogen and testosterone.153 DHEA has been used to treat aspects of adrenal insufficiency and postmenopausal changes including sexual dysfunction and symptoms of menopause.154, 155 Although largely shown to be ineffective in clinical trials for these indications, the safety profile is excellent, with no major adverse effects in a meta‐analysis of 1188 women taking DHEA, but there are reports of hair growth and acne.155, 156 DHEA has multiple biological actions in vivo including modulation of endothelial function, anti‐inflammatory effects, increasing insulin sensitivity, and neuroprotection.153 In PAH, DHEA promotes pulmonary vasodilation through reduction of intracellular calcium,157 normalizes the PASMC proliferation/apoptosis balance,158 and potentially improves RV function by mitigating oxidative stress.159 Clinically, lower serum levels of DHEA are associated with an increased risk of developing PAH in men.160 Furthermore, in men with PAH, higher serum levels of DHEA are associated with lower right atrial pressure and PVR.160
Several independent publications demonstrate that DHEA is efficacious in multiple preclinical models of PAH. In rats with chronic hypoxia (0.5 atm of pressure), treatment with DHEA (30 mg/kg every other day) can prevent and reverse pulmonary hypertension.157 DHEA reduces levels of intracellular calcium via increased expression and activity of the calcium‐activated potassium channel in PASMCs.157 In MCT rats, DHEA (10 mg/kg daily starting 18 days after monocrotaline) significantly reduces pulmonary vascular remodeling, mPAP, and RVH while normalizing treadmill walk distance.158 At the molecular level, DHEA decreases STAT3 (signal transducer and activator of transcription 3) activation, which restores the apoptosis/proliferation balance by reducing NFATc2 and survivin levels (Figure 2) and increasing BMPR2 levels (Figure 4).158 Finally, in SU‐5416 hypoxia rats (10% O2 for 3 weeks), treatment with DHEA (1% DHEA‐containing food) starting at return to normoxia for 5 weeks significantly blunts pulmonary vascular remodeling and RVH, improves RV function, and augments cardiac index.159 In the right ventricle, DHEA treatment lessens collagen expression and the number of cardiomyocytes undergoing apoptosis.159 Moreover, DHEA moderates oxidative stress, as demonstrated by a reduction in NADPH oxidase subunit gp91phox (Figure 2) and tissue NADPH levels.159 Thus, substantial evidence suggests that DHEA may affect multiple pathological processes in PAH.
DHEA is currently being investigated in the EDIPHY (Effects of DHEA in Pulmonary Hypertension) trial. EDIPHY is a crossover trial that will be conducted in 24 PAH patients who are on stable PAH‐directed therapy for at least 12 weeks (ClinicalTrials.gov identifier NCT03648385). Patients will be randomized to placebo or DHEA (50 mg daily) for 18 weeks, there will be a 4‐week wash‐out period, and then patients will be treated on the other arm of the trial. The primary outcome is change in RV longitudinal strain, as determined by cardiac MRI. Secondary outcomes will include RVEF, NT‐pro‐BNP, sex hormone levels, 6MWD, WHO functional class, symptom burden, and adverse effects.
Dichloroacetate
In PAH, PDK (pyruvate dehydrogenase kinase), a family of enzymes that negatively regulate glucose oxidation,66, 161 is activated through numerous mechanisms, including epigenetic activation of HIF‐1α.8 Activated PDK phosphorylates and inhibits mitochondrial PDH (pyruvate dehydrogenase), which suppresses oxidative metabolism, leading to a metabolic shift that favors uncoupled glycolysis (the Warburg phenomenon).66 In PAH, the Warburg effect, caused in part by PDK upregulation, occurs in both the pulmonary vasculature and the right ventricle (Figure 5).66 Warburg metabolism retains energetic stability (achieved by high rates of glycolytic flux) while minimizing basal rates of unstimulated apoptosis, which promotes cellular proliferation (in PASMCs, endothelial cells, and vascular fibroblasts) and hypertrophy (in RV cardiomyocytes).13
Dichloroacetate is a PDK inhibitor that has long been used to treat patients, particularly children with inherited mitochondrial disorders.162 The most common side effect of dichloroacetate is neuropathy, which is usually reversible with drug discontinuation.161 An extensive series of experiments shows dichloroacetate can prevent and regress PAH in multiple rodent models. Dichloroacetate's beneficial effects on the pulmonary vasculature and the right ventricle are achieved through several mechanisms. Dichloroacetate restores the hypoxia‐dependent whole‐cell potassium current in PASMCs.64 When given to chronically hypoxic rats (10% O2), dichloroacetate (70 mg/kg per day) partially reverses downregulation of voltage‐gated potassium channels, such as Kv1.5 and Kv2.1, in PASMCs. By retaining voltage‐gated potassium channel expression and function, dichloroacetate maintains the membrane potential of the PASMCs and thereby prevents activation of the large conductance voltage‐gated calcium channel. This reduces pulmonary vasoconstriction and results in less severe pulmonary vascular remodeling, lower pulmonary arterial pressures, and higher cardiac output in protocols for both prevention (dichloroacetate given day 1 of hypoxia) and regression (dichloroacetate initiated on day 10 of hypoxia).64 In MCT rats, dichloroacetate (80 mg/kg per day) increases expression of potassium channel Kv1.5 in lung extracts.12 This promotes vasodilation and apoptosis of PASMCs by normalization of potassium current regulation and membrane potential.12 Furthermore, dichloroacetate depolarizes mitochondria, which restores physiological H2O2 production and induces PASMC apoptosis.12 In a regression model, dichloroacetate reverses pulmonary vascular remodeling, leading to lower pulmonary pressures, normalization of RV thickness, and reduced mortality.12
In addition to the effects on the pulmonary vasculature, dichloroacetate (70 mg/kg per day) augments RV function via metabolic reprogramming, leading to greater glucose oxidation, which improves RV function in both PA‐banded (treatment starting day of PA banding) and MCT rats (10 days after monocrotaline).163 Also, dichloroacetate restores expression of potassium channels Kv1.2, Kv1.5, and Kv4.2 in the right ventricle, which normalizes cardiac repolarization—evident from a shortening of the QT interval on the ECG.163 The beneficial effects of dichloroacetate are more pronounced in MCT rats than in PA‐banded rats, suggesting that correction of the pulmonary vasculature plays a major role in beneficial effects of this drug on the right ventricle; importantly, however, there is a direct effect on RV function in isolated heart assessments.163 In fact, dichloroacetate (1 mM for 40 minutes) acutely increases monocrotaline RV contractility in the working heart model.163 Finally, in the fawn‐hooded rat model of PAH, dichloroacetate (0.75 g/L of drinking water for 6 months) decreases activation of FOXO1 (forkhead box protein O1), a transcription factor that upregulates RV PDK4 expression. The reduction in FOXO1 diminishes PDK4 levels and restores PDH activity, which improves glucose oxidation and contractile function in RV cardiomyocytes.164 In summary, dichloroacetate consistently reduces RVH and increases cardiac output and exercise capacity in multiple preclinical models of PAH.
The safety and efficacy of dichloroacetate were examined in ex vivo human PAH lungs and in PAH patients. In an ex vivo human lung perfusion system, dichloroacetate stimulates PDH activity and improves oxygen consumption in PAH lungs.165 However, the positive effects are not observed in all PAH patients’ lungs. In fact, the beneficial effects of dichloroacetate depend on the absence of polymorphisms in SIRT3 (sirtuin 3) or UCP2 (uncoupling protein 2), 2 proteins that regulate mitochondrial function in a PDK‐independent manner.166, 167, 168 In a 4‐month open‐label trial in 20 PAH patients, dichloroacetate reduced PVR and increased 6MWD. However, the greatest effects on PVR reduction and 6MWD changes were observed in patients with normal or low polymorphism scores or those with normal predicted function of SIRT3 and UCP2.165 Dichloroacetate doses up to 6.25 mg twice daily were well tolerated, but doses of 12.5 mg twice daily were associated with significant (but reversible) peripheral neuropathy.165 In summary, these data demonstrate that PAH patients, who are genetically susceptible to metabolic targeting with dichloroacetate, experience improvements in hemodynamics and exercise capacity. This trial lays the foundation for a personalized medicine approach using dichloroacetate in PAH patients who lack significant polymorphisms in the SIRT3 and UCP2 genes.
Metformin
Therapies that can target both the right ventricle and the pulmonary vasculature would be optimal for PAH treatment. Metformin, a biguanide that is frequently used to treat type 2 diabetes mellitus,169 has potential to promote pulmonary vasodilation by increasing endothelial nitric oxide synthase,170 to mitigate PASMC proliferation by inhibiting the pro‐proliferative MAPK (mitogen‐activated protein kinase),170 to negate the estrogen pathway via inhibition of aromatase transcription,171 and to combat pathological lipid deposition in the right ventricle.172 Clinically, metformin has a long safety history, but the most common side effect is gastrointestinal discomfort. A potentially dangerous adverse effect, lactic acidosis, can occur, particularly in patients with renal impairment.173 However, the reported incidence of metformin‐induced lactic acidosis is <10 per 100 000 patient‐years.173
In preclinical models, the beneficial effects of metformin are mediated through multiple molecular mechanisms. In hypoxic rats (inhaled oxygen tension 380 mm Hg for 21 days), metformin (100 mg/kg per day starting day 1 or 14 of hypoxia) prevents or slows progression of pulmonary hypertension. Metformin increases the amount of phosphorylated endothelial nitric oxide synthase in PA extracts and decreases Rho kinase activity, thereby reducing PASMC contractility.170 Moreover, metformin inhibits MAPK activity, which slows PASMC proliferation170 (Figure 2). In MCT rats (treatment starting at day of monocrotaline injection), metformin (100 mg/kg per day) curtails adverse pulmonary vascular remodeling, decreasing pulmonary pressures and RVH.170 In female SU‐5416 hypoxia rats (hypobaric: 412 mm Hg for 2 weeks, then return to room pressure), metformin (100 mg/kg per day) starting 3 weeks after return to room air pressure inhibits aromatase transcription, which subsequently lowers both lung and circulating estrogen levels.171 Moreover, metformin augments PASMC AMP‐kinase activity, which inhibits PASMC proliferation (Figure 2), slows pulmonary vascular remodeling, lowers pulmonary arterial pressures, and attenuates RVH.171 Finally, metformin also has potential to benefit the right ventricle directly by decreasing pathologic lipid deposition (Figure 6). In BMPR2 R899X transgenic mice, metformin (25 g/kg of high fat chow starting at 6 weeks of age and continuing for 6 weeks) reduces lipid deposition in the right ventricle.172 However, metformin does not significantly improve systolic or diastolic measures of RV function.172
Metformin use in PAH patients is currently being investigated in a phase 2 clinical trial (ClinicalTrials.gov identifier NCT01884051). The primary end points are measures of insulin resistance, urinary and plasma oxidant stress markers, RV lipid content and oxidative metabolism, and drug safety. Secondary end points will include lung metabolism, as quantified by 18F‐fluorodeoxyglucose uptake, BMPR2 expression in peripheral blood mononuclear cells, glucose and lipid metabolites, RVEF and RV volumes using MRI, insulin sensitivity indexes, and 6MWD.
Olaparib
Emerging evidence demonstrates that DNA damage is more abundant in PAH PASMCs than in healthy PASMCs.174 However, PAH PASMCs are able to proliferate despite accumulation of harmful DNA damage. The DNA repair enzyme PARP1 (poly[ADP‐ribose] polymerase 1) expression is increased in PASMCs from PAH patients,174 which may explain the paradox of proliferation despite compromised DNA integrity. Interestingly, markers of DNA damage and elevated PARP1 expression can be recapitulated in healthy PASMCs via incubation with TNF‐α, showing that inflammation promotes DNA damage in PAH.174 In PAH PASMCs, PARP1 antagonism reduces proliferation and promotes apoptosis (Figure 2).174 Olaparib is a PARP1 inhibitor that is currently approved for treatment of ovarian cancer associated with breast cancer susceptibility genes (BRCA1 [BRCA1, DNA repair associated] or BRCA2 [BRCA2, DNA repair associated]) and BRCA/HER2 (human epidermal growth factor receptor 2)–negative metastatic breast cancer.175 Importantly, olaparib improves progression‐free survival in BRCA mutation carriers with HER2‐negative metastatic breast cancer.176 Olaparib has substantial adverse effects including bone marrow suppression, abdominal pain, and nausea/vomiting.175
In PAH PASMCs, PARP1 inhibition normalizes miR‐204, which in‐turn decreases NFATc2 and HIF‐1α levels.174 Furthermore, PARP1 suppression reduces intracellular calcium concentration and normalizes mitochondrial membrane potential of PAH PASMCs.174 In whole‐animal experiments, olaparib reduces PAH severity in MCT and SU‐5416 hypoxia (10% O2 for 3 weeks) rats in regression approaches. Olaparib (6 mg/kg per day for 2 weeks) given either 14 days after monocrotaline or 5 weeks after SU‐5416 inhibits PASMC proliferation and increases PASMC apoptosis.174 This results in lower mPAP and less RVH in both MCT and SU‐5416 hypoxia rats.174
Olaparib is currently being investigated in a phase 1 open‐label clinical trial (ClinicalTrials.gov identifier NCT03251872). Six PAH patients who are WHO functional class II or III and on stable PAH therapy for at least 4 months will be treated with olaparib (400 mg twice daily) for 16 weeks, with the primary end point being change in PVR. Secondary end points will include change in invasive hemodynamics, RV function, volume, and mass quantified by cardiac MRI, WHO functional class, NT‐proBNP levels, and quality of life.
Paclitaxel
FOXO1 is one of the many proteins that regulate PASMC proliferation through its ability to induce apoptosis.177 In PAH patients and in MCT and SU‐5416 hypoxia rats, total FOXO1 protein levels are diminished and phosphorylated levels of FOXO1 are elevated in the pulmonary vasculature.177 The change in ratio of total FOXO1/phosphorylated FOXO1 is associated with development of pulmonary hypertension via promotion of PASMC proliferation.177
Paclitaxel, a microtubule stabilizer that is used as a chemotherapeutic agent for multiple types of cancer,178 has the ability to increase FOXO1 and inhibit phosphorylation of FOXO1.177 Paclitaxel, like many chemotherapeutic agents, has several significant side effects including myelosuppression, nausea, diarrhea, and peripheral neuropathy, limiting its widespread use.179 However, paclitaxel promotes nuclear localization and activation of FOXO1 in PASMCs. Activated FOXO1 slows cellular proliferation and induces apoptosis via alteration of expression of multiple cell‐cycle regulators and promotion of BMPR2 signaling in PASMCs177 (Figure 4). In disease regression studies of both MCT rats (5 mg/kg on days 21 and 28 after monocrotaline injection or aerosolized paclitaxel 1 mg/kg on days 21 and 28) and SU‐5416 hypoxia rats (10% O2 for 21 days and then paclitaxel 7 mg/kg on day 21 and 28), paclitaxel has significant therapeutic effects. Paclitaxel treatment activates BMPR2 signaling and induces PASMC apoptosis. These molecular and cellular changes result in reduction in RVSP, blunting of RVH and RV dilation, and improvement in RV function in both MCT and SU‐5416 hypoxia rats.177 In addition, paclitaxel may have beneficial effects via its ability to downregulate FOXM1 (forkhead box M1),180 an oncogene that is also implicated in pathological pulmonary vascular remodeling.181, 182, 183
Paclitaxel use in PAH is not being investigated, likely due to significant side effects. However, use of an aerosolized form of paclitaxel could limit systemic effects, and because this approach is efficacious in preclinical models,177 it may merit further investigation in patients. Paclitaxel has a scientific rigor score of 4 (Table 2), but the side effects suggest other therapies may take priority.
Ranolazine
The Randle cycle describes the reciprocal relationship between fatty acid oxidation (FAO) and glucose oxidation.184 In RVH, the Randle cycle is observed and is associated with inefficient metabolism and impaired RV function.185 Ranolazine is a partial inhibitor of FAO (Figure 5 and thus can combat the Randle cycle. Ranolazine is used to treat refractory angina pectoris in patients with coronary artery disease.186 In addition to the FAO effects, ranolazine may block sodium and potassium channels.186, 187 Ranolazine is generally well tolerated but can result in prolongation of the QT interval.186
In pulmonary artery–banded rats, ranolazine was used to exploit the Randle cycle to enhance RV function by improving RV energetics.185 Ranolazine (20 mg/day starting 3 weeks after pulmonary artery banding) reduces expression of Glut1, a glucose membrane transporter, and HK1 (hexokinase 1) and increases PDH activity,185 compatible with its reduction of uncoupled glycolysis. Ranolazine promotes glycolytic oxidation and thereby suppresses FAO in the right ventricle. This metabolic shift reverses RVH, improves RV function, and enhances exercise capacity,185 perhaps because glucose oxidation requires less oxygen per mole of ATP produced than does FAO. In MCT rats, ranolazine treatment (0.25 or 0.5% in chow starting 1 week after monocrotaline injection) regresses RVH and RV fibrosis, as demonstrated by histological assessment and a decrease in mRNA levels of collagen 1α1, CTGF, and TGF‐β in the right ventricle.188 Finally, ranolazine treatment renders isolated monocrotaline hearts less susceptible to stimulation‐induced ventricular tachycardia and ventricular fibrillation, likely because of improvements in cardiac repolarization.188
Two small clinical trials tested the utility of ranolazine in PAH patients. First, the effects of a 500‐mg dose at 6 hours (n=6 patients) and a 12‐week extension phase (n=4 patients at a dose of 500 mg/day) were examined in a placebo‐controlled trial. Acute administration of a single dose of 500 mg of ranolazine did not alter mPAP, PVR, or cardiac index; however, only 1 patient achieved the targeted therapeutic level of ranolazine at the 6‐hour time point.189 In the 12‐week extension study of 4 patients (500 mg daily), cardiopulmonary exercise testing, 6MWD, NT‐pro‐BNP, symptom burden, and RV function as quantified by echocardiography did not change significantly.189 Second, an open‐label study using a higher dose of ranolazine (1000 mg twice/day) in 11 PAH patients with evidence of RV dysfunction on echocardiography (RV fractional area change <35% or tricuspid annular plane systolic excursion <1.6 cm) yielded more encouraging results. Three months of ranolazine treatment significantly improved functional class, RV size, and peak RV strain during exercise without significantly altering pulmonary vascular disease severity.190
There is an ongoing multicenter, double‐blind, randomized (2:1 ranolazine:placebo) controlled trial that will examine whether 6 months of ranolazine (500–1000 mg twice a day) alters RV function. In this study, 24 patients with RV dysfunction (RVEF <45% on cardiac MRI) due to pulmonary hypertension caused by comorbidities other than left heart disease (ClinicalTrials.gov identifiers NCT01839110/NCT02829034) will be studied. The primary end point will be change in RVEF, as determined by cardiac MRI, with secondary end points to include 6MWD, functional class assessment, 4‐dimensional flow and T1 mapping from cardiac MRI, change in serum metabolites, microRNA changes in peripheral blood, quality of life as assessed by the 36‐Item Short Form (SF‐36) questionnaire, and adverse side effects.191
Rapamycin
As discussed earlier, mTORC can regulate PASMC proliferation. In addition, mTORC signaling can be directly inhibited by the mTOR inhibitor rapamycin. Rapamycin is used clinically as an immunosuppressing agent for solid organ transplant patients and is also used on drug‐eluting coronary stents to prevent in‐stent stenosis.192 However, rapamycin has several side effects including immune suppression, thrombocytopenia, hyperlipidemia, and impaired wound healing.192
The utility of rapamycin in preclinical PAH is controversial because multiple publications have shown divergent results. In a rat model that combined pneumonectomy and monocrotaline (7 days after pneumonectomy), rapamycin (2.5 mg/kg per day) reduces pulmonary vascular remodeling and RVH when given in a preventative model (2 days before monocrotaline) but not in a reversal model (given 15 days after pneumonectomy).193 In another prevention study, rapamycin (2 mg/kg/day) administration starting 1 day before monocrotaline injection slows pulmonary vascular remodeling resulting in blunted RVH.194 The beneficial effects of rapamycin may be related to the ability of rapamycin to increase levels of HO‐1 (hemoxygenase 1), a protein that promotes vasodilation.194 In chronic hypoxic (0.5 atm of pressure) male and female mice, rapamycin (3 mg/kg per day starting 3 weeks after hypoxia) attenuates pulmonary vascular remodeling and subsequent RVH.195 In a study of established PAH, treatment with rapamycin (2.5 mg/kg per day beginning 12 days after monocrotaline injection) reduced levels of phosphorylated S6 kinase in lung extracts, a downstream marker of the mTORC1 signaling. However, rapamycin treatment does not result in any differences in histological severity of pulmonary vascular remodeling, mPAP, cardiac index, or RVH.196 These results suggest that rapamycin may not regress pulmonary hypertension despite S6 kinase inhibition. Finally, the effects of higher dose rapamycin (5 mg/kg per day) in both prevention and regression was investigated in MCT rats. In a prevention strategy (given the day of monocrotaline injection) rapamycin reduces pulmonary vascular remodeling, PA pressures, and RVH.197 This is associated with an inhibition of S6 kinase signaling and Akt and GSK3 (glycogen synthase kinase 3) signaling, downstream effector kinases of the mTORC2 pathway (Figure 2).197 When administered in a regression protocol (21 days after monocrotaline injection for 21 days), rapamycin reduces pulmonary vascular disease severity and blunts mTORC1 and mTORC2 signaling.197
Nab‐rapamycin, an albumin‐bound version of rapamycin that has been studied in nonhematologic malignancies,198 is currently being studied in a phase 1 open‐label clinical trial. In total, 25 PAH patients who are WHO functional class III or IV on 2 or more PAH‐specific therapies with a 6MWD between 150 and 450 m will be treated with Nab‐rapamycin (ClinicalTrials.gov identifier NCT02587325). The primary end point is the number of patients with an adverse effect. The dosing protocol is not currently outlined. It is difficult to translate the preclinical doses of rapamycin to Nab‐rapamycin, so dosing studies may be needed.
Rituximab
PAH has a high prevalence of autoantibodies, particularly in the forms associated with scleroderma, and evidence of autoimmunity.20 These data provide biological plausibility for the use of rituximab, an anti‐CD20 antibody that targets B lymphocytes. CD20 is an antigen on the surface of B lymphocytes that gradually disappears as these cells mature into plasmocytes.199 Rituximab was first used to treat non‐Hodgkins B‐cell lymphoma but is now used to treat several autoimmune diseases.200 Rituximab is often used to treat conditions for which there is a clonal source of autoantibodies, such as rheumatoid arthritis and systemic lupus erythematosus.200 Rituximab can be used for extended periods of time, but excessive immunosuppression can become a problem, and in some patients, reactivation of infections, such as hepatitis B, may occur.201 Acute infusion of rituximab is often accompanied by infusion reaction, which includes fever, flushing, dyspnea, and chest pain. In fact, as many as 77% of patients experience a reaction during the first infusion.202
The therapeutic effects of an anti‐CD20 antibody were examined in a novel model of PAH generated by ovalbumin immunization and administration of SU‐5416. Ovalbumin immunization on days 1 and 7, combined with ovalbumin inhalation (1% ovalbumin in saline for 30 minutes twice a week for 4 weeks starting at day 14) and SU‐5416 injection (20 mg/kg weekly starting on day 15), causes pulmonary hypertension. An anti‐CD20 antibody (7 mg/kg on days 14, 17, and 21) reduces expression of HIF‐1α, VEGF (vascular endothelial growth factor), and IL6 in the pulmonary vasculature (Figure 3). The change in the inflammatory milieu slows proliferation of PASMCs, which leads to lower mPAP and less RVH.203
Rituximab is currently being investigated in systemic sclerosis–associated PAH. In this multicenter, double‐blind, randomized, placebo‐controlled trial (ClinicalTrials.gov identifier NCT01086540), 60 patients with systemic sclerosis–associated PAH will be randomized to placebo or rituximab (2 infusions of 1000 mg 14‐days apart) and followed for 48 weeks. The primary end point will be change in PVR at 24 weeks, with secondary end points including clinical worsening, carbon monoxide diffusing capacity, oxygen saturation, digital ulcers, severity of Raynaud phenomenon, 6MWD, biomarkers of disease progression, quality of life, and safety. An MRI substudy entitled Right Ventricular Response to Rituximab in Systemic Sclerosis–Associated Pulmonary Arterial Hypertension—A Magnetic Resonance Imaging Substudy, will quantify RV end‐diastolic volume index and stroke volume, as determined by cardiac MRI, 24 weeks after drug treatment. This trial has completed enrollment, and the results should be available soon.
Rosiglitazone/Pioglitazone
Both preclinical and human studies have demonstrated PPAR‐γ pathway dysregulation in PAH. For example, deletion of PPAR‐γ in smooth muscle cells causes a form of PAH in mice.204 Furthermore, in human PAH lungs, PPAR‐γ mRNA is reduced.205 Rosiglitazone and pioglitazone are thiazolidinediones that are PPAR‐γ agonists used to treat type 2 diabetes mellitus,206, 207 and they show promising effects in preclinical PAH models.
In a novel PAH model of male apolipoprotein E knockout mice fed a high‐fat diet, rosiglitazone treatment (10 mg/kg per day) improves insulin sensitivity, increases plasma adiponectin levels, and reverses pulmonary vascular remodeling, resulting in lower RVSP and less RVH.208 At the molecular level, rosiglitazone treatment promotes adiponectin‐mediated suppression of PDGF (platelet‐derived growth factor)–induced PASMC proliferation208 (Figure 2). In chronically hypoxic rats (12% O2 for 4 weeks), rosiglitazone (8 mg/kg for 5 days/week) starting at day 1 of hypoxia increases PPAR‐γ levels and decreases ET1 (endothelin 1) and VEGF levels, which slows pathologic pulmonary vascular remodeling and lowers PA pressures.209 In hypoxic mice (10% O2 for 3–5 weeks), rosiglitazone (10 mg/kg per day) treatment during the last 10 days of hypoxia has therapeutic effects. In hypoxic mice, rosiglitazone reduces PAH; the authors attribute this reduction to alteration of oxidative signaling (eg, reduced expression of Nox4 [NADPH oxidase 4]; Figure 1), decreased superoxide generation, blunted PDGF activation, and mitigation of PTEN (phosphatase and tensin homolog) downregulation in the lungs.210
In SU‐5416 hypoxia rats (3 weeks of 10% O2 followed by 6 weeks of normoxia), pioglitazone (20 mg/kg per day starting 1 week after return to normoxia) shows very promising results. Pioglitazone treatment nearly normalizes PA pressures via blunting of pulmonary vascular remodeling.211 These changes manifest as improvements in RV function, as quantified by cardiac MRI and echocardiography. Moreover, RV glucose uptake and RV fibrosis are reduced.211 At the organelle level, there is an improvement in mitochondrial structure and organization in RV cardiomyocytes. Furthermore, pioglitazone induces multiple genes in the FAO pathway (Figure 6), which improves utilization of fatty acids in neonatal rat cardiomyocytes. Finally, pioglitazone treatment reduces the levels of miR‐197 and miR‐146b in SU‐5416 RV tissue; these 2 microRNAs are also upregulated in human PAH right ventricles.211
Although the preclinical data are promising and grade well with a scientific rigor score of 8 (Table 1), use of rosiglitazone and pioglitazone for PAH must be considered cautiously because both increase the risk of heart failure in diabetic patients.212, 213 Moreover, the preclinical doses are significantly higher than those maximally used in patients (Table 2). No ongoing trials are currently attempting to translate the positive preclinical results into human PAH patients.
Tacrolimus
BMPR2 signaling is suppressed in all forms of PAH, not just in familial PAH60; therefore, compounds that promote BMPR2 signaling may be efficacious in PAH. In a high‐throughput assay that identified BMPR2 activators, tacrolimus, a calcineurin inhibitor used for immunosuppression in solid organ transplant patients,214 was the strongest promoter of BMPR2 signaling out of 3756 compounds screened.215 Although used frequently in solid organ transplant recipients, tacrolimus increases risk of infection, causes hypertension, and has a negative impact on renal function.216
Spiekerkoetter et al showed that tacrolimus stimulates BMPR2 signaling via calcineurin inhibition and sequestration of FK‐binding protein 2 (12 kDa FK506‐binding protein 2) (Figure 4) from the BMPR1 receptors ALK1 (activin receptor‐like kinase), ALK2, and ALK3. This triggers signaling by SMAD1/5 (SMAD family members 1–5) and MAPK, leading to increases in ID1 (inhibitor of differentiation) transcription.215 In human endothelial cells, tacrolimus induces expression of 2 downstream BMPR2 targets, ID1 and APLN (apelin), and improves endothelial function, as quantified by tube formation.215 Tacrolimus (0.05 mg/kg per day) reverses pulmonary vascular remodeling, lowers RVSP, and reduces RVH in a mouse model of chronic hypoxia (10% O2) superimposed on a BMPR2 endothelial cell knockout genotype.215 Tacrolimus is also effective in combating established pulmonary hypertension in MCT rats (when administered 21 days after monocrotaline injection) and in SU‐5416 hypoxia rats (10% O2 for 3 weeks, normoxia for 5 weeks, and then treatment with tacrolimus for 3 weeks).215
Tacrolimus was tested in human PAH patients in compassionate use and in a randomized placebo‐controlled trial. In 3 patients with end‐stage PAH, addition of tacrolimus to conventional PAH therapies was examined. In all 3 patients, tacrolimus treatment reduced symptomatic burden and improved exercise capacity.217 In peripheral blood mononuclear cells, tacrolimus increases BMPR2 mRNA levels, which is associated with induction of downstream BMPR2 signaling molecules ID1, SMURF1 (SMAD‐specific E3 ubiquitin ligase), and LIMK1 (Lim domain kinase 1).217 In a phase 2a randomized controlled trial, the effects of 16 weeks of tacrolimus at 3 different target serum levels (<2, 2–3, and 3–5 ng/mL) were investigated in 14 PAH patients and compared with 6 placebo controls.218 Overall, there was a heterogeneous response to tacrolimus regarding its ability to positively regulate mRNA levels of BMPR2 and ID1 in peripheral blood mononuclear cells.218 Patients who increased BMPR2 to a greater extent tended to have greater improvements in 6MWD and RV function (as quantified by RV fractional area change and global longitudinal strain).218 However, there were no significant differences in 6MWD and RV function when the pooled tacrolimus group was compared with the placebo group.218 The widespread utility of tacrolimus in PAH is unproved, and perhaps better biomarkers are needed to help identify patients who may respond to tacrolimus.
Tocilizumab
The IL6 pathway is another potential target for PAH treatment because animal models have implicated a role for IL6 in PAH. IL6 knockout mice exhibit a blunted response to hypoxia‐induced (10% O2) pulmonary vascular remodeling,219 whereas lung‐specific overexpression of IL6 accelerates pulmonary vascular disease progression under basal and hypoxic (10% O2) conditions.220 Furthermore, smooth muscle–specific ablation of the IL6 receptor protects mice from hypoxia‐induced (10% O2) pulmonary hypertension.62 In MCT and SU‐5416 hypoxia rats and in PAH patients, there is upregulation of the IL6 receptor in PASMCs.62 Moreover, there is an increased abundance of phosphorylated STAT3, the activated form of the downstream signaling protein of IL6, in PAH PASMCs. In human PASMCs, overexpression of the IL6 receptor has a proliferative effect, mediated via inhibition of apoptosis.62 In PAH patients, serum IL6 levels are higher than in healthy controls,22, 23, 221 and elevated serum IL6 levels are associated with increased risk of mortality.22, 222 In addition, RV dysfunction is more pronounced in PAH patients with higher IL6 levels despite having similar severity of pulmonary vascular disease,221 suggesting that IL6 may also have direct negative inotropic effects on the right ventricle.
Tocilizumab is an IL6 receptor–blocking antibody that is currently approved for the treatment of rheumatoid arthritis.223 Tocilizumab has a side‐effect profile that includes increased risk of infection, elevated levels of total cholesterol and low‐density lipoproteins, liver function abnormalities, and gastrointestinal side effects.224 The rationale for tocilizumab in PAH is strongly supported by a recent publication. In vitro, the use of an IL6 receptor–neutralizing antibody induces apoptosis in PASMCs incubated with recombinant IL662 (Figure 3). Use of ERBF (20S,21‐epoxy‐resibufogenin‐3‐formate), a nonpeptide IL6 receptor blocker at a dose of 0.5 mg/kg per day mitigates the severity of pulmonary hypertension in MCT rats in strategies for both prevention (given at time of monocrotaline) and regression (starting 1 week after monocrotaline). In SU‐5416 hypoxia rats (10% O2 for 3 weeks and return to normoxia for 5 weeks), ERBF (0.5 mg/kg per day starting 2 weeks after return to normoxia) significantly reduces PH severity.62 In both MCT and SU‐5416 hypoxia rats, ERBF reduces the abundance of phosphorylated STAT3 in PASMCs, which in turn promotes apoptosis.
Tocilizumab is currently being investigated in the TRANSFORM‐UK (Therapeutic Open‐Label Study of Tocilizumab in the Treatment of Pulmonary Arterial Hypertension) study (ClinicalTrials.gov identifier NCT02676947). TRANSFORM‐UK is a single‐arm study that will investigate whether 6 months of treatment with tocilizumab (8 mg/kg monthly) alters pulmonary vascular disease in 21 PAH patients (excluding PAH patients with systemic lupus erythematosus, rheumatoid arthritis, and mixed connective tissue disease) who are classified as NYHA functional class II to IV on stable PAH therapy for 1 month before enrollment.225 The primary end points will be change in PVR at 6 months and safety, with secondary end points including 6MWD, NT‐pro‐BNP, symptom burden, and quality of life.225 This trial has completed enrollment and results should be available soon.
Trimetazidine
As discussed earlier, inhibition of the Randle cycle may be a treatment option for RV dysfunction in PAH. Trimetazidine is an antianginal agent that partially inhibits FAO.226 In heart failure patients, trimetazidine improves LV ejection fraction and NYHA functional class and reduces hospitalization and mortality.227, 228, 229 Trimetazidine is used in heart failure patients with angina in Europe.230 Trimetazidine is generally well tolerated, with the most common side effect being gastrointestinal disturbances.231
For PAH, trimetazidine has the potential to improve RV function by activating the Randle cycle and increasing glucose oxidation (Figure 5). In PA‐banded rats, trimetazidine (0.7 g/L of drinking water) administration starting at the time of PA banding and continuing for 8 weeks normalizes levels of CPT1 (carnitine palmitoyltransferase 1; a key FAO enzyme), Glut1, and hexokinase.185 Moreover, trimetazidine activates PDH, reduces FAO, and increases glucose oxidation. The metabolic changes in the right ventricle augment cardiac output, dampen RVH, and improve exercise capacity.185
The use of trimetazidine in PAH is being investigated in a single‐center randomized controlled trial that will examine whether 3 months of trimetazidine treatment (35 mg twice a day) alters RV function in 25 PAH patients. The primary end point will be change in RV function, as quantified by cardiac MRI. Secondary end points will include changes in cardiac fibrosis quantified by T1 cardiac MRI mapping, functional class, and plasma levels of lactate dehydrogenase (ClinicalTrials.gov identifier NCT03273387).
TNF‐α Inhibitors
In PAH, the TNF‐α pathway is another potential therapeutic target. PAH patients have higher serum levels of TNF‐α than healthy controls,22 and elevated levels of TNF‐α are associated with increased bodily pain,232 suggesting that TNF‐α may contribute to systemic symptoms in PAH. Moreover, TNF‐α is linked to a reduction in BMPR2 protein abundance in PASMCs233 and thus has an important molecular target in the pulmonary vasculature.
TNF‐α inhibitors are immunomodulators that are used in a wide variety of rheumatological/autoimmune diseases including rheumatoid arthritis,234, 235 psoriasis/psoriatic arthritis,236 and inflammatory bowel disease.237 TNF‐α inhibitors are generally well tolerated, with side effects including injection site reaction, immunosuppression, and a trend for increased risk of nonmelanoma skin cancer.238 However, in heart failure patients, use of the TNF‐α inhibitor infliximab is associated with clinical worsening in patients with moderate to severe heart failure.239 In contrast, another TNF‐α inhibitor, etanercept, is proven to be safe in heart failure patients.240, 241
In MCT rats, treatment with etanercept (2.5 mg/kg twice a week) blunts pulmonary vascular remodeling and reduces PA pressures in protocols for prevention (starting the day following monocrotaline injection) and regression (14 days after monocrotaline injection).233 In pigs with acute endotoxin‐induced pulmonary hypertension, etanercept (25 mg) lowers pulmonary pressures and PVR index 4 hours after endotoxin administration.242 The mechanistic underpinnings of the protective effects of anti–TNF‐α therapy on adverse pulmonary vascular remodeling were recently identified. TNF‐α decreases BMPR2 protein expression and promotes intracellular accumulation of a cleaved and inactive form of BMPR2 in PASMCs (Figure 4). The reduction in BMPR2 signaling increases PASMC proliferation.55 Moreover, TNF‐α activates NOTCH2 (notch 2) signaling (Figures 2 and 3), which is pro‐proliferative, via SRC kinases.55 Etanercept (2.5 mg/kg twice weekly starting after week 5 of normoxia) in SU‐5416 hypoxia rats (3 weeks of 10% O2 followed by 8 weeks of normoxia) normalizes BMPR2, decreases NOTCH2, and restores activated SMAD in the pulmonary vasculature.55 These molecular changes result in less pulmonary vascular remodeling, lower RVSP, and moderation of RVH.55
No trial is currently under way to examine the effects of TNF‐α inhibitors in PAH patients. A strong scientific rigor score of 7 (Table 1), along with the safety profile, suggests that a trial could be considered in the future.
Verteporfin
YAP (Yes‐associated protein) is a Hippo signaling molecule that is activated by a stiff ECM and promotes cellular proliferation and survival.243 YAP induces a metabolic switch to glutaminolysis, an alternative metabolic pathway that utilizes glutamine as an energy substrate and promotes rapid cell growth and hypertrophy.63 Glutaminolysis is increased in cancer244 and in PAH19, 245 when cells are exposed to a stiff ECM. Glutaminolysis is also induced de novo in RVH.245 In MCT rats, primates with simian immunodeficient virus–associated PAH, and PAH patient samples, YAP protein levels are elevated in the pulmonary vasculature.19
Verteporfin is a YAP inhibitor that is used clinically in photodynamic therapy to treat neovascularization in age‐related macular degeneration.246 Verteporfin is not used frequently, but the side effects include injection site irritation and photosensitivity.247 In MCT rats, daily verteporfin (25 mg/kg per day) starting the day after monocrotaline decreases activity of lung lysyl oxidase, a collagen crosslinking enzyme that promotes ECM stiffness, and normalizes pulmonary arteriolar stiffness.19 In addition, verteporfin reduces mRNA levels of CCN2 and CCN1, 2 YAP‐regulated genes. Furthermore, verteporfin treatment blunts the activity of glutaminase, the enzyme that promotes glutaminolysis (Figure 5) in the lungs.19 Finally, verteporfin treatment mitigates pathological pulmonary vascular remodeling and decreases RVSP and RVH in MCT rats.
The clinical utility of verteporfin in PAH is not being investigated. It may be difficult to use verteporfin in PAH because it is given as an intravenous injection and it has a half‐life of only 5 hours.248 Moreover, it is photoactivated; therefore, it may cause systemic photosensitivity if given chronically. Finally, the preclinical doses greatly exceed those used clinically, and a scientific rigor score of 2 (Table 1) raise questions regarding translatability. Perhaps an inhaled formulation of verteporfin could circumvent the risk of systemic side effects to help realize the beneficial effects of YAP inhibition for PAH.
Conclusions
In this review, we discussed the scientific basis—at both the basic and clinical science levels—supporting the notion that 22 currently available medications may have the potential to improve outcomes in PAH. Furthermore, we highlighted the beneficial effects of anastrozole, dichloroacetate, ranolazine, and tacrolimus that were demonstrated in early phase clinical trials (Table 3). There is always a large barrier in translating data from preclinical models created in rather homogenous rodent populations to human disease in genetically heterogeneous patient populations with multiple comorbid conditions. In addition, studies in rodents often use higher doses of drug than are tolerated by patients (Table 2). Furthermore, many preclinical studies lack the rigor that randomized controlled trials in patients use, notably, absence of blinding, randomization, and careful surveillance for toxicity (Table 1).81 Moreover, preclinical studies are often brief in duration; this approach fails to mimic the use of the drug in PAH patients, which is often sustained for years. Consequently, preclinical studies may identify molecules that may not be beneficial in patients in clinical trials. These concerns should not lead us to abandon the testing of PAH candidate drugs in preclinical studies; rather, we should increase the rigor of preclinical testing, as recently highlighted.81 Nonetheless, the fact that these medications are currently used in humans, so the toxicities and drug–drug interactions are already known, could allow for proof‐of‐principle trials in PAH patients with less risk of adverse effects.
Table 3.
Summary of Current Indications, Side‐Effect Profiles, and Available Clinical Trial Data for Potentially Repurposed Drugs
| Drug | Current Indication in Patients | Side Effects | Completed Trial | Dose | Results | Ongoing Trials | Dose | Primary End Point |
|---|---|---|---|---|---|---|---|---|
| Aldosterone antagonist | Congestive heart failure (HFrEF), ascites, hypertension | Hyperkalemia, gynecomastia | No | NA | NA | Yes | 25–50 mg/d | 6MWD, VO2max, clinical worsening |
| Allopurinol | Gout, nephrolithiasis | Stevens‐Johnson syndrome, nausea | No | NA | NA | NA | NA | NA |
| Anakinra | Rheumatoid arthritis, refractory pericarditis | Headache, vomiting, immunosuppresion | Yes | 100 mg for 14 d | Decreased hs‐CRP and reduction in symptom burden | NA | NA | NA |
| Anastrozole | Adjuvant for breast cancer | Hot flashes, reduced bone mineral density | Yes | 1 mg/d | Increased 6MWD | Yes | 1 mg/d | 6MWD |
| Apabetalone | Coronary artery disease | Transaminase elevation | No | NA | NA | Yes | 100 mg twice daily | PVR |
| β‐Blockers | Congestive heart failure (HFrEF), angina, hypertension, variceal bleed prophylaxis in cirrhosis | Bradycardia, hypotension, fatigue | Yes |
Bisoprolol: Up to 10 mg/d Carvedilol: Up to 25 mg twice daily |
Bisoprolol: Decreased cardiac index and a trend towards reducedexercise capacity Carvedilol: Reduced RV glucose uptake but no change in cardiac output or exercise capacity |
No | NA | NA |
| Chloroquine | Rheumatological conditions, malaria | Vision disturbance, weakness, nausea | No | NA | NA | No | NA | NA |
| Colchicine | Gout, familial Mediterranean fever, chronic pericarditis | Diarrhea, peripheral neuropathy, bone marrow suppression | No | NA | NA | No | NA | NA |
| Dehydroepiandrosterone | Supplement, menopausal symptoms | Acne, excess hair growth | No | NA | NA | Yes | 50 mg | RV longitudinal strain on cardiac MRI |
| DHEA | Inherited mitochondrial disorders | Peripheral neuropathy, fatigue, confusion | Yes | Up to 6.25 mg twice daily | Reduced mPAP and PVR in susceptible patients | No | NA | NA |
| Metformin | Type 2 diabetes mellitus | Gastrointestinal disturbance, lactic acidosis, fatigue | No | NA | NA | Yes | Unknown | Insulin resistance,oxidant stress markers in urine and plasma, safety |
| Nab‐rapamycin | Multiple types of cancer | Thrombocytopenia, fatigue, rash, diarrhea, nausea | No | NA | NA | Yes | Unknown | Safety |
| Olaparib | Breast and ovarian cancer | Bone marrow suppression, abdominal pain, and nausea/vomiting | No | NA | NA | Yes | 400 mg twice daily | PVR |
| Paclitaxel | Multiple types of cancer | Diarrhea, bone marrow suppression, nausea, peripheral neuropathy | No | No | NA | No | NA | NA |
| Ranolazine | Refractory angina pectoris due to coronary artery disease | QT prolongation, nausea, dizziness | Yes |
500 mg daily 1000 mg twice daily |
Improved RV function at exercise with higher dose, no effect with lower dose | Yes | 500–1000 mg twice daily | RVEF via cardiac MRI |
| Rituximab |
Non‐Hodgkins lymphoma Rheumatological diseases |
Immunosuppression, fatigue, injection site reaction | No | NA | NA | Yes | 1000 mg 14 d a part | PVR |
| Rosiglitazone/pioglitazone | Type 2 diabetes mellitus | Increased risk of heart failure, joint pain, sore throat | No | NA | NA | No | NA | NA |
| Tacrolimus | Posttransplant immunosuppression | Immunosuppression, renal impairment, hypertension | Yes | Serum levels <2, 2–3, 3–5 ng/mL | Mixed results on 6MWD and RV function which depended on increases in BMPR2 activity | No | NA | NA |
| Tocilizumab | Rheumatoid arthritis | Immunosuppression hyperlipidemia, liver function abnormalities | No | NA | NA | Yes | 8 mg/kg monthly | PVR |
| Trimetazidine | Angina, congestive heart failure (HFrEF), approved in Europe but not North America | Gastrointestinal disturbance, tremor | No | NA | NA | Yes | 35 mg twice daily | RVEF via cardiac MRI |
| TNF‐α‐inhibitor | Rheumatological/autoimmune diseases | Immunosuppression, nonmelanoma skin cancer | No | NA | No | No | NA | NA |
| Verteporfin | Age‐related macular degeneration | Dry‐eye, injection site irritation, photosensitivity | No | No | NA | No | NA | NA |
BMPR indicates bone morphogenic protein receptor; DHEA, dehydroepiandrosterone; HFrEF, heart failure with reduced ejection fraction; hs‐CRP, high‐sensitivity C‐reactive protein; mPAP, mean pulmonary arterial pressure; MRI, magnetic resonance imaging; NA, not available; PVR, pulmonary vascular resistance; RV, right ventricular; RVEF, right ventricular ejection fraction; 6MWD, 6‐min walking distance; TNF‐α, tumor necrosis factor α; VO2max, maximal oxygen consumption.
In addition to hypothesis‐driven research, use of network analysis combined with patient data shows promise as an unbiased approach for repurposing medications. This method demonstrated a reduction in risk of coronary artery disease in patients treated with hydroxychloroquine.249 Consequently, this approach may promote discovery of even more drugs that could be repurposed for PAH and help circumvent the barriers that exist when attempting to translate preclinical data to patients by using patient data only.
Although repurposing medications seems to be a safe option, this method still needs to be approached with caution and scientific rigor because repurposing of approved drugs that appear promising in preclinical models can also fail to yield positive results when studied in PAH patients. For example, imatinib reduces the severity of pulmonary hypertension and improves survival in MCT rats250; in PAH patients it increases 6MWD and reduces PVR, but it is associated with an increased risk of subdural hematoma that prevents clinical approval.251 Moreover, although evidence suggests the preclinical efficacy of the selective serotonin reuptake inhibitor fluoxetine,252, 253, 254 the dopamine agonist and serotonin receptor antagonist terguride,255 and the ASK1 (apoptosis signal‐regulating kinase 1) inhibitor256 selonsertib, none of the clinical trials using these therapies have yielded positive results. Thus, we may need to more carefully choose the medications to repurpose and the patients in whom they are tested.
Perhaps precision medicine will help us identify PAH patients who will benefit from the 22 medications we have discussed. For instance, patients with specific genetic profiles respond to dichloroacetate, whereas those harboring polymorphisms in UCP2 and SIRT3 do not,165 suggesting that genotyping before enrolling in a trial may select a population of responders. Likewise, patients treated with anakinra or tocilizumab could be chosen based on the levels of IL1 and IL6 in peripheral blood rather than enrolling patients without an inflammatory phenotype. Moreover, DHEA may be most beneficial in patients with low serum DHEA levels, and thus these patients could be selected in a clinical trial. Finally, there is precedent for use of genomic profiling to identify PAH patients who may respond to therapy because mRNA levels of DSG2 (desmoglein 2), a desmosomal cadherin involved in Wnt/β‐catenin signaling, and RHOQ (ras homolog family member Q), a cytoskeletal protein involved in insulin signaling, in cultured lymphocytes can be used to identify patients who are vasoresponders.257 The use of genotyping and biomarker profiles are examples of a personalized approach that may enhance the success of translation from preclinical studies to human clinical trials.
Sources of Funding
Prins is funded by NIH K08 HL140100, Archer is supported by Canada Foundation for Innovation (229252 and 33012), NIH RO1 HL113003, a Tier 1 Canada Research Chair in Mitochondrial Dynamics and Translational Medicine (950‐229252), the Queen's Cardiopulmonary Unit (QCPU), and a grant from the William J Henderson Foundation. Thenappan is funded by AHA Scientist Development Grant 15SDG25560048.
Disclosures
Dr Thenappan receives modest contributions as an advisor to Gilead and Actelion. The remaining authors have no disclosures to report.
Acknowledgments
The authors would like to thank Cynthia Faraday from the Lillehei Heart Institute for assistance generating the figures.
J Am Heart Assoc. 2019;8:e011343 DOI: 10.1161/JAHA.118.011343.
References
- 1. Rabinovitch M, Bothwell T, Hayakawa BN, Williams WG, Trusler GA, Rowe RD, Olley PM, Cutz E. Pulmonary artery endothelial abnormalities in patients with congenital heart defects and pulmonary hypertension. A correlation of light with scanning electron microscopy and transmission electron microscopy. Lab Invest. 1986;55:632–653. [PubMed] [Google Scholar]
- 2. Rosenberg HC, Rabinovitch M. Endothelial injury and vascular reactivity in monocrotaline pulmonary hypertension. Am J Physiol. 1988;255:H1484–H1491. [DOI] [PubMed] [Google Scholar]
- 3. Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2012;122:4306–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Caruso P, Dunmore BJ, Schlosser K, Schoors S, Dos Santos C, Perez‐Iratxeta C, Lavoie JR, Zhang H, Long L, Flockton AR, Frid MG, Upton PD, D'Alessandro A, Hadinnapola C, Kiskin FN, Taha M, Hurst LA, Ormiston ML, Hata A, Stenmark KR, Carmeliet P, Stewart DJ, Morrell NW. Identification of MicroRNA‐124 as a major regulator of enhanced endothelial cell glycolysis in pulmonary arterial hypertension via PTBP1 (polypyrimidine tract binding protein) and pyruvate kinase M2. Circulation. 2017;136:2451–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Archer SL, Weir EK, Wilkins MR. Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation. 2010;121:2045–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, Loyd JE, Nichols WC, Trembath RC; Consortium IP . Heterozygous germline mutations in BMPR2, encoding a TGF‐beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–84. [DOI] [PubMed] [Google Scholar]
- 7. Deng Z, Haghighi F, Helleby L, Vanterpool K, Horn EM, Barst RJ, Hodge SE, Morse JH, Knowles JA. Fine mapping of PPH1, a gene for familial primary pulmonary hypertension, to a 3‐cM region on chromosome 2q33. Am J Respir Crit Care Med. 2000;161:1055–1059. [DOI] [PubMed] [Google Scholar]
- 8. Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, Dyck JR, Gomberg‐Maitland M, Thébaud B, Husain AN, Cipriani N, Rehman J. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation. 2010;121:2661–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Landsberg JW, Yuan JX. Calcium and TRP channels in pulmonary vascular smooth muscle cell proliferation. News Physiol Sci. 2004;19:44–50. [DOI] [PubMed] [Google Scholar]
- 10. Yu Y, Sweeney M, Zhang S, Platoshyn O, Landsberg J, Rothman A, Yuan JX. Pdgf stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am J Physiol Cell Physiol. 2003;284:C316–C330. [DOI] [PubMed] [Google Scholar]
- 11. Platoshyn O, Golovina VA, Bailey CL, Limsuwan A, Krick S, Juhaszova M, Seiden JE, Rubin LJ, Yuan JX. Sustained membrane depolarization and pulmonary artery smooth muscle cell proliferation. Am J Physiol Cell Physiol. 2000;279:C1540–C1549. [DOI] [PubMed] [Google Scholar]
- 12. McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, Michelakis ED. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res. 2004;95:830–840. [DOI] [PubMed] [Google Scholar]
- 13. Archer SL. Pyruvate kinase and warburg metabolism in pulmonary arterial hypertension: uncoupled glycolysis and the cancer‐like phenotype of pulmonary arterial hypertension. Circulation. 2017;136:2486–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Archer SL. Mitochondrial dynamics—mitochondrial fission and fusion in human diseases. N Engl J Med. 2013;369:2236–2251. [DOI] [PubMed] [Google Scholar]
- 15. Zhang H, Wang D, Li M, Plecitá‐Hlavatá L, D'Alessandro A, Tauber J, Riddle S, Kumar S, Flockton A, McKeon BA, Frid MG, Reisz JA, Caruso P, El Kasmi KC, Ježek P, Morrell NW, Hu CJ, Stenmark KR. Metabolic and proliferative state of vascular adventitial fibroblasts in pulmonary hypertension is regulated through a MicroRNA‐124/PTBP1 (polypyrimidine tract binding protein 1)/pyruvate kinase muscle axis. Circulation. 2017;136:2468–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bertero T, Cottrill KA, Lu Y, Haeger CM, Dieffenbach P, Annis S, Hale A, Bhat B, Kaimal V, Zhang YY, Graham BB, Kumar R, Saggar R, Wallace WD, Ross DJ, Black SM, Fratz S, Fineman JR, Vargas SO, Haley KJ, Waxman AB, Chau BN, Fredenburgh LE, Chan SY. Matrix remodeling promotes pulmonary hypertension through feedback mechanoactivation of the YAP/TAZ‐miR‐130/301 circuit. Cell Rep. 2015;13:1016–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guignabert C, Tu L, Girerd B, Ricard N, Huertas A, Montani D, Humbert M. New molecular targets of pulmonary vascular remodeling in pulmonary arterial hypertension: importance of endothelial communication. Chest. 2015;147:529–537. [DOI] [PubMed] [Google Scholar]
- 18. Todorovich‐Hunter L, Dodo H, Ye C, McCready L, Keeley FW, Rabinovitch M. Increased pulmonary artery elastolytic activity in adult rats with monocrotaline‐induced progressive hypertensive pulmonary vascular disease compared with infant rats with nonprogressive disease. Am Rev Respir Dis. 1992;146:213–223. [DOI] [PubMed] [Google Scholar]
- 19. Bertero T, Oldham WM, Cottrill KA, Pisano S, Vanderpool RR, Yu Q, Zhao J, Tai Y, Tang Y, Zhang YY, Rehman S, Sugahara M, Qi Z, Gorcsan J, Vargas SO, Saggar R, Wallace WD, Ross DJ, Haley KJ, Waxman AB, Parikh VN, De Marco T, Hsue PY, Morris A, Simon MA, Norris KA, Gaggioli C, Loscalzo J, Fessel J, Chan SY. Vascular stiffness mechanoactivates YAP/TAZ‐dependent glutaminolysis to drive pulmonary hypertension. J Clin Invest. 2016;126:3313–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res. 2014;115:165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Price LC, Wort SJ, Perros F, Dorfmüller P, Huertas A, Montani D, Cohen‐Kaminsky S, Humbert M. Inflammation in pulmonary arterial hypertension. Chest. 2012;141:210–221. [DOI] [PubMed] [Google Scholar]
- 22. Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, Trembath RC, Jennings S, Barker L, Nicklin P, Walker C, Budd DC, Pepke‐Zaba J, Morrell NW. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010;122:920–927. [DOI] [PubMed] [Google Scholar]
- 23. Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot‐Keros L, Duroux P, Galanaud P, Simonneau G, Emilie D. Increased interleukin‐1 and interleukin‐6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;151:1628–1631. [DOI] [PubMed] [Google Scholar]
- 24. Thenappan T, Prins KW, Pritzker MR, Scandurra J, Volmers K, Weir EK. The critical role of pulmonary arterial compliance in pulmonary hypertension. Ann Am Thorac Soc. 2016;13:276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lai YC, Potoka KC, Champion HC, Mora AL, Gladwin MT. Pulmonary arterial hypertension: the clinical syndrome. Circ Res. 2014;115:115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ryan JJ, Huston J, Kutty S, Hatton ND, Bowman L, Tian L, Herr JE, Johri AM, Archer SL. Right ventricular adaptation and failure in pulmonary arterial hypertension. Can J Cardiol. 2015;31:391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vonk Noordegraaf A, Westerhof BE, Westerhof N. The relationship between the right ventricle and its load in pulmonary hypertension. J Am Coll Cardiol. 2017;69:236–243. [DOI] [PubMed] [Google Scholar]
- 28. Forfia PR, Fisher MR, Mathai SC, Housten‐Harris T, Hemnes AR, Borlaug BA, Chamera E, Corretti MC, Champion HC, Abraham TP, Girgis RE, Hassoun PM. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med. 2006;174:1034–1041. [DOI] [PubMed] [Google Scholar]
- 29. van de Veerdonk MC, Kind T, Marcus JT, Mauritz GJ, Heymans MW, Bogaard HJ, Boonstra A, Marques KM, Westerhof N, Vonk‐Noordegraaf A. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol. 2011;58:2511–2519. [DOI] [PubMed] [Google Scholar]
- 30. Thenappan T, Shah SJ, Rich S, Tian L, Archer SL, Gomberg‐Maitland M. Survival in pulmonary arterial hypertension: a reappraisal of the NIH risk stratification equation. Eur Respir J. 2010;35:1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Benza RL, Miller DP, Gomberg‐Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB, Elliott CG, Liou TG, McGoon MD. Predicting survival in pulmonary arterial hypertension: insights from the registry to evaluate early and long‐term pulmonary arterial hypertension disease management (reveal). Circulation. 2010;122:164–172. [DOI] [PubMed] [Google Scholar]
- 32. Humbert M, Sitbon O, Yaïci A, Montani D, O'Callaghan DS, Jaïs X, Parent F, Savale L, Natali D, Günther S, Chaouat A, Chabot F, Cordier JF, Habib G, Gressin V, Jing ZC, Souza R, Simonneau G; Network FPAH . Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J. 2010;36:549–555. [DOI] [PubMed] [Google Scholar]
- 33. Wijeratne DT, Lajkosz K, Brogly SB, Lougheed MD, Jiang L, Housin A, Barber D, Johnson A, Doliszny KM, Archer SL. Increasing incidence and prevalence of World Health Organization groups 1 to 4 pulmonary hypertension: a population‐based cohort study in Ontario, Canada. Circ Cardiovasc Qual Outcomes. 2018;11:e003973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655–1665. [DOI] [PubMed] [Google Scholar]
- 35. Sitbon O, Humbert M, Jaïs X, Ioos V, Hamid AM, Provencher S, Garcia G, Parent F, Hervé P, Simonneau G. Long‐term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111:3105–3111. [DOI] [PubMed] [Google Scholar]
- 36. Rich JD, Rich S. Clinical diagnosis of pulmonary hypertension. Circulation. 2014;130:1820–1830. [DOI] [PubMed] [Google Scholar]
- 37. Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, Pulido T, Frost A, Roux S, Leconte I, Landzberg M, Simonneau G. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346:896–903. [DOI] [PubMed] [Google Scholar]
- 38. Galiè N, Olschewski H, Oudiz RJ, Torres F, Frost A, Ghofrani HA, Badesch DB, McGoon MD, McLaughlin VV, Roecker EB, Gerber MJ, Dufton C, Wiens BL, Rubin LJ; Ambrisentan in Pulmonary Arterial Hypertension Randomized, Double‐Blind, Placebo‐Controlled, Multicenter, Eficacy Studies (ARIES) Group . Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double‐blind, placebo‐controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117:3010–3019. [DOI] [PubMed] [Google Scholar]
- 39. Pulido T, Adzerikho I, Channick RN, Delcroix M, Galiè N, Ghofrani HA, Jansa P, Jing ZC, Le Brun FO, Mehta S, Mittelholzer CM, Perchenet L, Sastry BK, Sitbon O, Souza R, Torbicki A, Zeng X, Rubin LJ, Simonneau G; Investigators S . Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809–818. [DOI] [PubMed] [Google Scholar]
- 40. Galiè N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, Fleming T, Parpia T, Burgess G, Branzi A, Grimminger F, Kurzyna M, Simonneau G; Group SUiPAHSS . Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353:2148–2157. [DOI] [PubMed] [Google Scholar]
- 41. Galiè N, Brundage BH, Ghofrani HA, Oudiz RJ, Simonneau G, Safdar Z, Shapiro S, White RJ, Chan M, Beardsworth A, Frumkin L, Barst RJ; Pulmonary Arterial Hypertension and Response to Tadalafil (PHIRST) Study Group . Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119:2894–2903. [DOI] [PubMed] [Google Scholar]
- 42. Ghofrani HA, Galiè N, Grimminger F, Grünig E, Humbert M, Jing ZC, Keogh AM, Langleben D, Kilama MO, Fritsch A, Neuser D, Rubin LJ; PATENT‐1 Study Group . Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369:330–340. [DOI] [PubMed] [Google Scholar]
- 43. Sitbon O, Channick R, Chin KM, Frey A, Gaine S, Galiè N, Ghofrani HA, Hoeper MM, Lang IM, Preiss R, Rubin LJ, Di Scala L, Tapson V, Adzerikho I, Liu J, Moiseeva O, Zeng X, Simonneau G, McLaughlin VV; Investigators G . Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med. 2015;373:2522–2533. [DOI] [PubMed] [Google Scholar]
- 44. Jing ZC, Parikh K, Pulido T, Jerjes‐Sanchez C, White RJ, Allen R, Torbicki A, Xu KF, Yehle D, Laliberte K, Arneson C, Rubin LJ. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: a randomized, controlled trial. Circulation. 2013;127:624–633. [DOI] [PubMed] [Google Scholar]
- 45. McLaughlin VV, Benza RL, Rubin LJ, Channick RN, Voswinckel R, Tapson VF, Robbins IM, Olschewski H, Rubenfire M, Seeger W. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J Am Coll Cardiol. 2010;55:1915–1922. [DOI] [PubMed] [Google Scholar]
- 46. Barst RJ, Rubin LJ, Long WA, McGoon MD, Rich S, Badesch DB, Groves BM, Tapson VF, Bourge RC, Brundage BH, Koerner SK, Langleben D, Keller CA, Murali S, Uretsky BF, Clayton LM, Jöbsis MM, Blackburn SD, Shortino D, Crow JW; Primary Pulmonary Hypertension Study Group . A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334:296–301. [DOI] [PubMed] [Google Scholar]
- 47. Rich S, Haworth SG, Hassoun PM, Yacoub MH. Pulmonary hypertension: the unaddressed global health burden. Lancet Respir Med. 2018;6:577–579. [DOI] [PubMed] [Google Scholar]
- 48. Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium‐channel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327:76–81. [DOI] [PubMed] [Google Scholar]
- 49. Gomberg‐Maitland M, Maitland ML, Barst RJ, Sugeng L, Coslet S, Perrino TJ, Bond L, Lacouture ME, Archer SL, Ratain MJ. A dosing/cross‐development study of the multikinase inhibitor sorafenib in patients with pulmonary arterial hypertension. Clin Pharmacol Ther. 2010;87:303– 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Goldstein I, Lue TF, Padma‐Nathan H, Rosen RC, Steers WD, Wicker PA. Oral sildenafil in the treatment of erectile dysfunction. Sildenafil study group. N Engl J Med. 1998;338:1397–1404. [DOI] [PubMed] [Google Scholar]
- 51. Michelakis E, Tymchak W, Lien D, Webster L, Hashimoto K, Archer S. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: comparison with inhaled nitric oxide. Circulation. 2002;105:2398–2403. [DOI] [PubMed] [Google Scholar]
- 52. Sastry BK, Narasimhan C, Reddy NK, Raju BS. Clinical efficacy of sildenafil in primary pulmonary hypertension: a randomized, placebo‐controlled, double‐blind, crossover study. J Am Coll Cardiol. 2004;43:1149–1153. [DOI] [PubMed] [Google Scholar]
- 53. Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, Morrell NW. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105:1672–1678. [DOI] [PubMed] [Google Scholar]
- 54. McMurtry MS, Moudgil R, Hashimoto K, Bonnet S, Michelakis ED, Archer SL. Overexpression of human bone morphogenetic protein receptor 2 does not ameliorate monocrotaline pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;292:L872–L878. [DOI] [PubMed] [Google Scholar]
- 55. Hurst LA, Dunmore BJ, Long L, Crosby A, Al‐Lamki R, Deighton J, Southwood M, Yang X, Nikolic MZ, Herrera B, Inman GJ, Bradley JR, Rana AA, Upton PD, Morrell NW. TNFα drives pulmonary arterial hypertension by suppressing the BMP type‐II receptor and altering NOTCH signalling. Nat Commun. 2017;8:14079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, Ulrich S, Speich R, Huber LC. Interleukin‐6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3‐microRNA cluster 17/92 pathway. Circ Res. 2009;104:1184–1191. [DOI] [PubMed] [Google Scholar]
- 57. Mair KM, Wright AF, Duggan N, Rowlands DJ, Hussey MJ, Roberts S, Fullerton J, Nilsen M, Loughlin L, Thomas M, MacLean MR. Sex‐dependent influence of endogenous estrogen in pulmonary hypertension. Am J Respir Crit Care Med. 2014;190:456–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Long L, Yang X, Southwood M, Lu J, Marciniak SJ, Dunmore BJ, Morrell NW. Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type ii receptor degradation. Circ Res. 2013;112:1159–1170. [DOI] [PubMed] [Google Scholar]
- 59. Rudarakanchana N, Flanagan JA, Chen H, Upton PD, Machado R, Patel D, Trembath RC, Morrell NW. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet. 2002;11:1517–1525. [DOI] [PubMed] [Google Scholar]
- 60. Kuebler WM, Nicolls MR, Olschewski A, Abe K, Rabinovitch M, Stewart DJ, Chan SY, Morrell NW, Archer SL, Spiekerkoetter E. A pro‐con debate: current controversies in PAH pathogenesis at the American thoracic society international meeting in 2017. Am J Physiol Lung Cell Mol Physiol. 2018;315:L502–L516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Boucherat O, Vitry G, Trinh I, Paulin R, Provencher S, Bonnet S. The cancer theory of pulmonary arterial hypertension. Pulm Circ. 2017;7:285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tamura Y, Phan C, Tu L, Le Hiress M, Thuillet R, Jutant EM, Fadel E, Savale L, Huertas A, Humbert M, Guignabert C. Ectopic upregulation of membrane‐bound il6r drives vascular remodeling in pulmonary arterial hypertension. J Clin Invest. 2018;128:1956–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chan SY, Rubin LJ. Metabolic dysfunction in pulmonary hypertension: from basic science to clinical practice. Eur Respir Rev. 2017;26:170094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Michelakis ED, McMurtry MS, Wu XC, Dyck JR, Moudgil R, Hopkins TA, Lopaschuk GD, Puttagunta L, Waite R, Archer SL. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage‐gated potassium channels. Circulation. 2002;105:244–250. [DOI] [PubMed] [Google Scholar]
- 65. Ryan JJ, Archer SL. The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ Res. 2014;115:176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Archer SL, Fang YH, Ryan JJ, Piao L. Metabolism and bioenergetics in the right ventricle and pulmonary vasculature in pulmonary hypertension. Pulm Circ. 2013;3:144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Prins KW, Tian L, Wu D, Thenappan T, Metzger JM, Archer SL. Colchicine depolymerizes microtubules, increases junctophilin‐2, and improves right ventricular function in experimental pulmonary arterial hypertension. J Am Heart Assoc. 2017;6:e006195 DOI: 10.1161/JAHA.117.006195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Maron BA, Leopold JA. Aldosterone receptor antagonists: effective but often forgotten. Circulation. 2010;121:934–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Connell JM, Davies E. The new biology of aldosterone. J Endocrinol. 2005;186:1–20. [DOI] [PubMed] [Google Scholar]
- 70. Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized aldactone evaluation study investigators. N Engl J Med. 1999;341:709–717. [DOI] [PubMed] [Google Scholar]
- 71. Zannad F, McMurray JJ, Krum H, vanVeldhuisen DJ , Swedberg K, Shi H, Vincent J, Pocock SJ, Pitt B; Group E‐HS . Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med. 2011;364:11– 21. [DOI] [PubMed] [Google Scholar]
- 72. Te Riet L, van Esch JH, Roks AJ, van den Meiracker AH, Danser AH. Hypertension: renin‐angiotensin‐aldosterone system alterations. Circ Res. 2015;116:960–975. [DOI] [PubMed] [Google Scholar]
- 73. Moore KP, Aithal GP. Guidelines on the management of ascites in cirrhosis. Gut. 2006;55(suppl 6):vi1–vi12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Juurlink DN, Mamdani MM, Lee DS, Kopp A, Austin PC, Laupacis A, Redelmeier DA. Rates of hyperkalemia after publication of the randomized aldactone evaluation study. N Engl J Med. 2004;351:543–551. [DOI] [PubMed] [Google Scholar]
- 75. Navaneethan SD, Nigwekar SU, Sehgal AR, Strippoli GF. Aldosterone antagonists for preventing the progression of chronic kidney disease: a systematic review and meta‐analysis. Clin J Am Soc Nephrol. 2009;4:542–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Maron BA, Opotowsky AR, Landzberg MJ, Loscalzo J, Waxman AB, Leopold JA. Plasma aldosterone levels are elevated in patients with pulmonary arterial hypertension in the absence of left ventricular heart failure: a pilot study. Eur J Heart Fail. 2013;15:277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Maron BA, Zhang YY, White K, Chan SY, Handy DE, Mahoney CE, Loscalzo J, Leopold JA. Aldosterone inactivates the endothelin‐B receptor via a cysteinyl thiol redox switch to decrease pulmonary endothelial nitric oxide levels and modulate pulmonary arterial hypertension. Circulation. 2012;126:963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Maron BA, Oldham WM, Chan SY, Vargas SO, Arons E, Zhang YY, Loscalzo J, Leopold JA. Upregulation of steroidogenic acute regulatory protein by hypoxia stimulates aldosterone synthesis in pulmonary artery endothelial cells to promote pulmonary vascular fibrosis. Circulation. 2014;130:168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Aghamohammadzadeh R, Zhang YY, Stephens TE, Arons E, Zaman P, Polach KJ, Matar M, Yung LM, Yu PB, Bowman FP, Opotowsky AR, Waxman AB, Loscalzo J, Leopold JA, Maron BA. Up‐regulation of the mammalian target of rapamycin complex 1 subunit raptor by aldosterone induces abnormal pulmonary artery smooth muscle cell survival patterns to promote pulmonary arterial hypertension. FASEB J. 2016;30:2511–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Samokhin AO, Stephens T, Wertheim BM, Wang RS, Vargas SO, Yung LM, Cao M, Brown M, Arons E, Dieffenbach PB, Fewell JG, Matar M, Bowman FP, Haley KJ, Alba GA, Marino SM, Kumar R, Rosas IO, Waxman AB, Oldham WM, Khanna D, Graham BB, Seo S, Gladyshev VN, Yu PB, Fredenburgh LE, Loscalzo J, Leopold JA, Maron BA. Nedd9 targets. Sci Transl Med. 2018;10:eaap7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Provencher S, Archer SL, Ramirez FD, Hibbert B, Paulin R, Boucherat O, Lacasse Y, Bonnet S. Standards and methodological rigor in pulmonary arterial hypertension preclinical and translational research. Circ Res. 2018;122:1021–1032. [DOI] [PubMed] [Google Scholar]
- 82. Nozik‐Grayck E, Stenmark KR. Role of reactive oxygen species in chronic hypoxia‐induced pulmonary hypertension and vascular remodeling. Adv Exp Med Biol. 2007;618:101–112. [DOI] [PubMed] [Google Scholar]
- 83. Archer SL, Nelson DP, Weir EK. Detection of activated O2 species in vitro and in rat lungs by chemiluminescence. J Appl Physiol (1985). 1989;67:1912–1921. [DOI] [PubMed] [Google Scholar]
- 84. Pacher P, Nivorozhkin A, Szabó C. Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev. 2006;58:87–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Voelkel MA, Wynne KM, Badesch DB, Groves BM, Voelkel NF. Hyperuricemia in severe pulmonary hypertension. Chest. 2000;117:19–24. [DOI] [PubMed] [Google Scholar]
- 86. Bendayan D, Shitrit D, Ygla M, Huerta M, Fink G, Kramer MR. Hyperuricemia as a prognostic factor in pulmonary arterial hypertension. Respir Med. 2003;97:130–133. [DOI] [PubMed] [Google Scholar]
- 87. Spiekermann S, Schenk K, Hoeper MM. Increased xanthine oxidase activity in idiopathic pulmonary arterial hypertension. Eur Respir J. 2009;34:276. [DOI] [PubMed] [Google Scholar]
- 88. Bowers R, Cool C, Murphy RC, Tuder RM, Hopken MW, Flores SC, Voelkel NF. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med. 2004;169:764–769. [DOI] [PubMed] [Google Scholar]
- 89. Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn‐SOD (SOD1), Mn‐SOD (SOD2), and EC‐SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. 2002;33:337–349. [DOI] [PubMed] [Google Scholar]
- 90. Plimack ER, Kantarjian HM, Issa JP. Decitabine and its role in the treatment of hematopoietic malignancies. Leuk Lymphoma. 2007;48:1472–1481. [DOI] [PubMed] [Google Scholar]
- 91. Gliozzi M, Malara N, Muscoli S, Mollace V. The treatment of hyperuricemia. Int J Cardiol. 2016;213:23–27. [DOI] [PubMed] [Google Scholar]
- 92. Ngo TC, Assimos DG. Uric acid nephrolithiasis: recent progress and future directions. Rev Urol. 2007;9:17–27. [PMC free article] [PubMed] [Google Scholar]
- 93. Perez‐Ruiz F, Hernando I, Villar I, Nolla JM. Correction of allopurinol dosing should be based on clearance of creatinine, but not plasma creatinine levels: another insight to allopurinol‐related toxicity. J Clin Rheumatol. 2005;11:129–133. [DOI] [PubMed] [Google Scholar]
- 94. Hoshikawa Y, Ono S, Suzuki S, Tanita T, Chida M, Song C, Noda M, Tabata T, Voelkel NF, Fujimura S. Generation of oxidative stress contributes to the development of pulmonary hypertension induced by hypoxia. J Appl Physiol (1985). 2001;90:1299–1306. [DOI] [PubMed] [Google Scholar]
- 95. Jankov RP, Kantores C, Pan J, Belik J. Contribution of xanthine oxidase‐derived superoxide to chronic hypoxic pulmonary hypertension in neonatal rats. Am J Physiol Lung Cell Mol Physiol. 2008;294:L233–L245. [DOI] [PubMed] [Google Scholar]
- 96. Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7:27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Voelkel NF, Tuder RM, Bridges J, Arend WP. Interleukin‐1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am J Respir Cell Mol Biol. 1994;11:664–675. [DOI] [PubMed] [Google Scholar]
- 98. Pickworth J, Rothman A, Iremonger J, Casbolt H, Hopkinson K, Hickey PM, Gladson S, Shay S, Morrell NW, Francis SE, West JD, Lawrie A. Differential IL‐1 signaling induced by BMPR2 deficiency drives pulmonary vascular remodeling. Pulm Circ. 2017;7:768–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Mertens M, Singh JA. Anakinra for rheumatoid arthritis: a systematic review. J Rheumatol. 2009;36:1118–1125. [DOI] [PubMed] [Google Scholar]
- 100. Brucato A, Imazio M, Gattorno M, Lazaros G, Maestroni S, Carraro M, Finetti M, Cumetti D, Carobbio A, Ruperto N, Marcolongo R, Lorini M, Rimini A, Valenti A, Erre GL, Sormani MP, Belli R, Gaita F, Martini A. Effect of anakinra on recurrent pericarditis among patients with colchicine resistance and corticosteroid dependence: the AIRTRIP randomized clinical trial. JAMA. 2016;316:1906–1912. [DOI] [PubMed] [Google Scholar]
- 101. Trankle CR, Canada JM, Kadariya D, Markley R, Medina De Chazal H, Pinson J, Fox A, Van Tassell BW, Abbate A, Grinnan D. Interleukin‐1 (IL‐1) blockade reduces inflammation in pulmonary arterial hypertension and right ventricular failure: a single‐arm, open‐label, phase IB/II pilot study. Am J Respir Crit Care Med. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, Fishman AP, Goldring RM, Groves BM, Koerner SK. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107:216–223. [DOI] [PubMed] [Google Scholar]
- 103. Frost AE, Badesch DB, Barst RJ, Benza RL, Elliott CG, Farber HW, Krichman A, Liou TG, Raskob GE, Wason P, Feldkircher K, Turner M, McGoon MD. The changing picture of patients with pulmonary arterial hypertension in the United States: how reveal differs from historic and non‐us contemporary registries. Chest. 2011;139:128–137. [DOI] [PubMed] [Google Scholar]
- 104. McGoon MD, Benza RL, Escribano‐Subias P, Jiang X, Miller DP, Peacock AJ, Pepke‐Zaba J, Pulido T, Rich S, Rosenkranz S, Suissa S, Humbert M. Pulmonary arterial hypertension: epidemiology and registries. J Am Coll Cardiol. 2013;62:D51–D59. [DOI] [PubMed] [Google Scholar]
- 105. Prins KW, Thenappan T. World health organization group i pulmonary hypertension: epidemiology and pathophysiology. Cardiol Clin. 2016;34:363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Chen X, Austin ED, Talati M, Fessel JP, Farber‐Eger EH, Brittain EL, Hemnes AR, Loyd JE, West J. Oestrogen inhibition reverses pulmonary arterial hypertension and associated metabolic defects. Eur Respir J. 2017;50:1602337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Simpson ER, Clyne C, Rubin G, Boon WC, Robertson K, Britt K, Speed C, Jones M. Aromatase—a brief overview. Annu Rev Physiol. 2002;64:93–127. [DOI] [PubMed] [Google Scholar]
- 108. Burstein HJ, Griggs JJ, Prestrud AA, Temin S. American society of clinical oncology clinical practice guideline update on adjuvant endocrine therapy for women with hormone receptor‐positive breast cancer. J Oncol Pract. 2010;6:243–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Nabholtz JM. Long‐term safety of aromatase inhibitors in the treatment of breast cancer. Ther Clin Risk Manag. 2008;4:189–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wakeling AE, Dukes M, Bowler J. A potent specific pure antiestrogen with clinical potential. Cancer Res. 1991;51:3867–3873. [PubMed] [Google Scholar]
- 111. Hajri T, Han XX, Bonen A, Abumrad NA. Defective fatty acid uptake modulates insulin responsiveness and metabolic responses to diet in CD36‐null mice. J Clin Invest. 2002;109:1381–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kawut SM, Archer‐Chicko CL, DeMichele A, Fritz JS, Klinger JR, Ky B, Palevsky HI, Palmisciano AJ, Patel M, Pinder D, Propert KJ, Smith KA, Stanczyk F, Tracy R, Vaidya A, Whittenhall ME, Ventetuolo CE. Anastrozole in pulmonary arterial hypertension. A randomized, double‐blind, placebo‐controlled trial. Am J Respir Crit Care Med. 2017;195:360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov. 2003;2:205–213. [DOI] [PubMed] [Google Scholar]
- 114. Frump AL, Goss KN, Vayl A, Albrecht M, Fisher A, Tursunova R, Fierst J, Whitson J, Cucci AR, Brown MB, Lahm T. Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension: effects of endogenous and exogenous sex hormones. Am J Physiol Lung Cell Mol Physiol. 2015;308:L873–L890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Liu A, Philip J, Vinnakota KC, Van den Bergh F, Tabima DM, Hacker T, Beard DA, Chesler NC. Estrogen maintains mitochondrial content and function in the right ventricle of rats with pulmonary hypertension. Physiol Rep. 2017;5:e13157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Thenappan T, Ormiston ML, Ryan JJ, Archer SL. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ. 2018;360:j5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Meloche J, Potus F, Vaillancourt M, Bourgeois A, Johnson I, Deschamps L, Chabot S, Ruffenach G, Henry S, Breuils‐Bonnet S, Tremblay È, Nadeau V, Lambert C, Paradis R, Provencher S, Bonnet S. Bromodomain‐containing protein 4: the epigenetic origin of pulmonary arterial hypertension. Circ Res. 2015;117:525–535. [DOI] [PubMed] [Google Scholar]
- 118. Nicholls SJ, Ray KK, Johansson JO, Gordon A, Sweeney M, Halliday C, Kulikowski E, Wong N, Kim SW, Schwartz GG. Selective bet protein inhibition with apabetalone and cardiovascular events: a pooled analysis of trials in patients with coronary artery disease. Am J Cardiovasc Drugs. 2018;18:109–115. [DOI] [PubMed] [Google Scholar]
- 119. Nootens M, Kaufmann E, Rector T, Toher C, Judd D, Francis GS, Rich S. Neurohormonal activation in patients with right ventricular failure from pulmonary hypertension: relation to hemodynamic variables and endothelin levels. J Am Coll Cardiol. 1995;26:1581–1585. [DOI] [PubMed] [Google Scholar]
- 120. Bristow MR, Minobe W, Rasmussen R, Larrabee P, Skerl L, Klein JW, Anderson FL, Murray J, Mestroni L, Karwande SV. Beta‐adrenergic neuroeffector abnormalities in the failing human heart are produced by local rather than systemic mechanisms. J Clin Invest. 1992;89:803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. López‐Sendón J, Swedberg K, McMurray J, Tamargo J, Maggioni AP, Dargie H, Tendera M, Waagstein F, Kjekshus J, Lechat P, Torp‐Pedersen C; Cardiology TFB‐BotESo . Expert consensus document on beta‐adrenergic receptor blockers. Eur Heart J. 2004;25:1341–1362. [DOI] [PubMed] [Google Scholar]
- 122. Ge PS, Runyon BA. The changing role of beta‐blocker therapy in patients with cirrhosis. J Hepatol. 2014;60:643–653. [DOI] [PubMed] [Google Scholar]
- 123. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2013;128:e240–e327. [DOI] [PubMed] [Google Scholar]
- 124. Chatterjee S, Biondi‐Zoccai G, Abbate A, D'Ascenzo F, Castagno D, Van Tassell B, Mukherjee D, Lichstein E. Benefits of β blockers in patients with heart failure and reduced ejection fraction: network meta‐analysis. BMJ. 2013;346:f55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. MERIT‐HF Study Group . Effect of metoprolol CR/XL in chronic heart failure: metoprolol cr/xl randomised intervention trial in congestive heart failure (MERIT‐HF). Lancet. 1999;353:2001–2007. [PubMed] [Google Scholar]
- 126. CIBIS‐II Investigators and Committees . The cardiac insufficiency bisoprolol study II (CIBIS‐II): a randomised trial. Lancet. 1999;353:9–13. [PubMed] [Google Scholar]
- 127. Packer M, Fowler MB, Roecker EB, Coats AJ, Katus HA, Krum H, Mohacsi P, Rouleau JL, Tendera M, Staiger C, Holcslaw TL, Amann‐Zalan I, DeMets DL; Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) Study Group . Effect of carvedilol on the morbidity of patients with severe chronic heart failure: results of the carvedilol prospective randomized cumulative survival (COPERNICUS) study. Circulation. 2002;106:2194–2199. [DOI] [PubMed] [Google Scholar]
- 128. Bogaard HJ, Natarajan R, Mizuno S, Abbate A, Chang PJ, Chau VQ, Hoke NN, Kraskauskas D, Kasper M, Salloum FN, Voelkel NF. Adrenergic receptor blockade reverses right heart remodeling and dysfunction in pulmonary hypertensive rats. Am J Respir Crit Care Med. 2010;182:652–660. [DOI] [PubMed] [Google Scholar]
- 129. de Man FS, Handoko ML, van Ballegoij JJ, Schalij I, Bogaards SJ, Postmus PE, van der Velden J, Westerhof N, Paulus WJ, Vonk‐Noordegraaf A. Bisoprolol delays progression towards right heart failure in experimental pulmonary hypertension. Circ Heart Fail. 2012;5:97–105. [DOI] [PubMed] [Google Scholar]
- 130. Ishikawa M, Sato N, Asai K, Takano T, Mizuno K. Effects of a pure alpha/beta‐adrenergic receptor blocker on monocrotaline‐induced pulmonary arterial hypertension with right ventricular hypertrophy in rats. Circ J. 2009;73:2337–2341. [DOI] [PubMed] [Google Scholar]
- 131. Wenzel D, Knies R, Matthey M, Klein AM, Welschoff J, Stolle V, Sasse P, Roll W, Breuer J, Fleischmann BK. Beta(2)‐adrenoceptor antagonist ICI 118,551 decreases pulmonary vascular tone in mice via a G(i/o) protein/nitric oxide‐coupled pathway. Hypertension. 2009;54:157–163. [DOI] [PubMed] [Google Scholar]
- 132. Piao L, Fang YH, Parikh KS, Ryan JJ, D'Souza KM, Theccanat T, Toth PT, Pogoriler J, Paul J, Blaxall BC, Akhter SA, Archer SL. GRK2‐mediated inhibition of adrenergic and dopaminergic signaling in right ventricular hypertrophy: therapeutic implications in pulmonary hypertension. Circulation. 2012;126:2859–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Perros F, de Man FS, Bogaard HJ, Antigny F, Simonneau G, Bonnet S, Provencher S, Galiè N, Humbert M. Use of β‐blockers in pulmonary hypertension. Circ Heart Fail. 2017;10:e003703. [DOI] [PubMed] [Google Scholar]
- 134. Ghosh S, Gupta M, Xu W, Mavrakis DA, Janocha AJ, Comhair SA, Haque MM, Stuehr DJ, Yu J, Polgar P, Naga Prasad SV, Erzurum SC. Phosphorylation inactivation of endothelial nitric oxide synthesis in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2016;310:L1199–L1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Thenappan T, Roy SS, Duval S, Glassner‐Kolmin C, Gomberg‐Maitland M. Beta‐blocker therapy is not associated with adverse outcomes in patients with pulmonary arterial hypertension: a propensity score analysis. Circ Heart Fail. 2014;7:903–910. [DOI] [PubMed] [Google Scholar]
- 136. So PP, Davies RA, Chandy G, Stewart D, Beanlands RS, Haddad H, Pugliese C, Mielniczuk LM. Usefulness of beta‐blocker therapy and outcomes in patients with pulmonary arterial hypertension. Am J Cardiol. 2012;109:1504–1509. [DOI] [PubMed] [Google Scholar]
- 137. Bandyopadhyay D, Bajaj NS, Zein J, Minai OA, Dweik RA. Outcomes of beta‐blocker use in pulmonary arterial hypertension: a propensity‐matched analysis. Eur Respir J. 2015;46:750–760. [DOI] [PubMed] [Google Scholar]
- 138. Grinnan D, Bogaard HJ, Grizzard J, Van Tassell B, Abbate A, DeWilde C, Priday A, Voelkel NF. Treatment of group I pulmonary arterial hypertension with carvedilol is safe. Am J Respir Crit Care Med. 2014;189:1562–1564. [DOI] [PubMed] [Google Scholar]
- 139. van Campen JS, de Boer K, van de Veerdonk MC, van der Bruggen CE, Allaart CP, Raijmakers PG, Heymans MW, Marcus JT, Harms HJ, Handoko ML, de Man FS, Vonk Noordegraaf A, Bogaard HJ. Bisoprolol in idiopathic pulmonary arterial hypertension: an explorative study. Eur Respir J. 2016;48:787–796. [DOI] [PubMed] [Google Scholar]
- 140. Farha S, Saygin D, Park MM, Cheong HI, Asosingh K, Comhair SA, Stephens OR, Roach EC, Sharp J, Highland KB, DiFilippo FP, Neumann DR, Tang WHW, Erzurum SC. Pulmonary arterial hypertension treatment with carvedilol for heart failure: a randomized controlled trial. JCI Insight. 2017;2:95240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Ma XL, Yue TL, Lopez BL, Barone FC, Christopher TA, Ruffolo RR Jr, Feuerstein GZ. Carvedilol, a new beta adrenoreceptor blocker and free radical scavenger, attenuates myocardial ischemia‐reperfusion injury in hypercholesterolemic rabbits. J Pharmacol Exp Ther. 1996;277:128–136. [PubMed] [Google Scholar]
- 142. Mochizuki M, Yano M, Oda T, Tateishi H, Kobayashi S, Yamamoto T, Ikeda Y, Ohkusa T, Ikemoto N, Matsuzaki M. Scavenging free radicals by low‐dose carvedilol prevents redox‐dependent Ca2+ leak via stabilization of ryanodine receptor in heart failure. J Am Coll Cardiol. 2007;49:1722–1732. [DOI] [PubMed] [Google Scholar]
- 143. Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, Shenoy SK, Lefkowitz RJ. A unique mechanism of beta‐blocker action: carvedilol stimulates beta‐arrestin signaling. Proc Natl Acad Sci USA. 2007;104:16657–16662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Noma T, Lemaire A, Naga Prasad SV, Barki‐Harrington L, Tilley DG, Chen J, Le Corvoisier P, Violin JD, Wei H, Lefkowitz RJ, Rockman HA. Beta‐arrestin‐mediated beta1‐adrenergic receptor transactivation of the EGFR confers cardioprotection. J Clin Invest. 2007;117:2445–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Al‐Bari MA. Chloroquine analogues in drug discovery: new directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J Antimicrob Chemother. 2015;70:1608–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Terkeltaub RA, Furst DE, Bennett K, Kook KA, Crockett RS, Davis MW. High versus low dosing of oral colchicine for early acute gout flare: twenty‐four‐hour outcome of the first multicenter, randomized, double‐blind, placebo‐controlled, parallel‐group, dose‐comparison colchicine study. Arthritis Rheum. 2010;62:1060–1068. [DOI] [PubMed] [Google Scholar]
- 147. Dinarello CA, Wolff SM, Goldfinger SE, Dale DC, Alling DW. Colchicine therapy for familial mediterranean fever. A double‐blind trial. N Engl J Med. 1974;291:934–937. [DOI] [PubMed] [Google Scholar]
- 148. Imazio M, Brucato A, Cemin R, Ferrua S, Maggiolini S, Beqaraj F, Demarie D, Forno D, Ferro S, Maestroni S, Belli R, Trinchero R, Spodick DH, Adler Y; Investigators I . A randomized trial of colchicine for acute pericarditis. N Engl J Med. 2013;369:1522–1528. [DOI] [PubMed] [Google Scholar]
- 149. Lee FY, Lu HI, Zhen YY, Leu S, Chen YL, Tsai TH, Chung SY, Chua S, Sheu JJ, Hsu SY, Chang HW, Sun CK, Yip HK. Benefit of combined therapy with nicorandil and colchicine in preventing monocrotaline‐induced rat pulmonary arterial hypertension. Eur J Pharm Sci. 2013;50:372–384. [DOI] [PubMed] [Google Scholar]
- 150. Slobodnick A, Shah B, Krasnokutsky S, Pillinger MH. Update on colchicine, 2017. Rheumatology (Oxford). 2018;57:i4–i11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Deftereos S, Giannopoulos G, Panagopoulou V, Bouras G, Raisakis K, Kossyvakis C, Karageorgiou S, Papadimitriou C, Vastaki M, Kaoukis A, Angelidis C, Pagoni S, Pyrgakis V, Alexopoulos D, Manolis AS, Stefanadis C, Cleman MW. Anti‐inflammatory treatment with colchicine in stable chronic heart failure: a prospective, randomized study. JACC Heart Fail. 2014;2:131–137. [DOI] [PubMed] [Google Scholar]
- 152. Quick AP, Landstrom AP, Wehrens XH. Junctophilin‐2 at the intersection of arrhythmia and pathologic cardiac remodeling. Heart Rhythm. 2016;13:753–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Traish AM, Kang HP, Saad F, Guay AT. Dehydroepiandrosterone (DHEA)—a precursor steroid or an active hormone in human physiology. J Sex Med. 2011;8:2960–2982; quiz 2983. [DOI] [PubMed] [Google Scholar]
- 154. Scheffers CS, Armstrong S, Cantineau AE, Farquhar C, Jordan V. Dehydroepiandrosterone for women in the peri‐ or postmenopausal phase. Cochrane Database Syst Rev. 2015;1:CD011066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Elraiyah T, Sonbol MB, Wang Z, Khairalseed T, Asi N, Undavalli C, Nabhan M, Altayar O, Prokop L, Montori VM, Murad MH. Clinical review: the benefits and harms of systemic dehydroepiandrosterone (DHEA) in postmenopausal women with normal adrenal function: a systematic review and meta‐analysis. J Clin Endocrinol Metab. 2014;99:3536–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Morales AJ, Haubrich RH, Hwang JY, Asakura H, Yen SS. The effect of six months treatment with a 100 mg daily dose of dehydroepiandrosterone (DHEA) on circulating sex steroids, body composition and muscle strength in age‐advanced men and women. Clin Endocrinol (Oxf). 1998;49:421–432. [DOI] [PubMed] [Google Scholar]
- 157. Bonnet S, Dumas‐de‐La‐Roque E, Bégueret H, Marthan R, Fayon M, Dos Santos P, Savineau JP, Baulieu EE. Dehydroepiandrosterone (DHEA) prevents and reverses chronic hypoxic pulmonary hypertension. Proc Natl Acad Sci USA. 2003;100:9488–9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Paulin R, Meloche J, Jacob MH, Bisserier M, Courboulin A, Bonnet S. Dehydroepiandrosterone inhibits the Src/STAT3 constitutive activation in pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol. 2011;301:H1798–H1809. [DOI] [PubMed] [Google Scholar]
- 159. Alzoubi A, Toba M, Abe K, O'Neill KD, Rocic P, Fagan KA, McMurtry IF, Oka M. Dehydroepiandrosterone restores right ventricular structure and function in rats with severe pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol. 2013;304:H1708–H1718. [DOI] [PubMed] [Google Scholar]
- 160. Ventetuolo CE, Baird GL, Barr RG, Bluemke DA, Fritz JS, Hill NS, Klinger JR, Lima JA, Ouyang P, Palevsky HI, Palmisciano AJ, Krishnan I, Pinder D, Preston IR, Roberts KE, Kawut SM. Higher estradiol and lower dehydroepiandrosterone‐sulfate levels are associated with pulmonary arterial hypertension in men. Am J Respir Crit Care Med. 2016;193:1168–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic‐targeting therapy for cancer. Br J Cancer. 2008;99:989–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Stacpoole PW, Kurtz TL, Han Z, Langaee T. Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev. 2008;60:1478–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Piao L, Fang YH, Cadete VJ, Wietholt C, Urboniene D, Toth PT, Marsboom G, Zhang HJ, Haber I, Rehman J, Lopaschuk GD, Archer SL. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. J Mol Med (Berl). 2010;88:47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Piao L, Sidhu VK, Fang YH, Ryan JJ, Parikh KS, Hong Z, Toth PT, Morrow E, Kutty S, Lopaschuk GD, Archer SL. Foxo1‐mediated upregulation of pyruvate dehydrogenase kinase‐4 (PDK4) decreases glucose oxidation and impairs right ventricular function in pulmonary hypertension: therapeutic benefits of dichloroacetate. J Mol Med (Berl). 2013;91:333–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Michelakis ED, Gurtu V, Webster L, Barnes G, Watson G, Howard L, Cupitt J, Paterson I, Thompson RB, Chow K, O'Regan DP, Zhao L, Wharton J, Kiely DG, Kinnaird A, Boukouris AE, White C, Nagendran J, Freed DH, Wort SJ, Gibbs JSR, Wilkins MR. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci Transl Med. 2017;9:eaao4583. [DOI] [PubMed] [Google Scholar]
- 166. Paulin R, Dromparis P, Sutendra G, Gurtu V, Zervopoulos S, Bowers L, Haromy A, Webster L, Provencher S, Bonnet S, Michelakis ED. Sirtuin 3 deficiency is associated with inhibited mitochondrial function and pulmonary arterial hypertension in rodents and humans. Cell Metab. 2014;20:827–839. [DOI] [PubMed] [Google Scholar]
- 167. Dromparis P, Paulin R, Sutendra G, Qi AC, Bonnet S, Michelakis ED. Uncoupling protein 2 deficiency mimics the effects of hypoxia and endoplasmic reticulum stress on mitochondria and triggers pseudohypoxic pulmonary vascular remodeling and pulmonary hypertension. Circ Res. 2013;113:126–136. [DOI] [PubMed] [Google Scholar]
- 168. Pak O, Sommer N, Hoeres T, Bakr A, Waisbrod S, Sydykov A, Haag D, Esfandiary A, Kojonazarov B, Veit F, Fuchs B, Weisel FC, Hecker M, Schermuly RT, Grimminger F, Ghofrani HA, Seeger W, Weissmann N. Mitochondrial hyperpolarization in pulmonary vascular remodeling. Mitochondrial uncoupling protein deficiency as disease model. Am J Respir Cell Mol Biol. 2013;49:358–367. [DOI] [PubMed] [Google Scholar]
- 169. Rojas LB, Gomes MB. Metformin: an old but still the best treatment for type 2 diabetes. Diabetol Metab Syndr. 2013;5:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Agard C, Rolli‐Derkinderen M, Dumas‐de‐La‐Roque E, Rio M, Sagan C, Savineau JP, Loirand G, Pacaud P. Protective role of the antidiabetic drug metformin against chronic experimental pulmonary hypertension. Br J Pharmacol. 2009;158:1285–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Dean A, Nilsen M, Loughlin L, Salt IP, MacLean MR. Metformin reverses development of pulmonary hypertension via aromatase inhibition. Hypertension. 2016;68:446–454. [DOI] [PubMed] [Google Scholar]
- 172. Hemnes AR, Brittain EL, Trammell AW, Fessel JP, Austin ED, Penner N, Maynard KB, Gleaves L, Talati M, Absi T, Disalvo T, West J. Evidence for right ventricular lipotoxicity in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med. 2014;189:325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. DeFronzo R, Fleming GA, Chen K, Bicsak TA. Metformin‐associated lactic acidosis: current perspectives on causes and risk. Metabolism. 2016;65:20–29. [DOI] [PubMed] [Google Scholar]
- 174. Meloche J, Pflieger A, Vaillancourt M, Paulin R, Potus F, Zervopoulos S, Graydon C, Courboulin A, Breuils‐Bonnet S, Tremblay E, Couture C, Michelakis ED, Provencher S, Bonnet S. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation. 2014;129:786–797. [DOI] [PubMed] [Google Scholar]
- 175. Bitler BG, Watson ZL, Wheeler LJ, Behbakht K. Parp inhibitors: clinical utility and possibilities of overcoming resistance. Gynecol Oncol. 2017;147:695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, Delaloge S, Li W, Tung N, Armstrong A, Wu W, Goessl C, Runswick S, Conte P. Olaparib for metastatic breast cancer in patients with a germline brca mutation. N Engl J Med. 2017;377:523–533. [DOI] [PubMed] [Google Scholar]
- 177. Savai R, Al‐Tamari HM, Sedding D, Kojonazarov B, Muecke C, Teske R, Capecchi MR, Weissmann N, Grimminger F, Seeger W, Schermuly RT, Pullamsetti SS. Pro‐proliferative and inflammatory signaling converge on FoXO1 transcription factor in pulmonary hypertension. Nat Med. 2014;20:1289–1300. [DOI] [PubMed] [Google Scholar]
- 178. Weaver BA. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014;25:2677–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Marupudi NI, Han JE, Li KW, Renard VM, Tyler BM, Brem H. Paclitaxel: a review of adverse toxicities and novel delivery strategies. Expert Opin Drug Saf. 2007;6:609–621. [DOI] [PubMed] [Google Scholar]
- 180. Khongkow P, Gomes AR, Gong C, Man EP, Tsang JW, Zhao F, Monteiro LJ, Coombes RC, Medema RH, Khoo US, Lam EW. Paclitaxel targets FOXM1 to regulate KIF20A in mitotic catastrophe and breast cancer paclitaxel resistance. Oncogene. 2016;35:990–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Dai J, Zhou Q, Tang H, Chen T, Li J, Raychaudhuri P, Yuan JX, Zhou G. Smooth muscle cell‐specific FOXM1 controls hypoxia‐induced pulmonary hypertension. Cell Signal. 2018;51:119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Dai Z, Zhu MM, Peng Y, Jin H, Machireddy N, Qian Z, Zhang X, Zhao YY. Endothelial and smooth muscle cell interaction via FOXM1 signaling mediates vascular remodeling and pulmonary hypertension. Am J Respir Crit Care Med. 2018;198:788–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Bourgeois A, Lambert C, Habbout K, Ranchoux B, Paquet‐Marceau S, Trinh I, Breuils‐Bonnet S, Paradis R, Nadeau V, Paulin R, Provencher S, Bonnet S, Boucherat O. FOXM1 promotes pulmonary artery smooth muscle cell expansion in pulmonary arterial hypertension. J Mol Med (Berl). 2018;96:223–235. [DOI] [PubMed] [Google Scholar]
- 184. Randle PJ, Priestman DA, Mistry SC, Halsall A. Glucose fatty acid interactions and the regulation of glucose disposal. J Cell Biochem. 1994;55(suppl):1–11. [DOI] [PubMed] [Google Scholar]
- 185. Fang YH, Piao L, Hong Z, Toth PT, Marsboom G, Bache‐Wiig P, Rehman J, Archer SL. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: exploiting randle's cycle. J Mol Med (Berl). 2012;90:31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Chaitman BR. Ranolazine for the treatment of chronic angina and potential use in other cardiovascular conditions. Circulation. 2006;113:2462–2472. [DOI] [PubMed] [Google Scholar]
- 187. Rayner‐Hartley E, Sedlak T. Ranolazine: a contemporary review. J Am Heart Assoc. 2016;5:e003196 DOI: 10.1161/JAHA.116.003196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Liles JT, Hoyer K, Oliver J, Chi L, Dhalla AK, Belardinelli L. Ranolazine reduces remodeling of the right ventricle and provoked arrhythmias in rats with pulmonary hypertension. J Pharmacol Exp Ther. 2015;353:480–489. [DOI] [PubMed] [Google Scholar]
- 189. Gomberg‐Maitland M, Schilz R, Mediratta A, Addetia K, Coslet S, Thomeas V, Gillies H, Oudiz RJ. Phase I safety study of ranolazine in pulmonary arterial hypertension. Pulm Circ. 2015;5:691–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Khan SS, Cuttica MJ, Beussink‐Nelson L, Kozyleva A, Sanchez C, Mkrdichian H, Selvaraj S, Dematte JE, Lee DC, Shah SJ. Effects of ranolazine on exercise capacity, right ventricular indices, and hemodynamic characteristics in pulmonary arterial hypertension: a pilot study. Pulm Circ. 2015;5:547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Han Y, Forfia PR, Vaidya A, Mazurek JA, Park MH, Ramani G, Chan SY, Waxman AB. Rationale and design of the ranolazine PH‐RV study: a multicentred randomised and placebo‐controlled study of ranolazine to improve RV function in patients with non‐group 2 pulmonary hypertension. Open Heart. 2018;5:e000736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Li J, Kim SG, Blenis J. Rapamycin: one drug, many effects. Cell Metab. 2014;19:373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Nishimura T, Faul JL, Berry GJ, Veve I, Pearl RG, Kao PN. 40‐o‐(2‐hydroxyethyl)‐rapamycin attenuates pulmonary arterial hypertension and neointimal formation in rats. Am J Respir Crit Care Med. 2001;163:498–502. [DOI] [PubMed] [Google Scholar]
- 194. Zhou H, Liu H, Porvasnik SL, Terada N, Agarwal A, Cheng Y, Visner GA. Heme oxygenase‐1 mediates the protective effects of rapamycin in monocrotaline‐induced pulmonary hypertension. Lab Invest. 2006;86:62–71. [DOI] [PubMed] [Google Scholar]
- 195. Paddenberg R, Stieger P, von Lilien AL, Faulhammer P, Goldenberg A, Tillmanns HH, Kummer W, Braun‐Dullaeus RC. Rapamycin attenuates hypoxia‐induced pulmonary vascular remodeling and right ventricular hypertrophy in mice. Respir Res. 2007;8:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. McMurtry MS, Bonnet S, Michelakis ED, Haromy A, Archer SL. Statin therapy, alone or with rapamycin, does not reverse monocrotaline pulmonary arterial hypertension: the rapamcyin‐atorvastatin‐simvastatin study. Am J Physiol Lung Cell Mol Physiol. 2007;293:L933–L940. [DOI] [PubMed] [Google Scholar]
- 197. Houssaini A, Abid S, Mouraret N, Wan F, Rideau D, Saker M, Marcos E, Tissot CM, Dubois‐Randé JL, Amsellem V, Adnot S. Rapamycin reverses pulmonary artery smooth muscle cell proliferation in pulmonary hypertension. Am J Respir Cell Mol Biol. 2013;48:568–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Gonzalez‐Angulo AM, Meric‐Bernstam F, Chawla S, Falchook G, Hong D, Akcakanat A, Chen H, Naing A, Fu S, Wheler J, Moulder S, Helgason T, Li S, Elias I, Desai N, Kurzrock R. Weekly nab‐Rapamycin in patients with advanced nonhematologic malignancies: final results of a phase I trial. Clin Cancer Res. 2013;19:5474–5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008;112:1570–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Gürcan HM, Keskin DB, Stern JN, Nitzberg MA, Shekhani H, Ahmed AR. A review of the current use of rituximab in autoimmune diseases. Int Immunopharmacol. 2009;9:10–25. [DOI] [PubMed] [Google Scholar]
- 201. Kasi PM, Tawbi HA, Oddis CV, Kulkarni HS. Clinical review: serious adverse events associated with the use of rituximab—a critical care perspective. Crit Care. 2012;16:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Levin AS, Otani IM, Lax T, Hochberg E, Banerji A. Reactions to rituximab in an outpatient infusion center: a 5‐year review. J Allergy Clin Immunol Pract. 2017;5:107–113.e101. [DOI] [PubMed] [Google Scholar]
- 203. Mizuno S, Farkas L, Al Husseini A, Farkas D, Gomez‐Arroyo J, Kraskauskas D, Nicolls MR, Cool CD, Bogaard HJ, Voelkel NF. Severe pulmonary arterial hypertension induced by su5416 and ovalbumin immunization. Am J Respir Cell Mol Biol. 2012;47:679–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Hansmann G, de Jesus Perez VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, Schellong S, Urashima T, Wang L, Morrell NW, Rabinovitch M. An antiproliferative BMP‐2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest. 2008;118:1846–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Ameshima S, Golpon H, Cool CD, Chan D, Vandivier RW, Gardai SJ, Wick M, Nemenoff RA, Geraci MW, Voelkel NF. Peroxisome proliferator‐activated receptor gamma (PPARgamma) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ Res. 2003;92:1162–1169. [DOI] [PubMed] [Google Scholar]
- 206. Kahn BB, McGraw TE. Rosiglitazone, PPARγ, and type 2 diabetes. N Engl J Med. 2010;363:2667–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi‐Benedetti M, Moules IK, Skene AM, Tan MH, Lefèbvre PJ, Murray GD, Standl E, Wilcox RG, Wilhelmsen L, Betteridge J, Birkeland K, Golay A, Heine RJ, Korányi L, Laakso M, Mokán M, Norkus A, Pirags V, Podar T, Scheen A, Scherbaum W, Schernthaner G, Schmitz O, Skrha J, Smith U, Taton J; PROactive Investigators . Secondary prevention of macrovascular events in patients with type 2 diabetes in the proactive study (prospective pioglitazone clinical trial in macrovascular events): a randomised controlled trial. Lancet. 2005;366:1279–1289. [DOI] [PubMed] [Google Scholar]
- 208. Hansmann G, Wagner RA, Schellong S, Perez VA, Urashima T, Wang L, Sheikh AY, Suen RS, Stewart DJ, Rabinovitch M. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator‐activated receptor‐gamma activation. Circulation. 2007;115:1275–1284. [DOI] [PubMed] [Google Scholar]
- 209. Kim EK, Lee JH, Oh YM, Lee YS, Lee SD. Rosiglitazone attenuates hypoxia‐induced pulmonary arterial hypertension in rats. Respirology. 2010;15:659–668. [DOI] [PubMed] [Google Scholar]
- 210. Nisbet RE, Bland JM, Kleinhenz DJ, Mitchell PO, Walp ER, Sutliff RL, Hart CM. Rosiglitazone attenuates chronic hypoxia‐induced pulmonary hypertension in a mouse model. Am J Respir Cell Mol Biol. 2010;42:482–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Legchenko E, Chouvarine P, Borchert P, Fernandez‐Gonzalez A, Snay E, Meier M, Maegel L, Mitsialis SA, Rog‐Zielinska EA, Kourembanas S, Jonigk D, Hansmann G. Pparγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci Transl Med. 2018;10:eaao0303. [DOI] [PubMed] [Google Scholar]
- 212. Mannucci E, Monami M, Di Bari M, Lamanna C, Gori F, Gensini GF, Marchionni N. Cardiac safety profile of rosiglitazone: a comprehensive meta‐analysis of randomized clinical trials. Int J Cardiol. 2010;143:135–140. [DOI] [PubMed] [Google Scholar]
- 213. Lincoff AM, Wolski K, Nicholls SJ, Nissen SE. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta‐analysis of randomized trials. JAMA. 2007;298:1180–1188. [DOI] [PubMed] [Google Scholar]
- 214. Fung JJ. Tacrolimus and transplantation: a decade in review. Transplantation. 2004;77:S41–S43. [DOI] [PubMed] [Google Scholar]
- 215. Spiekerkoetter E, Tian X, Cai J, Hopper RK, Sudheendra D, Li CG, El‐Bizri N, Sawada H, Haghighat R, Chan R, Haghighat L, de Jesus Perez V, Wang L, Reddy S, Zhao M, Bernstein D, Solow‐Cordero DE, Beachy PA, Wandless TJ, Ten Dijke P, Rabinovitch M. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest. 2013;123:3600–3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Naesens M, Kuypers DR, Sarwal M. Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol. 2009;4:481–508. [DOI] [PubMed] [Google Scholar]
- 217. Spiekerkoetter E, Sung YK, Sudheendra D, Bill M, Aldred MA, van de Veerdonk MC, Vonk Noordegraaf A, Long‐Boyle J, Dash R, Yang PC, Lawrie A, Swift AJ, Rabinovitch M, Zamanian RT. Low‐dose FK506 (tacrolimus) in end‐stage pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;192:254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Spiekerkoetter E, Sung YK, Sudheendra D, Scott V, Del Rosario P, Bill M, Haddad F, Long‐Boyle J, Hedlin H, Zamanian RT. Randomised placebo‐controlled safety and tolerability trial of FK506 (tacrolimus) for pulmonary arterial hypertension. Eur Respir J. 2017;50:1602449. [DOI] [PubMed] [Google Scholar]
- 219. Savale L, Tu L, Rideau D, Izziki M, Maitre B, Adnot S, Eddahibi S. Impact of interleukin‐6 on hypoxia‐induced pulmonary hypertension and lung inflammation in mice. Respir Res. 2009;10:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin‐6 overexpression induces pulmonary hypertension. Circ Res. 2009;104:236–244, 228p following 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Prins KW, Archer SL, Pritzker M, Rose L, Weir EK, Sharma A, Thenappan T. Interleukin‐6 is independently associated with right ventricular function in pulmonary arterial hypertension. J Heart Lung Transplant. 2018;37:376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Heresi GA, Aytekin M, Hammel JP, Wang S, Chatterjee S, Dweik RA. Plasma interleukin‐6 adds prognostic information in pulmonary arterial hypertension. Eur Respir J. 2014;43:912–914. [DOI] [PubMed] [Google Scholar]
- 223. Yazici Y, Curtis JR, Ince A, Baraf H, Malamet RL, Teng LL, Kavanaugh A. Efficacy of tocilizumab in patients with moderate to severe active rheumatoid arthritis and a previous inadequate response to disease‐modifying antirheumatic drugs: the rose study. Ann Rheum Dis. 2012;71:198–205. [DOI] [PubMed] [Google Scholar]
- 224. Navarro G, Taroumian S, Barroso N, Duan L, Furst D. Tocilizumab in rheumatoid arthritis: a meta‐analysis of efficacy and selected clinical conundrums. Semin Arthritis Rheum. 2014;43:458–469. [DOI] [PubMed] [Google Scholar]
- 225. Hernández‐Sánchez J, Harlow L, Church C, Gaine S, Knightbridge E, Bunclark K, Gor D, Bedding A, Morrell N, Corris P, Toshner M. Clinical trial protocol for TRANSFORM‐UK: a therapeutic open‐label study of tocilizumab in the treatment of pulmonary arterial hypertension. Pulm Circ. 2018;8:2045893217735820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Kantor PF, Lucien A, Kozak R, Lopaschuk GD. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long‐chain 3‐ketoacyl coenzyme a thiolase. Circ Res. 2000;86:580–588. [DOI] [PubMed] [Google Scholar]
- 227. Gao D, Ning N, Niu X, Hao G, Meng Z. Trimetazidine: a meta‐analysis of randomised controlled trials in heart failure. Heart. 2011;97:278–286. [DOI] [PubMed] [Google Scholar]
- 228. Zhang L, Lu Y, Jiang H, Sun A, Zou Y, Ge J. Additional use of trimetazidine in patients with chronic heart failure: a meta‐analysis. J Am Coll Cardiol. 2012;59:913–922. [DOI] [PubMed] [Google Scholar]
- 229. Zhou X, Chen J. Is treatment with trimetazidine beneficial in patients with chronic heart failure? PLoS One. 2014;9:e94660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, Falk V, González‐Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GMC, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P; ESC Scientific Document Group . 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37:2129–2200. [DOI] [PubMed] [Google Scholar]
- 231. Chrusciel P, Rysz J, Banach M. Defining the role of trimetazidine in the treatment of cardiovascular disorders: some insights on its role in heart failure and peripheral artery disease. Drugs. 2014;74:971–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Matura LA, Ventetuolo CE, Palevsky HI, Lederer DJ, Horn EM, Mathai SC, Pinder D, Archer‐Chicko C, Bagiella E, Roberts KE, Tracy RP, Hassoun PM, Girgis RE, Kawut SM. Interleukin‐6 and tumor necrosis factor‐α are associated with quality of life‐related symptoms in pulmonary arterial hypertension. Ann Am Thorac Soc. 2015;12:370–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Zhang LL, Lu J, Li MT, Wang Q, Zeng XF. Preventive and remedial application of etanercept attenuate monocrotaline‐induced pulmonary arterial hypertension. Int J Rheum Dis. 2016;19:192–198. [DOI] [PubMed] [Google Scholar]
- 234. Weinblatt ME, Kremer JM, Bankhurst AD, Bulpitt KJ, Fleischmann RM, Fox RI, Jackson CG, Lange M, Burge DJ. A trial of etanercept, a recombinant tumor necrosis factor receptor: Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med. 1999;340:253–259. [DOI] [PubMed] [Google Scholar]
- 235. Moreland LW, Schiff MH, Baumgartner SW, Tindall EA, Fleischmann RM, Bulpitt KJ, Weaver AL, Keystone EC, Furst DE, Mease PJ, Ruderman EM, Horwitz DA, Arkfeld DG, Garrison L, Burge DJ, Blosch CM, Lange ML, McDonnell ND, Weinblatt ME. Etanercept therapy in rheumatoid arthritis. A randomized, controlled trial. Ann Intern Med. 1999;130:478–486. [DOI] [PubMed] [Google Scholar]
- 236. Mease PJ. Tumour necrosis factor (TNF) in psoriatic arthritis: pathophysiology and treatment with TNF inhibitors. Ann Rheum Dis. 2002;61:298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Ford AC, Sandborn WJ, Khan KJ, Hanauer SB, Talley NJ, Moayyedi P. Efficacy of biological therapies in inflammatory bowel disease: systematic review and meta‐analysis. Am J Gastroenterol. 2011;106:644–659, quiz 660. [DOI] [PubMed] [Google Scholar]
- 238. Lis K, Kuzawińska O, Bałkowiec‐Iskra E. Tumor necrosis factor inhibitors—state of knowledge. Arch Med Sci. 2014;10:1175–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT; Anti‐TNF Therapy Against Congestive Heart Failure Investigators . Randomized, double‐blind, placebo‐controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor‐alpha, in patients with moderate‐to‐severe heart failure: results of the anti‐TNF therapy against congestive heart failure (ATTACH) trial. Circulation. 2003;107:3133–3140. [DOI] [PubMed] [Google Scholar]
- 240. Bozkurt B, Torre‐Amione G, Warren MS, Whitmore J, Soran OZ, Feldman AM, Mann DL. Results of targeted anti‐tumor necrosis factor therapy with etanercept (ENBREL) in patients with advanced heart failure. Circulation. 2001;103:1044–1047. [DOI] [PubMed] [Google Scholar]
- 241. Mann DL, McMurray JJ, Packer M, Swedberg K, Borer JS, Colucci WS, Djian J, Drexler H, Feldman A, Kober L, Krum H, Liu P, Nieminen M, Tavazzi L, van Veldhuisen DJ, Waldenstrom A, Warren M, Westheim A, Zannad F, Fleming T. Targeted anticytokine therapy in patients with chronic heart failure: results of the randomized etanercept worldwide evaluation (renewal). Circulation. 2004;109:1594–1602. [DOI] [PubMed] [Google Scholar]
- 242. Mutschler D, Wikström G, Lind L, Larsson A, Lagrange A, Eriksson M. Etanercept reduces late endotoxin‐induced pulmonary hypertension in the pig. J Interferon Cytokine Res. 2006;26:661–667. [DOI] [PubMed] [Google Scholar]
- 243. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. Role of YAP/TAZ In mechanotransduction. Nature. 2011;474:179–183. [DOI] [PubMed] [Google Scholar]
- 244. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2:e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Piao L, Fang YH, Parikh K, Ryan JJ, Toth PT, Archer SL. Cardiac glutaminolysis: a maladaptive cancer metabolism pathway in the right ventricle in pulmonary hypertension. J Mol Med (Berl). 2013;91:1185–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Verteporfin In Photodynamic Therapy Study Group . Verteporfin therapy of subfoveal choroidal neovascularization in age‐related macular degeneration: two‐year results of a randomized clinical trial including lesions with occult with no classic choroidal neovascularization—verteporfin in photodynamic therapy report 2. Am J Ophthalmol. 2001;131:541–560. [DOI] [PubMed] [Google Scholar]
- 247. Meads C, Hyde C. Photodynamic therapy with verteporfin is effective, but how big is its effect? Results of a systematic review. Br J Ophthalmol. 2004;88:212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Schmidt‐Erfurth U, Hasan T. Mechanisms of action of photodynamic therapy with verteporfin for the treatment of age‐related macular degeneration. Surv Ophthalmol. 2000;45:195–214. [DOI] [PubMed] [Google Scholar]
- 249. Cheng F, Desai RJ, Handy DE, Wang R, Schneeweiss S, Barabási AL, Loscalzo J. Network‐based approach to prediction and population‐based validation of in silico drug repurposing. Nat Commun. 2018;9:2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, Grimminger F. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115:2811–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Hoeper MM, Barst RJ, Bourge RC, Feldman J, Frost AE, Galié N, Gómez‐Sánchez MA, Grimminger F, Grünig E, Hassoun PM, Morrell NW, Peacock AJ, Satoh T, Simonneau G, Tapson VF, Torres F, Lawrence D, Quinn DA, Ghofrani HA. Imatinib mesylate as add‐on therapy for pulmonary arterial hypertension: results of the randomized impres study. Circulation. 2013;127:1128–1138. [DOI] [PubMed] [Google Scholar]
- 252. Zhai FG, Zhang XH, Wang HL. Fluoxetine protects against monocrotaline‐induced pulmonary arterial hypertension: potential roles of induction of apoptosis and upregulation of kv1.5 channels in rats. Clin Exp Pharmacol Physiol. 2009;36:850–856. [DOI] [PubMed] [Google Scholar]
- 253. Guignabert C, Raffestin B, Benferhat R, Raoul W, Zadigue P, Rideau D, Hamon M, Adnot S, Eddahibi S. Serotonin transporter inhibition prevents and reverses monocrotaline‐induced pulmonary hypertension in rats. Circulation. 2005;111:2812–2819. [DOI] [PubMed] [Google Scholar]
- 254. Wang HM, Wang Y, Liu M, Bai Y, Zhang XH, Sun YX, Wang HL. Fluoxetine inhibits monocrotaline‐induced pulmonary arterial remodeling involved in inhibition of rhoa‐rho kinase and akt signalling pathways in rats. Can J Physiol Pharmacol. 2012;90:1506–1515. [DOI] [PubMed] [Google Scholar]
- 255. Dumitrascu R, Kulcke C, Königshoff M, Kouri F, Yang X, Morrell N, Ghofrani HA, Weissmann N, Reiter R, Seeger W, Grimminger F, Eickelberg O, Schermuly RT, Pullamsetti SS. Terguride ameliorates monocrotaline‐induced pulmonary hypertension in rats. Eur Respir J. 2011;37:1104–1118. [DOI] [PubMed] [Google Scholar]
- 256. Budas GR, Boehm M, Kojonazarov B, Viswanathan G, Tian X, Veeroju S, Novoyatleva T, Grimminger F, Hinojosa‐Kirschenbaum F, Ghofrani HA, Weissmann N, Seeger W, Liles JT, Schermuly RT. ASK1 inhibition halts disease progression in preclinical models of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2018;197:373–385. [DOI] [PubMed] [Google Scholar]
- 257. Hemnes AR, Trammell AW, Archer SL, Rich S, Yu C, Nian H, Penner N, Funke M, Wheeler L, Robbins IM, Austin ED, Newman JH, West J. Peripheral blood signature of vasodilator‐responsive pulmonary arterial hypertension. Circulation. 2015;131:401–409; discussion 409. [DOI] [PMC free article] [PubMed] [Google Scholar]