

Abstract
Background
Neutrophil gelatinase‐associated lipocalin (NGAL) or lipocalin 2 may promote atherosclerosis and plaque instability leading to increased risk of cardiac events. We investigated the relationships between plasma NGAL, cardiovascular disease biomarkers, and long‐term cardiac events.
Methods and Results
The study population consisted of 1131 ambulant older white women (mean age 75 years) without clinical coronary heart disease (CHD) and measures of plasma NGAL in the Perth Longitudinal Study of Ageing Women with 14.5‐year CHD and heart failure hospitalizations or death (events) captured using linked records. Over 14.5 years, 256 women had CHD events, while 118 had heart failure events. Per SD increase in log‐transformed NGAL there was a 35% to 37% increase in relative hazards for CHD and heart failure events in unadjusted analyses, which remained significant after adjustment for conventional risk factors for CHD events (hazard ratio 1.29, 95% CI 1.13–1.48, P<0.001) but not heart failure (P>0.05). Women in the highest 2 quartiles of NGAL had higher relative hazards for CHD events compared with women in the lowest quartile hazard ratio 1.61, 95% CI 1.08–2.39, P=0.019 and hazard ratio 1.97, 95% CI 1.33–3.93, P=0.001, respectively. These associations were independent of high‐sensitivity cardiac troponin I, homocysteine, and estimated renal function. NGAL correctly reclassified 1 in 4 women who sustained a CHD event up in risk and 1 in 10 women without CHD events down in risk.
Conclusions
NGAL was associated with increased risk of long‐term CHD events, independent of conventional risk factors and biomarkers. These findings provide mechanistic insight into the role of NGAL with cardiac events.
Keywords: coronary heart disease, elderly, lipocalin 2, myocyte necrosis, neutrophil gelatinase‐associated lipocalin
Subject Categories: Cardiovascular Disease, Chronic Ischemic Heart Disease, Epidemiology, Mortality/Survival, Biomarkers
Clinical Perspective
What Is New?
Neutrophil gelatinase‐associated lipocalin is a recently identified marker of inflammation.
Older women with elevated neutrophil gelatinase‐associated lipocalin levels had a higher risk of coronary heart disease hospitalization and death, independent of conventional risk factors and other cardiac biomarkers.
What Are the Clinical Implications?
This association between elevated neutrophil gelatinase‐associated lipocalin and coronary heart disease events was in addition to cardiac troponin I levels and may represent a future risk stratification tool to identify older patients with atypical symptoms who will benefit most from invasive coronary angiography.
Introduction
An increasing number of inflammation‐related proteins are being assessed as biomarkers of coronary artery disease (CAD) progression and risk prediction. The data from recent studies point to neutrophil gelatinase‐associated lipocalin (NGAL) as a previously underappreciated factor in CAD development and progression. NGAL belongs to the lipocalin superfamily, proteins that specialized in binding and transporting small hydrophobic molecules, and exists in 3 molecular forms in blood and urine: a monomer, a homodimer, and a heterodimer, covalently bound with matrix metalloproteinase‐9. NGAL is highly expressed in kidney tubule cells in response to injury and hence, a known marker of acute kidney injury. Recent evidence from both mouse and human studies demonstrates that NGAL is highly expressed in cardiomyocytes and atherosclerotic plaques.1, 2, 3, 4 In these sites, NGAL modulates a range of cellular responses, such as proliferation, differentiation, and apoptosis1 associated with vascular function and atherosclerosis5 and also in vascular remodeling.6, 7 NGAL has also been found to be associated with obesity in humans, in particular, cholesterol mobilization to peripheral tissues.8 Studies of animal diet–induced obesity suggest that NGAL may be an important marker of inflammation leading to endothelial dysfunction and elevated blood pressure.9
To investigate these relationships further, we analyzed the predictive role of NGAL on clinical outcomes in a large cohort study of older women. In addition, to better understand the potential pathophysiological role of plasma NGAL in cardiovascular disease, we studied the relationship between NGAL and high‐sensitivity cardiac troponin I (hs‐cTnI) , a validated marker of acute coronary syndrome and mortality previously studied in this and other populations.10, 11 Second, given the evidence that elevated homocysteine promotes inflammatory monocyte differentiation, activates pro‐inflammatory pathways, and accelerates the progression of atherosclerosis,12 we also investigated this potential pathological pathway.
Methods
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure. This material will be available on request and approval at the Perth Longitudinal Study of Ageing Women website.13
Ethics Statement
The Human Ethics Committee of the University of Western Australia approved the study and written informed consents were obtained from all participants. Human ethics approval for the use of linked data for the project was provided by the Human Research Ethics Committee of the Western Australian Department of Health, project number #2009/24.
Study Population
The participants were recruited in 1998 to a 5‐year prospective, randomized, controlled trial of oral calcium supplements to prevent osteoporotic fractures, the CAIFOS (Calcium Intake Fracture Outcome) study as described previously.14 Briefly, 1500 women were recruited from the Western Australian general population by mail using the electoral roll. All participants were ambulant with an expected survival beyond 5 years and were not receiving any medication (including hormone replacement therapy) known to affect bone metabolism. Baseline disease burden, cardiovascular disease risk, and medications were comparable between these participants and the general population of similar age, although these participants were more likely to be from higher socioeconomic groups.14 At the conclusion of CAIFOS, participants were subsequently included in additional follow‐up studies for an additional 10 years as the Perth Longitudinal Study of Aging in Women (PLSAW) http://www.lsaw.com.au. The randomized controlled trial was commenced and completed before the introduction of clinical trial registries; hence the trial was retrospectively registered in the Australian New Zealand Clinical Trials Registry ACTRN12615000750583.
Baseline Risk Factors
Baseline medical history including the presence of diabetes mellitus, hypertension, smoking history (current smokers/former smokers or nonsmokers), and medications was obtained from all participants as described previously.11
Biochemistry
Venous blood samples were collected between 0830 and 1030 hours after overnight fasting at baseline (1998) and were stored at −80°C until assessment. Total cholesterol, high‐density lipoprotein cholesterol (HDLC), and triglyceride concentrations were determined using a Hitachi 917 auto analyzer (Roche Diagnostics). Low‐density lipoprotein cholesterol was calculated using Friedewald's method.15 hs‐cTnI was measured in 2013 in baseline serum stored at −80°C using the Abbott ARCHITECT i2000SR STAT hs‐TnI assay as described previously.11 Plasma that had not been previously defrosted was used to measure NGAL using a 2‐step chemiluminescent microparticle monoclonal immunoassay on an automated platform (Abbott Diagnostics, Longford, Ireland). The interassay coefficient of variation over a 3‐week period was 6.07% and 2.63% at concentrations of 21 and 1194 ng/L, respectively. Baseline creatinine was measured using an isotope dilution mass spectrometry traceable Jaffe kinetic assay in 2003 on a Hitachi 917 analyzer (Roche Diagnostics GmbH, Mannheim Germany). Estimated glomerular filtration rate (eGFR) using creatinine was calculated using the Chronic Kidney Disease Epidemiology Collaboration (CKD‐EPI) equation. The plasma total l‐homocysteine was measured using a Fluorescence Polarization Immuno‐assay on an Abbott IMx Analyzer (Abbott Laboratories, Abbott Park, IL). The interassay coefficient of variation for homocysteine measurement was <5%. The total l‐homocysteine includes free monomeric homocysteine, free dimeric homocysteine, protein‐bound forms, and mixed dimeric low molecular mass forms.
Cardiac Disease Hospitalizations and Deaths
The first episode of cardiac hospitalization or death was identified using data from the Hospital Morbidity Data Collection and WA Deaths Registry, linked via the Western Australian Data Linkage System as described previously.11
Statistical Analysis
Baseline data are presented as either mean±SD, median and interquartile range (IQR), or number and (%) where appropriate. Non‐normally distributed baseline factors including NGAL were transformed using their natural logarithm (ln‐NGAL) to reduce skewness and improve approximation of normal distribution. The associations between baseline covariates on ln‐NGAL were determined using univariate linear regression analysis. Forward stepwise multivariable regression analysis of variables significant in the univariate analysis was used to determine variables independently associated with NGAL. As NGAL, homocysteine, and hs‐TnI were not normally distributed, Spearman's correlations were undertaken for unadjusted analyses. Unadjusted and multivariable‐adjusted Cox proportional hazards regression analyses were undertaken to test the association of NGAL with coronary heart disease (CHD) hospitalizations and deaths. No violations of the Cox proportional hazards assumptions were detected. Multivariable‐adjustments included the variables used in the Framingham risk equation (model 1: age, body mass index [BMI], current smoker, prescription of antihypertensive medication, systolic blood pressure, history of diabetes mellitus) plus treatment code (calcium or placebo). Further analyses including replacing BMI with HDLC and total cholesterol (model 2) as well as further adjustments for potential mediators of disease including estimated glomerular filtration rate (eGFR), hs‐c‐TnI, and homocysteine were also undertaken. Additionally, given the advanced age of these women, we also performed competing‐risks analyses based on Fine and Gray's proportional subdistribution hazards model16 to account for the competing risk of noncardiac mortality (all‐other causes of death). The discriminative performance were calculated using the Harrell's C‐statistic17 with discrimination considered to be poor (<0.6), moderate (0.6–0.8), or good (≥0.8). Because of the low sensitivity of these analyses, net reclassification and integrated discrimination improvement indices were also performed.18 In order to do so, the first model included all the variables in the general Framingham risk model using BMI and treatment code (referent model), and the second model included all variables in the general Framingham cardiovascular risk model using BMI plus treatment code with the addition of NGAL (ln‐NGAL, quartiles of NGAL, or median NGAL). The calibration was tested by separating participants into deciles of predicted risk compared with the actual risk. For net reclassification improvement, we used a category‐free approach (no P value applied) as there are no suggested cut points for cardiovascular risk in the elderly. All analyses were undertaken using IBM SPSS Statistics Version 21 (2012, IBM Corp, Armonk, NY), STATA (version 13; StataCorp LP, College Station, TX), or SAS (Version 9.4; SAS Institute Inc, Chicago, IL). P values of <0.05 in 2‐tailed testing were considered statistically significant.
Results
Baseline Characteristics
Participant selection is outlined in Figure 1. To investigate the associations and predictive utility of NGAL, we excluded these 114 individuals with prevalent CHD at baseline from all subsequent analysis. NGAL levels were significantly higher in the excluded cohort of 114 women with prevalent CHD compared with those included in the study cohort (82.3 [IQR 66.1–101.8] versus 75.9 [IQR 61.5–94.0], P=0.042). Baseline characteristics of the remaining 1131 older women included in the study are presented in Table 1.
Figure 1.
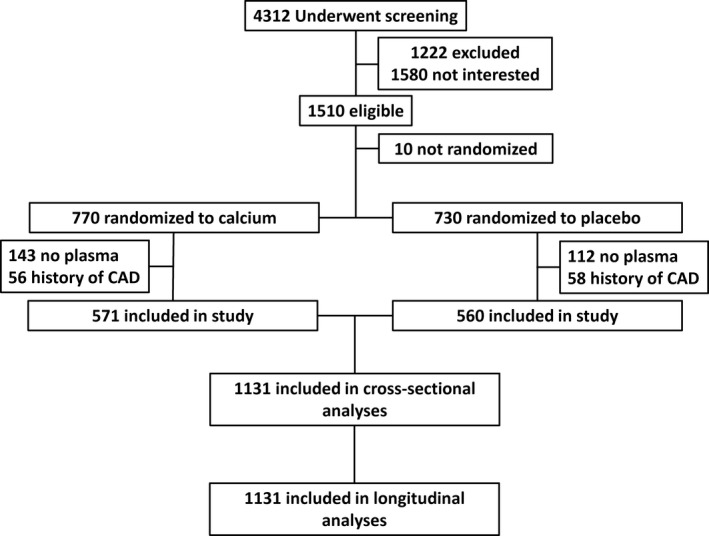
Overview of the study participants. All participants had hospital (1980–2013) and mortality (1998–2013) records available from the Western Australian Data linkage system. CAD indicates coronary artery disease.
Table 1.
Baseline Characteristics of the Cohort by Quartiles of NGAL
| Baseline Characteristics | Whole Cohort (n=1131) | Quartile 1 (n=280) | Quartile 2 (n=284) | Quartile 3 (n=284) | Quartile 4 (n=283) |
|---|---|---|---|---|---|
| NGAL, median [IQR], ng/mL | 76 [62–95] | 54 [46–58] | 69 [65–72] | 85 [80–89] | 113 [102–136] |
| Age, mean±SD, y | 75.2±2.7 | 74.8±2.75† | 75.1±2.6† | 75.3±2.9† | 75.8±2.8† |
| BMI, mean±SD, kg/m2 | 27.0±4.7 | 25.7±4.3† | 26.6±4.5† | 27.0±4.3† | 28.9±5.0† |
| Smoking history | |||||
| Never, N (%) | 708 (62.9) | 175 (62.7) | 181 (64.0) | 175 (62.3) | 177 (62.5) |
| Previous, N (%) | 413 (36.7) | 102 (36.6) | 101 (35.7) | 104 (37.0) | 106 (37.5) |
| Current, N (%) | 5 (0.4) | 2 (0.7) | 1 (0.4) | 2 (0.7) | 0 (0.0) |
| Diabetes mellitus, N (%) | 61 (5.4) | 16 (5.7) | 10 (3.5) | 12 (4.2) | 23 (8.1) |
| Antihypertensive medication, N (%) | 476 (42.1) | 91 (32.5)† | 110 (38.7)† | 117 (41.2)† | 158 (55.8)† |
| Statin medication, N (%) | 186 (16.4) | 44 (15.7) | 46 (16.2) | 47 (16.5) | 49 (17.3) |
| Low‐dose aspirin, N (%) | 201 (17.8) | 42 (15.0) | 46 (16.2) | 58 (20.4) | 55 (19.4) |
| Alcohol intake, median [IQR], g/d | 1.9 [0.3–10.1] | 2.8 [0.3–11.9]† | 2.1 [0.3–11.3]† | 2.1 [0.3–10.0]† | 1.1 [0.3–6.2]† |
| Exercise, median [IQR], h/wk | 4.0 [1.0–7.0] | 4.0 [2.0–7.0]† | 4.0 [1.0–8.0]† | 4.0 [0.3–7.0]† | 3.0 [0.0–6.0]† |
| Blood pressure | |||||
| SBP, mean±SD, mm Hg | 138±18 | 138±18 | 138±19 | 137±19 | 137±18 |
| DBP, mean±SD, mm Hg | 73±11 | 73±12 | 73±11 | 73±11 | 73±12 |
| MAP, mean±SD, mm Hg | 95±12 | 95±12 | 95±12 | 94±12 | 94±12 |
| Lipid profiles (n=807) | |||||
| Total cholesterol, mean±SD, mg/dL | 227±43 | 225±47 | 225±40 | 234±41 | 225±42 |
| LDLC, mean±SD, mg/dL | 143±39 | 140±41 | 142±36 | 150±38 | 141±39 |
| HDLC, mean±SD, mg/dL | 56±15 | 60±15† | 56±15† | 56±13† | 53±15† |
| Triglycerides, mean±SD, mg/dL | 137±63 | 123±56† | 129±60† | 141±62† | 154±70† |
| Cardiovascular biomarkers | |||||
| hs‐cTnI, median [IQR], pg/mL | 4.6 [3.6–6.1] | 4.1 [3.4–5.4]† | 4.4 [3.6–5.9]† | 4.7 [3.7–6.4]† | 5.1 [4.0–6.5]† |
| Homocysteine, median [IQR], μmol/L* | 11.3 [9.3–13.5] | 9.9 [8.5–12.1]† | 10.7 [8.8–13.1]† | 11.9 [9.8–14.2]† | 12.5 [10.6–15.4]† |
| eGFR, mean±SD, mL/min per 1.73 m2 | 66.3±13.0 | 72.3±10.6† | 68.3±11.3† | 64.3±12.6† | 60.6±13.0† |
Results are mean±SD or number and (%). BMI indicates body mass index; DBP, diastolic blood pressure; eGFR, estimated glomerular filtration rate; HDLC, high‐density lipoprotein cholesterol; hs‐cTnI, high‐sensitivity cardiac troponin I; IQR, interquartile range; LDLC, low‐density lipoprotein cholesterol; MAP, mean arterial pressure; NGAL, neutrophil gelatinase‐associated lipocalin; SBP, systolic blood pressure.
Measured in 905 women.
Significant across group differences using univariate ANOVA, x2 test or Kruskal‐Wallis test where appropriate.
Correlations Between Plasma NGAL and Baseline Variables
The correlations between ln‐NGAL levels and these demographic, clinical, lifestyle, and laboratory variables are shown in Table 2. Using forward stepwise linear regression, the most parsimonious model for ln‐NGAL that explained 18.6% of the circulating levels included eGFR (P<0.001), BMI (P<0.001), ln‐homocysteine (P<0.001), age (P=0.004), and HDLC (P=0.016).
Table 2.
Correlations and Multivariate Associations Between ln‐NGAL, Clinical Variables, and Cardiac Biomarkers
| Univariate Model | Multivariable Model | |||
|---|---|---|---|---|
| r | P Value | Standardized β | P Value | |
| Age, y | 0.142 | <0.001 | 0.104 | 0.004 |
| BMI, kg/m2 | 0.267 | <0.001 | 0.213 | <0.001 |
| Diabetes mellitus (yes) | 0.069 | 0.021 | ||
| Antihypertensive medications (yes) | 0.175 | <0.001 | ||
| Statin medications (yes) | 0.003 | 0.926 | ||
| Low‐dose aspirin (yes) | 0.046 | 0.120 | ||
| Systolic blood pressure, mm Hg | −0.030 | 0.327 | ||
| Diastolic blood pressure, mm Hg | −0.016 | 0.604 | ||
| Current smoking (yes) | −0.016 | 0.580 | ||
| Total cholesterol, mg/dL | 0.025 | 0.470 | ||
| High‐density lipoprotein cholesterol, mg/dL | −0.157 | <0.001 | −0.091 | 0.016 |
| Low‐density lipoprotein cholesterol, mg/dL | 0.030 | 0.401 | ||
| Triglycerides, mg/dL | 0.174 | <0.001 | ||
| eGFR, mL/min per 1.73 m2 | −0.320 | <0.001 | −0.194 | <0.001 |
| Ln‐high‐sensitivity cardiac troponin I | 0.160 | <0.001 | ||
| Ln‐homocysteine | 0.284 | <0.001 | 0.144 | <0.001 |
Multivariable model r 2=0.186. BMI indicates body mass index; eGFR, estimated glomerular filtration rate; NGAL, neutrophil gelatinase‐associated lipocalin.
NGAL Levels and CHD Events
Over 14.5 years, 256/1131 (22.6%) women sustained CHD hospitalization or death. For every 1 SD increase in ln‐NGAL, there was a 33% increase in the relative hazards for CHD hospitalizations or death after adjusting for age and treatment (hazard ratio [HR] 1.33, 95% CI 1.18–1.51, P<0.001). This estimate remained similar after adjusting for conventional cardiovascular risk factors used in the Framingham risk equations (Table 3). Using a quartile approach, the relationship between baseline NGAL and CHD hospitalization and death is shown in Figure 2, with the adjusted HR presented in Table 4. Compared with those in the lowest quartile of NGAL, those in the highest 2 quartiles had increased relative hazards for CHD hospitalizations or deaths in unadjusted analysis and multivariable‐adjusted models. Similar results were seen when replacing the BMI with total cholesterol and HDLC in the subset of 807 women with these data available (Table 4).
Table 3.
Multivariable‐Adjusted HR for Cardiac Events Per SD Increase in the Natural Logarithm of Circulating NGAL
| Coronary Heart Disease Events (n=256) | Heart Failure Event (n=118) | |||
|---|---|---|---|---|
| HR (95% CI) | P Value | HR (95% CI) | P Value | |
| Model 1 (n=1131)* | 1.29 (1.13–1.48) | <0.001 | 1.21 (0.99–1.48) | 0.060 |
| +eGFR | 1.25 (1.07–1.45) | 0.004 | 1.30 (1.04–1.63) | 0.019 |
| +ln‐hs‐cTnI | 1.27 (1.11–1.46) | 0.001 | 1.18 (0.97–1.44) | 0.106 |
| +ln‐homocysteine | 1.21 (1.03–1.42) | 0.027 | 1.11 (0.88–1.40) | 0.379 |
| +eGFR, ln‐hs‐cTnI, and ln–homocysteine | 1.21 (1.02–1.44) | 0.032 | 1.17 (0.91–1.51) | 0.232 |
| Model 2 (n=807)† | 1.49 (1.29–1.53) | <0.001 | 1.22 (0.98–1.52) | 0.076 |
| +eGFR | 1.37 (1.16–1.62) | <0.001 | 1.26 (0.98–1.61) | 0.066 |
| +ln‐hs‐cTnI | 1.40(1.20–1.64) | <0.001 | 1.45 (1.20–1.74) | 0.133 |
| +ln‐homocysteine | 1.35 (1.13–1.62) | 0.001 | 1.14 (0.54–2.72) | 0.305 |
| +eGFR, ln‐hs‐cTnI and ln‐homocysteine | 1.34 (1.11–1.62) | 0.003 | 1.17 (0.89–1.54) | 0.249 |
Cox proportional hazard regression models were adjusted for conventional cardiovascular risk factors (Model 1 including; age, BMI, current smoking, systolic blood pressure, prescription of antihypertensive medications, diabetes mellitus, and treatment code [calcium or placebo] or Model 2 including; age, total cholesterol, high‐density lipoprotein cholesterol, current smoking, systolic blood pressure, prescription of antihypertensive medications, diabetes mellitus, and treatment code [calcium or placebo]). BMI indicates body mass index; eGFR, estimated glomerular filtration rate; HR, hazard ratio; hs‐cTnI, high‐sensitivity cardiac troponin I; NGAL, neutrophil gelatinase‐associated lipocalin.
Homocysteine was measured in 905 women.
Homocysteine was measured in 700 women.
Figure 2.
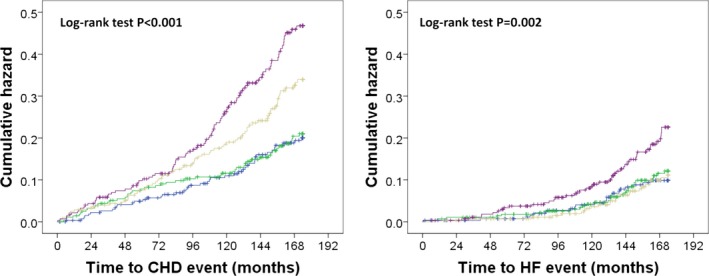
Kaplan–Meier hazard plots for CHD (n=256) and HF hospitalizations and deaths (n=118) by quartiles of NGAL, blue line quartile 1 (<61.5 ng/mL), green line quartile 2 (61.5–75.8 ng/mL), beige line quartile 3 (75.9–94.8 ng/mL), and purple line quartile 4 (≥94.9 ng/mL). CHD indicates coronary heart disease; HF, heart failure; NGAL, neutrophil gelatinase‐associated lipocalin.
Table 4.
Multivariable‐Adjusted HR for Cardiac Events by Quartile of NGAL After Adjustment of Other Risk Factors
| Number of Events (%) | Model 1* HR (95% CI) | P Value | Model 2* HR (95% CI) | P Value | |
|---|---|---|---|---|---|
| Whole cohort (n=1131) | |||||
| CHD events (n=256) | |||||
| Quartile 1 (<61.5 ng/mL) | 44 (15.7) | 1.00 (referent) | 1.00 (referent) | ||
| Quartile 2 (61.5–75.8 ng/mL) | 48 (16.9) | 1.02 (0.68–1.54) | 0.909 | 1.03 (0.67–1.59) | 0.887 |
| Quartile 3 (75.9–94.8 ng/mL) | 72 (25.4) | 1.62 (1.11–2.35) | 0.012 | 1.61 (1.08–2.39) | 0.019 |
| Quartile 4 (≥94.9 ng/mL) | 92 (32.5) | 2.14 (1.49–3.08) | <0.001 | 1.97 (1.33–2.93) | 0.001 |
| P for trend | <0.001 | <0.001 | |||
| Heart failure events (n=118) | |||||
| Quartile 1 (<61.5 ng/mL) | 22 (7.9) | 1.00 (referent) | 1.00 (referent) | ||
| Quartile 2 (61.5–75.8 ng/mL) | 27 (9.5) | 1.13 (0.65–1.99) | 0.664 | 1.22 (0.66–1.91) | 0.528 |
| Quartile 3 (75.9–94.8 ng/mL) | 24 (8.5) | 1.01 (0.57–1.81) | 0.971 | 1.01 (0.54–1.88) | 0.985 |
| Quartile 4 (≥94.9 ng/mL) | 61 (15.9) | 1.97 (1.17–3.30) | 0.002 | 1.65 (0.92–2.96) | 0.093 |
| P for trend | 0.004 | 0.079 | |||
| Those with lipids (n=807) | |||||
| CHD events (n=170) | |||||
| Quartile 1 (<61.5 ng/mL) | 24 (12.0) | 1.00 (referent) | 1.00 (referent) | ||
| Quartile 2 (61.5–75.8 ng/mL) | 32 (15.2) | 1.23 (0.71–1.60) | 0.442 | 1.28 (0.73–2.23) | 0.384 |
| Quartile 3 (75.9–94.8 ng/mL) | 52 (25.2) | 2.16 (1.33–3.50) | 0.002 | 2.15 (1.29–3.58) | 0.003 |
| Quartile 4 (≥94.9 ng/mL) | 62 (32.6) | 2.81 (1.75–4.52) | <0.001 | 2.64 (1.59–4.38) | <0.001 |
| P for trend | <0.001 | <0.001 | |||
| Heart failure events (n=83) | |||||
| Quartile 1 (<61.5 ng/mL) | 15 (7.5) | 1.00 (referent) | 1.00 (referent) | ||
| Quartile 2 (61.5–75.8 ng/mL) | 20 (9.5) | 1.18 (0.61–2.31) | 0.624 | 1.28 (0.63–2.60) | 0.494 |
| Quartile 3 (75.9–94.8 ng/mL) | 19 (9.2) | 1.16 (0.59–2.28) | 0.669 | 1.31 (0.64–2.66) | 0.462 |
| Quartile 4 (≥94.9 ng/mL) | 29 (15.3) | 1.88 (1.00–3.53) | 0.049 | 1.63 (0.82–3.23) | 0.161 |
| P for trend | 0.034 | 0.167 | |||
CHD indicates coronary heart disease; HR, hazard ratio; NGAL, neutrophil gelatinase‐associated lipocalin.
Cox regression models were adjusted for conventional cardiovascular risk factors (Model 1 including age, and treatment code [calcium or placebo] or Model 2 including age, BMI or total cholesterol and high‐density lipoprotein cholesterol, current smoking, systolic blood pressure, prescription of antihypertensive medications, diabetes mellitus, and treatment code [calcium or placebo]).
NGAL Levels and Heart Failure Events
Over 14.5 years, 118/1131 (10.4%) women sustained HF hospitalization or death. For every 1 SD increase in ln‐NGAL, there was a 30% increase in the relative hazards for HF hospitalizations or death after adjusting for age and treatment (HR 1.30, 95% CI 1.08–1.56, P=0.005), which was no longer statistically significant (P=0.060) after adjusting for conventional cardiovascular risk factors (Table 3). Using a quartile approach, the relationship between baseline NGAL and HF hospitalization and death is shown in Figure 2. The values for the HRs are shown in Table 4. Compared with those in the lowest quartile of NGAL, those in the fourth quartile had increased relative hazards for HF hospitalizations or deaths in unadjusted analysis but not in multivariable‐adjusted models (Table 4). Similar results were seen when replacing the BMI with total cholesterol and HDLC in the subset of 807 women with these data available (Table 4).
NGAL Levels and Cardiac Risk Prediction
As circulating NGAL levels were not associated with HF events in multivariable‐adjusted models, no further analyses were performed to determine whether the addition of NGAL improved risk prediction in addition to conventional risk factors. To study the effects of NGAL as a risk predictor for CHD, we compared it individually with conventional risk factors used in the Framingham Risk Score (Table 5). Ln‐NGAL alone had poor discrimination quantified by the area under the receiver operating characteristic curve (c‐statistic) for CHD (0.594, 95% CI 0.558–0.629). The addition of NGAL did not improve the C‐statistic for 14.5‐year cardiac events compared with conventional cardiovascular risk factors. However, newer measures of discrimination such as the Net Reclassification Index and the Integrated Discrimination Index suggested that the addition of NGAL may lead to substantial improvements. This was particularly evident in models incorporating HDLC and total cholesterol, with large proportions of women having a CHD event moved correctly up in risk and women who did not sustain a CHD event moved down in risk (Table 5).
Table 5.
Area Under the Curve, NRI, and IDI
| c‐Statistic | P Value* | Category Free NRI (95% CI) | Correctly Reclassified Higher (%) | Correctly Reclassified Lower (%) | IDI | P Value* | |
|---|---|---|---|---|---|---|---|
| Model 1 (n=1131) | |||||||
| CHD events (n=256) | |||||||
| Conventional risk factors | 0.625 | ||||||
| +ln‐NGAL | 0.637 | 0.175 | 0.258 | 14.9 | 11.0 | 0.013 | <0.001 |
| +Quartiles of NGAL | 0.641 | 0.135 | 0.329 | 23.2 | 9.8 | 0.017 | <0.001 |
| +Above the median NGAL | 0.639 | 0.146 | 0.371 | 30.6 | 6.5 | 0.015 | <0.001 |
| Model 2 (n=807) | |||||||
| CHD events (n=170) | |||||||
| Conventional risk factors | 0.637 | ||||||
| +ln‐NGAL | 0.660 | 0.064 | 0.366 | 18.3 | 18.3 | 0.029 | <0.001 |
| +Quartiles of NGAL | 0.662 | 0.078 | 0.451 | 31.7 | 13.4 | 0.029 | <0.001 |
| +Above the median NGAL | 0.658 | 0.149 | 0.471 | 36.6 | 10.5 | 0.027 | <0.001 |
BMI indicates body mass index; CHD, coronary heart disease; IDI, integrated discrimination improvement; NGAL, neutrophil gelatinase‐associated lipocalin; NRI, net reclassification improvement.
Compared with conventional risk factors (Model 1 including age, BMI, current smoking, systolic blood pressure, prescription of antihypertensive medications, diabetes mellitus, and treatment code [calcium or placebo] or Model 2 including age, total cholesterol, high‐density lipoprotein cholesterol, current smoking, systolic blood pressure, prescription of antihypertensive medications, diabetes mellitus, and treatment code [calcium or placebo]).
Putative Pathophysiological Mechanisms of NGAL With Cardiac Outcomes
Given the lack of information on the mechanisms of adverse effects of circulating NGAL on coronary atherosclerosis, we sought to investigate whether NGAL was acting as a surrogate marker of kidney function (eGFR), cardiac inflammation (homocysteine), and myocardial ischemia (hs‐cTnI) or was independent of these markers of cardiovascular disease by including these biomarkers individually or collectively into the multivariable‐adjusted models (Table 6). The Kaplan–Meier hazard plots for CHD and HF hospitalizations and deaths separated by above and below the median NGAL and hs‐cTnI are presented in Figure 3. For all analyses with the CHD outcome with the biomarkers individually and NGAL, both remained significantly associated with CHD outcomes. When all 3 biomarkers were added, NGAL remained significantly associated with CHD events.
Table 6.
Multivariable‐Adjusted HR for Coronary Events by NGAL and Biomarkers of Potential Pathological Mechanisms*
| Number of Events (%) | Model 1* HR (95% CI) | P Value | Number of Events (%) | Model 2* HR (95% CI) | P Value | |
|---|---|---|---|---|---|---|
| NGAL and hs‐cTnI | ||||||
| ↓NGAL and ↓hs‐cTnI, n=319/229 | 42 (13.2) | 1.00 (referent) | 23 (10.0) | 1.00 (referent) | ||
| ↓NGAL and ↑hs‐cTnI, n=239/171 | 49 (20.5) | 1.57 (1.01–2.43) | 0.043 | 31 (19.3) | 1.92 (1.09–3.37) | 0.026 |
| ↑NGAL and ↓hs‐cTnI, n=240/177 | 49 (20.4) | 1.51 (0.97–2.35) | 0.069 | 32 (18.1) | 1.76 (0.99–3.15) | 0.056 |
| ↑NGAL and ↑hs‐cTnI, n=322/219 | 115 (35.7) | 2.72 (1.84–4.03) | <0.001 | 81 (37.0) | 3.73 (2.24–6.22) | <0.001 |
| NGAL and homocysteine | ||||||
| ↓NGAL and ↓homocysteine, n=273/216 | 39 (14.3) | 1.00 (referent) | 30 (13.2) | 1.00 (referent) | ||
| ↓NGAL and ↑homocysteine, n=175/138 | 32 (18.3) | 1.25 (0.77–2.03) | 0.365 | 19 (13.8) | 0.85 (0.46–1.56) | 0.602 |
| ↑NGAL and ↓homocysteine, n=172/135 | 38 (22.1) | 1.47 (0.92–2.34) | 0.104 | 30 (22.4) | 1.56 (0.93–2.63) | 0.095 |
| ↑NGAL and ↑homocysteine, n=285/211 | 88 (30.9) | 1.97 (1.32–2.94) | 0.001 | 64 (30.3) | 1.85 (1.17–2.95) | 0.009 |
| NGAL and chronic kidney disease | ||||||
| ↓NGAL and no CKD, n=403/306 | 61 (15.1) | 1.00 (referent) | 38 (12.4) | 1.00 (referent) | ||
| ↓NGAL and CKD, n=100/80 | 22 (22.0) | 1.54 (0.92–2.59) | 0.102 | 16 (20.0) | 1.56 (0.84–2.90) | 0.161 |
| ↑NGAL and no CKD, n=290/205 | 66 (22.8) | 1.57 (1.09–2.24) | 0.014 | 45 (22.0) | 1.78 (1.15–2.77) | 0.010 |
| ↑NGAL and CKD, n=223/165 | 88 (39.5) | 2.99 (2.14–4.18) | <0.001 | 89 (39.4) | 3.22 (2.11–4.92) | <0.001 |
Cox regression models were adjusted for conventional cardiovascular risk factors (Model 1 including age, and treatment code [calcium or placebo] or Model 2 including age, total cholesterol, high‐density lipoprotein cholesterol, current smoking, systolic blood pressure, prescription of antihypertensive medications, diabetes mellitus, and treatment code [calcium or placebo]). ↓=Less than the median value and ↑ greater than or equal to the median value. CKD indicates chronic kidney disease; HR, hazard ratio; hs‐cTnI, high‐sensitivity cardiac troponin I; NGAL, neutrophil gelatinase‐associated lipocalin.
Homocysteine only measured in 905 women.
Figure 3.
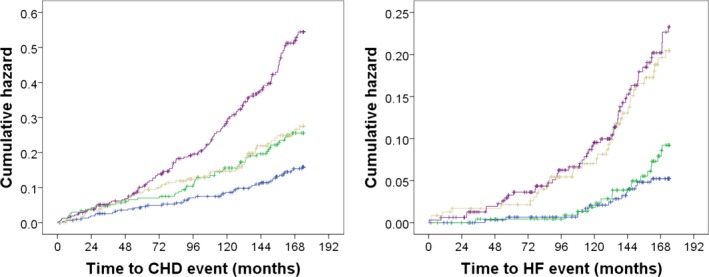
Kaplan–Meier hazard plots for CHD (n=256) and HF hospitalizations and deaths (n=118) by ↑ or ↓ median NGAL and ↑ or ↓ median hs‐TnI. Blue line=↓ median NGAL and ↓ median hs‐TnI, green line=↑ median NGAL and ↓ median hs‐TnI, beige line=↓ median NGAL and ↑ median hs‐TnI and purple line=↑ median NGAL and ↑ median hs‐TnI. CHD indicates coronary heart disease; HF, heart failure; hs‐TnI, high‐sensitivity cardiac troponin I; NGAL, neutrophil gelatinase‐associated lipocalin.
Further Analyses
Interaction testing between NGAL and cardiovascular risk factors for CHD events is presented in Table 7. This testing suggested that NGAL may be more strongly associated with CHD events when HDLC is normal. To explore the extent of reverse causality bias, we excluded cardiac events occurring within the first 2 years as well as undertaking competing risks regression analyses with deaths as a competing risk, both of which did not substantially alter the findings (Table 8). Additionally, we tested the association of NGAL with myocardial infarctions and/or CHD death (hard CHD) that occurred in 172/1131 (15.2%) of the women. Per SD increase in ln‐NGAL was associated with increased relative hazards of hard CHD (unadjusted HR 1.39, 95% CI 1.20–1.61, P<0.001 and multivariable‐adjusted HR 1.30, 95% CI 1.10–1.53, P=0.002). Similarly, in the quartile analysis, women in the highest quartile of NGAL had the highest relative hazards of hard CHD (quartile 4 versus quartile 1 unadjusted HR 2.28, 95% CI 1.49–3.50, P<0.001 and multivariable‐adjusted HR 1.84, 95% CI 1.16–2.94, P=0.010).
Table 7.
Age‐Adjusted HR for CHD Events Per SD Increase in ln‐NGAL
| Coronary Heart Disease Events | |||
|---|---|---|---|
| N (%) | HR (95% CI), P Value* | P Value Inter. | |
| Framingham risk scores | |||
| <20% (n=577) | 100 (17) | 1.21 (0.98–1.49), 0.079 | 0.268 |
| ≥20% (n=513) | 142 (28) | 1.41 (1.20–1.65), <0.001 | |
| Age, y | |||
| <75 y (n=515) | 100 (19) | 1.38 (1.13–1.69), 0.002 | 0.788 |
| ≥75 y (n=616) | 156 (25) | 1.31 (1.12–1.53), 0.001 | |
| BMI | |||
| <25 kg/m2 (n=401) | 80 (20) | 1.29 (0.98–1.69), 0.067 | |
| 25–29 kg/m2 (n=473) | 102 (22) | 1.45 (1.20–1.75), <0.001 | 0.479 |
| ≥30 kg/m2 (n=255) | 74 (29) | 1.12 (0.90–1.39), 0.317 | |
| Hypertension | |||
| No (n=496) | 82 (17) | 1.08 (0.85–1.37), 0.548 | 0.101 |
| Yes (n=635) | 174 (27) | 1.38 (1.20–1.59), <0.001 | |
| Elevated total cholesterol | |||
| <200 mg/dL (n=194) | 35 (18) | 1.27 (0.93–1.74), 0.137 | 0.248 |
| ≥200 mg/dL (n=613) | 135 (22) | 1.57 (1.31–1.83), <0.001 | |
| Low HDLC | |||
| <60 mg/dL (n=514) | 123 (24) | 1.64 (1.37–1.95), <0.001 | 0.030 |
| ≥60 mg/dL (n=293) | 47 (16) | 1.14 (0.86–1.51), 0.360 | |
| Smoking history | |||
| Never (n=708) | 143 (20) | 1.29 (1.09–1.53), 0.003 | 0.837 |
| Yes (n=418) | 112 (27) | 1.36 (1.14–1.64), 0.001 | |
| History of diabetes mellitus | |||
| No (n=1070) | 238 (22) | 1.34 (1.17–1.52), <0.001 | 0.830 |
| Yes (n=61) | 18 (30) | 1.16 (0.78–1.73), 0.459 | |
| Lipid‐lowering therapy | |||
| No (n=945) | 214 (23) | 1.32 (1.15–1.50), <0.001 | 0.091 |
| Yes (n=186) | 42 (23) | 1.44 (1.02–2.04), 0.038 | |
| Low‐dose aspirin | |||
| No (n=930) | 200 (22) | 1.35 (1.17–1.56), <0.001 | 0.523 |
| Yes (n=201) | 56 (28) | 1.26 (0.98–1.61), 0.072 | |
| Physical activity | |||
| <median (n=538) | 127 (24) | 1.29 (1.09–1.53), 0.004 | 0.514 |
| ≥median (n=593) | 129 (22) | 1.37 (1.15–1.64), <0.001 | |
| eGFR <60 mL/min per 1.73 m2 | |||
| Yes (n=323) | 110 (34) | 1.33 (1.10–1.61), 0.003 | 0.446 |
| No (n=694) | 127 (18) | 1.18 (0.97–1.43), 0.085 | |
| hs‐cTnI | |||
| <median (n=558) | 91 (16) | 1.38 (1.11–1.70), 0.004 | 0.442 |
| ≥median (n=562) | 164 (29) | 1.24 (1.06–1.44), 0.006 | |
| Homocysteine† | |||
| <median (n=445) | 77 (17) | 1.26 (0.99–1.61), 0.061 | 0.829 |
| ≥median (n=460) | 120 (26) | 1.30 (1.08–1.56), 0.005 | |
BMI indicates body mass index; eGFR, estimated glomerular filtration rate; HDLC, high‐density lipoprotein cholesterol; HR, hazard ratio; hs‐cTnI, high‐sensitivity cardiac troponin I; NGAL, neutrophil gelatinase‐associated lipocalin.
P value from age and treatment (calcium or placebo)‐adjusted Cox regression. P value per inter. from the interaction between per SD increase in ln‐NGAL with baseline variables with significance set at (P<0.1).
Homocysteine was measured in a subset of 905 women.
Table 8.
Multivariable‐Adjusted Competing Risks Analysis (Noncardiac Deaths) for Cardiac Events Per Quartile of NGAL
| SHR (95% CI)* | P Value | |
|---|---|---|
| Whole cohort (n=1131) | ||
| CHD events (n=256) | ||
| Quartile 1 (<61.5 ng/mL) | 1.00 (referent) | |
| Quartile 2 (61.5–75.8 ng/mL) | 1.03 (0.67–1.59) | 0.887 |
| Quartile 3 (75.9–94.8 ng/mL) | 1.61 (1.08–2.39) | 0.019 |
| Quartile 4 (≥94.9 ng/mL) | 1.97 (1.33–2.93) | 0.001 |
| Heart failure events (n=118) | ||
| Quartile 1 (<61.5 ng/mL) | 1.00 (referent) | |
| Quartile 2 (61.5–75.8 ng/mL) | 1.22 (0.66–1.91) | 0.528 |
| Quartile 3 (75.9–94.8 ng/mL) | 1.01 (0.54–1.88) | 0.985 |
| Quartile 4 (≥94.9 ng/mL) | 1.65 (0.92–2.96) | 0.093 |
| Those with lipids (n=807) | ||
| CHD events (n=170) | ||
| Quartile 1 (<61.5 ng/mL) | 1.00 (referent) | |
| Quartile 2 (61.5–75.8 ng/mL) | 1.28 (0.73–2.23) | 0.384 |
| Quartile 3 (75.9–94.8 ng/mL) | 2.15 (1.29–3.58) | 0.003 |
| Quartile 4 (≥94.9 ng/mL) | 2.64 (1.59–4.38) | <0.001 |
| Heart failure events (n=83) | ||
| Quartile 1 (<61.5 ng/mL) | 1.00 (referent) | |
| Quartile 2 (61.5–75.8 ng/mL) | 1.28 (0.63–2.60) | 0.494 |
| Quartile 3 (75.9–94.8 ng/mL) | 1.31 (0.64–2.66) | 0.462 |
| Quartile 4 (≥94.9 ng/mL) | 1.63 (0.82–3.23) | 0.161 |
BMI indicates body mass index; CHD, coronary heart disease; NGAL, neutrophil gelatinase‐associated lipocalin; SHR, subdistribution hazard ratio.
Models were adjusted for conventional cardiovascular risk factors (Model 1 including age, BMI, current smoking, systolic blood pressure, prescription of antihypertensive medications, diabetes mellitus, and treatment code [calcium or placebo] or Model 2 including age, total cholesterol, high‐density lipoprotein cholesterol, current smoking, systolic blood pressure, prescription of antihypertensive medications, diabetes mellitus; and treatment code [calcium or placebo]).
Discussion
To our knowledge, this is the first prospective study to investigate the associations between plasma NGAL and the cardiac ischemia biomarker hs‐cTnI. Additionally, we investigated NGAL as a predictor of CHD and HF in older women with no previous clinical CHD. Higher levels of plasma NGAL were independently associated with a worse CHD risk profile, elevated levels of hs‐cTnI and homocysteine, and a 2‐ to 3‐fold increase in the relative hazards for CHD hospitalizations and death in the highest quartile of NGAL women compared with those in the lowest quartile. These findings are consistent with other prospective cohort studies in middle‐aged19 and older community‐dwelling adults20 that have shown independent associations between NGAL and CHD events. These associations were robust and independent of hs‐TnI, homocysteine, and eGFR. To our knowledge, this is the first study to show that NGAL is associated with CHD events, independent of hs‐TnI.
All women had detectable NGAL, with levels correlating with hs‐cTnI. Cardiac troponin levels are now the standard of care to identify myocyte necrosis and for cardiovascular risk stratification. Serum cardiac troponin levels directly relate to the risk of future cardiovascular events in both young and elderly populations and improve risk prediction in addition to current established risk factors.11, 21 Therefore, the establishment of NGAL as a novel biomarker with added predictive value above and beyond cardiac troponins has implications for provision of cardiovascular care and associated health costs. Similarly, NGAL levels correlated with homocysteine concentrations (r=0.3, P<0.001), supporting a previous report in a small cohort of 141 patients who had advanced carotid atherosclerotic lesions and who underwent carotid endarterectomy for vascular repair, r=0.4, P<0.001.22 Taken together, these findings suggest that a relationship between NGAL and homocysteine exists and warrants further study.
While the women in our study did not have pre‐existing clinical CHD at baseline, NGAL was associated with several traditional cardiovascular risk factors including age, hypertension, renal impairment, and low HDL levels. Similar findings were reported by Elneihoum et al,23 where plasma levels of NGAL were higher in older hypertensive women compared with younger normotensive individuals. We and others have demonstrated the relationship between increased circulating NGAL and high creatinine/reduced eGFR.24 In the present study, the strongest relationship between NGAL and CHD risk was seen in women with established cardiovascular risk factors, suggesting that plasma NGAL provides additive predictive value above cardiac troponin I in women at the highest risk. This is demonstrated in Figure 3, showing that women with elevated NGAL but not hs‐TnI and vice versa had similar long‐term prognosis while women with elevated levels of both had the highest CHD risk. Given that hs‐cTnIs are emerging as valuable markers of long‐term cardiovascular risk in people without a history of cardiovascular disease,25 these findings are particularly noteworthy.
Regarding determination of clinical limits for NGAL levels and associated cardiovascular risk assessment, there are published reports using a variety of in‐house or commercial assays for quantification,20, 26, 27, 28 but the agreement between these assays and the specificity of these assays to detect different circulating forms remains to be demonstrated. Given these issues, we used 3 different models based on the distribution with Framingham risk scores plus ln‐NGAL (continuous), median NGAL, and quartiles of NGAL to provide similar data to previous studies investigating broader composite cardiovascular disease events or deaths.19, 20 Similar to these studies, we found modest improvements to the c‐statistic; however, in our study these were nonsignificant, presumably because of the smaller sample size and fewer events. We did, however, see relatively large improvements to reclassification with the addition of NGAL to Framingham risk scores. The discrepancy between the findings for the c‐statistic and the net reclassification may reflect previous observations that the use of receiver operating characteristic may be less sensitive and less clinically relevant than reclassification indices.29, 30
Circulating NGAL has been related to local production in many tissues and thus may reflect local tissue inflammation. To date the majority of human studies investigating the association of circulating NGAL with heart disease have been cross‐sectional studies, in patients with pre‐existing disease. Such studies make causality harder to infer. Importantly, these studies do suggest that NGAL may be related to the presence and severity of CAD.3, 23, 27, 31 Indeed, 2 prospective studies of patients undergoing cardiac surgery for CAD have shown that pre/postoperative plasma NGAL is independently associated with all‐cause mortality,28 cardiovascular deaths, or subsequent cardiac events.26 In a prospective study of younger men and women without a history of clinical cardiovascular disease, plasma NGAL was independently associated with 10‐year cardiovascular outcomes and modestly improved the c‐statistic and net reclassification when added to the Framingham risk scores.19 Daniels et al20 followed elderly men and women without a history of clinical cardiovascular disease for a maximum of 16.2 years and found that plasma NGAL was independently associated with a composite end point of myocardial infarction, revascularization, or cardiovascular disease death, providing incremental value to the well‐established biomarkers N‐terminal pro‐brain natriuretic peptide and C‐reactive protein. It is reassuring that the adjusted HR for the composite outcome (HR 1.33) in this study was very similar to that seen in this cohort (HR 1.29), suggesting that these findings may be generalizable to other similarly aged populations. However, before this present study, the association of NGAL with markers of ongoing CAD, plaque rupture, or myocyte damage (such as with cardiac troponin elevation) has not been investigated.
The recent CANTOS trial showed that a human monoclonal antibody targeting interleukin‐1β lowered rates of recurrent cardiovascular events in patients with myocardial infarction and elevated high‐sensitivity C‐reactive protein, independent of low‐density lipoprotein cholesterol lowering.32 This has reinvigorated discussions on direct roles for chronic inflammation in CAD. Similarly, there may be a chronic inflammatory role for NGAL. Indeed, NGAL is induced by interleukin‐1β in murine adipocytes,33 human hepatic stellate cells and hepatocytes,34 vascular smooth muscle cells,35 and isolated neonatal rat cardiomyocytes.36 Significantly, one study has reported a close association between NGAL and high‐sensitivity C‐reactive protein, with concomitant reductions in both high‐sensitivity C‐reactive protein (−40%) and NGAL (−32%) following 8‐week treatment with rosiglitazone,8 an insulin‐sensitizer with interleukin‐1β antagonism effects.37, 38 However, it should be noted that this study was nonrandomized and had no control group as well as a limited sample size. Therefore, further investigations are needed in this area.
Aside from the role of NGAL in inflammation, studies suggest that NGAL is involved in iron homeostasis. Given that iron overload and deficiencies have been associated with impaired cardiac function and hypercoagulation, NGAL may therefore play a key role in development of certain cardiomyopathies. In addition, NGAL may play a role in regulating aldosterone‐mediated hypertension, with genetic inactivation of NGAL in mice preventing mineralocorticoid‐induced blood pressure increases. Furthermore, in vitro studies suggest that NGAL may influence the proliferation of fibroblasts. Given a key role of proliferation, particularly by myofibroblasts, in the mechanisms of remodeling, the proliferative effects of NGAL may also be involved in pathological cardiac remodeling.
With an aging population, the proportion of elderly patients undergoing percutaneous coronary intervention has increased considerably.39 The elderly are also at higher risk for procedure‐related complications and peri‐interventional mortality following percutaneous coronary intervention, despite appearing to derive significant benefits in terms of quality of life.39, 40 Furthermore, CHD screening investigations such as CT coronary angiography and exercise stress tests are less useful in this elderly population because of poor exercise capacity and coronary artery calcification–related artifact.41 NGAL levels in addition to cTnI levels in elderly patients may represent a future method to select elderly patients with atypical symptoms who would benefit the most from invasive coronary angiography.
This study has several limitations. First, blood samples were frozen at −80°C for 15 years before measurement of NGAL levels. Unfortunately, no studies have assessed the long‐term stability of NGAL at −80°C. However, the levels of plasma NGAL observed and the strong association between NGAL and outcomes in our study argue that there is sufficient stability to preserve the analyte. Second, NGAL levels were only measured at 1 time point (baseline). Serial measurements of NGAL may have provided additional risk stratification. Third, there is the potential for some misclassification of hospitalizations within the first 18 months of follow‐up because of International Classification of Diseases (ICD9) coding. Finally, this study was limited to older women aged 70 years and over and may not be generalizable to younger populations or older men. Nevertheless, this study has several strengths including being a well‐designed, longitudinal community‐based study with 14.5‐year follow‐up of clinical outcomes independent of self‐report, which is one of the largest studies with measurements of plasma NGAL and long‐term follow‐up.
Conclusions
In conclusion, we have demonstrated that circulating NGAL is independently associated with long‐term CHD outcomes in older women and may improve risk prediction in high‐risk older women.
Author Contributions
The authors had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: Chong, Lewis, Prince. Acquisition of data: Prince, Lim, Lewis, Byrnes. Analysis and interpretation of data: Lewis, Prince, Chong. Drafting of the manuscript: Chong, Prince, Lewis. Critical revision of the manuscript for important intellectual content: All authors. Statistical analysis: Lewis. Obtained funding: Lim, Prince, Lewis. Administrative, technical, and material support: Prince, Lewis, Chong. Study supervision: Prince, Lewis, Chong.
Sources of Funding
The study was supported by Healthway Health Promotion Foundation of Western Australia, Sir Charles Gairdner Hospital Research Advisory Committee Grant and by project grants 254627, 303169, and 572604 from the National Health and Medical Research Council of Australia. The salary of Dr Chong is supported by a Future Leader Fellowship (ID 100463) from the National Heart Foundation of Australia and Sydney Medical School Foundation Fellowship. The salary of Dr Lewis is supported by a National Health and Medical Research Council (Australia) Career Development Fellowship (ID 1107474). The salary of Dr Ooi is supported by a Future Leader Fellowship (ID 100422) from the National Heart Foundation of Australia. None of the funding agencies had any role in the conduct of the study; collection, management, analysis, or interpretation of the data; or preparation, review, or approval of the manuscript.
Disclosures
None.
Acknowledgments
The authors wish to thank the staff at the Western Australian Data Linkage Branch; Hospital Morbidity Data Collection; Registry of Births, Deaths and Marriages; Victorian Department of Justice and Regulation; and the National Coronial Information System for their work on providing the data for this study.
(J Am Heart Assoc. 2019;8:e011028 DOI: 10.1161/JAHA.118.011028)
References
- 1. Schmidt‐Ott KM, Mori K, Li JY, Kalandadze A, Cohen DJ, Devarajan P, Barasch J. Dual action of neutrophil gelatinase‐associated lipocalin. J Am Soc Nephrol. 2007;18:407–413. [DOI] [PubMed] [Google Scholar]
- 2. Hemdahl A‐L, Gabrielsen A, Zhu C, Eriksson P, Hedin U, Kastrup J, Thorén P, Hansson GK. Expression of neutrophil gelatinase–associated lipocalin in atherosclerosis and myocardial infarction. Arterioscler Thromb Vasc Biol. 2006;26:136–142. [DOI] [PubMed] [Google Scholar]
- 3. Cruz DN, Gaiao S, Maisel A, Ronco C, Devarajan P. Neutrophil gelatinase‐associated lipocalin as a biomarker of cardiovascular disease: a systematic review. Clin Chem Lab Med. 2012;50:1533–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eilenberg W, Stojkovic S, Piechota‐Polanczyk A, Kaun C, Rauscher S, Groger M, Klinger M, Wojta J, Neumayer C, Huk I, Demyanets S. Neutrophil gelatinase‐associated lipocalin (NGAL) is associated with symptomatic carotid atherosclerosis and drives pro‐inflammatory state in vitro. Eur J Vasc Endovasc Surg. 2016;51:623–631. [DOI] [PubMed] [Google Scholar]
- 5. Wu G, Li H, Zhou M, Fang Q, Bao Y, Xu A, Jia W. Mechanism and clinical evidence of lipocalin‐2 and adipocyte fatty acid‐binding protein linking obesity and atherosclerosis. Diabetes Metab Res Rev. 2014;30:447–456. [DOI] [PubMed] [Google Scholar]
- 6. Oberoi R, Bogalle EP, Matthes LA, Schuett H, Koch AK, Grote K, Schieffer B, Schuett J, Luchtefeld M. Lipocalin (LCN) 2 mediates pro‐atherosclerotic processes and is elevated in patients with coronary artery disease. PLoS One. 2015;10:e0137924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tarjus A, Martinez‐Martinez E, Amador C, Latouche C, El Moghrabi S, Berger T, Mak TW, Fay R, Farman N, Rossignol P, Zannad F, Lopez‐Andres N, Jaisser F. Neutrophil gelatinase‐associated lipocalin, a novel mineralocorticoid biotarget, mediates vascular profibrotic effects of mineralocorticoids. Hypertension. 2015;66:158–166. [DOI] [PubMed] [Google Scholar]
- 8. Wang Y, Lam KS, Kraegen EW, Sweeney G, Zhang J, Tso AW, Chow WS, Wat NM, Xu JY, Hoo RL, Xu A. Lipocalin‐2 is an inflammatory marker closely associated with obesity, insulin resistance, and hyperglycemia in humans. Clin Chem. 2007;53:34–41. [DOI] [PubMed] [Google Scholar]
- 9. Song E, Fan P, Huang B, Deng HB, Cheung BM, Feletou M, Vilaine JP, Villeneuve N, Xu A, Vanhoutte PM, Wang Y. Deamidated lipocalin‐2 induces endothelial dysfunction and hypertension in dietary obese mice. J Am Heart Assoc. 2014;3:e000837 DOI: 10.1161/JAHA.114.000837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Neumann JT, Havulinna AS, Zeller T, Appelbaum S, Kunnas T, Nikkari S, Jousilahti P, Blankenberg S, Sydow K, Salomaa V. Comparison of three troponins as predictors of future cardiovascular events—prospective results from the FINRISK and BiomaCaRE studies. PLoS One. 2014;9:e90063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lewis JR, Lim WH, Wong G, Abbs S, Zhu K, Lim EM, Thompson PL, Prince RL. Association between high‐sensitivity cardiac troponin I and cardiac events in elderly women. J Am Heart Assoc. 2017;6:e004174 DOI: 10.1161/JAHA.116.004174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ganguly P, Alam SF. Role of homocysteine in the development of cardiovascular disease. Nutr J. 2015;14:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Longitudinal Study of Ageing Women. 2016. http://www.lsaw.com.au/pages/view/about
- 14. Prince RL, Devine A, Dhaliwal SS, Dick IM. Effects of calcium supplementation on clinical fracture and bone structure: results of a 5‐year, double‐blind, placebo‐controlled trial in elderly women. Arch Intern Med. 2006;166:869–875. [DOI] [PubMed] [Google Scholar]
- 15. Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low‐density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18:499–502. [PubMed] [Google Scholar]
- 16. Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999;94:496–509. [Google Scholar]
- 17. Pencina MJ, D'Agostino RB Sr, Song L. Quantifying discrimination of Framingham risk functions with different survival C statistics. Stat Med. 2012;31:1543–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pencina MJ, D'Agostino RB Sr, D'Agostino RB Jr, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27:157–172; discussion 207‐12. [DOI] [PubMed] [Google Scholar]
- 19. Lindberg S, Jensen JS, Mogelvang R, Pedersen SH, Galatius S, Flyvbjerg A, Magnusson NE. Plasma neutrophil gelatinase‐associated lipocalinin in the general population: association with inflammation and prognosis. Arterioscler Thromb Vasc Biol. 2014;34:2135–2142. [DOI] [PubMed] [Google Scholar]
- 20. Daniels LB, Barrett‐Connor E, Clopton P, Laughlin GA, Ix JH, Maisel AS. Plasma neutrophil gelatinase‐associated lipocalin is independently associated with cardiovascular disease and mortality in community‐dwelling older adults: the Rancho Bernardo Study. J Am Coll Cardiol. 2012;59:1101–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zeller T, Tunstall‐Pedoe H, Saarela O, Ojeda F, Schnabel RB, Tuovinen T, Woodward M, Struthers A, Hughes M, Kee F, Salomaa V, Kuulasmaa K, Blankenberg S; MORGAM Investigators . High population prevalence of cardiac troponin I measured by a high‐sensitivity assay and cardiovascular risk estimation: the MORGAM Biomarker Project Scottish Cohort. Eur Heart J. 2014;35:271–281. [DOI] [PubMed] [Google Scholar]
- 22. Giaginis C, Zira A, Katsargyris A, Klonaris C, Theocharis S. Clinical implication of plasma neutrophil gelatinase‐associated lipocalin (NGAL) concentrations in patients with advanced carotid atherosclerosis. Clin Chem Lab Med. 2010;48:1035–1041. [DOI] [PubMed] [Google Scholar]
- 23. Elneihoum AM, Falke P, Hedblad B, Lindgarde F, Ohlsson K. Leukocyte activation in atherosclerosis: correlation with risk factors. Atherosclerosis. 1997;131:79–84. [DOI] [PubMed] [Google Scholar]
- 24. Lim WH, Lewis JR, Wong G, Teo R, Lim EM, Byrnes E, Prince RL. Plasma neutrophil gelatinase‐associated lipocalin and kidney function decline and kidney disease‐related clinical events in older women. Am J Nephrol. 2015;41:156–164. [DOI] [PubMed] [Google Scholar]
- 25. Willeit P, Welsh P, Evans JDW, Tschiderer L, Boachie C, Jukema JW, Ford I, Trompet S, Stott DJ, Kearney PM, Mooijaart SP, Kiechl S, Di Angelantonio E, Sattar N. High‐sensitivity cardiac troponin concentration and risk of first‐ever cardiovascular outcomes in 154,052 participants. J Am Coll Cardiol. 2017;70:558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lindberg S, Pedersen SH, Mogelvang R, Jensen JS, Flyvbjerg A, Galatius S, Magnusson NE. Prognostic utility of neutrophil gelatinase‐associated lipocalin in predicting mortality and cardiovascular events in patients with ST‐segment elevation myocardial infarction treated with primary percutaneous coronary intervention. J Am Coll Cardiol. 2012;60:339–345. [DOI] [PubMed] [Google Scholar]
- 27. Zografos T, Haliassos A, Korovesis S, Giazitzoglou E, Voridis E, Katritsis D. Association of neutrophil gelatinase‐associated lipocalin with the severity of coronary artery disease. Am J Cardiol. 2009;104:917–920. [DOI] [PubMed] [Google Scholar]
- 28. Moledina DG, Parikh CR, Garg AX, Thiessen‐Philbrook H, Koyner JL, Patel UD, Devarajan P, Shlipak MG, Coca SG; TRIBE‐AKI Consortium . Association of perioperative plasma neutrophil gelatinase‐associated lipocalin levels with 3‐year mortality after cardiac surgery: a prospective observational cohort study. PLoS One. 2015;10:e0129619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cook NR, Buring JE, Ridker PM. The effect of including C‐reactive protein in cardiovascular risk prediction models for women. Ann Intern Med. 2006;145:21–29. [DOI] [PubMed] [Google Scholar]
- 30. Cook NR. Use and misuse of the receiver operating characteristic curve in risk prediction. Circulation. 2007;115:928–935. [DOI] [PubMed] [Google Scholar]
- 31. Choi KM, Lee JS, Kim EJ, Baik SH, Seo HS, Choi DS, Oh DJ, Park CG. Implication of lipocalin‐2 and visfatin levels in patients with coronary heart disease. Eur J Endocrinol. 2008;158:203–207. [DOI] [PubMed] [Google Scholar]
- 32. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 33. Sommer G, Weise S, Kralisch S, Lossner U, Bluher M, Stumvoll M, Fasshauer M. Lipocalin‐2 is induced by interleukin‐1β in murine adipocytes in vitro. J Cell Biochem. 2009;106:103–108. [DOI] [PubMed] [Google Scholar]
- 34. Borkham‐Kamphorst E, Drews F, Weiskirchen R. Induction of lipocalin‐2 expression in acute and chronic experimental liver injury moderated by pro‐inflammatory cytokines interleukin‐1β through nuclear factor‐κB activation. Liver Int. 2011;31:656–665. [DOI] [PubMed] [Google Scholar]
- 35. Bu D‐x, Hemdahl A‐L, Gabrielsen A, Fuxe J, Zhu C, Eriksson P, Yan Z‐Q. Induction of neutrophil gelatinase‐associated lipocalin in vascular injury via activation of nuclear factor‐κB. Am J Pathol. 2006;169:2245–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yndestad A, Landrø L, Ueland T, Dahl CP, Flo TH, Vinge LE, Espevik T, Frøland SS, Husberg C, Christensen G. Increased systemic and myocardial expression of neutrophil gelatinase‐associated lipocalin in clinical and experimental heart failure. Eur Heart J. 2009;30:1229–1236. [DOI] [PubMed] [Google Scholar]
- 37. Moulin D, Bianchi A, Boyault S, Sebillaud S, Koufany M, Francois M, Netter P, Jouzeau JY, Terlain B. Rosiglitazone induces interleukin‐1 receptor antagonist in interleukin‐1β–stimulated rat synovial fibroblasts via a peroxisome proliferator–activated receptor β/δ–dependent mechanism. Arthritis Rheumatol. 2005;52:759–769. [DOI] [PubMed] [Google Scholar]
- 38. Burrage PS, Schmucker AC, Ren Y, Sporn MB, Brinckerhoff CE. Retinoid X receptor and peroxisome proliferator‐activated receptor‐gamma agonists cooperate to inhibit matrix metalloproteinase gene expression. Arthritis Res Ther. 2008;10:R139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bauer T, Mollmann H, Weidinger F, Zeymer U, Seabra‐Gomes R, Eberli F, Serruys P, Vahanian A, Silber S, Wijns W, Hochadel M, Nef HM, Hamm CW, Marco J, Gitt AK. Predictors of hospital mortality in the elderly undergoing percutaneous coronary intervention for acute coronary syndromes and stable angina. Int J Cardiol. 2011;151:164–169. [DOI] [PubMed] [Google Scholar]
- 40. Kaehler J, Koester R, Hamm CW, Meinertz T. Quality of life following percutaneous coronary interventions in octogenarians. Dtsch Med Wochenschr. 2005;130:639–643. [DOI] [PubMed] [Google Scholar]
- 41. Janowitz WR. Measurement of coronary artery calcium in elderly patients. Am J Geriatr Cardiol. 1999;8:215–224. [PubMed] [Google Scholar]