

Abstract
Background
Cardiac fibrosis is a core pathological process associated with heart failure. The recruitment and differentiation of primitive fibroblast precursor cells of bone marrow origin play a critical role in pathological interstitial cardiac fibrosis. The KC a3.1 channels are expressed in both ventricular fibroblasts and circulating mononuclear cells in rats and are upregulated by angiotensin II. We hypothesized that KC a3.1 channels mediate the inflammatory microenvironment in the heart, promoting the infiltrated bone marrow–derived circulating mononuclear cells to differentiate into myofibroblasts, leading to myocardial fibrosis.
Methods and Results
We established a cardiac fibrosis model in rats by infusing angiotensin II to evaluate the impact of the specific KC a3.1 channel blocker TRAM‐34 on cardiac fibrosis. At the same time, mouse CD4+ T cells and rat circulating mononuclear cells were separated to investigate the underlying mechanism of the TRAM‐34 anti–cardiac fibrosis effect. TRAM‐34 significantly attenuated cardiac fibrosis and the inflammatory reaction and reduced the number of fibroblast precursor cells and myofibroblasts. Inhibition of KC a3.1 channels suppressed angiotensin II–stimulated expression and secretion of interleukin‐4 and interleukin‐13 in CD4+ T cells and interleukin‐4– or interleukin‐13–induced differentiation of monocytes into fibrocytes.
Conclusions
KC a3.1 channels facilitate myocardial inflammation and the differentiation of bone marrow‐derived monocytes into myofibroblasts in cardiac fibrosis caused by angiotensin II infusion.
Keywords: angiotensin II, cardiac remodeling, differentiation, inflammation, K‐channel
Subject Categories: Fibrosis, Inflammation, Ion Channels/Membrane Transport, Pathophysiology, Mechanisms
Clinical Perspective
What Is New?
Our study demonstrated that KCa3.1 channels facilitate myocardial inflammation and differentiation of bone marrow–derived monocytes into myofibroblasts, thereby promoting cardiac fibrosis caused by angiotensin II infusion.
We found an interaction between KCa3.1 and TRPV4/TRPC6 channels in cardiac fibroblasts that contributed to the differentiation of fibroblasts into myofibroblasts.
What Are the Clinical Implications?
These findings elucidate the role and mechanism of KCa3.1 channels in the development and progression of cardiac fibrosis.
The study also provides valuable insights into the KCa3.1 channel as a therapeutic target to prevent cardiac fibrosis.
Introduction
Cardiac fibrosis is a common pathological process in numerous forms of heart disease and manifests as excessive deposition of extracellular matrix (ECM), increase of collagen content, and imbalanced synthesis and arrangement of various types of ECM proteins.1 Cardiac myofibroblasts, differentiated from resident or circulation‐derived fibroblasts, are not only principal ECM producers but also responders to proinflammatory cytokines including tumor necrosis factor‐α (TNF‐α), interleukin (IL)‐1, IL‐6, transforming growth factor‐β (TGF‐β); vasoactive peptide angiotensin II (Ang II), endothelin‐1; and hormones such as norepinephrine.2 Ang II, the key effector of the renin‐angiotensin system, plays a central role in cardiac fibrosis through hemodynamic‐dependent and ‐independent mechanisms, which evoke an inflammatory microenvironment in the hypertensive heart and promote the differentiation of infiltrated monocytes into myofibroblasts.3 A thorough understanding of the underlying mechanism of Ang II–induced cardiac fibrosis may help in fibrosis prevention and treatment.
Intermediate‐conductance Ca2+‐activated K+ (KCa3.1) channels regulate the membrane potential and calcium entry into cells, thereby modulating calcium signaling–regulated processes. Previous studies have shown that KCa3.1 channels are closely involved in regulation of cell cycle, cell phenotypic modulation, proliferation, and migration.4, 5, 6, 7 KCa3.1 channels also contribute to activation of inflammatory cells.8 With regard to organ fibrosis, KCa3.1 channels play a pivotal role in renal fibrosis.9 KCa3.1 channels are also regarded as a potential therapeutic target in idiopathic pulmonary fibrosis.10 However, the role and molecular mechanism of KCa3.1 channels in cardiac fibrosis remain unclear.
The differentiation of cardiac fibroblasts to more active myofibroblasts is the hallmark of cardiac fibrosis, as characterized by increased collagen production and expression of α‐smooth muscle actin (α‐SMA).11 There is a growing list of transient receptor potential (TRP) cation channels associated with cellular differentiation including the conversion of fibroblasts into myofibroblasts. Most of the TRP channels are activated by a variety of ligands, the mechanical environment, and/or changes in internal Ca2+ stores. Cardiac fibroblasts utilize a Ca2+ signal from these channels to transform quiescent fibroblasts into myofibroblasts. Of those channels TRPV4 and TRPC6 have been linked to myofibroblast differentiation.12 KCa3.1 channels can regulate cell membrane potential and then influence Ca2+ entry. Whether KCa3.1 channels interact with TRPV4 and TRPC6 channels and modulate the differentiation of fibroblasts into myofibroblasts needs to be investigated.
We previously showed that, in fibroblasts isolated from rat hearts, both mRNA and protein expression of KCa3.1 channels are upregulated when treatment with Ang II activates Ang II type I receptors. Inhibition of KCa3.1 channels with the specific blocker TRAM‐34 or small interfering RNA (siRNA) suppresses Ang II–stimulated fibroblast proliferation and collagen synthesis and secretion.13 Furthermore, treatment with TRAM‐34 in vivo effectively attenuates cardiac fibrosis induced by abdominal aortic constriction and reduces the expression of leukocyte differentiation antigens CD45 and CD3, macrophage‐specific antigen F4/80, TNF‐α, and monocyte chemotactic protein 1 (MCP‐1) in the myocardium.14 In addition, our in vitro investigation demonstrates that blockade of KCa3.1 channels abrogates Ang II–induced monocyte transendothelium migration and reduces the synthesis and release of MCP‐1 by endothelial cells.14 These findings suggest that KCa3.1 channels are involved in Ang II–mediated inflammatory and fibrotic signaling.
In the present study, we hypothesized that KCa3.1 channels could facilitate cardiac fibrosis through mediating the recruitment of bone marrow–derived monocytes to the myocardium, where they differentiate into myofibroblasts under an inflammatory microenvironment induced by Ang II. Using the established cardiac fibrosis model in rats by Ang II infusion, we observed that treatment with TRAM‐34 significantly attenuated cardiac fibrosis and the inflammatory reaction as well as the number of fibroblast precursor cells and myofibroblasts. By separating circulating mononuclear cells from male adult healthy rats and CD4+ T cells from male adult healthy mice, we observed that inhibition of KCa3.1 channels suppressed Ang II–stimulated expression and secretion of IL‐4 and IL‐13 in CD4+ T cells and IL‐4– or IL‐13–induced differentiation of monocytes into fibrocytes. Our results provide novel evidence for the role and mechanism of KCa3.1 channels in cardiac fibrosis.
Methods
The data, analytic methods, and study materials will be available from the corresponding author on reasonable request to other researchers for purposes of reproducing the results or replicating the procedure.
Rat Model of Ang II Infusion–Induced Cardiac Fibrosis
All animals were obtained from the Laboratory Animal Centre of Xi'an Jiaotong University (Xi'an, China). Experimental protocols were approved by the Institutional Animal Care and Use Committee of Xi'an Jiaotong University and conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. Male Sprague Dawley rats (180‐200 g) were operated on for subcutaneous implantation of osmotic minipumps loaded with saline solution (control group, n=18) or Ang II (0.8 mg/[kg·d]). Two groups of rats were treated with combined infusion with Ang II and either Miglyol 812 neutral oil (TRAM‐34 solvent, 1 mL/[kg·d] given by intraperitoneal injection, Ang II group, n=18), or TRAM‐34 (80 mg/[kg·d] intraperitoneally, Ang II+TRAM‐34 group, n=18). The dosages of Ang II and TRAM‐34 referenced our previous study.14, 15 The treatment with TRAM‐34 or its solvent was for 4 weeks and began at 12 hours after minipump implantation.
During the study period the systolic blood pressure (SBP) was measured weekly using a tail‐cuff system (BP‐300A; Chengdu Taimeng Company, Sichuan, China). Four weeks after Ang II infusion the rats were anesthetized with sodium pentobarbital (45 mg/kg intraperitoneally), and blood was collected from the abdominal aorta. The hearts were isolated and weighed to calculate the heart‐weight/body‐weight ratio. The concentration of hydroxyproline in the myocardium of the left ventricle (LV) was measured by enzyme‐linked immunosorbent assay (ELISA) following the manufacturer's protocol. In brief, 20‐mg LV tissue samples in 0.2 mL PBS were homogenized with a tissue homogenizer, and the homogenates were centrifuged at 3000g for 10 minutes at 4°C. The supernate was collected to assay hydroxyproline level immediately.
Echocardiography
Transthoracic echocardiography was performed as described previously by using a Vevo 2100 ultrasound machine (Visualsonics Inc, Toronto, Canada) with a 21‐MHz transducer.16 Animals were maintained under light anesthesia by inhalation of 1.8% isoflurane and placed on a heating pad. A 2‐dimensional image of the LV was obtained at a level of the papillary muscles, and a 2‐dimensional guided M‐mode trace crossing the anterior and posterior walls was recorded at sweep speed of 100 mm/s. The following parameters were measured in a blinded fashion on the M‐mode tracings using the leading‐edge technique: LV internal dimensions (LVID) at diastole and systole, LV mass, and ejection fraction. Fractional shortening was calculated as (LVIDdiastole−LVIDsystole)/LVIDdiastole. Measurements were taken from 5 cardiac cycles, and the averages were used.
Isolation of Cardiac Fibroblasts and Electrophysiology
After 4 weeks’ intervention, the hearts from each group of rats were quickly excised and placed in cold PBS. Ventricular fibroblasts were isolated by standard collagenase digestion methods, as we previously described.13 Cells were cultured in DMEM with 5 mmol/L glucose (Gibco, ThermoFisher, Walthm, MA)) with 10% fetal bovine serum (Invitrogen, Carlsbad, CA), penicillin (100 U/mL), and streptomycin (100 mg/mL) incubated at 37°C in a humidified atmosphere with 5% CO2. After growing to the fusion status, cells were detached and seeded in CC2 glass slides for immunofluorescent double staining or 1‐mL cell chambers for electrophysiology. Membrane current was recorded with the whole‐cell patch‐clamp technique at room temperature (22°C to 23°C) as described previously.17
Mouse CD4+ T‐Cell Isolation and Culture
Splenocytes were isolated according to the protocol of Ismahil et al.18 Briefly, male C57BL/6 mice (n=30) were anesthetized with sodium pentobarbital (50 mg/kg intraperitoneally), and spleens were harvested. A splenic cell suspension was prepared by grinding spleens with a push rod of a 2‐mL syringe in PBS and passed through a 100‐μm nylon cell strainer (BD Falcon, Durham, NC). Red blood cell lysis was conducted using red blood cell lysate buffer (Gibco). Splenic CD4+ T cells were separated and collected using the mouse CD4+ T‐cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany) following the manufacturer's protocol.
The sorted CD4+ T cells were seeded in 6‐well plates at 106 cells per well in completed RPMI 1640 medium containing 10% fetal bovine serum, 50 μmol/L 2‐mercaptoethanol, 2 mmol/L glutamine, 100 U/mL penicillin, and 100 mg/mL streptomycin as well as 2 μg/mL anti‐CD3 (16‐0031; eBioscience, San Diego, CA) and 2 μg/mL anti‐CD28 (16‐0281; eBioscience) for activation. The CD4+ T cells were divided into 3 groups: a control group without treatment, an Ang II group treated with 0.1 μmol/L Ang II, and an Ang II+TRAM‐34 group treated with 0.1 μmol/L Ang II plus 1 μmol/L TRAM‐34. After 72 hours of incubation at 37°C in a humidified atmosphere with 5% CO2, the supernatants were harvested to measure IL‐4 and IL‐13 concentrations by ELISA, and the total protein of CD4+ T cells was extracted to detect IL‐4 and IL‐13 expression by Western blotting analysis.
RNA Interference
Specific siRNA molecules targeted to KCa3.1 channels were synthesized by Gene Pharma Company (Shanghai, China). The siRNA sequences of mice KCa3.1 channels were as follows19: sense, 5′‐CGAUAAAUCACCUCAAGAUtt‐3′; antisense, 5′‐AUCUUGAGGUGAUUUAUCGtg‐3′. The siRNA sequences of rat KCa3.1 channels were as follows: sense, 5′‐GCCACUGGUUCGUGGCCAAACUAUA‐3′; antisense, 5′‐UAUAGUUUGGCCACGAACCAGUGGC‐3′. The siRNA molecules at 150 and 100 nmol/L were separately transfected into the mouse CD4+ T cells and rat peripheral blood monocytes (PBMCs) using Lipofectamine 2000 reagent (Invitrogen) for 6 hours according to the manufacturer's instructions. Transfected CD4+ T cells were used for IL‐4 and IL‐13 secretion and protein expression measurement. Transfected PBMCs were used for studying IL‐4– or IL‐13–induced fibrocyte differentiation.
Rat Peripheral Blood Monocyte Isolation and Fibrocyte Differentiation
Peripheral blood anticoagulated with dipotassium EDTA was collected from healthy adult male rats. PBMCs were isolated by Ficoll‐Hypaque gradient centrifugation (Hao‐Yang, Tianjin, China) following the manufacturer's protocol and incubated in serum‐free medium, which consists of FibroLife (LM‐0001, Lifeline Cell Technology, Walkersville, MD) supplemented with 20 mmol/L HEPES, 2×ITS+3, 2×nonessential amino acids, 2 mmol/L sodium pyruvate, 4 mmol/L glutamine (Invitrogen, Carlsbad, CA), 100 U/mL penicillin, and 100 μg/mL streptomycin.20 PBMCs were cultured in CC2 glass slides at 2.5×105 cells/mL with or without 3 ng/mL rat recombinant IL‐4 or IL‐13 (Peprotech, Rocky Hill, NJ) and 3 ng/mL rat recombinant IL‐4 or IL‐13 plus 1 μmol/L TRAM‐34 in a humidified incubator containing 5% CO2 at 37°C for 5 days. Then the detachable upper medium chamber was removed, and cells were stained by immunocytochemistry to identify the differentiated fibrocytes.
Immunohistochemistry and Immunocytochemistry
Myocardial tissue was fixed in formalin and then embedded in paraffin. Deparaffined sections (5 μm in thickness) were stained with Masson trichrome reagent. Images were digitized (×400 magnification, 6 fields per heart) under a microscope (DP72; Olympus, Tokyo, Japan). The expression of CD45, CD3, CD68, TNF‐α, MCP‐1, collagen I, and collagen III in the myocardium was immunolocalized using anti‐CD45 antibody (1:300), anti‐CD3 antibody (1:200), anti‐CD68 antibody (1:200), anti–TNF‐α antibody (1:300), anti–MCP‐1 antibody (1:200), anti–collagen I antibody (1:200), and anti–collagen III antibody (1:200), respectively, and visualized with diaminobenzidine. Negative control was performed by incubating the tissue with anti‐IgG antibody without primary antibody.
PBMCs cultured on 8‐well glass microscope slides were air dried for at least 60 minutes before fixation and permeabilization in acetone for 15 minutes. Nonspecific binding was blocked by incubation in PBS‐bovine serum albumin for 60 minutes. Slides were incubated with different primary antibodies, anti–collagen I (1:200), anti–discoidin domain receptor 2 (DDR‐2) (1:200), or anti‐CD34 (1:200) for 60 minutes and stained with diaminobenzidine. Anti‐IgG antibody without primary antibody was used as negative control.
Immunofluorescent Double Staining
For immunofluorescent double staining, deparaffinized sections were fixed in 100% cold methanol, and nonspecific proteins were blocked with 1% bovine serum albumin. The sections were incubated with rabbit anti–rat CD45 antibody (1:200) and mouse anti–rat CD34 antibody (1:200), rabbit anti–rat DDR‐2 antibody (1:200), and mouse anti–rat CD11b antibody (1:200) or rabbit anti–rat DDR‐2 antibody (1:200) and mouse anti–rat α‐SMA antibody (1:200) at 4°C overnight. After the incubation with appropriate fluorescence‐labeled secondary antibody at room temperature for 1 hour, the sections were observed under a fluorescence microscope (DP72; Olympus, Tokyo, Japan).
Isolated cardiac fibroblasts were seeded in 8‐well glass slides for 1 night; after adherence, the chamber on the slides was removed, and cells were stained by immunofluorescent double staining. Cells were fixed in 100% cold methanol, and nonspecific proteins were blocked with 1% bovine serum albumin. Then slides were incubated with mouse anti–rat KCa3.1 antibody (1:200) and goat anti–rat TRPV4 antibody (1:200) or mouse anti–rat KCa3.1 antibody (1:200) and goat anti–rat TRPC6 antibody (1:200) at 4°C overnight. After the incubation with appropriate fluorescence‐labeled secondary antibody at room temperature for 1 hour, the slides were observed under a confocal microscope (A1R; Nikon, Chiyoda, Japan).
Electrophysiology
Membrane current was recorded in primary rat cardiac fibroblasts, which were isolated from 4‐week‐intervened rats, with the whole‐cell patch‐clamp technique. The detached cardiac fibroblasts were placed into a 1‐mL cell chamber mounted on an inverted microscope (Diaphot; Nikon, Japan), allowed to settle to the bottom (2 hours), and then superfused with Tyrode solution (pH 7.4) that contained (mmol/L) 136 NaCl, 5.4 KCl, 1 MgCl2, 1.8 CaCl2, 10 glucose, and 10 HEPES. Borosilicate glass electrodes (1.2‐mm outer diameter) were pulled with a Brown Flaming puller (Model P‐97; Sutter Instrument Co, Novato, CA), and had a resistance of 2 to 3 MΩ when filled with pipette solution that contained (mmol/L) 120 K‐aspartate, 20 KCl, 1 MgCl2, 5 EGTA, 5 Na‐phosphocreatine, 5 Mg‐ATP, 0.1 GTP, 10 HEPES, 800 nmol/L free Ca2+ with pH adjusted to 7.2 with KOH. The tip potentials were zeroed before the pipette contacted the cell. After a gigaohm seal was obtained by negative suction, the cell membrane was ruptured by a gentle suction to establish a whole‐cell configuration. Membrane currents were recorded with an Axopatch 200B amplifier (Axon Instruments, Union City, CA), filtered at 2 kHz using a low‐pass Bessel filter, digitized at 10 kHz, and stored in the hard disk of an IBM‐compatible computer for subsequent analysis with Clampfit 9.0 (Molecular Devices, Sunnyvale, CA). All current recording experiments were conducted at room temperature (22°C to 23°C).
Western Blotting Analysis
The expression of inflammation‐ and fibrosis‐related markers in the myocardium and the expression of IL‐4 and IL‐13 in CD4+ T cells were determined by Western blotting analysis with the procedure as described previously.21 Immunodetection was performed by overnight incubation with primary antibodies for anti–collagen I (1:500), anti–collagen III (1:500), anti–connective tissue growth factor (CTGF) (1:500), anti–TGF‐β (1:500), anti‐CD45 (1:500), anti‐CD3 (1:1000), anti‐CD68 (1:1000), anti–TNF‐α (1:500), anti–MCP‐1 (1:1000), anti–IL‐4 (1:500), and anti‐IL‐13 (1:500) at 4°C and then incubated with horseradish peroxidase–conjugated secondary antibodies (1:5000) for 1 hour at room temperature. The bound antibodies were detected with an enhanced chemiluminescence detection system (Bio‐Rad, Hercules, CA) and quantified by densitometry using the Chemi‐Genius Bio‐Imaging System (Syngene, Cambridge, UK). To ensure equal sample loading, the ratio of band intensity to β‐actin was obtained to quantify the relative protein expression level.
Statistical Analysis
All data obeyed normal distribution characteristics and were presented as means±SEM. Statistical comparisons among control, Ang II, and TRAM‐34 groups were performed by 1‐way ANOVA with Bonferroni posttest using SPSS Statistics 19 (IBM, Armonk, NY). Two‐way ANOVA for repeated measures followed by the Bonferroni post hoc test was used to analyze SBP differences over time between control and Ang II with or without TRAM‐34 treatment. A value of P<0.05 was considered to have significant difference.
Materials
Antibodies against collagen I, collagen III, CD45, CD3, CD68, TNF‐α, and MCP‐1 were provided by Abcam (London, UK). Antibodies for TGF‐β, IL‐4, IL‐13, and DDR‐2 were purchased from Bioss Biotechnology (Beijing, China). Antibodies against CTGF, CD34, KCa3.1, TRPV4, and TRPC6 were provided by Santa Cruz Biotechnology (Dallas, TX). Alzet osmotic minipumps (model 2ML4) were obtained from Durect (Cupertino, CA). TRAM‐34 was from Shanghai Yuan‐Ding Chemical Technologies, Ltd (Shanghai, China). Miglyol 812 neutral oil was purchased from Spectrum Chemicals (Gardena, CA). Ang II and other chemicals used in this study were obtained from Sigma‐Aldrich (St. Louis, MO). ELISA kits for Ang II, hydroxyproline, and CTGF were purchased from Bio‐Swanp (Wuhan, China). MCP‐1, TNF‐α, and TGF‐β ELISA kits were from R&D Systems (Minneapolis, MN).
Results
KCa3.1 Channel Inhibition Suppresses Cardiac Fibrosis by Ang II
To ensure the establishment of cardiac remodeling induced by Ang II, we evaluated the alterations of blood pressure, heart‐weight/body‐weight ratio, and cardiac function of rats. One week after Ang II infusion, rats exhibited a significant rise of SBP, and the increment remained to the end of the experiment. Three‐week treatment with 80 mg/kg TRAM‐34, a specific KCa3.1 channel blocker, attenuated the increase of SBP induced by Ang II, and this effect of TRAM‐34 was maintained during the experiment (Figure 1A). The SBP‐lowering effect of TRAM‐34 was possibly attributable to the reduction of renin, angiotensinogen, and endogenous Ang II levels in plasma and myocardium in accord with our previous study.14 Compared with the control group, the heart‐weight/body‐weight ratio was significantly increased after Ang II infusion for 4 weeks, which was prevented by treatment with TRAM‐34 (Figure 1B). Figure 1C shows typical echocardiographic tracing images in rats from different group. Group data confirmed the decrease of ejection fraction and fractional shortening and the increase of LVIDdiastole, LVIDsystole, and LV mass by Ang II infusion, and the effects of Ang II were attenuated by TRAM‐34 treatment (Figure 1D).
Figure 1.
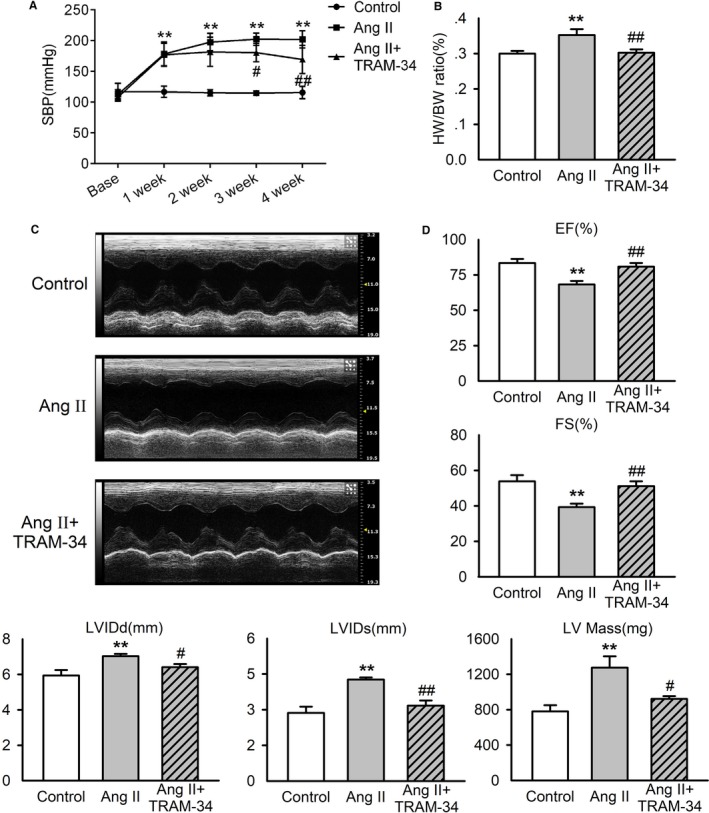
Effects of TRAM‐34 on blood pressure and cardiac function in Ang II–infusion rats. Rats received saline solution (control group) or Ang II infusion by osmotic minipump subdermal implantation for 4 weeks, and Ang II–infusion rats were treated with Miglyol 812 neutral oil, the solvent of TRAM‐34 (Ang II group), or 80 mg/kg per day TRAM‐34 (Ang II+TRAM‐34 group). A, Systolic blood pressure measured with a tail‐cuff system (n=10). B, Left ventricle weight–to–body weight ratio (n=7). C, Representative M‐mode echocardiographic tracings from short‐axis left ventricle 2‐dimensional images of rats. D, Grouped functional results of rat left ventricle (n=6). Data are presented as mean±SEM, **P<0.01 vs control, # P<0.05, ## P<0.01 vs Ang II. Ang II indicates angiotensin II; EF, ejection fraction; FS, fractional shortening; HW/BW, heart weight–to–body weight ratio; LVIDd, left ventricular inner dimensions in diastole; LVIDs, left ventricular inner dimensions in systole; SBP, stolic blood pressure.
Expression of collagen I and collagen III in the LV myocardium was determined by immunohistochemistry staining and Western blotting analysis. The levels of collagen I and collagen III were increased significantly in the Ang II group compared with that of the control group and were reduced dramatically by administration of TRAM‐34 (Figure 2A and 2B). Masson trichrome staining revealed that Ang II infusion caused significant collagen deposition in both interstitial and perivascular regions, which was reduced by TRAM‐34 (Figure 2C). Consistent with the histological findings, TRAM‐34 attenuated the increase of hydroxyproline concentration in the LV of the rats subjected to Ang II infusion for 4 weeks (Figure 2D).
Figure 2.
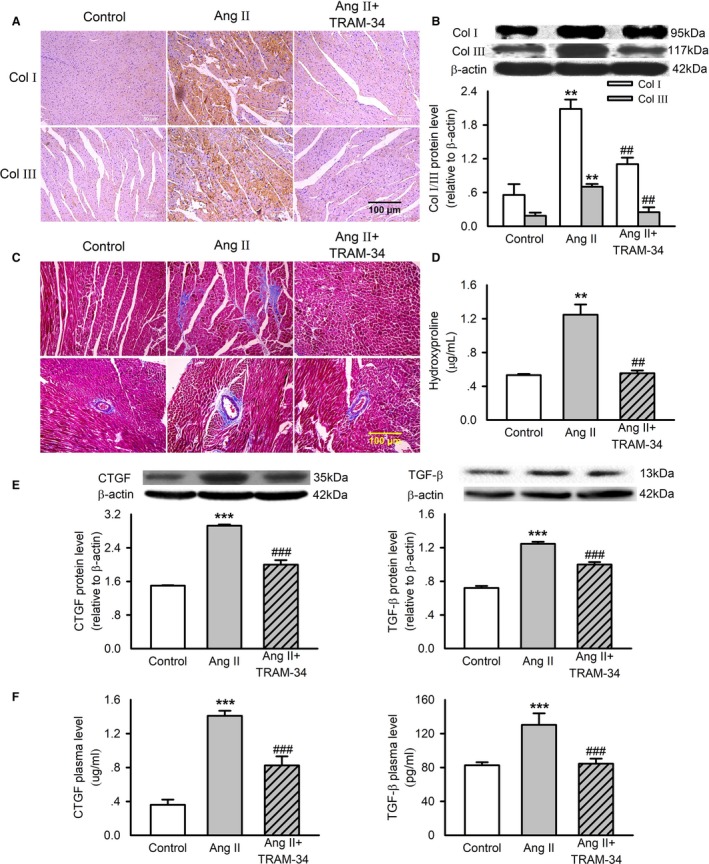
Effects of TRAM‐34 on left ventricular fibrosis in Ang II‐infusion rats. A, Immunohistochemistry staining of collagen I and collagen III in rat left ventricles (n=6, ×200). B, Representative Western blotting images and mean values of collagen I and collagen III in the myocardium. C, Masson trichrome staining of left ventricles (n=6, ×200). D, Mean values of hydroxyproline content in rat myocardium measured by ELISA. E, Representative Western blotting images and mean values of CTGF and TGF‐β in the myocardium. F, Mean values of CTGF and TGF‐β concentration in rat plasma measured by ELISA. Data are presented as mean±SEM, n=6, **P<0.01, ***P<0.001 vs control, ## P<0.01, ### P<0.001 vs Ang II. Ang II indicates angiotensin II; Col I, collagen I; Col III, collagen III; CTGF, connective tissue growth factor; TGF‐β, transforming growth factor‐β.
CTGF and TGF‐β are implicated as key molecules for fibrogenesis. Ang II infusion in rats significantly increased cardiac and plasma levels of CTGF and TGF‐β, and this effect was partly reversed by TRAM‐34 treatment (Figure 2E and 2F). These findings indicate that KCa3.1 channels contribute to cardiac fibrosis induced by Ang II infusion.
KCa3.1 Channels Contribute to Ang II–Mediated Cardiac Inflammation
We then assessed whether KCa3.1 channels were involved in the process of hypertensive cardiac inflammation evoked by Ang II. Using immunohistochemistry and Western blotting, we observed increased expressions of MCP‐1 and TNF‐α as well as inflammatory cell markers, including CD45 (leukocyte), CD3 (T lymphocyte), and CD68 (macrophage) in the myocardium of Ang II–infusion rats (Figure 3A and 3B). Treatment with TRAM‐34 reversed the increased expression of these proinflammatory factors and cell markers. Figure 3C displays the levels of MCP‐1 and TNF‐α measured by ELISA in the plasma of the rats. Ang II infusion increased the plasma concentration of MCP‐1 but not the TNF‐α, and this action was abolished by treatment with TRAM‐34. These results confirmed the role of KCa3.1 channels in Ang II–evoked cardiac inflammation.
Figure 3.
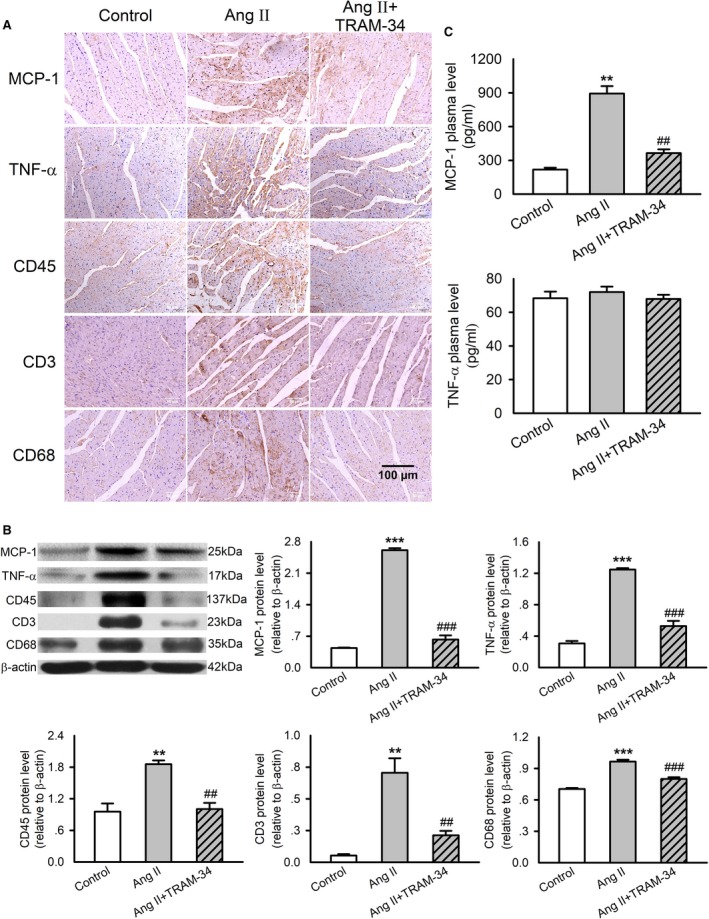
Effects of TRAM‐34 on expression and secretion of proinflammatory cytokines and leukocyte markers in myocardium and plasma in Ang II–infusion rats. A, Immunohistochemistry staining of MCP‐1, TNF‐α, CD45, CD3, and CD68 in rat left ventricles (n=6, ×200). B, Representative Western blotting images and mean values of MCP‐1, TNF‐α, CD45, CD3, and CD68 in the myocardium. C, The concentrations of MCP‐1 and TNF‐α in rat plasma measured by ELISA. Data are presented as mean±SEM, n=6, **P<0.01, ***P<0.001 vs control, ## P<0.01, ### P<0.001 vs Ang II. Ang II indicates angiotensin II; MCP‐1, monocyte chemotactic protein 1; TNF‐α, tumor necrosis factor‐α.
Effect of KCa3.1 Channels on the Recruitment and Differentiation of Fibrocytes in Myocardium
Figure 4 illustrates the location of fibrocytes and their differentiation in rat myocardium by immunofluorescence double staining. Fibrocytes expressed cell markers of CD45 (leukocyte marker), CD34 (hematopoietic stem cell marker) (Figure 4A), CD11b (monocyte marker), and DDR‐2 (fibroblast marker) (Figure 4B), indicating that they were differentiated from bone marrow–derived monocytes. The myocardial density of these fibrocytes identified by the double staining was significantly higher in the Ang II–treated hearts and was reduced markedly, by ≈65%, by administration of TRAM‐34. The immunofluorescent double staining of myofibroblast markers (α‐SMA and DDR‐2) in the rat myocardium was also enhanced by Ang II infusion and similarly reversed by TRAM‐34 treatment (Figure 4C). The results implicate a role of KCa3.1 channels in promoting fibrocyte recruitment into the myocardium and subsequent differentiation into myofibroblasts, thereby contributing to Ang II–induced fibrosis.
Figure 4.
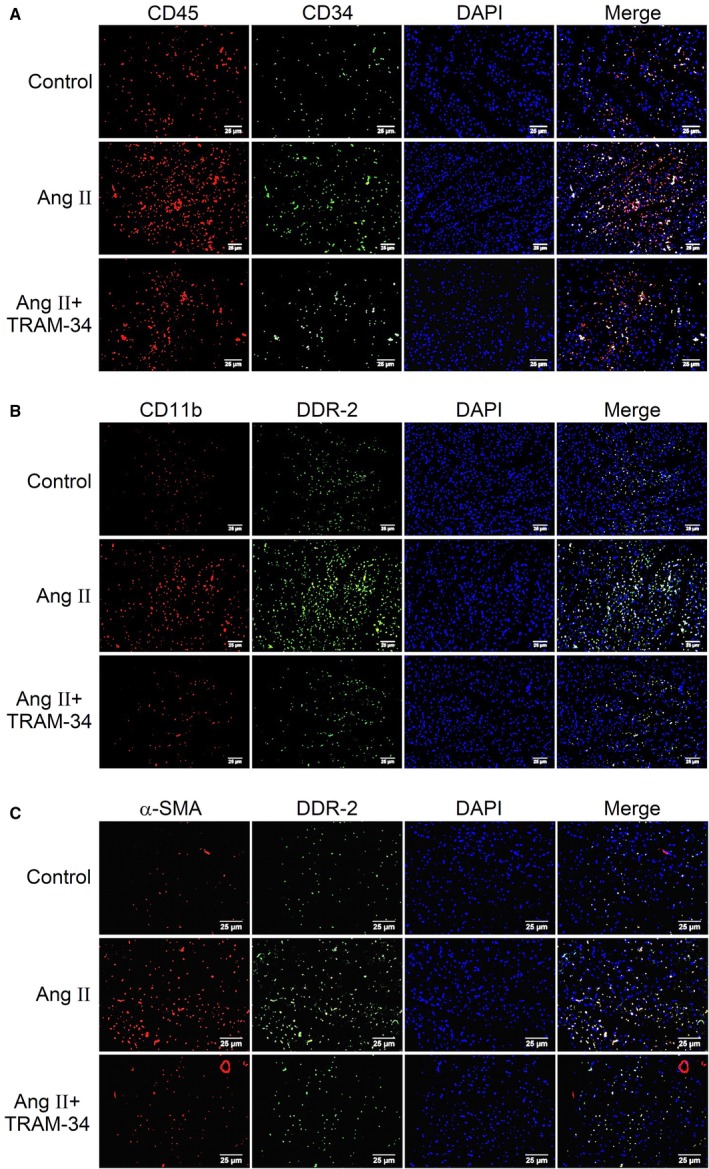
Effect of TRAM‐34 on Ang II‐induced differentiation of bone marrow–derived monocyte into myofibrocyte in rat myocardium. A, Immunofluorescent double staining of fibrocyte markers CD45 (red) and CD34 (green) in rat left ventricular myocardium (n=6, ×400). B, Immunofluorescent double staining of fibrocyte markers CD11b (red) and DDR‐2 (green) in rat left ventricular myocardium (n=6, ×400). C, Immunofluorescent double staining of myofibroblast markers α‐SMA (red) and DDR‐2 (green) in rat left ventricular myocardium (n=6, ×400). α‐SMA indicates α‐smooth muscle actin; Ang II, angiotensin II; DDR‐2, discoidin domain receptor 2.
Effect of KCa3.1 Channels on Ang II–Induced IL‐4 and IL‐13 Expression and Secretion
Using Western blotting and ELISA, we examined the role of KCa3.1 channels in IL‐4 and IL‐13 expression and secretion in Ang II–infusion rat plasma and isolated mouse CD4+ T cells. Ang II infusion significantly increased the plasma levels of IL‐4 and IL‐13, which were suppressed by treatment with TRAM‐34 (Figure 5A). These changes of T helper (Th)‐2 cytokines indicated KCa3.1 channel dependence of Ang II–induced activation of Th‐2 cells. We then isolated CD4+ T cells from healthy male mice to further investigate the role of KCa3.1 in Ang II–induced activation of Th‐2 cells. In cultured murine CD4+ T cells, both expression and secretion of IL‐4 and IL‐13 in CD4+ T cells were significantly increased by Ang II stimulation at 0.1 μmol/L, and the effects of Ang II were prevented by TRAM‐34 at 1 μmol/L (Figure 5B and 5C). Similarly, KCa3.1 siRNA (150 nmol/L) also suppressed the enhanced expression and secretion of IL‐4 and IL‐13 in CD4+ T cells evoked by Ang II (Figure 5D and 5E). The concentration of Ang II and its stimulation time as well as the concentration of KCa3.1 siRNA were chosen according to the results in Data S1 and in Figures S1 and S2. These findings further solidify the role of KCa3.1 channels in mediating activation of Th‐2 cells.
Figure 5.
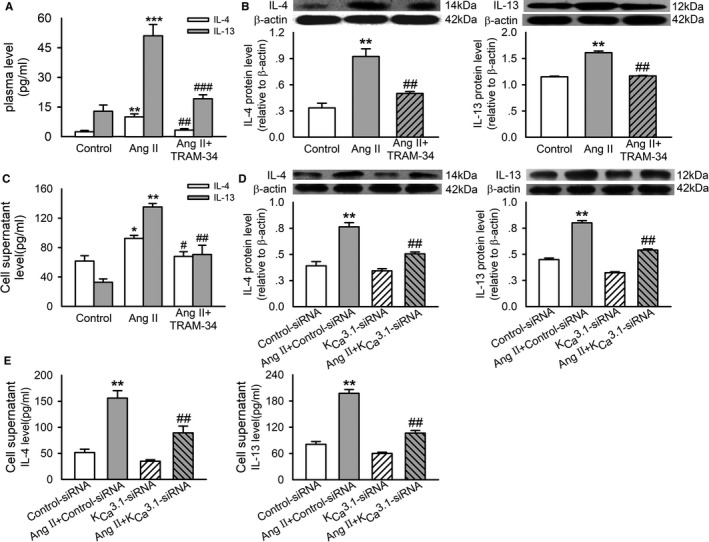
Role of KC a3.1 channels in Ang II–induced IL‐4 and IL‐13 expression and secretion. A, The concentration of IL‐4 and IL‐13 in rat plasma measured by ELISA. B, Representative Western blotting images and quantitative analysis of IL‐4 and IL‐13 in CD4+ T cells incubated without (control) or with 0.1 μmol/L Ang II or Ang II plus 1 μmol/L TRAM‐34 for 72 hours. C, The concentration of IL‐4 and IL‐13 in the supernatant of cultured CD4+ T cells treated with the interventions as described in B. D, Representative Western blotting images and quantitative analysis of IL‐4 and IL‐13 in CD4+ T cells transfected with 150 nmol/L control siRNA or KC a3.1 siRNA and treated without or with 0.1 μmol/L Ang II for 72 hours. E, The concentration of IL‐4 and IL‐13 in the supernatant of cultured CD4+ T cells treated with the interventions as described in D. Data are presented as mean±SEM, n=6, *P<0.05, **P<0.01, ***P<0.001 vs control, # P<0.05, ## P<0.01, ### P<0.001 vs Ang II. Ang II indicates angiotensin II; IL‐4 indicates interleukin‐4; IL‐13, interleukin‐13; siRNA, small interfering RNA.
KCa3.1 Channels and IL‐4– or IL‐13–Induced Differentiation of Monocytes Into Fibrocytes
To investigate whether KCa3.1 channels were involved in the differentiation of monocytes into fibrocytes mediated by IL‐4 and IL‐13, PBMCs from peripheral blood of healthy adult rats were isolated and cultured in serum‐free medium. Fibrocytes were defined by morphology (adherent spindle‐shaped cells with an oval nucleus) and by coexpression of CD34, collagen I, and DDR‐2. As shown in Figure 6A and 6B, IL‐4 and IL‐13 facilitated the differentiation of monocytes into fibrocytes, and such action was reduced to 55% and 50% by blockade of KCa3.1 channels with 1 μmol/L TRAM‐34 and downregulation of KCa3.1 channels with 100 nmol/L KCa3.1 siRNA, respectively (Figure 6C). The images of DDR‐2 immunohistochemistry staining illustrated the effect of KCa3.1 siRNA on differentiation of monocytes into fibrocytes in myocardium (Figure S3).
Figure 6.
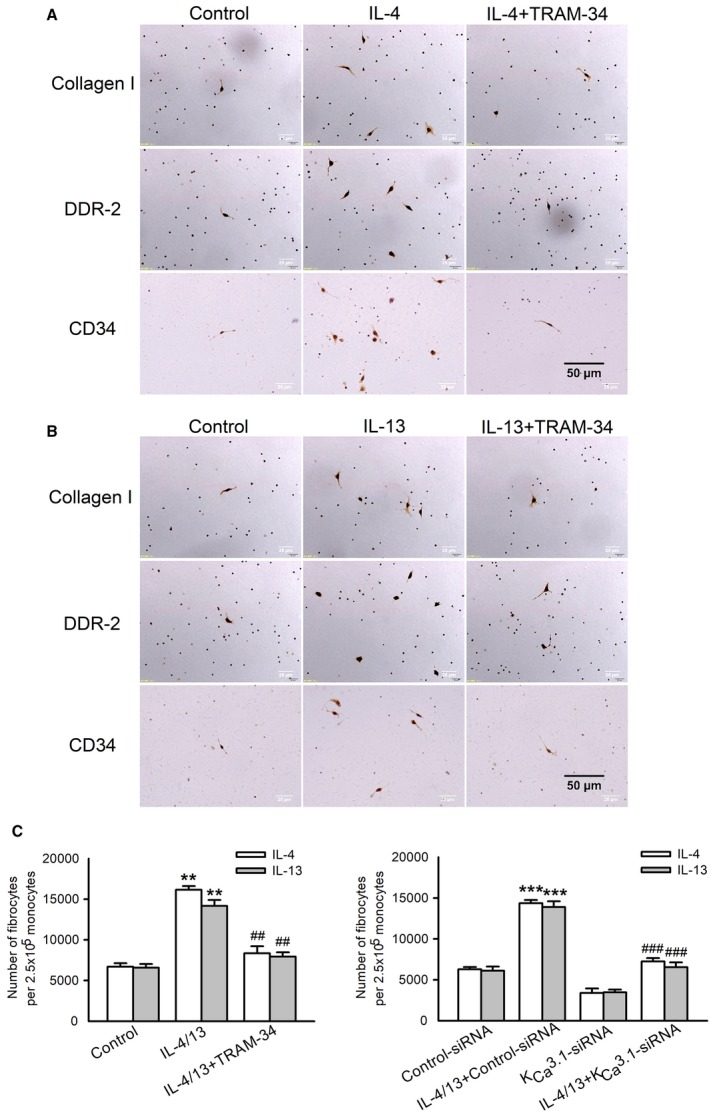
Role of KC a3.1 channels in IL‐4/IL‐13–induced differentiation of PBMCs into fibrocytes. A and B, Immunocytochemistry staining of collagen I, DDR‐2, and CD34 in PBMCs differentiated into fibrocytes incubated without (control) or with 3 ng/mL IL‐4/IL‐13, and IL‐4 or IL‐13 plus 1 μmol/L TRAM‐34 for 5 days (n=6, ×400). C, Mean values for number of differentiated fibrocytes per 2.5×105 monocytes in each group. Data are presented as mean±SEM, n=6, **P<0.01, ***P<0.001 vs control, ## P<0.01, ### P<0.001 vs IL‐4/13. DDR‐2 indicates discoidin domain receptor 2; IL‐4 indicates interleukin‐4; IL‐13, interleukin‐13; PBMC, peripheral blood monocyte; siRNA, small interfering RNA.
KCa3.1 Current in Primary Cardiac Fibroblasts Isolated From Ang II–Infusion Rats
We then investigated whether chronic Ang II stimulation altered KCa3.1 channel activity in rat cardiac fibroblasts. Using whole‐cell patch clamp, we recorded the membrane currents in isolated rat cardiac fibroblasts with 300‐millisecond voltage steps to between −100 and +100 mV from a holding potential of −70 mV (inset) (Figure 7A). The membrane current with weak inward rectification at more positive potentials was found to be inhibited by the KCa3.1 channel blocker TRAM‐34 (1 μmol/L), suggesting that the KCa3.1 current was 1 of the predominant potassium‐channel currents in primary cardiac fibroblasts. Importantly, the membrane current and the scale of its inhibition by TRAM‐34 were significantly increased in primary cardiac fibroblasts prepared from rats receiving Ang II infusion. Treatment with TRAM‐34 in vivo similarly reversed this facilitated effect of Ang II on the KCa3.1 current in primary cardiac fibroblasts. Figure 7B illustrates the mean values of the current‐voltage relationships of the TRAM‐34–sensitive current obtained by digital subtraction of the current before TRAM‐34 from the current after addition of TRAM‐34 to primary cardiac fibroblasts. The current‐voltage curves of TRAM‐34–sensitive current showed a weak inward rectification, a typical characteristic of the KCa3.1 channel current. The current density at −30 to +100 mV was enhanced in cells isolated from Ang II–infusion rats and significantly decreased by TRAM‐34 treatment.
Figure 7.
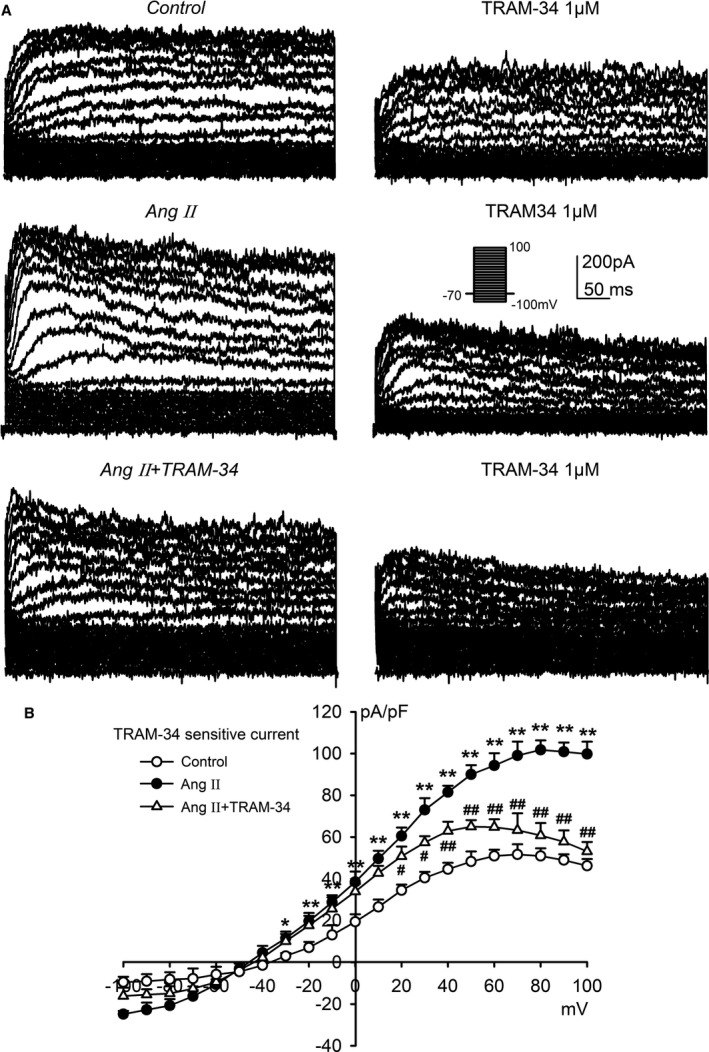
KC a3.1 currents in primary cardiac fibroblasts isolated from Ang II–infusion rats. A, Membrane currents recorded with the protocol as shown in the inset in isolated primary cardiac fibroblasts from rats in different groups before and after application of TRAM‐34 (1 μmol/L). B, Current‐voltage relationships of the mean values of TRAM‐34–sensitive current obtained by digital subtraction of the current before TRAM‐34 application by the current after TRAM‐34 application. Data are presented as mean±SEM, n=6, *P<0.05, **P<0.01 vs control, # P<0.05, ## P<0.01 vs Ang II. Ang II indicates angiotensin II.
Interaction Between KCa3.1 and TRPV4/TRPC6 Channels in Rat Myocardium and Isolated Primary Cardiac Fibroblasts
TRP channels have emerged as the primary ion channels mediating Ca2+ signals in cardiac fibroblasts.2 We then explored the possibility that an interaction between KCa3.1 and TRPV4 or TRPC6 channels exists in fibroblasts that regulates progression of cardiac fibrosis. This was examined by immunofluorescence double staining for expression and colocation of KCa3.1 with TRPV4 or TRPC6 channels in rat LV myocardium (Figure 8A and 8B). Ang II infusion for 4 weeks significantly increased the expression of all 3 channels of KCa3.1, TRPV4, and TRPC6, and this was reversed by treatment with TRAM‐34 in vivo. To determine whether the interaction among these channels appeared in fibroblasts, we stained the primary cardiac fibroblasts isolated from rat ventricle in each group. As shown in Figure 8C and 8D, the expressions of these channels in cardiac fibroblasts were comparable among different groups. The result from coimmunoprecipitation in healthy rat cardiac fibroblasts further demonstrated the physical interaction between KCa3.1 and TRPV4 or TRPC6 channels (Figure S4).
Figure 8.
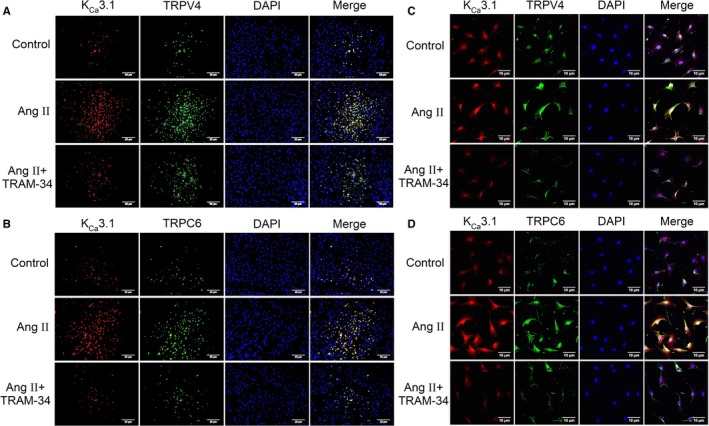
Colocalization of KC a3.1 and TRP channels in left ventricle and isolated primary cardiac fibroblasts from Ang II–infusion rats. A, Immunofluorescent double staining of KC a3.1 channels (red) and TRPV4 channels (green) in the myocardium (n=6, ×400). B, Immunofluorescent double staining of KC a3.1 channels (red) and TRPC6 channels (green) in the myocardium (n=6, ×400). C, Immunofluorescent double staining of KC a3.1 channels (red) and TRPV4 channels (green) in primary cardiac fibroblasts (n=6, ×400). D, Immunofluorescent double staining of KC a3.1 channels (red) and TRPC6 channels (green) in primary cardiac fibroblasts (n=6, ×400). Ang II indicates angiotensin II; TRPV4, transient receptor potential V4; TRPC6, transient receptor potential C6.
Discussion
In the present study we demonstrated for the first time that blockade of KCa3.1 channels in rats subjected to chronic Ang II infusion inhibited cardiac fibrosis by alleviating myocardial inflammation and differentiation of bone marrow–derived monocytes to myofibroblasts in vivo. Ex vivo experiments further revealed that blockade or downregulation of KCa3.1 channels in CD4+ T cells suppressed Ang II–stimulated expression of IL‐4 and IL‐13 and halted IL‐4– and IL‐13–mediated differentiation of monocytes to fibrocytes.
Ang II has been shown to induce cardiac fibrosis and remodeling through induction of hypertension and direct activation of its receptors localized at the myocardium.22, 23, 24 In the current study on a rat model of cardiac fibrosis through chronic Ang II infusion, we observed typical changes of myocardial hypertrophy and fibrosis, manifested as increased heart weight and collagen synthesis and deposition as well as activation of collagen synthesis signaling. We confirmed the increment of SBP and decline in LV systolic function following Ang II treatment. Importantly, our data showed that all these changes were largely prevented by concomitant treatment with the KCa3.1 selective inhibitor TRAM‐34, suggesting that KCa3.1 channels play an essential role in the development of cardiac fibrosis in this model.
Inflammatory signaling is closely involved in the progression of cardiac fibrosis.25 Ang II has been known to possess proinflammatory action in the setting of cardiovascular diseases by inducing vascular damage, upregulating adhesion molecules or cytokines, and increasing inflammatory cell infiltration as well as regulating repair processes.26, 27 We previously showed, in the rat model of cardiac fibrosis as a consequence of abdominal aorta constriction, that enhanced expression of inflammatory cytokines and infiltration of inflammatory cells were efficiently attenuated by the treatment with TRAM‐34.14 In the current study on Ang II–infusion rats, we further confirmed the increase of TNF‐α and MCP‐1 expression in the myocardium and observed the increment of MCP‐1 level in plasma. TNF‐α, a pleiotropic cytokine, can facilitate the necrosis and apoptosis of cardiomyocytes and increase synthesis of ECM proteins, leading to fibrosis.28 There are studies showing that TNF‐α also enhances the generation of monocytic fibroblast precursors in the heart.29 MCP‐1 plays a key role in regulating recruitment of monocytes and T lymphocytes.3, 30 Haudek et al reported that cardiac fibrosis induced by Ang II infusion is dependent on the induction of MCP‐1, which modulates the uptake and differentiation of a subpopulation of bone marrow–derived monocyte fibroblast precursor cells.31, 32 Our recent investigation showed that an Ang II–induced increase of MCP‐1 secretion in endothelium promoted the transendothelium migration of monocytes.14 Therefore, the increment of MCP‐1 plasma level in Ang II–treated rats was, at least in part, released from endothelial cells and likely contributed to the leukocyte recruitment into the myocardium. Hypertension induced by Ang II infusion facilitates the infiltration of leukocytes into the heart. The interactions among macrophages, T cells, and monocytic fibroblast precursor cells regulate the imbalance of proinflammatory and anti‐inflammatory factors.33, 34, 35, 36 In this study we found that the expressions of surface markers of leukocytes (CD45), T cells (CD3), and macrophages (CD68) were increased in the myocardium of Ang II–treated rats, implying that Ang II infusion promoted T lymphocyte and monocyte infiltration into the myocardium. The myocardial inflammation in Ang II–treated rats was alleviated by TRAM‐34, which suggests that KCa3.1 channels are involved in myocardial inflammation induced by Ang II infusion.
Excessive proliferation of cardiac fibroblasts accompanies the development of cardiac fibrosis. Resident cardiac fibroblasts activated by inflammatory responses are traditionally thought to play a major role in organ fibrosis.3 Recent studies underscore an important role of circulating fibrocytes differentiated from bone marrow–derived monocytes, which typically express CD34 and CD45 in cardiac fibrosis induced by Ang II or other pathological stimuli.32, 37 Ang II infusion results in myocardial accumulation of bone marrow–derived CD34+/CD45+ fibroblast precursor cells that express collagen I, CD11b, as well as the cardiac fibroblast marker DDR‐2, making them differ from residential fibroblasts (CD34−/CD45−).31, 38 We previously demonstrated that blockade of KCa3.1 channels with TRAM‐34 attenuated pressure overload–induced cardiac fibrosis in vivo and prevented Ang II–stimulated fibroblast proliferation and monocyte transendothelium migration in vitro.13, 14 However, whether KCa3.1 channels affect the Ang II infusion–induced increase of fibrocytes remains unexplored. In the present study we furthered previous findings by documenting that KCa3.1 channels play a key role in Ang II–induced increase of fibrocytes in myocardium.
The differentiation of bone marrow–derived monocytes to fibrocytes is regulated by cytokines. Pilling et al reported that Th‐2 cytokines such as IL‐4 and IL‐13 facilitate the differentiation of CD14+ PBMCs to fibrocytes.39 KCa3.1 channels are expressed extensively on T cells, and previous studies have demonstrated the critical roles for KCa3.1 channels in T cell receptor‐stimulated Ca2+ flux and proliferation of preactivated CD4+ T cells.8, 40 Ang II infusion has been shown to activate NADPH oxidase subunits, reactive oxygen species, and activity of T lymphocytes.41, 42 In the current study, chronic Ang II infusion increased plasma levels of IL‐4 and IL‐13, and treatment with TRAM‐34 abolished the increment of IL‐4 and IL‐13. Moreover, our in vitro experiments showed that both TRAM‐34 and KCa3.1 siRNA similarly alleviated the increase of IL‐4 and IL‐13 expression and secretion induced by Ang II in mouse CD4+ T cells. These findings strongly indicate the significance of KCa3.1 channels in regulating the expression and secretion of Th‐2 cytokines IL‐4 and IL‐13 in CD4+ T cells on Ang II stimulation.
Differentiated fibrocytes are characterized by a spindle shape and expression of CD34, collagen I, and DDR‐2 but not of CD14 in cell markers.39 In our current research we provided evidence for KCa3.1 channels in promoting fibrocyte differentiation induced by IL‐4 and IL‐13 with cellular morphological alteration to elongated, spindle‐shape. TRAM‐34 and KCa3.1 siRNA can significantly decrease the number of spindle‐shaped fibrocytes.
A critical event leading to cardiac fibrosis is the differentiation of cardiac fibroblasts into myofibroblasts. Myofibroblasts express contractile proteins such as α‐SMA and have enhanced expression and secretion of ECM proteins.43, 44 Recent studies have revealed that profibrotic mediators such as TGF‐β and Ang II induce expression of TRPC6 in ventricular fibroblasts via p38 MAPK/serum‐responsive factor. This in turn activates calcineurin/nuclear factor of activated T‐cell signaling cascades that are involved in fibroblast proliferation and α‐SMA expression.45, 46 TRPV4 is also implicated in myofibroblast activation, which is governed by orchestrated signals from secreted growth factors (such as TGF‐β) and mechanosensitive stimuli through a rho‐rho–associated protein kinase signaling pathway.47 The Ca2+ influx through TRP channels is greatly influenced by cell membrane potential, which is controlled by K+ channels. KCa3.1 channels are found to be expressed in nonexcitable cells and to be associated only with non–voltage‐gated Ca2+ channels, such as TRP channels. The activation of KCa3.1 channels in fibroblasts and myofibroblasts induces a hyperpolarization, which increases Ca2+ entry through TRP channels by enhancing the electrochemical gradient for Ca2+ and leads to elevated intracellular Ca2+ concentration. In this case KCa3.1 channels play a crucial role in this low–energy‐cost and positive feedback loop. Interestingly, in Ang II–treated rat hearts we found an increased density of myofibroblasts that express KCa3.1 and TRPV4 or TRPC6 channels at higher levels. These cells also exhibit interaction between KCa3.1 and TRPV4 or TRPC6 channels, indicating that these channel proteins form channel complexes after chronic Ang II stimulation. Importantly, treatment with TRAM‐34 reversed this phenomenon in vivo. Moreover, in fibroblasts isolated from hearts of rats subjected to Ang II infusion, the current density of KCa3.1 channels was increased, which was again attenuated by TRAM‐34 treatment. These results strongly indicate that KCa3.1 channels couple with TRPV4 or TRPC6 channels and promote differentiation of myofibroblasts.
In conclusion, we have demonstrated that enhanced KCa3.1 channel activity was evident in fibroblasts from Ang II–treated hearts and that KCa3.1 channels in different types of cells play a critical role in the development of cardiac fibrosis. This action is mediated through its proinflammatory effect that facilitates the differentiation of bone marrow–derived monocytes into fibrocytes and then through coupling with TRPV4 or TRPC6 channels to promote the differentiation of fibroblasts into myofibroblasts (Figure 9). Treatment with the KCa3.1 channel inhibitor TRAM‐34 alleviates cardiac fibrosis by Ang II infusion. Our findings imply that KCa3.1 channels may provide a potential therapeutic strategy to prevent or reverse cardiac fibrosis.
Figure 9.
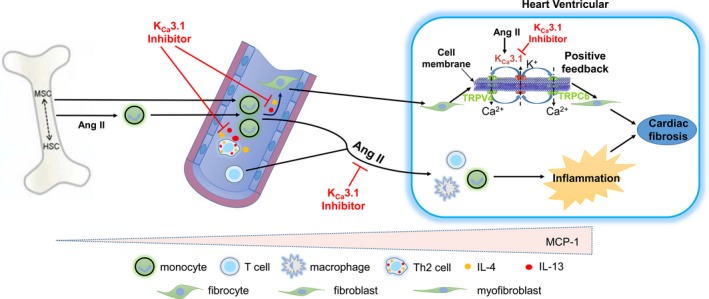
Schematic presentation of proposed mechanisms of KC a3.1‐mediated cardiac fibrosis in Ang II–treated rats. KC a3.1 channels promote cardiac fibrosis in the following 2 ways: (1) KC a3.1 channels facilitate the secretion of Th2 cytokines IL‐4 and IL‐13 and IL‐4/13–mediated bone marrow–derived monocyte differentiation into fibrocytes, which get into ventricular myocardium and then mature into fibroblasts. Furthermore, KC a3.1 channels promote the differentiation of fibroblasts into myofibroblasts through its interaction with TRPV4 and TRPC6 on the cell membrane. (2) KC a3.1 channels facilitate monocyte infiltration into myocardium and their subsequent differentiation into macrophages, which further induce myocardium inflammation and promote cardiac fibrosis ultimately. Ang II indicates angiotensin II; HSC, hematopoietic stem cell; IL‐4, interleukin‐4; IL‐13, interleukin‐13; MSC, mesenchymal stem cell; Th2, T helper cell 2; TRPC6, transient receptor potential C6; TRPV4, transient receptor potential V4.
Sources of Funding
This work was supported by the National Natural Science Foundation of China (grants number 81570214, 81370191, and 81600656).
Disclosures
None.
Supporting information
Data S1. Supplemental Methods
Figure S1. Concentration‐dependent and time‐dependent effects of Ang II on cell viability and IL‐4/13 expression in mice CD4+ T cells.
Figure S2. KCa3.1 expression in CD4+ T cells treated with KCa3.1‐siRNA.
Figure S3. The role of KCa3.1 in IL‐4/IL‐13–induced differentiation of monocytes into fibrocytes.
Figure S4. KCa3.1 channels interact with TRPV4 and TRPC6 channels in rat cardiac fibroblasts.
(J Am Heart Assoc. 2019;8:e010418 DOI: 10.1161/JAHA.118.010418)
References
- 1. Nguyen TP, Qu Z, Weiss JN. Cardiac fibrosis and arrhythmogenesis: the road to repair is paved with perils. J Mol Cell Cardiol. 2014;70:83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yue Z, Zhang Y, Xie J, Jiang J, Yue L. Transient receptor potential (TRP) channels and cardiac fibrosis. Curr Top Med Chem. 2013;13:270–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jia L, Li Y, Xiao C, Du J. Angiotensin II induces inflammation leading to cardiac remodeling. Front Biosci (Landmark Ed). 2012;17:221–231. [DOI] [PubMed] [Google Scholar]
- 4. Zhao LM, Zhang W, Wang LP, Li GR, Deng XL. Advanced glycation end products promote proliferation of cardiac fibroblasts by upregulation of KCa3.1 channels. Pflugers Arch. 2012;464:613–621. [DOI] [PubMed] [Google Scholar]
- 5. Deng XL, Lau CP, Lai K, Cheung KF, Lau GK, Li GR. Cell cycle‐dependent expression of potassium channels and cell proliferation in rat mesenchymal stem cells from bone marrow. Cell Prolif. 2007;40:656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Su XL, Wang Y, Zhang W, Zhao LM, Li GR, Deng XL. Insulin‐mediated upregulation of K(Ca)3.1 channels promotes cell migration and proliferation in rat vascular smooth muscle. J Mol Cell Cardiol. 2011;51:51–57. [DOI] [PubMed] [Google Scholar]
- 7. Tharp DL, Wamhoff BR, Turk JR, Bowles DK. Upregulation of intermediate‐conductance Ca2+‐activated K+ channel (IKCA1) mediates phenotypic modulation of coronary smooth muscle. Am J Physiol Heart Circ Physiol. 2006;291:H2493–H2503. [DOI] [PubMed] [Google Scholar]
- 8. Di L, Srivastava S, Zhdanova O, Sun Y, Li Z, Skolnik EY. Nucleoside diphosphate kinase B knock‐out mice have impaired activation of the K+ channel KCa3.1, resulting in defective T cell activation. J Biol Chem. 2010;285:38765–38771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grgic I, Kiss E, Kaistha BP, Busch C, Kloss M, Sautter J, Muller A, Kaistha A, Schmidt C, Raman G, Wulff H, Strutz F, Grone HJ, Kohler R, Hoyer J. Renal fibrosis is attenuated by targeted disruption of KCa3.1 potassium channels. Proc Natl Acad Sci USA. 2009;106:14518–14523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cruse G, Singh SR, Duffy SM, Doe C, Saunders R, Brightling CE, Bradding P. Functional KCa3.1 K+ channels are required for human fibrocyte migration. J Allergy Clin Immunol. 2011;128:1303–1309.e1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yong KW, Li YH, Huang GY, Lu TJ, Safwani WKZW, Pingguan‐Murphy B, Xu F. Mechanoregulation of cardiac myofibroblast differentiation: implications for cardiac fibrosis and therapy. Am J Physiol Heart Circ Physiol. 2015;309:H532–H542. [DOI] [PubMed] [Google Scholar]
- 12. Stempien‐Otero A, Kim DH, Davis J. Molecular networks underlying myofibroblast fate and fibrosis. J Mol Cell Cardiol. 2016;97:153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang LP, Wang Y, Zhao LM, Li GR, Deng XL. Angiotensin II upregulates K(Ca)3.1 channels and stimulates cell proliferation in rat cardiac fibroblasts. Biochem Pharmacol. 2013;85:1486–1494. [DOI] [PubMed] [Google Scholar]
- 14. Zhao LM, Wang LP, Wang HF, Ma XZ, Zhou DX, Deng XL. The role of KCa3.1 channels in cardiac fibrosis induced by pressure overload in rats. Pflugers Arch. 2015;467:2275–2285. [DOI] [PubMed] [Google Scholar]
- 15. Gu HP, Lin S, Xu M, Yu HY, Du XJ, Zhang YY, Yuan G, Gao W. Up‐regulating relaxin expression by G‐quadruplex interactive ligand to achieve antifibrotic action. Endocrinology. 2012;153:3692–3700. [DOI] [PubMed] [Google Scholar]
- 16. Du XJ, Gao XM, Wang B, Jennings GL, Woodcock EA, Dart AM. Age‐dependent cardiomyopathy and heart failure phenotype in mice overexpressing beta(2)‐adrenergic receptors in the heart. Cardiovasc Res. 2000;48:448–454. [DOI] [PubMed] [Google Scholar]
- 17. Zhao LM, Wang Y, Yang Y, Guo R, Wang NP, Deng XL. Metformin restores intermediate‐conductance calcium‐activated K(+) channel‐ and small‐conductance calcium‐activated K(+) channel‐mediated vasodilatation impaired by advanced glycation end products in rat mesenteric artery [corrected]. Mol Pharmacol. 2014;86:580–591. [DOI] [PubMed] [Google Scholar]
- 18. Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ Res. 2014;114:266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tao R, Lau CP, Tse HF, Li GR. Regulation of cell proliferation by intermediate‐conductance Ca2+‐activated potassium and volume‐sensitive chloride channels in mouse mesenchymal stem cells. Am J Physiol Cell Physiol. 2008;295:C1409–C1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pilling D, Vakil V, Gomer RH. Improved serum‐free culture conditions for the differentiation of human and murine fibrocytes. J Immunol Methods. 2009;351:62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Y, Sun HY, Liu YG, Song Z, She G, Xiao GS, Wang Y, Li GR, Deng XL. Tyrphostin AG556 increases the activity of large conductance Ca(2+) ‐activated K(+) channels by inhibiting epidermal growth factor receptor tyrosine kinase. J Cell Mol Med. 2017;21:1826–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gonzalez A, Lopez B, Querejeta R, Diez J. Regulation of myocardial fibrillar collagen by angiotensin II. A role in hypertensive heart disease? J Mol Cell Cardiol. 2002;34:1585–1593. [DOI] [PubMed] [Google Scholar]
- 23. Mann DL. Angiotensin II as an inflammatory mediator: evolving concepts in the role of the renin angiotensin system in the failing heart. Cardiovasc Drugs Ther. 2002;16:7–9. [DOI] [PubMed] [Google Scholar]
- 24. Sekiguchi K, Li X, Coker M, Flesch M, Barger PM, Sivasubramanian N, Mann DL. Cross‐regulation between the renin‐angiotensin system and inflammatory mediators in cardiac hypertrophy and failure. Cardiovasc Res. 2004;63:433–442. [DOI] [PubMed] [Google Scholar]
- 25. Suthahar N, Meijers WC, Sillje HHW, de Boer RA. From inflammation to fibrosis‐molecular and cellular mechanisms of myocardial tissue remodelling and perspectives on differential treatment opportunities. Curr Heart Fail Rep. 2017;14:235–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van Thiel BS, van der Pluijm I, te Riet L, Essers J, Danser AH. The renin‐angiotensin system and its involvement in vascular disease. Eur J Pharmacol. 2015;763:3–14. [DOI] [PubMed] [Google Scholar]
- 27. Simoes ESAC, Teixeira MM. ACE inhibition, ACE2 and angiotensin‐(1‐7) axis in kidney and cardiac inflammation and fibrosis. Pharmacol Res. 2016;107:154–162. [DOI] [PubMed] [Google Scholar]
- 28. Duerrschmid C, Crawford JR, Reineke E, Taffet GE, Trial J, Entman ML, Haudek SB. TNF receptor 1 signaling is critically involved in mediating angiotensin‐II‐induced cardiac fibrosis. J Mol Cell Cardiol. 2013;57:59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frangogiannis NG, Entman ML. Chemokines in myocardial ischemia. Trends Cardiovasc Med. 2005;15:163–169. [DOI] [PubMed] [Google Scholar]
- 30. Kuras Z, Yun YH, Chimote AA, Neumeier L, Conforti L. KCa3.1 and TRPM7 channels at the uropod regulate migration of activated human T cells. PLoS One. 2012;7:e43859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Haudek SB, Xia Y, Huebener P, Lee JM, Carlson S, Crawford JR, Pilling D, Gomer RH, Trial J, Frangogiannis NG, Entman ML. Bone marrow‐derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc Natl Acad Sci USA. 2006;103:18284–18289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Haudek SB, Cheng J, Du J, Wang Y, Hermosillo‐Rodriguez J, Trial J, Taffet GE, Entman ML. Monocytic fibroblast precursors mediate fibrosis in angiotensin‐II‐induced cardiac hypertrophy. J Mol Cell Cardiol. 2010;49:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Han YL, Li YL, Jia LX, Cheng JZ, Qi YF, Zhang HJ, Du J. Reciprocal interaction between macrophages and T cells stimulates IFN‐γ and MCP‐1 production in Ang II‐induced cardiac inflammation and fibrosis. PLoS One. 2012;7:e35506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ma F, Li Y, Jia L, Han Y, Cheng J, Li H, Qi Y, Du J. Macrophage‐stimulated cardiac fibroblast production of IL‐6 is essential for TGF β/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS One. 2012;7:e35144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Qi G, Jia L, Li Y, Bian Y, Cheng J, Li H, Xiao C, Du J. Angiotensin II infusion‐induced inflammation, monocytic fibroblast precursor infiltration, and cardiac fibrosis are pressure dependent. Cardiovasc Toxicol. 2011;11:157–167. [DOI] [PubMed] [Google Scholar]
- 36. Ren J, Yang M, Qi G, Zheng J, Jia L, Cheng J, Tian C, Li H, Lin X, Du J. Proinflammatory protein CARD9 is essential for infiltration of monocytic fibroblast precursors and cardiac fibrosis caused by angiotensin II infusion. Am J Hypertens. 2011;24:701–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sopel M, Falkenham A, Oxner A, Ma I, Lee TD, Legare JF. Fibroblast progenitor cells are recruited into the myocardium prior to the development of myocardial fibrosis. Int J Exp Pathol. 2012;93:115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xu J, Lin SC, Chen J, Miao Y, Taffet GE, Entman ML, Wang Y. CCR2 mediates the uptake of bone marrow‐derived fibroblast precursors in angiotensin II‐induced cardiac fibrosis. Am J Physiol Heart Circ Physiol. 2011;301:H538–H547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shao DD, Suresh R, Vakil V, Gomer RH, Pilling D. Pivotal advance: Th‐1 cytokines inhibit, and Th‐2 cytokines promote fibrocyte differentiation. J Leukoc Biol. 2008;83:1323–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Srivastava S, Panda S, Li Z, Fuhs SR, Hunter T, Thiele DJ, Hubbard SR, Skolnik EY. Histidine phosphorylation relieves copper inhibition in the mammalian potassium channel KCa3.1. Elife. 2016;5:e16093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hoch NE, Guzik TJ, Chen W, Deans T, Maalouf SA, Gratze P, Weyand C, Harrison DG. Regulation of T‐cell function by endogenously produced angiotensin II. Am J Physiol Regul Integr Comp Physiol. 2009;296:R208–R216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Frangogiannis NG, Michael LH, Entman ML. Myofibroblasts in reperfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain (SMemb). Cardiovasc Res. 2000;48:89–100. [DOI] [PubMed] [Google Scholar]
- 44. Shinde AV, Humeres C, Frangogiannis NG. The role of α‐smooth muscle actin in fibroblast‐mediated matrix contraction and remodeling. Biochim Biophys Acta. 2017;1863:298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nishida M, Onohara N, Sato Y, Suda R, Ogushi M, Tanabe S, Inoue R, Mori Y, Kurose H. Gα12/13‐mediated up‐regulation of TRPC6 negatively regulates endothelin‐1‐induced cardiac myofibroblast formation and collagen synthesis through nuclear factor of activated T cells activation. J Biol Chem. 2007;282:23117–23128. [DOI] [PubMed] [Google Scholar]
- 46. Davis J, Burr AR, Davis GF, Birnbaumer L, Molkentin JD. A TRPC6‐dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev Cell. 2012;23:705–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Adapala RK, Thoppil RJ, Luther DJ, Paruchuri S, Meszaros JG, Chilian WM, Thodeti CK. TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J Mol Cell Cardiol. 2013;54:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental Methods
Figure S1. Concentration‐dependent and time‐dependent effects of Ang II on cell viability and IL‐4/13 expression in mice CD4+ T cells.
Figure S2. KCa3.1 expression in CD4+ T cells treated with KCa3.1‐siRNA.
Figure S3. The role of KCa3.1 in IL‐4/IL‐13–induced differentiation of monocytes into fibrocytes.
Figure S4. KCa3.1 channels interact with TRPV4 and TRPC6 channels in rat cardiac fibroblasts.