

Short abstract
Individuals with sickle cell disease have severe anemia due to the production of abnormal hemoglobin S, chronic red blood cell hemolysis, and increased oxidative stress leading to endothelial cell dysfunction, vasculopathy, and progressive organ damage. The transcription factor NRF2 (erythroid-derived 2)-like 2) is a master regulator of antioxidant proteins; under low oxidative stress, NRF2 is sequestered in the cytoplasm by Kelch-like ECH-associated protein 1, β-transducin repeat-containing protein or HRD1, and directed to the proteasome for degradation. When cells are exposed to oxidative stress, NRF2 is released from these repressor proteins, translocates to the nucleus, and activates antioxidant genes to suppress cellular reactive oxidant species and inflammation. In erythroid progenitors, NRF2 also modulates fetal hemoglobin expression through direct binding in the γ-globin promoter and modification of chromatin structure in the β-globin locus. In sickle erythroid cells, NRF2 provides unique benefits through fetal hemoglobin induction to inhibit hemoglobin S polymerization and protection against oxidative stress due to chronic hemolysis. Thus, development of small chemical molecules that activate NRF2 has the potential to ameliorate the clinical severity of sickle cell disease. In this review, we discuss progress towards understanding NRF2 regulation and strategies to develop agents for the treatment of sickle cell disease.
Impact statement
Sickle cell disease (SCD) is a group of inherited blood disorders caused by mutations in the human β-globin gene, leading to the synthesis of abnormal hemoglobin S, chronic hemolysis, and oxidative stress. Inhibition of hemoglobin S polymerization by fetal hemoglobin holds the greatest promise for treating SCD. The transcription factor NRF2, is the master regulator of the cellular oxidative stress response and activator of fetal hemoglobin expression. In animal models, various small chemical molecules activate NRF2 and ameliorate the pathophysiology of SCD. This review discusses the mechanisms of NRF2 regulation and therapeutic strategies of NRF2 activation to design the treatment options for individuals with SCD.
Keywords: Sickle cell disease, fetal hemoglobin, KEAP1, NRF2, oxidative stress
Introduction
Sickle cell disease (SCD) is a group of inherited blood disorders caused by mutations in the human β-globin gene, which encodes the β-chain subunit of adult hemoglobin A. The A to T mutation in codon 6 of β-globin leads to the synthesis of hemoglobin S (HbS), which under deoxygenated conditions undergoes polymerization. Red blood cells (RBCs) containing polymerized HbS become sickled shape causing chronic hemolysis, anemia, and vascular occlusion in vital organs including the brain, lungs, and kidney producing complications such as stroke and acute chest syndrome among others. The pathophysiology of organ damage involves the generation of inflammation and oxidative stress by sickle RBC formation.1
Hemolysis and oxidative stress in SCD pathophysiology
The hallmark of SCD is the high levels of plasma-free hemoglobin and heme from chronic hemolysis leading to increased serum lactate dehydrogenase and bilirubin. Intravascular hemolysis is associated with splenic dysfunction, leg ulcers, and priapism. Haptoglobin is a cyto-protective protein that neutralizes the toxic effects of heme; however, this protein becomes overwhelmed due to high heme levels,2 producing vascular endothelial dysfunction, nitric oxide (NO) depletion, and hypoxia–reperfusion injury.3
The bioavailability of NO in SCD is impaired due to excess free radical superoxide, and its consumption by cell-free heme generated during hemolysis. The limited bioavailability of NO impairs the scavenging of arginine normally metabolized to ornithine and urea by arginase. Several mechanisms contribute to the high oxidative burden in SCD including: (1) autoxidation of HbS; (2) RBC sickling and hemolysis; (3) hypoxia/reperfusion injury, (4) recurrent vaso-occlusion tissue injury; and (5) chronic inflammation. Reactive oxygen species (ROS) and reactive nitrogen species counter balance the sophisticated antioxidant protein systems to maintain redox homeostasis in cells. Many classes of antioxidant proteins exist including reduced glutathione, vitamins C and E, bilirubin and urate; the non-catalytic antioxidant proteins, such as thioredoxin, glutaredoxin, and metallothioneins; and the superoxide dismutase, catalase, peroxiredoxin, and glutathione peroxidase enzymes. Ultimately, redox reactions in cells are enabled from oxidized and reduced nicotinamide adenine dinucleotide phosphate (NADP+/NADPH) and nicotinamide adenine dinucleotide (NAD+/NADH), respectively. NADPH reduces oxidized thioredoxin and glutathione (GSH) through the actions of thioredoxin and glutathione reductase, respectively.4
The pathophysiology of SCD involves activation of multiple antioxidant genes to accomplish cellular protective mechanisms. For instance, high plasma-free heme stimulates heme oxygenase 1 (HMOX1)5 expression to convert heme to carbon monoxide and biliverdin producing vasodilation and cytoprotection.6 The high oxidant burden in SCD activates NAD(P)H quinone oxidoreductase 1 (NQO1) expression as a scavenger of superoxide anion to prevent sickle vasculopathy.7–9
Central role of nuclear factor NRF2 (erythroid-derived 2)-like 2) in controlling oxidative stress
The transcription factor NRF2 was originally identified as a DNA binding protein in the β-like globin gene (HBB) locus and later characterized as the major regulator of oxidative stress.10 Systematic and complementary genome wide analyses showed NRF2 binds the consensus antioxidant response element (ARE) 5′-TGACnnnGC-3′ in the proximal promoters of a wide variety of genes (Figure 1(a)).11 The ARE motif resembles the DNA-binding elements of other basic leucine zipper domain proteins such as AP-1, NFE2, and MAF.12 Protein–protein interaction studies demonstrated that NRF2 heterodimerizes with small MAF proteins to bind DNA and activate gene transcription (Figure 1(b)). Moreover, NRF2 competes with the transcription factor BACH1 (tBTB Domain and CNC Homolog 1),13– a member of cap ‘n’ collar protein, for binding in the ARE motifs of antioxidant genes to regulate the cellular oxidative stress levels.16,17
Figure 1.

NRF2 as the master regulator of cytoprotective responses. (a) Shown is the NRF2 consensus binding sequences, known as the antioxidant response element (ARE). (b) NRF2 heterodimerizes with MAF family proteins through the basic leucine zipper (bZip) domain to bind the ARE of target genes. (c) NRF2 regulates a wide variety of genes involved in redox metabolism, inflammation, and protein hemostasis among other cellular processes. ACC1: acetyl-coenzyme A carboxylase 1; CD36: cluster of differentiation 36; FAS: fatty acid synthase; G6PD: glucose-6-phosphate dehydrogenase; Gpx8: glutathione peroxidase 8; HMOX1: heme oxygenase-1; GCLC: glutamate-cysteine ligase catalytic subunit; GCLM: glutamate-cysteine ligase: modifier subunit; HMOX1: heme oxygenase 1; IDH1: isocitrate dehydrogenase 1; IL6: interleukin 6; IL17D: Interleukin 17D; NQO1: NAD(P)H dehydrogenase (quinone) 1; ME1: malic enzyme 1; MTHFD2: methylenetetrahydrofolate dehydrogenase 2; PGD: phosphogluconate dehydrogenase; PPAT: phosphoribosyl pyrophosphate amidotransferase; SCD1: stearoyl-CoA desaturase; SQSTM: equestosome-1. (A color version of this figure is available in the online journal.)
The multi-functional transcription factor NRF2 possesses complementary and overlapping characteristics to control gene expression (Figure 1(c)). For example, NRF2 regulates the enzymes involved in GSH production including glutamate–cysteine ligase complex modifier subunit (GCLM), GCL catalytic subunit (GCLC), cysteine/glutamate transporter XCT, and glutathione reductase.18 Other proteins required to synthesize GSH such as glutathione S-transferases and glutathione peroxidase 2 are also regulated by NRF2.11 In addition, several genes involved in the production, regeneration, and utilization of thioredoxin such as thioredoxin reductase 1 and peroxiredoxin 1 are regulated by NRF2.19,20 Enzymes required for NADPH production including glucose-6-phosphate dehydrogenase, phosphoglycerate dehydrogenase, malic enzyme 1, and isocitrate dehydrogenase 1 are targets of NRF2 regulation,21 along with the major antioxidant response proteins NQO1, HMOX1, and ferritin.22 NRF2 also activates the expression of a large number of genes necessary for purine and lipid metabolism, inflammation, and proteostasis discussed below.21
Regulation of NRF2
Role of the ubiquitin proteasome system in NRF2 regulation
One of the key mechanisms controlling NRF2 expression is protein stability, involving three signaling pathways comprising KEAP1 (Kelch-like ECH-associated protein 1), HRD1, and β-TrCP (β-transducin repeat-containing protein). These proteins facilitate NRF2 proteasomal degradation by different mechanisms. The structure of NRF2 contains six conserved domains, Neh1–Neh6 (Figure 2(a)).23 The Neh1 domain is a basic leucine zipper motif critical for NRF2 dimerization with small MAF proteins and facilitates DNA binding. KEAP1 interacts in the Neh2 domain containing the DLG and ETGE motifs. The Neh4 and Neh5 domains are important for the transactivation activity of NRF2 and serve as the site for HRD1 binding. The Neh6 domain of NRF2 is a serine-rich region containing the DSGIS and DSAPGS motifs that interact with β-TrCP.
Figure 2.

Regulation of NRF2 stability by different E3 ubiquitin ligase complexes. (a) Shown is a schematic structure of the NRF2 protein domains such as Neh1 involved in small MAF heterodimerization and the transactivation domains Neh4 and Neh5 for interacting the co-activators CBP/p300. Also depicted are the individual domains in NRF2 required for interaction with KEAP1, HRD1, and β-transducin repeat-containing protein (β-TrCP). (b) KEAP1-mediated NRF2 repression. Under basal conditions, NRF2 activity is repressed through ubiquitination (Ub) via cullin-dependant E3 ubiquitin ligase Cul3, bound to Kelch-like ECH associated protein 1 (KEAP1). Multiple cysteines residues in KEAP1 including C151, C273 and C288 are required for its interaction with the DLG and ETGE motifs in the Neh2 domain of NRF2. When cysteine residues are modified by electrophiles or ROS, the conformation structure of KEAP1 is altered and interactions with NRF2 disrupted; the net result is reversal of NRF2 proteasomal degradation. (c) HRD1-mediated NRF2 repression. HRD1 is the E3 ubiquitin ligase, which resides in the ER. HRD1 ubiquitylates NRF2 after interacted with the Neh4 and Neh5 domains. (d) β-TrCP-mediate NRF2 repression. Shown is a schematic of NRF2 repression by β-TrCP, which interacts in the Neh6 domain of NRF2 comprising DSGIS and DSAPGS. The DSGIS motif contains a functional GSK-3 phosphorylation site that mediates NRF2 degradation. (e) Mechanism of KEAP1 inactivation that induce γ-globin gene expression. Several cysteine residues in KEAP1 are critical for interaction with NRF2; once modified through various mechanisms, disruption of interaction between KEAP1 and NRF2 occurs, which inhibits polyubiquitination of NRF2. Stabilized NRF2 undergoes nuclear translocation to activate target genes containing the antioxidant response element (ARE) including the antioxidant and γ-globin genes among others. (A color version of this figure is available in the online journal.)
Canonical KEAP1–Cul3–Rbx1 E3 ubiquitin ligase
The KEAP1–Cul3–Rbx1 E3 complex is the major regulator of NRF2 degradation (Figure 2(b)). Under normal cellular stress conditions, NRF2 is sequestered in the cytoplasm by KEAP1 and directed for ubiquitination by the E3 ligase complex. RING box protein 1 recruits the catalytic function of ubiquitin-conjugating enzyme E2 after binding the C terminal region of Cul3. The polyubiquitination of NRF2 on the lysine residues in the Neh2 domain is catalyzed by E2 and KEAP1, which promotes rapid NRF2 degradation. Several cysteine residues including C151, C273, and C288 of KEAP1 are critical for interaction with NRF2. Modifications of these critical cysteine sites by electrophiles lead to conformational changes in KEAP1 disrupting interaction with NRF2, thus inhibiting its silencing effects.
Cysteine residues in KEAP1 are modified by endogenous metabolites such as itaconate and succinate. Itaconate regulates macrophage function and activates NRF2 to produce an anti-inflammatory effect by modifying KEAP1 cysteine residues via alkylation. Once activated, NRF2 stimulates the expression of downstream antioxidant and anti-inflammatory genes.24 Cysteine residues in KEAP1 are also modified through succination to enhance NRF2 protein stability.25,26 Other mechanisms of KEAP1-inactivation involve autophagy-related protein p6227,28 which is a scaffold protein that sequesters KEAP1 in the autophagosome to inhibit its function.29
HRD1 E3 ubiquitin ligase in endoplasmic reticulum
The protein synoviolin is an endoplasmic reticulum (ER) membrane-associated E3 ubiquitin ligase recently identified as a negative regulator of NRF2 during cirrhosis.30 Cirrhotic livers are characterized by increased ROS production and ER stress, causing perturbation of the unfolded protein response.31 The XBP1–HRD1 arm of the ER stress pathway when activated, upregulates HRD1 expression, producing enhanced NRF2 ubiquitylation and degradation to attenuate the antioxidant response (Figure 2(c)). Interaction of HRD1 with the Neh4 and Neh5 domains of NRF2 mediates degradation, which is independent of KEAP1 promoting high ROS level in cirrohosis.30
GSK-3β/β-TrCP–Skp1–Cul1–Rbx1 E3 ubiquitin ligase
The discovery of a redox-insensitive degron in the Neh6 domain of NRF2 led to the identification of a new E3 ubiquitin ligase complex involved in degradation.32 The Neh6 domain is recognized by the F box WD40 substrate adaptor β-TrCP33; phosphorylation of β-TrCP by GSK3β greatly increases its affinity for NRF2 (Figure 2(d)).34,35 Once activated, β-TrCP binds to the SKP1-CUL1-RBX1 E3 ubiquitin ligase complex and ubiquitinates NRF2 in a KEAP1-independent manner to promote degradation.33,34,36
Although three pathways play a role in NRF2 post-translational regulation, KEAP1 has been investigated most extensively for its role in erythroid differentiation and hemoglobin switching (Figure 2(e)). Small chemical compounds such as tert-Butylhydroquinone (tBHQ), diethylmaleate, and 2-cyano-3,12-dioxo-oleana-1,9(11)-dien-28-oic acid (CDDO-Im) modify KEAP1 cysteine residues,37 to disrupt interactions with NRF2. Once stabilized, NRF2 translocates to the nucleus to activate downstream genes containing ARE motif such as γ-globin and the antioxidant genes.
Regulation of NRF2 at the protein translation levels by microRNA genes
The miRNA genes encode small non-coding RNA molecules that function in mRNA silencing and post-transcriptional regulation. Several miRNA genes are involved in NRF2 silencing including miR-153, miR-27a, miR-142-5p, miR-144, miR-28, and miR-34a.38–42 Related to erythroid development, miR-144 decreases NRF2 expression in K562 cells, human erythroid progenitors, and SCD reticulocytes.43 The net effect of increased miR-144 expression and reduction of NRF2 protein levels is the altered expression of GSH44 and the antioxidant response capacity.45,46 Narasimhan et al.47 demonstrated that ectopic expression of miR-144, miR-153, miR-27a, and miR-142-5p decreased NRF2 levels in neuronal cells, leading to reduced GSH. Other miRNAs such as miR-27b repressed NRF2 expression during intracerebral hemorrhage observed in rat brain injury.48 These findings suggest a possible role for these miRNA genes in controlling cellular oxidative stress through NRF2 regulation.
Regulation of NRF2 gene transcription
The transcriptional regulation of NRF2 involves the oncogenes K-Ras, B-Raf, and Myc49 although the precise mechanism is unknown. Recently, two groups demonstrated activation of NRF2 by K-rat sarcoma through a 12-O-tetradecanoylphorbol-13-acetate-responsive element in the NRF2 gene promoter.50,51 In addition, there are three xenobiotic response-like elements in the NRF2 gene that are bound by the aryl hydrocarbon receptor. NRF2 transcription is activated by aryl hydrocarbon receptor binding, while siRNA-mediated gene silencing of the receptor reduced NRF2 mRNA levels and activation of downstream antioxidant genes.52,53
NRF2 in SCD mouse models
NRF2 loss exacerbated the SCD phenotype
Since NRF2 activation inhibits age-related progression of intravascular hemolysis, vascular inflammation, and pulmonary damage in mice, it follows that NRF2 deficiency would accelerate these complications in SCD. To test this hypothesis, Ofori-Acquah and colleagues54 created chimeric SCD mice lacking NRF2 expression in non-hematopoietic tissues that caused early death. In addition, these mice had high plasma hemoglobin, heme, and lactate dehydrogenase levels contributing to progressive intravascular hemolysis,55 and pulmonary edema to exacerbate the SCD phenotype.
To elucidate the role of NRF2 in erythropoiesis, our group established an NRF2 knockout SCD mouse model56 where impaired in utero survival of SCD pups and fetal hemoglobin (HbF) expression was observed in embryonic day 13.5- and 18.5-day fetal liver, adult spleen, and bone marrow. As expected, NRF2 loss led to an increase of ROS and RBC sickling under hypoxic conditions and greater splenomegaly with red pulp expansion.56 In addition, NRF2 loss in SCD mice reduced the expression of antioxidant proteins NQO1, HMOX1, and catalase, causing increased pro-inflammatory cytokines IL6, IL1β, and TNFα, and the adhesion molecules ICAM1 and VCAM-1 levels. These observations demonstrated a role of NRF2 in the developmental regulation of γ-globin and its ability to control the oxidative stress and phenotypic severity of SCD.
Genetic and chemical NRF2 activation in the SCD mouse model
Recently, KEAP1 ablation to produce constitutive NRF2 activation was achieved in the SCD mouse model.57 KEAP1 ablation improved the SCD phenotype as demonstrated by a decrease in pro-inflammatory cytokines and adhesion molecules levels. Notably, after KEAP1 ablation, heme levels were reduced and oxidative stress was inhibited. The inflammatory cytokines such as interleukin-6 and interleukin-1β were suppressed, while liver fibrosis was reversed. Moreover, when SCD mice were treated with the NRF2 inducer CDDO-Im, a reduction of inflammation was observed along with improved organ function.57 Similarly, Ghosh et al.55 demonstrated that D3T (3H-1, 2-dithiole-3-thione), another small chemical inducer of NRF2, decreased acute chest syndrome and mortality in SCD mice. Our group treated SCD mice with the NRF2 inducer, dimethyl fumarate (DMF), which induced HbF expression and reduced RBC sickling under hypoxia conditions.58 Chronic DMF administration induced expression of NRF2-dependent antioxidant genes to detoxify heme and suppressed inflammation. These results support the notion of NRF2 induction as a therapeutic approach to induce HbF expression, reduce oxidative stress, and preserve organ function in SCD.57
Mechanisms of globin gene regulation by NRF2
NRF2 binds the HBB locus to alter chromatin structure and γ-globin gene expression
Multiple transcription factors are involved in the regulation of the five major globin genes located in the HBB locus on chromosome 11. To elucidate the mechanisms of drug-mediated γ-globin activation, studies conducted by Lowrey and colleagues59 demonstrated enhanced NRF2 binding in the γ-globin promoter after tBHQ and simvastatin treatment. Deletion of a critical region 100 bp upstream of the γ-globin transcription start site, 5′-TGACAAGGC-3′, abolished the HbF induction by these agents. Our group investigated the ability of DMF to activate γ-globin expression; we demonstrated HbF induction in human erythroid progenitors through NRF2 binding in the γ-globin gene ARE.60 These small chemical compounds alter NRF2 protein stability by different mechanisms to induce HbF expression in erythroid progenitors. Through a JASPAR61 software search, we identified 23 NRF2 consensus ARE motifs – TGAnnnnGC in the HBB locus (Figure 3). Subsequently, NRF2 was demonstrated to bind the HBB locus control region and γ-globin promoter (Figure 4), which correlates with gene transcription through long-range chromatin looping to regulate globin gene expression during hemoglobin switching.60
Figure 3.

Predicted NRF2 binding sites across the human β-like globin (HBB) locus. ENCODE ChIP-seq data generated using K562 cells were downloaded from the UCSC server to discover NRF2 consensus binding motif across the HBB locus (https://genome.ucsc.edu/cgi-bin/hg).61 The location for human β-like globin genes (ε-, Gγ-, Aγ-, δ- and β-globin) and locus control region (LCR) are shown. The histone active chromatin marks H3K4Me3 (histone 3 lysine 4 trimethylation) and H3K27Ac (lysine 27 acetylation) and the repressive marks H3K9Me3 (histone 3 lysine 9 trimethylation) and H3K27Me3 (lysine 27 trimethylation) are shown by the black and gray horizontal lines. The blue peaks represent DNaseI hypersensitivity sites (DNaseI HS). The ENCODE data were modified with predicted antioxidant response element (ARE) motifs (black bar) with the general consensus sequence 5′-TGACnnnGC-3′. (A color version of this figure is available in the online journal.)
Figure 4.

Proposed beneficial effects of NRF2 function in SCD. Shown is a model of the role of oxidative stress in SCD. High reactive oxygen species occur in SCD due to HbS polymerization, RBC sickling, chronic hemolysis, and high reactive oxygen species. The net result is inactivation of KEAP1, HRD1 or β-TrCP, which promote NRF2 stabilization and nuclear translocation. Once bound to the ARE, NRF2 regulates a wide variety of genes such as γ-globin to induce HbF and the antioxidant genes (e.g. HMOX1, NQO1) to suppress oxidative stress and ameliorate the pathophysiology of SCD. Under NRF2 knockout conditions, preclinical data generated in SCD mice demonstrated exacerbation of oxidative stress and γ-globin gene silencing, leading to increased severity of the pathophysiology of SCD. (A color version of this figure is available in the online journal.)
Indirect effects of NRF2 in globin gene regulation
Other than direct regulation in the HBB locus, NRF2 and drug inducers alter the expression of the transcription factors KLF1 and BCL11A.62 KLF1 plays a major role in developmentally regulated globin gene switching and activates adult β-globin expression, while BCL11A is the major repressor of γ-globin during erythropoiesis. Treatment with tBHQ induced HbF expression via NRF2 activation with significant downregulation of KLF1 and BCL11A. In addition, using HOMER software, Lessard et al.63 illustrated that NRF2 binding sites are enriched near CpGs hypomethylated dinucleotides in adult erythroblasts with an increase of hydroxymethylcytosine marks during commitment to the erythroid lineage. These studies support the direct and indirect regulation of globin gene expression by NRF2 through DNA binding in the γ-globin promoter and locus control region, and silencing of the major repressors KLF1 and BCL11A, and epigenetic DNA methylation.
NRF2 regulation of multiple cellular protein targets influence SCD phenotype
Heme biosynthesis
A major contributor to the pathophysiology of SCD is the elevated plasma heme levels due to chronic RBC hemolysis, and as a consequence, there is a significant increase in HMOX1 levels in adults with SCD.64 HMOX1 is the rate limiting enzyme in the degradation of heme, converting heme to biliverdin, with the release of iron and carbon monoxide, which is regulated by NRF2 in multiple cellular systems.16 In SCD mouse models, NRF2 activation significantly increased HMOX1 expression,57 while studies from our group showed NRF2 knockout silenced HMOX1 expression and increased severity of the SCD phenotype (Figure 4).56 Moreover, in SCD mice treated with DMF,58 HMOX1 expression was activated and amelioration of the SCD phenotype was observed. In a Phase I clinical trial, treatment with broccoli sprout homogenate containing the NRF2 activator, sulforaphane, increased the peripheral blood mRNA levels for HMOX1 in SCD patients.65
There are other proteins regulated by NRF2 involved in heme biosynthesis such as the ATP binding cassette subfamily B member 6 (ABCB6)66 and ferrochelatase.11 ABCB6 imports porphyrins, such as coproporphyrinogen III, from the cytosol to the mitochondria to support heme synthesis, while ferrochelatase participates in the final step of heme biosynthesis.67 By microarray analysis, it was demonstrated that ABCB6 gene is transcriptionally regulated by NRF2 in arsenic exposed human lymphoblastoid cells68 and hepatocytes cells.66 Ferrochelatase catalyzes the insertion of ferrous iron into protoporphyrin IX to produce heme and is regulated by NRF2 in a gene dose-response model.69,70 While NRF2 controls the biological utilization of oxygen and iron by facilitating the transcription of genes that incorporate iron into heme, NRF2 also regulates the mobilization of iron during hemoglobin degradation. Expression of the heme-responsive transporter gene was decreased in NRF2 knockout mice, while drug-mediated activation of NRF2 increased its expression at the mRNA and protein levels.71–73
Apart from heme-bound iron, cells maintain a pool of labile iron to accomplish other biosynthetic processes such as iron–sulfur cluster generation. Individuals with SCD on chronic transfusion regimens suffer hemosiderosis from iron overload due to inability of the body to excrete iron. This leads to the formation of oxygen-derived free radicals, such as hydroxyl radical, through the Fenton reaction. While upregulation of ferritin gene transcription by NRF2 alters iron homeostasis by increasing storage and decreasing labile iron, NRF2 also buffers labile iron by altering its flux across cell membranes.74,75
NRF2 regulates GSH synthesis
NRF2 controls the expression of enzymes responsible for producing GSH, which is the most abundant antioxidant cofactor in cells.76 NRF2 drives the expression of two GSH subunits including the GCL complex comprising GCLM and catalytic GCLC components. GCL catalyzes the reaction of glutamate with cysteine to form the dipeptide gamma-glutamylcysteine, which is the rate-limiting step in GSH synthesis. In addition, NRF2 controls the abundance of cysteine in cells through activation of the solute carrier family 7 members 11, which encodes the cysteine/glutamate transporter XCT.77
NRF2 involved in glutamine metabolism
Glutamine is a nonessential amino acid converted to glutamate to provide anti-oxidative activity that plays an important role in cellular energy homeostasis. Glutamine is converted to α-ketoglutarate, a citric acid cycle intermediate that is a cofactor for dioxygenase proteins such as ten-eleven translocation enzymes,78 and prolyl hydroxylases among others.79 Moreover, glutamine is required for the synthesis of nicotinamide adenine dinucleotide in response to ROS stress in sickle RBCs.80,81 Recent clinical trials demonstrated the beneficial effects of glutamine in preventing SCD vaso-occlusive crises.81 SCD patients treated with L-glutamine showed significant increases in NADH and NAD redox potential. Furthermore, L-glutamine decreased the endothelial adhesion of sickle RBCs in vitro, suggesting the clinical improvement most likely occurred through oxidative stress mechanisms. The regulation of glutamine by NRF2 was also investigated in cancer cells where NRF2 activation increased dependency on exogenous glutamine through enhanced glutamate consumption to achieve GSH synthesis.82,83 The ability of NRF2 to regulate glutamine metabolism is another reason to support the development of small molecule activators for the treatment of SCD.
NRF2 regulation by microRNA genes during erythropoiesis
Recently, the miR-144 and miR-451 (miR-144/451) gene locus was demonstrated to play a major role in erythroid differentiation, homeostasis, and fine-tuning gene expression in erythroid cells.84,85 Interestingly, in adults with SCD increased miR-144 expression decreased NRF2 and GSH levels, and was associated with severe anemia.46 Moreover, low NRF2 expression was associated with a lack of the antioxidant proteins GCLC/M and superoxide dismutase 1.86 We performed genome-wide analysis to discover miRNA genes associated with HbF expression in individuals with SCD. We observed an 8-fold up-regulation of miR-144 in patients with low HbF group compared to those with high HbF.87 Functional studies in adult erythroid cells showed NRF2 gene silencing by miR-144 and concomitant repression of γ-globin expression; by contrast, treatment with antagomir that inhibits miR-144 reversed its silencing effects.88 Additional studies have shown miR-28 inhibits NRF2 through a KEAP1-indepenedent mechanism and similarly miR-153, miR-27a, and miR-142-5p down-regulate NRF2 expression.38–40 These studies support miRNA-mediated mechanisms of NRF2 regulation in γ-globin expression.
Development of NRF2 drug inducers to activate fetal hemoglobin expression
Various pharmacologic agents increase NRF2 levels via inactivation of KEAP1 resulting in NRF2 translocation into the nucleus and transcriptional activation of antioxidant genes.89 Among these agents, several small chemical compounds have been explored to induce HbF expression in erythroid progenitors, SCD mice, and individuals with SCD through NRF2 activation (Table 1).
Table 1.
Summary of drugs that induce HbF through the Keap1/NRF2 pathway.
| Drug | Structure | Effect | References |
|---|---|---|---|
| Sulforaphane (SFN) |

|
Induce HbF expression in primary erythroid progenitors; increase mRNA levels of NRF2 target HMOX1 gene in SCD patients | Doss et al.65 |
| Dimethyl fumarate (DMF) |
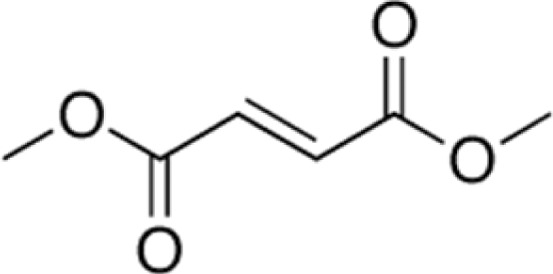
|
Induce HbF in normal and sickle erythroid progenitors; induce HbF in SCD mice; decrease inflammation factor expression in SCD mice. | Zhu et al.60 Krishnamoorthy et al.58 Belcher et al.90 |
| Tertiary butylhydroquinone (tBHQ) |
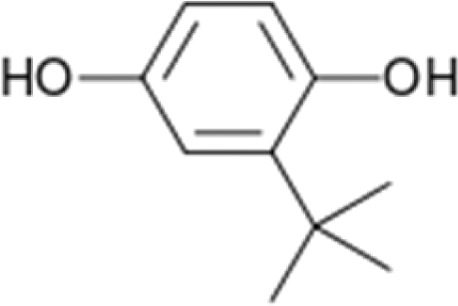
|
Induces NRF2 activation and binding to γ-globin promoter; Increase γ-globin expression and HbF level in K562 and primary erythroid progenitors; increased NOx levels in SCD patients | Macari et al.62 Hoppe et al.91 |
| Simvastatin |
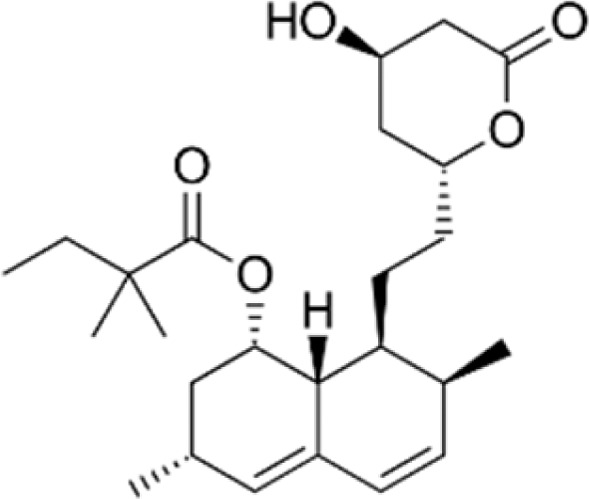
|
Increase γ-globin expression and HbF level in K562 and primary erythroid progenitors by NRF2 activation and inhibits β-globin transcription by suppression KLF1 and BCL11A | Macari and Lowrey59 |
| MLN9708 |
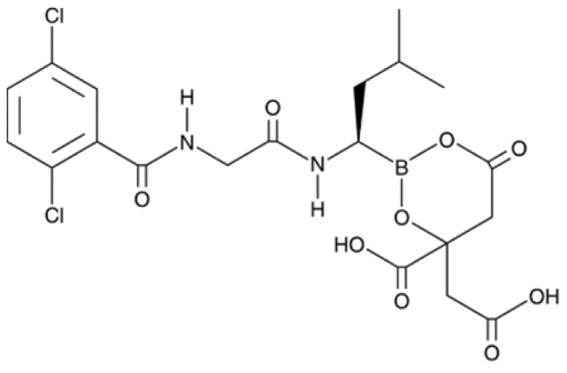
|
Inhibits proteasome mediated ubiquitination and degradation of NRF2; induces ROS generation and antioxidant response in B-CFU cells from SCD patient; induces NRF2 nuclear localization; Increases HbF level in K562 cells | Kupperman et al.92 Pullarkat et al.93 |
| N-acetylcysteine (NAC) |
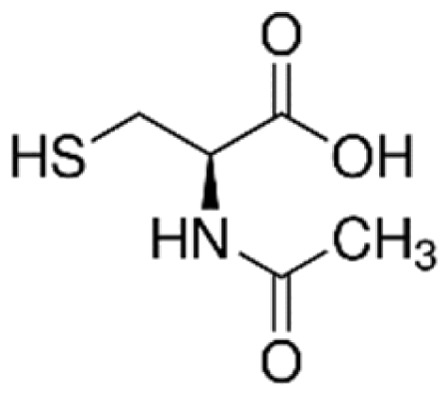
|
Induce GSH biosynthesis; | Dodd et al.94 |
| 3H-1,2-dithiole-3-thione (D3T) |
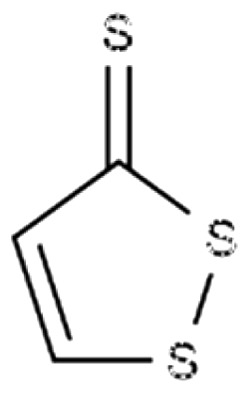
|
Induce GSH biosynthesis; modulate inflammatory response in SCD mouse | Manandhar et al.95 Ghosh et al.54 |
NRF2: Nuclear factor (erythroid-derived 2)-like 2; SCD: sickle cell disease; HbF: fetal hemoglobin.
Sulforaphane is an organosulfur compound obtained from cruciferous vegetables such as broccoli sprouts and cabbages. Treatment of SCD erythroid progenitors with this agent activates NRF2 expression and improved GSH levels.96 In a recent Phase I trial completed in adults with SCD,65 sulforaphane increased the HMOX1 mRNA levels and produced a trend toward inducing HbF expression.
DMF is a methyl ester of fumarate which was originally approved as oral therapy for psoriasis97 and later multiple sclerosis.98 The primary metabolite of DMF is monomethyl fumarate that activates NRF2 and several antioxidant genes.99 Our group demonstrated the ability of DMF to induce HbF in normal and sickle erythroid progenitors and SCD mice via NRF2 activation.60 SCD mice treated with DMF displayed less vaso-occlusion with improved oxidative stress tolerance. DMF increased nuclear translocation of NRF2 and transcription of NRF2-responsive genes in SCD mouse livers and kidneys.58 DMF also increased the expression of genes associated with oxidative stress including HMOX1, haptoglobin, hemopexin, and ferritin heavy chain and decreased markers of inflammation, such as nuclear factor-kappa B phospho-p65, and toll-like receptor 4. Chronic DMF administration decreased the hepatic necrosis in SCD mice, and the levels of inflammatory cytokines,97 supporting the development of DMF for the treatment of SCD.
The synthetic aromatic organic derivative of hydroquinone tBHQ induces the expression of NRF2, HMOX1, GCLC, and NQO192 and inhibits the NFκB DNA binding in peripheral blood mononuclear cells.100 In normal erythroid progenitors, tBHQ induces HbF expression through NRF2 binding to the γ-globin ARE.62 Moreover, tBHQ silences the adult β-globin gene, KLF1, and BCL11A when combined with simvastatin via the PI3K/AKT signaling pathway.62,90
Simvastatin (Zocor) is a statin drug developed to decrease cholesterol levels in humans by inhibiting the function of HMG-CoA reductase.101 This agent modulates ROS levels, NRF2 activation, and PI3K/AKT signaling. It was also demonstrated using human erythroid progenitors that simvastatin treatment increased NRF2 binding in the HBB locus control region.59 A recent Phase I/II clinical trial with short-term simvastatin treatment in SCD patients showed increased NO levels, and decreased C-reactive protein and interleukin-6 expression. However, no effects on vascular endothelial growth factor, vascular cell adhesion molecule-1 or tissue factor protein levels were observed.91
MLN9708 is a second-generation oral proteasome inhibitor of MLN2239. This prodrug is hydrolyzed in plasma to the biologically active form, which inhibits a subunit of the 20S proteasome.92 The oral bioavailable prodrug of MLN9708 regulates the NRF2/KEAP1/Cul3-Rbx1 pathway to induce HbF. To confirm the ability of this agent to induce HbF via NRF2 activation, erythroid progenitors from SCD patients were treated with NRF2 siRNA, which silenced HbF expression.93
Another potent antioxidant agent, N-acetylcysteine increases GSH94 and NRF2 levels to promote nuclear translocation and activation of downstream target genes.102 N-acetylcysteine is currently under investigation in adults with SCD to determine its effect on blood coagulation, specifically von Willebrands factor and oxidative stress (NCT01849016). Compared to steady-state levels, the concentration of cysteine and GSH in blood increased rapidly after N-acetylcysteine infusion with a decrease in RBC fragments and dense cells.103,104 Since a limited number of SCD patients participated in this trial, additional data are needed to validate these findings.
The sulfur-containing compound dithiolethione D3T found in cruciferous vegetables was shown to induce NRF2 and GSH biosynthesis.95 In addition, D3T modulates inflammatory diseases including endo-toxemic shock, multiple sclerosis, and light-induced retinal dysfunctions in animal models.54 Although NRF2 pathway activation was discovered by D3T treatment,95 this agent has not been investigated as an HbF inducer.
Summary and future directions
James Herrick reported the first case of sickle cell anemia in 1910, followed many years later by discovery of the genetic defect by Linus Pauling in 1949. After several decades of research, the first HbF inducing agent hydroxyurea was FDA-approved in 1998 for the treatment of vaso-occlusive pain in adults with SCD.105 Almost two decades later, in 2017, Endari (L-glutamine) became the second FDA-approved drug for the treatment of pain episodes in children and adults with SCD.81 Despite slow progress, the number of studies to develop new therapies for SCD has exploded, with over 40 active clinical trials registered in Clinical Trials.Gov (https://clinicaltrials.gov/). A wide variety of small molecules are under development that target the downstream detrimental effects of sickle RBC formation including chronic hemolysis, NO depletion, and vasculopathy.55,57–59,65 For example, the Phase II SUSTAIN trial of crizanlizumab (SelG1) tested a humanized anti-P-selectin antibody that prolonged the time to first pain crisis in SCD patients106; subsequently, SelG1 was shown to decrease the number of sickle cell pain crises over time.107 Several other agents should emerge from these widespread clinical efforts establishing a repertoire of effective agents to develop combination therapy for SCD.
Historically, the most successful treatment for SCD involved HbF induction and inhibition of HbS polymerization in RBCs to ameliorate the clinical severity. Two major classes of agents including histone deacetylase108 and DNA methyl transferase109 inhibitors are under development. For example, the histone deacetylase inhibitor arginine butyrate induced HbF in SCD and β-thalassemia patients; however, butyrate requires intravenous administration hindering further clinical development. The short chain fatty acid derivative 2,2-dimethylbutyrate (HQK-1001) induced HbF in individuals with β-thalassemia,110 but failed to induce in SCD patients.111 Currently, the most promising HbF inducer is the DNA methyl transferase inhibitor decitabine; however, clinical development was hampered by drug inactivation when given by oral administration. The recently completed Phase II trial demonstrated decitabine combined with tetrahydrouridine effectively blocks the metabolism to mediate HbF induction in SCD patients.112
Our group and others have focused on the development of chemical activators of NRF2 to produce antioxidant effects and HbF induction to ameliorate the clinical severity of SCD. Although transcription factors are difficult to target for clinical treatment, NRF2 is unique since it is sequestered in the cytoplasm by KEAP1, β-TrCP, and HRD1, allowing the development of agents that inactivate these negative regulatory proteins. For example, DMF modifies cysteine 151 in KEAP1 to inactivate the enzyme and mediate NRF2 stabilization and translocation to the nucleus where γ-globin gene activation occurs.59,60 Tecfidera® (DMF) is FDA-approved and used routinely to treat multiple sclerosis where it mediates potent immune modulation and anti-inflammatory effects. Future clinical trials are required to establish whether DMF induces HbF in SCD patients.
Another, NRF2 activators sulforaphane was tested for in vivo effects in a Phase I clinical trial.65 HbF induction was not observed in SCD patients; however, the lack of response might be due to low concentrations of sulforaphane in homogenized broccoli sprouts. Small chemicals that directly inhibit KEAP1 such as DMF might be more efficacious. Other potential agents for development as HbF inducers include CDDO-Im, a cancer chemotherapy agent113 and D3T under preclinical development.55 Both agents are potent NRF2 activators. Several miRNA genes that target NRF2 have the potential for targeted treatment of SCD. We recently demonstrated NRF2 activation and HbF induction by miR-144 antagomir in sickle erythroid progenitors88; other miRNA genes such as miR-153, miR-27a, miR-142-5p, miR-28, and miR-34a38–42,114, have the potential for clinical development.
The recent explosion of clinical trials to develop additional effective therapies for SCD has challenged patient recruitment efforts in major medical centers. In an effort to address this hindrance to moving forward new therapies for SCD, the American Society of Hematology (ASH) will establish the ASH Research Collaborative to accelerate progress in hematology. One such collaborative will be comprised of sickle cell disease treatment programs across the United States. We are hopeful that several new therapies including NRF2 activators will achieve FDA-approval to make possible combination drug therapies for SCD.
Authors’ contributions
XZ, ARO, LHN and BL conducted the literature search and contributed to writing the paper. BSP executed the conception and wrote this paper.
DECLARATION OF CONFLICTING INTERESTS
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
FUNDING
This work was supported by grant HL69234 from the National Heart, Lung, and Blood Institute to BSP. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Stuart MJ, Nagel RL. Sickle-cell disease. Lancet 2004; 364:1343–60 [DOI] [PubMed] [Google Scholar]
- 2.Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA 2005; 293:1653–62 [DOI] [PubMed] [Google Scholar]
- 3.Rees DC, Williams TN, Gladwin MT. Sickle-cell disease". Lancet 2010; 376:2018–31 [DOI] [PubMed] [Google Scholar]
- 4.Sies H, Berndt C, Jones DP. Oxidative stress. Annu Rev Biochem 2017; 86:715–48 [DOI] [PubMed] [Google Scholar]
- 5.Nath KA, Grande JP, Haggard JJ, Croatt AJ, Katusic ZS, Solovey A, Hebbel RP. Oxidative stress and induction of heme oxygenase-1 in the kidney in sickle cell disease. Am J Pathol 2001; 158:893–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev 2006; 86:583–650 [DOI] [PubMed] [Google Scholar]
- 7.Siegel D, Gustafson DL, Dehn DL, Han JY, Boonchoong P, Berliner LJ, Ross D. NAD(P)H:quinone oxidoreductase 1: role as a superoxide scavenger. Mol Pharmacol 2004; 65:1238–47 [DOI] [PubMed] [Google Scholar]
- 8.Voskou S, Aslan M, Fanis P, Phylactides M, Kleanthous M. Oxidative stress in β-thalassaemia and sickle cell disease. Redox Biol 2015; 6:226–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghosh S, Tan F, Yu T, Li Y, Adisa O, Mosunjac M, Ofori-Acquah SF. Global gene expression profiling of endothelium exposed to heme reveals an organ-specific induction of cytoprotective enzymes in sickle cell disease. PLoS One 2011; 6:e18399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen T, Nioi P, Pickett CB. The NRF2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem 2009; 284:13291–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F, Xue P, Pi J, Kleeberger SR, Bell DA. Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucl Acids Res 2012; 40:7416–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rushmore TH, Morton MR, Pickett CB. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem 1991; 266:11632–9 [PubMed] [Google Scholar]
- 13.Sun J, Brand M, Zenke Y, Tashiro S, Groudine M, Igarashi K. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc Natl Acad Sci U S A 2004; 101:1461–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dhakshinamoorthy S, Jain AK, Bloom DA, Jaiswal AK. Bach1 competes with NRF2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J Biol Chem 2005; 280:16891–900 [DOI] [PubMed] [Google Scholar]
- 15.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 2012; 24:981–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma Q. Role of NRF2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 2013; 53:401–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reddy NM, Kleeberger SR, Yamamoto M, Kensler TW, Scollick C, Biswal S, Reddy SP. Genetic dissection of the NRF2-dependent redox signaling-regulated transcriptional programs of cell proliferation and cytoprotection. Physiol Genom 2007; 32:74–81 [DOI] [PubMed] [Google Scholar]
- 18.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the KEAP1-NRF2-ARE pathway. Annu Rev Pharmacol Toxicol 2007; 47:89–116 [DOI] [PubMed] [Google Scholar]
- 19.Chen ZH, Saito Y, Yoshida Y, Sekine A, Noguchi N, Niki E. 4-Hydroxynonenal induces adaptive response and enhances PC12 cell tolerance primarily through induction of thioredoxin reductase 1 via activation of NRF2. J Biol Chem 2005; 280:41921–7 [DOI] [PubMed] [Google Scholar]
- 20.Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. Transcription factor NRF2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem 2000; 275:16023–9 [DOI] [PubMed] [Google Scholar]
- 21.Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, Motohashi H. NRF2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012; 22:66–79 [DOI] [PubMed] [Google Scholar]
- 22.Lee JM, Chan K, Kan YW, Johnson JA. Targeted disruption of NRF2 causes regenerative immune-mediated hemolytic anemia. Proc Natl Acad Sci U S A 2004; 101:9751–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. KEAP1 represses nuclear activation of antioxidant responsive elements by NRF2 through binding to the amino-terminal Neh2 domain. Genes Dev 1999; 13:76–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, Jedrychowski MP, Costa ASH, Higgins M, Hams E, Szpyt J, Runtsch MC, King MS, McGouran JF, Fischer R, Kessler BM, McGettrick AF, Hughes MM, Carroll RG, Booty LM, Knatko EV, Meakin PJ, Ashford MLJ, Modis LK, Brunori G, Sévin DC, Fallon PG, Caldwell ST, Kunji ERS, Chouchani ET, Frezza C, Dinkova-Kostova AT, Hartley RC, Murphy MP, O'Neill LA. Itaconate is an anti-inflammatory metabolite that activates NRF2 via alkylation of KEAP1. Nature 2018; 556:113–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ooi A, Dykema K, Ansari A, Petillo D, Snider J, Kahnoski R, Anema J, Craig D, Carpten J, Teh BT, Furge KA. CUL3 and NRF2 mutations confer an NRF2 activation phenotype in a sporadic form of papillary renal cell carcinoma. Cancer Res 2013; 73:2044–51 [DOI] [PubMed] [Google Scholar]
- 26.Adam J, Hatipoglu E, O'Flaherty L, Ternette N, Sahgal N, Lockstone H, Baban D, Nye E, Stamp GW, Wolhuter K, Stevens M, Fischer R, Carmeliet P, Maxwell PH, Pugh CW, Frizzell N, Soga T, Kessler BM, El-Bahrawy M, Ratcliffe PJ, Pollard PJ. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and NRF2 signaling. Cancer Cell 2011; 20:524–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor NRF2 through inactivation of KEAP1. Nat Cell Biol 2010; 12:213–23 [DOI] [PubMed] [Google Scholar]
- 28.Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, White E, Zhang DD. A noncanonical mechanism of NRF2 activation by autophagy deficiency: direct interaction between KEAP1 and p62. Mol Cell Biol 2010; 30:3275–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang T, Harder B, Rojo de la Vega M, Wong PK, Chapman E, Zhang DD. p62 links autophagy and NRF2 signaling. Free Radic Biol Med 2015; 88:199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu T, Zhao F, Gao B, Tan C, Yagishita N, Nakajima T, Wong PK, Chapman E, Fang D, Zhang DD. Hrd1 suppresses NRF2-mediated cellular protection during liver cirrhosis. Genes Dev 2014; 28:708–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meakin PJ, Chowdhry S, Sharma RS, Ashford FB, Walsh SV, McCrimmon RJ, Dinkova-Kostova AT, Dillon JF, Hayes JD, Ashford ML. Susceptibility of NRF2-null mice to steatohepatitis and cirrhosis upon consumption of a high-fat diet is associated with oxidative stress, perturbation of the unfolded protein response, and disturbance in the expression of metabolic enzymes but not with insulin resistance. Mol Cell Biol 2014; 34:3305–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Redox-regulated turnover of NRF2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J Biol Chem 2004; 279:31556–67 [DOI] [PubMed] [Google Scholar]
- 33.Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, Hayes JD. NRF2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013; 32:3765–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the NRF2 transcription factor in a KEAP1-independent manner. Mol Cell Biol 2011; 31:1121–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salazar M, Rojo AI, Velasco D, de Sagarra RM, Cuadrado A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor NRF2. J Biol Chem 2006; 281:14841–51 [DOI] [PubMed] [Google Scholar]
- 36.Gameiro I, Michalska P, Tenti G, Cores Á, Buendia I, Rojo AI, Georgakopoulos ND, Hernández-Guijo JM, Teresa Ramos M, Wells G, López MG, Cuadrado A, Menéndez JC, León R. Discovery of the first dual GSK3β inhibitor/NRF2 inducer. A new multitarget therapeutic strategy for Alzheimer's disease. Sci Rep 2017; 7:45701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keleku-Lukwete N, Suzuki M, Yamamoto M. An overview of the advantages of KEAP1-NRF2 system activation during inflammatory disease treatment. Antioxid Redox Signal 2018; 29:1746–55 [DOI] [PubMed] [Google Scholar]
- 38.Zhang XS, Ha S, Wang XL, Shi YL, Duan SS, Li ZA. Tanshinone IIA protects dopaminergic neurons against 6-hydroxydopamine-induced neurotoxicity through miR-153/NF-E2-related factor 2/antioxidant response element signaling pathway. Neuroscience 2015; 303:489–502 [DOI] [PubMed] [Google Scholar]
- 39.Xue WL, Bai X, Zhang L. rhTNFR:Fc increases NRF2 expression via miR-27a mediation to protect myocardium against sepsis injury. Biochem Biophys Res Commun 2015; 464:855–61 [DOI] [PubMed] [Google Scholar]
- 40.Wang N, Zhang L, Lu Y, Zhang M, Zhang Z, Wang K, Lv J. Down-regulation of microRNA-142-5p attenuates oxygen-glucose deprivation and reoxygenation-induced neuron injury through up-regulating NRF2/ARE signaling pathway. Biomed Pharmacother 2017; 89:1187–95 [DOI] [PubMed] [Google Scholar]
- 41.Yang M, Yao Y, Eades G, Zhang Y, Zhou Q. MiR-28 regulates NRF2 expression through a KEAP1-independent mechanism. Breast Cancer Res Treat 2011; 129:983–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang X, Gao Y, Qin J, Lu S. The role of miR-34a in the hepatoprotective effect of hydrogen sulfide on ischemia/reperfusion injury in young and old rats. PLoS One 2014; 9:e113305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou S, Ye W, Zhang Y, Yu D, Shao Q, Liang J, Zhang M. miR-144 reverses chemoresistance of hepatocellular carcinoma cell lines by targeting NRF2-dependent antioxidant pathway. Am J Transl Res 2016; 8:2992–3002 [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou C, Zhao L, Zheng J, Wang K, Deng H, Liu P, Chen L, Mu H. MicroRNA-144 modulates oxidative stress tolerance in SH-SY5Y cells by regulating nuclear factor erythroid 2-related factor 2-glutathione axis. Neurosci Lett 2017; 655:21–7 [DOI] [PubMed] [Google Scholar]
- 45.Yu M, Liu Y, Zhang B, Shi Y, Cui L, Zhao X. Inhibiting microRNA-144 abates oxidative stress and reduces apoptosis in hearts of streptozotocin-induced diabetic mice. Cardiovasc Pathol 2015; 24:375–81 [DOI] [PubMed] [Google Scholar]
- 46.Sangokoya C, Telen MJ, Chi JT. microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood 2010; 116:4338–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Narasimhan M, Riar AK, Rathinam ML, Vedpathak D, Henderson G, Mahimainathan L. Hydrogen peroxide responsive miR153 targets NRF2/ARE cytoprotection in paraquat induced dopaminergic neurotoxicity. Toxicol Lett 2014; 228:179–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu W, Li F, Liu Z, Xu Z, Sun B, Cao J, Liu Y. MicroRNA-27b inhibition promotes NRF2/ARE pathway activation and alleviates intracerebral hemorrhage-induced brain injury. Oncotarget 2017; 8:70669–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeNicola GM1, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, Scrimieri F, Winter JM, Hruban RH, Iacobuzio-Donahue C, Kern SE, Blair IA, Tuveson DA. Oncogene-induced NRF2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011; 475:106–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The KEAP1-NRF2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol 2013; 1:45–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tao S, Wang S, Moghaddam SJ, Ooi A, Chapman E, Wong PK, Zhang DD. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res 2014; 74:7430–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miao W, Hu L, Scrivens PJ, Batist G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: direct cross-talk between phase I and II drug-metabolizing enzymes. J Biol Chem 2005; 280:20340–8 [DOI] [PubMed] [Google Scholar]
- 53.Shin S, Wakabayashi N, Misra V, Biswal S, Lee GH, Agoston ES, Yamamoto M, Kensler TW. NRF2 modulates aryl hydrocarbon receptor signaling: influence on adipogenesis. Mol Cell Biol 2007; 27:7188–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ghosh S, Ihunnah CA, Hazra R, Walker AL, Hansen JM, Archer DR, Owusu-Ansah AT, Ofori-Acquah SF. Nonhematopoietic NRF2 dominantly impedes adult progression of sickle cell anemia in mice. JCI Insight 2016; 1:pii:e81090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ghosh S, Hazra R, Ihunnah CA, Weidert F, Flage B, Ofori-Acquah SF. Augmented NRF2 activation protects adult sickle mice from lethal acute chest syndrome. Br J Haematol 2018; 182:271–5 [DOI] [PubMed] [Google Scholar]
- 56.Zhu X, Xi C, Thomas B, Pace BS. Loss of NRF2 function exacerbates the pathophysiology of sickle cell disease in a transgenic mouse model. Blood 2018; 131:558–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Keleku-Lukwete N, Suzuki M, Otsuki A, Tsuchida K, Katayama S, Hayashi M, Naganuma E, Moriguchi T, Tanabe O, Engel JD, Imaizumi M, Yamamoto M. Amelioration of inflammation and tissue damage in sickle cell model mice by NRF2 activation. Proc Natl Acad Sci U S A 2015; 112:12169–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krishnamoorthy S, Pace B, Gupta D, Sturtevant S, Li B, Makala L, Brittain J, Moore N, Vieira B, Thullen T, Stone I, Li H, Hobbs WE, Light DR. Dimethyl fumarate increases fetal hemoglobin, provides vascular protection and heme detoxification and corrects anemia in sickle cell disease. JCI Insight 2017; 2:pii:96409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Macari ER, Lowrey CH. Induction of human fetal hemoglobin via the NRF2 antioxidant response signaling pathway. Blood 2011; 117:5987–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu X, Li B, Pace BS. NRF2 mediates γ-globin gene regulation and fetal hemoglobin induction in human erythroid progenitors. Haematologica 2017; 102:e285–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, Bessy A, Chèneby J, Kulkarni SR, Tan G, Baranasic D, Arenillas DJ, Sandelin A, Vandepoele K, Lenhard B, Ballester B, Wasserman WW, Parcy F, Mathelier A. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res 2018; 46:D1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Macari ER, Schaeffer EK, West RJ, Lowrey CH. Simvastatin and t-butylhydroquinone suppress KLF1 and BCL11A gene expression and additively increase fetal hemoglobin in primary human erythroid cell. Blood 2013; 121:830–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lessard S, Beaudoin M, Benkirane K, Lettre G. Comparison of DNA methylation profiles in human fetal and adult red blood cell progenitors. Genome Med 2015; 7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lanaro C, Franco-Penteado CF, Albuqueque DM, Saad ST, Conran N, Costa FF. Altered levels of cytokines and inflammatory mediators in plasma and leukocytes of sickle cell anemia patients and effects of hydroxyurea therapy. J Leukoc Biol 2009; 85:235–42 [DOI] [PubMed] [Google Scholar]
- 65.Doss JF, Jonassaint JC, Garrett ME, Ashley-Koch AE, Telen MJ, Chi JT. Phase 1 study of a sulforaphane-containing broccoli sprout homogenate for sickle cell disease. PLoS One 2016; 11:e0152895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chavan H, Oruganti M, Krishnamurthy P. The ATP-binding cassette transporter ABCB6 is induced by arsenic and protects against arsenic cytotoxicity. Toxicol Sci 2011; 120:519–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ferreira GC, Franco R, Lloyd SG, Moura I, Moura JJ, Huynh BH. Structure and function of ferrochelatase. J Bioenerg Biomembr 1995; 27:221–9 [DOI] [PubMed] [Google Scholar]
- 68.Cordova EJ, Valenzuela OL, Sánchez-Peña LC, Escamilla-Guerrero G, Hernández-Zavala A, Orozco L, Del Razo LM. Nuclear factor erythroid 2-related factor gene variants and susceptibility of arsenic-related skin lesions. Hum Exp Toxicol 2014; 33:582–9 [DOI] [PubMed] [Google Scholar]
- 69.MacLeod AK, Mcmahon M, Plummer SM, Higgins LG, Penning TM, Igarashi K, Hayes JD. Characterization of the cancer chemopreventive NRF2-dependent gene battery in human keratinocytes: demonstration that the KEAP1-NRF2 pathway, and not the BACH1-NRF2 pathway, controls cytoprotection against electrophiles as well as redox-cycling compounds. Carcinogenesis 2009; 30:1571–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu KC, Cui JY, Klaassen CD. Beneficial role of NRF2 in regulating NADPH generation and consumption. Toxicol Sci 2011; 123:590–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rajagopal A, Rao AU, Amigo J, Tian M, Upadhyay SK, Hall C, Uhm S, Mathew MK, Fleming MD, Paw BH, Krause M, Hamza I. Haem homeostasis is regulated by the conserved and concerted functions of HRG-1 proteins. Nature 2008; 453:1127–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Winterbourn CC. The biological chemistry of hydrogen peroxide. Meth Enzymol 2013; 528:3–25 [DOI] [PubMed] [Google Scholar]
- 73.Campbell MR, Karaca M, Adamski KN, Chorley BN, Wang X, Bell DA. Novel hematopoietic target genes in the NRF2-mediated transcriptional pathway. Oxid Med Cell Longev 2013; 2013:120305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest 2017; 127:750–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D. Activation of the p62-KEAP1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016; 63:173–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Forman HJ, Zhang H, Rinna A. Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med 2009; 30:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ye P, Mimura J, Okada T, Sato H, Liu T, Maruyama A, Ohyama C, Itoh K. NRF2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol Cell Biol 2014; 34:3421–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kaelin WG, Jr, McKnight SL. Influence of metabolism on epigenetics and disease. Cell 2013; 153:56–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, Zhao S, Ye D, Xiong Y, Guan KL. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 2012; 26:1326–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Niihara Y, Zerez CR, Akiyama DS, Tanaka KR. Oral L-glutamine therapy for sickle cell anemia: I. Subjective clinical improvement and favorable change in red cell NAD redox potential. Am J Hematol 1998; 58:117–21 [DOI] [PubMed] [Google Scholar]
- 81.Niihara Y, Miller ST, Kanter J, Lanzkron S, Smith WR, Hsu LL, Gordeuk VR, Viswanathan K, Sarnaik S, Osunkwo I, Guillaume E, Sadanandan S, Sieger L, Lasky JL, Panosyan EH, Blake OA, New TN, Bellevue R, Tran LT, Razon RL, Stark CW, Neumayr LD, Vichinsky EP. Investigators of the phase 3 trial of l-glutamine in sickle cell disease. A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med 2018; 379:226–35 [DOI] [PubMed] [Google Scholar]
- 82.Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, Karakousi TR, Ellis DC, Bhutkar A, Sánchez-Rivera FJ, Subbaraj L, Martinez B, Bronson RT, Prigge JR, Schmidt EE, Thomas CJ, Goparaju C, Davies A, Dolgalev I, Heguy A, Allaj V, Poirier JT, Moreira AL, Rudin CM, Pass HI, Vander Heiden MG, Jacks T, Papagiannakopoulos T. KEAP1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med 2017; 23:1362–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sayin VI, LeBoeuf SE, Singh SX, Davidson SM, Biancur D, Guzelhan BS, Alvarez SW, Wu WL, Karakousi TR, Zavitsanou AM, Ubriaco J, Muir A, Karagiannis D, Morris PJ, Thomas CJ, Possemato R, Vander Heiden MG, Papagiannakopoulos T. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife 2017; 6:pii:e28083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yu D, dos Santos CO, Zhao G, Jiang J, Amigo JD, Khandros E, Dore LC, Yao Y, D'Souza J, Zhang Z, Ghaffari S, Choi J, Friend S, Tong W, Orange JS, Paw BH, Weiss MJ. miR-451 protects against erythroid oxidant stress by repressing 14-3-3zeta. Genes Dev 2010; 24:1620–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim M, Tan YS, Cheng WC, Kingsbury TJ, Heimfeld S, Civin CI. MIR144 and MIR451 regulate human erythropoiesis via RAB14. Br J Haematol 2015; 168:583–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yang H, Magilnick N, Lee C, Kalmaz D, Ou X, Chan JY, Lu SC. Nrf1 and NRF2 regulate rat glutamate-cysteine ligase catalytic subunit transcription indirectly via NF-kappaB and AP-1. Mol Cell Biol 2005; 25:5933–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li B, Torres CM, Takezaki M, Neunert C, Kutlar A, Pace BS. Micro rna profiling to identify regulator of fetal hemoglobin expression in sickle cell disease. Blood 2014; 124:4924797299 [Google Scholar]
- 88.Li B, Zhu X, Ward CM, Starlard-Davenport A, Takezaki M, Berry A, Ward A, Wilder C, Neunert C, Kutlar A, Pace BS. MIR-144-mediated NRF2 gene silencing inhibits fetal hemoglobin expression in sickle cell disease. Exp Hematol 2018;in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Abed DA, Goldstein M, Albanyan H, Jin H, Hu L. Discovery of direct inhibitors of KEAP1-NRF2 protein-protein interaction as potential therapeutic and preventive agents. Acta Pharm Sin B 2015; 5:285–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Belcher JD, Chen C, Nguyen J, Zhang P, Abdulla F, Nguyen P, Killeen T, Xu P, O'Sullivan G, Nath KA, Vercellotti GM. Control of oxidative stress and inflammation in sickle cell disease with the NRF2 activator dimethyl fumarate. Antioxid Redox Signal 2017; 26:748–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hoppe C, Kuypers F, Larkin S, Hagar W, Vichinsky E, Styles L. A pilot study of the short-term use of simvastatin in sickle cell disease: effects on markers of vascular dysfunction. Br J Haematol 2011; 153:655–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, Yu J, Yang Y, Hales P, Bruzzese F, Liu J, Blank J, Garcia K, Tsu C, Dick L, Fleming P, Yu L, Manfredi M, Rolfe M, Bolen J. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res 2010; 70:1970–80 [DOI] [PubMed] [Google Scholar]
- 93.Pullarkat V, Meng Z, Tahara SM, Johnson CS, Kalra VK. Proteasome inhibition induces both antioxidant and hb f responses in sickle cell disease via the NRF2 pathway. Hemoglobin 2014; 38:188–95 [DOI] [PubMed] [Google Scholar]
- 94.Dodd S, Dean O, Copolov DL, Malhi GS, Berk M. N.- acetylcysteine for antioxidant therapy: pharmacology and clinical utility. Expert Opin Biol Ther 2008; 8:1955–62 [DOI] [PubMed] [Google Scholar]
- 95.Manandhar S, Cho JM, Kim JA, Kensler TW, Kwak MK. Induction of NRF2-regulated genes by 3H-1, 2-dithiole-3-thione through the ERK signaling pathway in murine keratinocytes. Eur J Pharmacol 2007; 577:17–27 [DOI] [PubMed] [Google Scholar]
- 96.Zhang Y, Talalay P, Cho CG, Posner GH. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc Natl Acad Sci U S A 1992; 89:2399–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mrowietz U, Christophers E, Altmeyer P. Treatment of psoriasis with fumaric acid esters: results of a prospective multicentre study. German Multicentre Study. Br J Dermatol 1998; 138:456–60 [DOI] [PubMed] [Google Scholar]
- 98.Stangel M, Linker RA. Dimethyl fumarate (BG-12) for the treatment of multiple sclerosis. Expert Rev Clin Pharmacol 2013; 6:355–62 [DOI] [PubMed] [Google Scholar]
- 99.Satoh T, McKercher SR, Lipton SA. NRF2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic Biol Med 2013; 65:645–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Turley AE1, Zagorski JW2, Rockwell CE3. The NRF2 activator tBHQ inhibits T cell activation of primary human CD4 T cells. Cytokine 2015; 71:289–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Duane WC, Hunninghake DB, Freeman ML, Pooler PA, Schlasner LA, Gebhard RL. Simvastatin, a competitive inhibitor of HMG-CoA reductase, lowers cholesterol saturation index of gallbladder bile. Hepatology 1988; 8:1147–50 [DOI] [PubMed] [Google Scholar]
- 102.Li N, Alam J, Venkatesan MI, Eiguren-Fernandez A, Schmitz D, Di Stefano E, Slaughter N, Killeen E, Wang X, Huang A, Wang M, Miguel AH, Cho A, Sioutas C, Nel AE. NRF2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J Immunol 2004; 173:3467–81 [DOI] [PubMed] [Google Scholar]
- 103.Nur E, Brandjes DP, Teerlink T, Otten HM, Oude Elferink RP, Muskiet F, Evers LM, ten Cate H, Biemond BJ, Duits AJ, Schnog JJ. Curama Study Group N-acetylcysteine reduces oxidative stress in sickle cell patients. Ann Hematol 2012; 91:1097–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Joep WRS, Fijnvandraat K, Rijneveld AW, Boom MB, Kerkhoffs J-L, van Meurs AH, De Groot MR, Heijboer H, Dresse M-F, Ferster A, Hermans P, Vanderfaeillie A, Van Den Neste EW, Samantha Benghiat F, Howard J, Kesse-Adu R, Delannoy A, Efira A, Azerad M-A, de Borgie CAJM, Biemond BJ. N-acetylcysteine in patients with sickle cell disease: a randomized controlled trial. Blood 2016; 128:123 [Google Scholar]
- 105.Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, McMahon RP, Bonds DR. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the multicenter study of hydroxyurea in sickle cell anemia. N Engl J Med 1995; 332:1317–22 [DOI] [PubMed] [Google Scholar]
- 106.Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, Guthrie TH, Knight-Madden J, Alvarez OA, Gordeuk VR, Gualandro S, Colella MP, Smith WR, Rollins SA, Stocker JW, Rother RP. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med 2017; 376:429–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kutlar A, Kanter J, Liles DK, Alvarez OA, Cançado RD, Friedrisch JR, Knight-Madden JM, Bruederle A, Shi M, Zhu Z, Ataga KI. Effect of crizanlizumab on pain crises in subgroups of patients with sickle cell disease: a SUSTAIN study analysis. Am J Hematol 2019; 94:55–61 [DOI] [PubMed] [Google Scholar]
- 108.Atweh GF, Sutton M, Nassif I, Boosalis V, Dover GJ, Wallenstein S, Wright E, McMahon L, Stamatoyannopoulos G, Faller DV, Perrine SP. Sustained induction of fetal hemoglobin by pulse butyrate therapy in sickle cell disease. Blood 1999; 93:1790–7 [PMC free article] [PubMed] [Google Scholar]
- 109.Saunthararajah Y, Hillery CA, Lavelle D, Molokie R, Dorn L, Bressler L, Gavazova S, Chen YH, Hoffman R, DeSimone J. Effects of 5-aza-2'-deoxycytidine on fetal hemoglobin levels, red cell adhesion, and hematopoietic differentiation in patients with sickle cell disease. Blood 2003; 102:3865–70 [DOI] [PubMed] [Google Scholar]
- 110.Kutlar A, Reid ME, Inati A, Taher AT, Abboud MR, El-Beshlawy A, Buchanan GR, Smith H, Ataga KI, Perrine SP, Ghalie RG. A dose-escalation phase IIa study of 2,2-dimethylbutyrate (HQK-1001), an oral fetal globin inducer, in sickle cell disease. Am J Hematol 2013; 88:E255–60 [DOI] [PubMed] [Google Scholar]
- 111.Reid ME, El Beshlawy A, Inati A, Kutlar A, Abboud MR, Haynes J, Jr, Ward R, Sharon B, Taher AT, Smith W, Manwani D, Ghalie RG. A double-blind, placebo-controlled phase II study of the efficacy and safety of 2,2-dimethylbutyrate (HQK-1001), an oral fetal globin inducer, in sickle cell disease. Am J Hematol 2014; 89:709–13 [DOI] [PubMed] [Google Scholar]
- 112.Molokie R, Lavelle D, Gowhari M, Pacini M, Krauz L, Hassan J, Ibanez V, Ruiz MA, Ng KP, Woost P, Radivoyevitch T, Pacelli D, Fada S, Rump M, Hsieh M, Tisdale JF, Jacobberger J, Phelps M, Engel JD, Saraf S, Hsu LL, Gordeuk V, DeSimone J. Saunthararajah Oral tetrahydrouridine and decitabine for non-cytotoxic epigenetic gene regulation in sickle cell disease: a randomized phase 1 study. PLoS Med 2017; 14:e1002382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang YY, Yang YX, Zhe H, He ZX, Zhou SF. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: an update on its pharmacokinetic and pharmacodynamic properties. Drug Des Devel Ther 2014; 8:2075–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ward CM, Li B, Pace BS. Stable expression of miR-34a mediates fetal hemoglobin induction in K562 cells. Exp Biol Med 2016; 241:719–29 [DOI] [PMC free article] [PubMed] [Google Scholar]