

Abstract
Dysregulation of dopaminergic system induced by HIV-1 Tat protein-mediated direct inhibition of the dopamine transporter (DAT) has been implicated as a mediating factor of HIV-1 associated neurocognitive disorders. We have reported that single point mutations on human DAT (hDAT) at tyrosine88 (Y88F), lysine92 (K92M), and histidine547 (H547A) differentially regulate basal dopamine uptake but diminish Tat-induced inhibition of dopamine uptake by changing dopamine transport process. This study evaluated the effects of double (Y88F/H547A) and triple (Y88F/K92M/H547A) mutations on basal dopamine uptake, Tat-induced inhibition of DAT function, and dynamic transport process. Compared to wild-type hDAT, the Vmax values of [3H]Dopamine uptake were increased by 96% in Y88F/H547A but decreased by 97% in Y88F/K92M/H547A. [3H]WIN35,428 binding sites were not altered in Y88F/H547A but decreased in Y88F/K92M/H547A. Y88F/H547A mutant attenuated Tat-induced inhibition of dopamine uptake observed in wild-type hDAT. Y88F/H547A displayed an attenuation of zinc-augmented [3H]WIN35,428 binding, increased basal dopamine efflux, and reduced amphetamine-induced dopamine efflux, indicating this mutant alters transporter conformational transitions. These findings further demonstrate that both tyrosine88 and histidine547 on hDAT play a key role in stabilizing basal dopamine transport and Tat-DAT integration. This study provides mechanistic insights into developing small molecules to block multiple sites in DAT for Tat binding.
Introduction
About thirty-seven million people are currently living with HIV-1 infection worldwide, leading to a significant global public health problem. While the effective antiretroviral therapies significantly reduced the mortality rate in the patients with HIV-1 infection, nearly 50% of HIV-1 infected patients have various degrees of neurological complications that are referred to as HIV-1-associated neurocognitive disorders (HAND)1. The continuous exposure of the central nerves system to HIV-1 viral proteins, inflammation, and antiretroviral agents results in neuropathological and neurocognitive deficits observed in the patients with HAND2–7. Transactivator of transcription (Tat) protein, one of seven HIV-1 viral proteins, has been shown to play a critical role in HIV-1 viral replication as well as the development of HAND2,8, which can be exacerbated by concurrent cocaine abuse9. Thus, developing an intervention strategy in the early stage of HIV-1 infection would prevent the development of neurocognitive dysfunction in HIV-1 infected individuals.
Normal dopaminergic transmission is important for maintaining different brain activities including attention, learning, memory10,11, and motivation12,13. Dopamine (DA) transporter (DAT) is a presynaptic membrane protein that reuptakes the released DA from the synaptic cleft back into cytosol, maintaining a stable DA homeostasis. The DAT activity is directly inhibited by HIV-1 Tat protein and cocaine, which synergistically enhances synaptic DA levels9. The dysregulation of DA system is a mediating factor of HAND as well as a factor in cocaine abuse14,15. Using computational modeling and experimental approach, we have identified several key residues on human DAT (hDAT), which are crucial for Tat-hDAT interaction and dynamic transport process9. Furthermore, we have demonstrated that in vitro exposure to Tat reduced reuptake of DA via hDAT in cells16–18 and rat striatal synaptosomes19. The inhibitory effect of Tat on DAT function results from Tat directly interacting with DAT16,20,21. Single point mutations of hDAT at tyrosine88 (to phenylalanine, Y88F), lysine 92 (to methionine, K92M), histidine547 (to alanine, H547A) differentially alter basal DA uptake but attenuate the Tat inhibitory effects on DA transport17,18. For example, DA uptake is decreased in K92M and increased in H547A, respectively; however Y88F mutant preserves basal DA uptake17,18. Notably, the mutational effects on normal DA uptake and Tat inhibitory effect on DAT function are associated with alterations of transporter conformational transitions9.
We have demonstrated that Tat protein inhibits DAT function in an allosteric modulatory mechanism19,22. Recent studies have demonstrated that novel SRI-compounds exhibit a partial antagonistic role in DAT function as allosteric modulators23–25. We have reported that SRI-30827, one of the novel allosteric modulators, blocks Tat interaction with DAT26. Thus, identifying the specific binding sites on hDAT for Tat and its role in DA transport process could be beneficial to attenuation of the inhibitory effect of Tat on DAT-mediated dopaminergic transmission. On the other hand, through an allosteric modulatory mechanism, the inhibitory effect of Tat on DAT function can also be diminished by targeting the specific DAT residues that are distinct from Tat binding sites. However, based on our computational structural models for Tat binding with hDAT, the interaction of Tat with hDAT involves multiple residues of DAT9,21 and our previous results obtained from single point mutations of DAT only present the role of a particular residue in Tat-DAT interaction. Therefore, this study investigated the mutational effects of Y88F/H547A and Y88F/K92M/H547A on basal DAT function, Tat inhibitory effect on DA uptake, and dynamic DA transport process.
Results
Computational modeling: Impact of Tyr88/His547 and Tyr88/Lys92/His547 on functional relevance of human DAT
Based on the constructed hDAT-Tat binding model in our previous work20, hDAT residues Y88, K92, and H547 could form hydrogen bonds with residues K19, P18, and R49 of HIV-1 Tat, respectively. According to our previous computational and experimental results, single mutation of either hDAT Y88 (Y88F), K92 (K92M) or H547 (H547A) could significantly attenuate the binding between DAT and Tat17,18. As shown in Fig. 1A,B, hDAT Y88, K92 and H547 residues independently interact with Tat, suggesting an effect for double or triple mutation on disturbing DAT-Tat binding. For example, two hydrogen bonds in the DAT-Tat interface were eliminated by double mutation of Y88F/H547A on hDAT (Fig. 1C), and three hydrogen bonds in the DAT-Tat interface were eliminated by triple mutation of Y88F/K92M/H547A on hDAT (Fig. 1D). Therefore, one may expect that Y88F/H547A and Y88F/K92M/H547A mutants are more effective in inhibiting DAT-Tat binding than single point mutations of Y88F, K92M and H547A.
Figure 1.

Key residues, D-Y88, D-K92 and D-H547 involved in the HIV-1 Tat-DAT binding. (A) Typical Tat-DAT binding complex from MD trajectory. Tat and DAT are represented as gold and cyan ribbons, respectively. The dashed box indicates the binding surface between Tat and DAT. (B) Tat residues, T-P18, T-K19 and T-R49, are represented as ball-stick style and colored in yellow. DAT residues, D-Y88, D-K92 and D-H547, are represented as ball-stick style and colored in green. Dashed lines represent inter-molecular hydrogen bonds with labeled distances. (The prefix T- and D- indicates residues of Tat and DAT, respectively). (C) Double mutation D-Y88F/D-H547A on hDAT-TAT structure. D-H547A mutation eliminates one hydrogen bond with T-R49, and D-Y88F mutation eliminates the hydrogen bond with T-K19. (D) Triple mutation D-Y88F/D-K92M/D-H547A on hDAT-TAT structure. Addition of a triple mutation of D-Y88F, D-K92M, and D-H547A, also eliminates the hydrogen bond with T-P18.
Mutations of Y88/H547 and Y88/K92/H547 differentially alter DA uptake kinetics and DAT binding
We have demonstrated that single point mutations on hDAT at His547 (H547A) and Lys92 (K92M) displayed a 196% increase and a 71% decrease in the Vmax of [3H]DA uptake, respectively, while Y88F preserved normal DA uptake17,18. The current study determined the multiple mutations of these residues on DA uptake and DAT binding sites. As shown in Table 1 and Fig. 2A, compared to WT hDAT (12.52 ± 0.68 pmol/min/105 cells), the Vmax values were increased by 92% in Y88F/H547A (24.06 ± 4.32 pmol/min/105 cells, t(7) = 2.33, p < 0.05) and dramatically decreased in Y88F/H547A/K92M (0.5 ± 0.31 pmol/min/105 cells, t(6) = 16.2, p < 0.001), respectively. Additionally, compared to WT hDAT (1.21 ± 0.17 µM), the Km value in Y88F/H547A was increased by 250% (4.25 ± 1.05 µM, t(7) = 2.54, p < 0.05) but not altered in Y88F/K92F/H547A.
Table 1.
Kinetic properties and inhibitory activities in [3H]DA uptake in WT and mutated hDAT.
| Vmax (pmol/min/105 cells) | Km (µM) | IC50 (nM) | ||||
|---|---|---|---|---|---|---|
| DA | Cocaine | GBR12909 | AMPH | |||
| WT hDAT | 12.52 ± 0.68 | 1.21 ± 0.17 | 2063 ± 582.5 | 350 ± 49.4 | 511 ± 56.6 | 926 ± 167.4 |
| Y88F/H547A | 24.06 ± 4.32* | 4.25 ± 1.05* | 7918 ± 637.6* | 131 ± 13.9* | 253 ± 40.4* | 1279 ± 200.5 |
| Y88F/ K92M/H547A | 0.5 ± 0.31* | 1.89 ± 1.25 | ND | ND | ND | ND |
Data are presented as mean ± S.E.M. values from four to six independent experiments performed in duplicates. *p < 0.05 compared with WT hDAT (unpaired Student’s t test). ND. Not determined.
Figure 2.
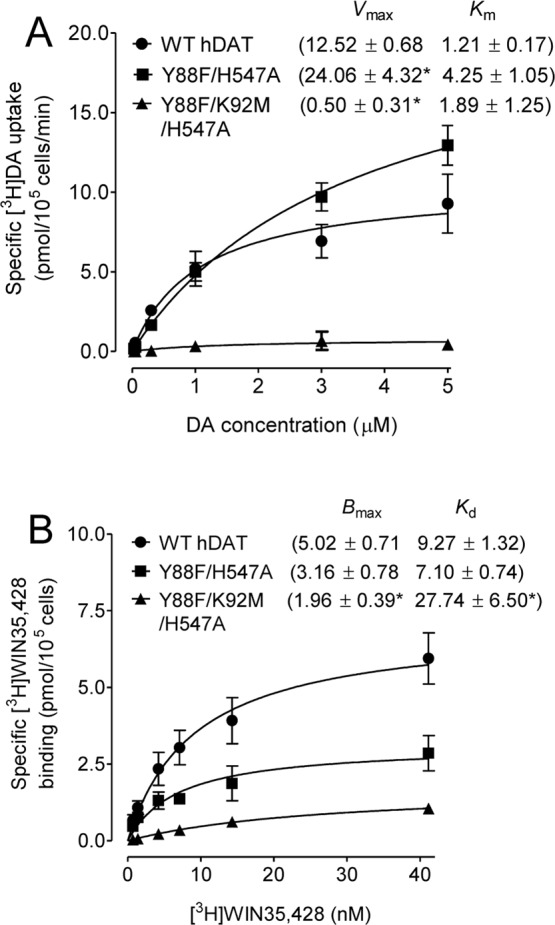
Effects of Y88F/H547A and Y88F/K92M/H547A on [3H]DA uptake and [3H]WIN35,428 binding. (A) Kinetic parameters (Vmax and Km) of [3H]DA uptake in WT hDAT and mutants. [3H]DA uptake assay was conducted in PC12 cells transfected with WT hDAT and mutants by incubation with one of 6 mixed concentrations of the [3H]DA as the total rate of DA uptake. Nonspecific uptake of [3H]DA was determined by a parallel incubation of each mixed DA concentrations with nomifensine (10 µM, final concentration). Specific [3H]DA uptake (Vmax) was calculated by subtraction of nonspecific DA uptake from total DA uptake. The parameter values were presented as means ± S.E.M. *p < 0.05 compared to WT hDAT value (unpaired Student’s t test) (n = 4–5/group). (B) Saturation analysis of [3H]WIN35,428 binding in WT hDAT and mutants. Bmax and Kd values were estimated by fitting one-site binding and presented as means ± S.E.M. *p < 0.05 compared to WT hDAT value (unpaired Student’s t test) (n = 4–12/group).
In the hDAT, the [3H]WIN35,428 binding sites share pharmacological identity with the DA uptake site and is also part of the cocaine binding domain27,28. Kinetic analysis of [3H]WIN35,428 binding was conducted to determine the effects of multiple mutations of hDAT on the DA binding site. As shown in Table 2 and Fig. 2B, in comparison to WT hDAT, neither Bmax nor Kd values of [3H]WIN35,428 were altered in Y88F/H547A, whereas Y88F/K92M/H547A displayed a 61% decrease in the Bmax value (t(14) = 2.4, p < 0.05) and a 199% increase in the Kd value (t(14) = 4.4, p < 0.001). Thus, due to the significant reduction on DA uptake for Y88F/K92M/H547A, only the Y88F/H547A mutant was included for the rest of experiments.
Table 2.
Kinetic properties and inhibitory activities in [3H]WIN 35,428 binding in WT and mutated hDAT.
| Bmax(pmol/105 cells) | Kd (nM) | IC50 (nM) | |||
|---|---|---|---|---|---|
| DA | Cocaine | GBR12909 | |||
| WT hDAT | 5.02 ± 0.71 | 9.27 ± 1.32 | 2291 ± 487.3 | 235 ± 53.3 | 485 ± 49.4 |
| Y88F/H547A | 3.16 ± 0.78 | 7.10 ± 0.74 | 7236 ± 1825* | 1494 ± 333.0* | 452 ± 113.0 |
| Y88F/K92M/H547A | 1.96 ± 0.39* | 27.74 ± 6.50* | ND | ND | ND |
Data are presented as mean ± S.E.M. of IC50 values from 4–12 independent experiments performed in duplicates. *p < 0.05 compared with WT hDAT (unpaired student’s t test). ND. Not determined.
Mutation of Y88/H547 alters the inhibition potency of DA uptake and DAT binding by substrates and inhibitors
To characterize the influence of Y88F/H547A on DA transport and the specific binding site in hDAT for DA, cocaine, GBR12909, and amphetamine (AMPH), we determined the ability of these DAT inhibitors or substrate to inhibit [3H] DA uptake in WT hDAT and Y88F/H547A (Table 1). In comparison to WT hDAT, Y88F/H547A reduced the apparent affinity (IC50) for DA [WT hDAT: 2063 ± 582.5 nM; Y88F/H547A: 7918 ± 637.6 nM; t(10) = 6.78, p < 0.0001]. In contrast, compared to WT hDAT, Y88F/ H547A displayed an increase in IC50 for cocaine [WT hDAT: 350 ± 49.4 nM; H547A/Y88F: 131 ± 13.9 nM; t(6) = 4.28, p < 0.01] and GBR12909 [WT hDAT: 511 ± 56.6 nM; Y88F/H547A: 253 ± 40.4 nM; t(8) = 3.71, p < 0.01]. We also evaluated the mutational effect of Y88F/H547A on the IC50 of DA, cocaine, and GBR12909 for inhibiting [3H]WIN35,428 binding (Table 2). Compared to WT hDAT, Y88F/H547A increased the IC50 values for DA (WT hDAT: 2291 ± 487.3 nM; Y88F/H547A: 7236 ± 1825 nM) and cocaine (WT hDAT: 235 ± 53.3; Y88F/H547A: 1494 ± 333.0 nM, t(12) = 3.73, p < 0.001]. No difference in IC50 GBR12909 inhibiting [3H]WIN35,428 binding was observed between WT hDAT and Y88F/H547A.
Effect of Y88F/H547A on basal PKC-mediated regulation of DAT function
The activation of PKC by phorbol 12-myristate 13-acetate (PMA), a PKC activator, decreases DA uptake through DAT29,30. We have demonstrated that the addition of 1 µM PMA produced a 40% and 60% decrease in the Vmax of DA uptake in WT hDAT and H547A mutant, respectively, compared to the respective control, suggesting that H547A displays increased sensitivity to PMA-induced DAT phosphorylation18. In the current study, we also found that Y88F/H547A displayed increased the Vmax relative to WT hDAT. We then determined whether Y88F/H547A-upregulated DA uptake is also mediated by alteration of sensitivity to PMA-induced DAT phosphorylation (Fig. 3). A two way ANOVA analysis on the Vmax values of [3H]DA uptake revealed a significant main effect of PMA [F(1, 20) = 9.40, p < 0.01]. No significant mutation × PMA treatment interaction [F(1, 20) = 2.62; p = 0.076] was found. Addition of PMA reduced the Vmax values in WT hDAT by 51% [t(5) = 2.79, p < 0.05] and Y88F/H547A by 60% [t(5) = 3.90, p < 0.05], respectively, compared to their respective vehicle controls. Similarly, two-way ANOVA analysis on the Km values revealed significant main effects of mutation [F(1, 20) = 5.52, p < 0.05] and PMA treatment [F(1, 20) = 10.71, p < 0.001]. No significant interaction of mutation × PMA treatment was observed. The addition of 1 µM PMA reduced Km values by 53% in WT hDAT [t(5) = 3.31, p < 0.05] and 63% in Y88F/H547A [t(5) = 2.54, p < 0.05], respectively. These results suggest that the double mutation of Tyr88 and His547 attenuates the single mutant H547A-increased sensitivity of PMA-induced decrease in DA uptake.
Figure 3.
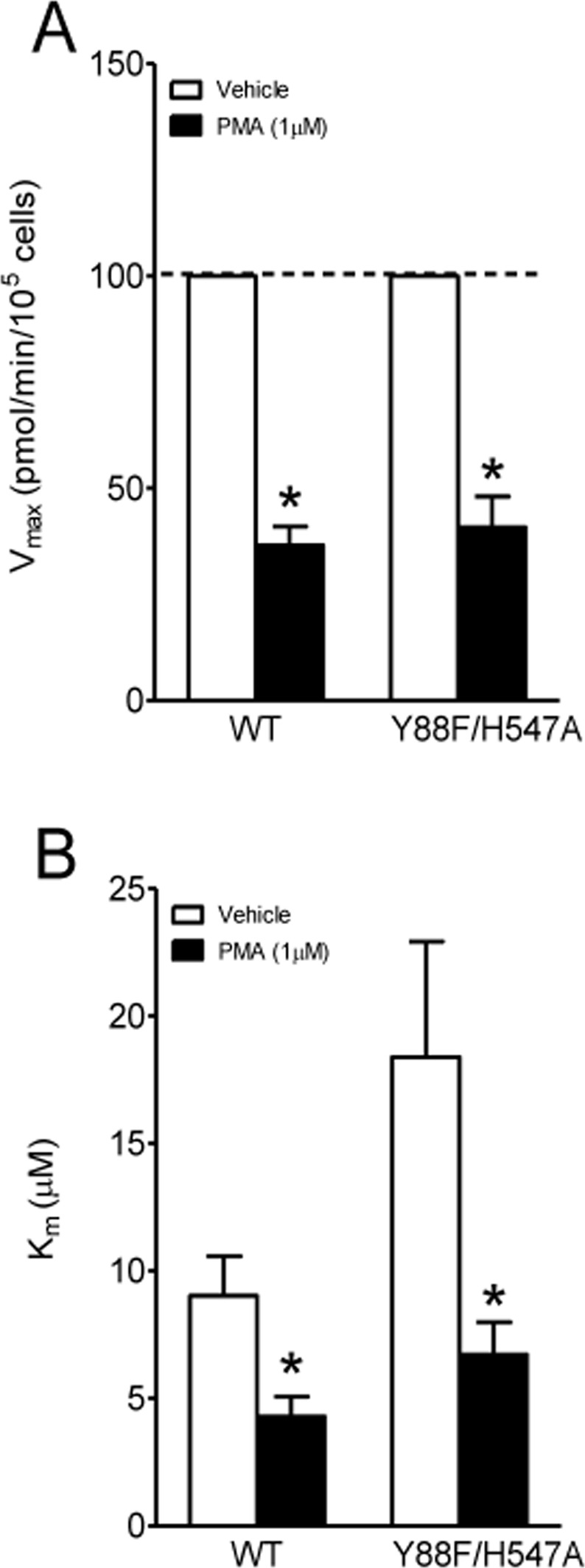
Effects of PMA on basal PKC-regulated DAT function in WT hDAT and Y88F/H547A mutant. Kinetic analysis of [3H]DA uptake was conducted in the presence or absence of PMA (PKC activator, 1 µM). (A) Vmax and (B) Km. Data were presented as means ± S.E.M. (n = 6/group). *p < 0.05 compared to their respective controls (paired t-test after two-way ANOVA analyses).
Double Mutation of H547/Y88 attenuate Tat-induced inhibition of DAT
Our previous studies demonstrated that single point mutation of Tyr88 (Y88F) or His547 (H547A) attenuated Tat-induced inhibition of [3H]DA uptake observed in WT hDAT17,18. Since the triple mutant (Y88F/K92M/H547A) displayed an extremely low Vmax of [3H]DA uptake, only the double mutant (Y88F/H547A) was tested for its effect on Tat-induced inhibition of DA uptake. Based on the computational modeling predictions (Fig. 1), the double mutation of Tyr88 and His547 would eliminate two pairs of hydrogen bonds between D-Y88-T-K19 and D-H547-T-R49 and impair Tat binding on hDAT, thereby attenuating Tat-induced inhibition of DA uptake. Due to the difference in the specific [3H]DA uptake between WT hDAT and Y88F/H547A mutants (Fig. 2), the inhibitory effects of Tat on DAT function in WT and Y88F/H547A were presented as % of Tat-mediated [3H]DA uptake relative to their respective controls (in the absence of Tat). As shown in Fig. 4, the application of 140 nM recombinant Tat1–86 (rTat1–86) significantly reduced the specific [3H] DA uptake in WT hDAT by 31% relative to its control [t(4) = 11.54, p < 0.001]. However, the Tat-induced reduction of [3H] DA uptake in WT hDAT was attenuated in Y88F/H547A compared to its control [t(4) = 0.42, p > 0.05], suggesting that the double mutation of Tyr88 and His547 attenuates Tat’s inhibitory effect on DA uptake.
Figure 4.
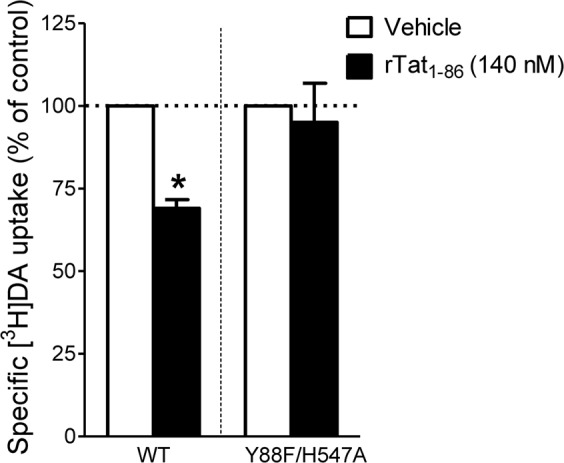
Effects of Tat on kinetic analysis of [3H]DA uptake. PC12 cells transfected with WT hDAT or Y88F/H547A were preincubated with vehicle or recombinant Tat1–86 (rTat1–86) at room temperature for 20 min followed by [3H]DA application. Data were calculated as mean ± S.E.M. of specific [3H]DA uptake relative to respective controls as 100% for individual experiment (WT hDAT: 1109.30 ± 230.27 dpm and Y88F/H547A: 400.13 ± 81.75 dpm and n = 5/group). *p < 0.05 compared to their respective controls (paired t test).
Effects of double mutation of Y88/H547 on zinc regulation of DAT conformational transitions and basal DA efflux
Tat protein has been shown to regulate DA transport allosterically, which may lead to alteration of transporter conformational transitions19,22. We have previously demonstrated that His547 residue plays a critical role in allosteric modulation of Tat-DAT interaction18. To determine whether the His547 residue still acts as a potential site for the regulation of Tat-DAT in Y88F/H547A, we examined the effects of Y88F/H547A on zinc modulation of [3H]DA uptake and [3H]WIN35,428 binding. Addition of Zn2+ is able to partially reverse an inward-facing state to an outward-facing state31,32. On the basis of this principle, the addition of Zn2+ to WT hDAT would inhibit DA uptake, whereas in a functional mutation of DAT, Zn2+ might diminish the preference for the inward-facing conformation and thus enhance DA uptake. As shown in Fig. 5A, two-way ANOVA analysis on the specific [3H]DA uptake in WT hDAT and Y88F/H547A revealed significant main effects of mutation [F(1, 12) = 55.34, p < 0.0001], zinc [F(3, 36) = 39.15, p < 0.0001], and mutation × zinc interaction [F(3, 36) = 3.70, p < 0.05]. The addition of zinc significantly decreased the [3H]DA uptake in WT hDAT and its mutant in a concentration dependent manner. Compared to the respective controls (in absence of Zn2+), the addition of Zn2+ (10 and 100 µM) produced a similar reduction of [3H]DA uptake in WT (~40%, ps < 0.01) but a greater reductions (60% and 71% at 10 and 100 µM, respectively) in Y88F/H547A (ps < 0.01). This suggests that the double mutation of Y88/H547 does not affect zinc-induced regulation of DA uptake. In contrast, as shown in Fig. 5B, two-way ANOVA analysis of the specific [3H]WIN35,428 binding revealed significant main effects of mutation [F(1, 16) = 5.82, p < 0.05], zinc [F(3, 48) = 13.50, p < 0.0001], and mutation × zinc interaction [F(3, 48) = 9.94, p < 0.0001]. Compared to the respective controls (in absence of Zn2+), addition of Zn2+ (10 and 100 µM) significantly increased [3H] WIN35,428 binding in WT hDAT (49% at 10 µM and 69% at 100 µM, ps < 0.05, Bonferroni t-test). The Zn2+ (10 and 100 µM)-induced increase in [3H]WIN 35,428 binding in WT hDAT was significantly diminished in Y88F/H547A [1 µM, t(16) = 2.37, p < 0.05; 10 µM, t(16) = 3.23, p < 0.01; 100 µM, t(16) = 3.57, p < 0.01], suggesting that Y88F/H547A attenuates zinc-induced increase in [3H]WIN35,428 of hDAT by altering transporter conformational transitions.
Figure 5.

Effects of Y88F/H547A mutant on transporter conformational transitions. (A) [3H]DA uptake and (B) [3H]WIN 35,428 binding were conducted in PC12 cells transfected with WT hDAT or Y88F/H547A which were incubated with one of the concentrations of ZnCl2 (1, 10, 100 µM, final concentration) or KRH buffer (control) followed by addition of a fixed concentration of [3H]DA uptake or [3H]WIN 35,428 binding. The histogram shows [3H]DA uptake and [3H]WIN 35,428 binding expressed as mean ± S.E.M. of the respective controls set as 100% for individual experiment. *p < 0.05 compared to the respective controls (Bonferroni t-test) in [3H]DA uptake (n = 7/group) and [3H]WIN 35,428 binding (n = 9/group). #p < 0.05 compared to WT hDAT with same concentration of ZnCl2 application (unpaired student t-test). (C) DAT-mediated basal DA efflux properties of WT hDAT and mutant. Functional DA efflux was conducted in PC12 cells transfected WT hDAT or Y88F/H547A by preloading with a fixed concentration of [3H]DA (0.05 μM, final concentration) at room temperature for 20 min followed by replacing with fresh buffer at indicated time points. Finally, the buffer in each well was separated from cells, and radioactivity in the buffer and remaining in the cells was counted. Each fractional efflux value of [3H]DA in WT hDAT and Y88F/H547A was expressed as percentage of total [3H]DA in the cells at the start of the experiment. Fractional efflux values of [3H]DA at 1, 10, 20, 30 and 40 min are expressed as the percentage of total [3H]DA with preloading with 0.05 μM (WT hDAT: 11044 ± 241 dpm and Y88F/H547A: 660 ± 123 dpm) present in the cells at the start of the experiment (n = 5/group). ×p < 0.05 compared to WT hDAT (Bonferroni t-test).
To further determine the effect of Tyr88/His547 mutation on transporter conformational transitions, we examined basal efflux levels of [3H]DA in PC12 cells expressing WT hDAT and Y88F/H547A (Fig. 5C). twenty min after preloading with 0.05 µM [3H]DA, cells were washed and fractional DA efflux samples were collected at the indicated time. Two-way ANOVA revealed significant main effects of mutation [F(1, 8) = 59.38, p < 0.0001], time [F(5, 40) = 116.35, p < 0.0001], and mutation × time interaction [F(5, 40) = 8.16, p < 0.0001]. Post hoc analyses showed that compared to WT hDAT, DA efflux levels were significantly elevated at 1 and 10 min (ps < 0.05, Bonferroni t test).
Effects of Y88/H547 mutation on amphetamine-regulated DA efflux and uptake
AMPH acts as a substrate for DAT which subsequently reverses the transporter leading to DA efflux33,34. AMPH-stimulated [3H] DA efflux in PC12 cells transfected with WT hDAT and its mutants was determined under a stable baseline of DA efflux (Fig. 6A), showing no difference on basal [3H]DA efflux in control groups (F(4, 27) = 1.92, p = 0.14, one-way ANOVA). As shown in Fig. 6B, AMPH induced [3H]DA efflux in WT hDAT and mutants in a concentration dependent manner. Two-way ANOVA analysis on the Vmax values revealed significant main effects of mutation [F(4, 26) = 4.63, p < 0.01], AMPH [F(5, 130) = 76.81, p < 0.0001] and mutation × AMPH interaction [F(20, 130) = 2.34, p < 0.01]. As shown in Table 3, the Vmax values of [3H]DA efflux in response to AMPH were reduced in H547A (0.22 ± 0.04) and Y88F/H547A (0.20 ± 0.03) compared to WT hDAT (0.42 ± 0.03, ps < 0.05, Bonferroni’s multiple comparison test). No difference in Km was found among WT hDAT and all mutants.
Figure 6.

Effects of AMPH on the kinetic analysis of DA efflux and uptake. PC12 cells transfected with WT hDAT and its mutants were washed, and preloaded with KRH buffer containing 50 nM [3H]DA for 20 min at room temperature. After three quick washes with KRH, aliquots from each well were collected and replaced with fresh KRH every 5 min for 15 min to establish basal efflux. At 20 min, AMPH (5–250 µM) was added, and the same aliquots were taken after 5 min as described in Material and Methods. (A) Baseline of [3H]DA efflux before addition of AMPH. Data were presented as means ± S.E.M. of the ratios of efflux to [3H]DA in cells (efflux dmp/cellular dmp) in WT hDAT and mutants (n = 4–13). (B) AMPH-mediated efflux is expressed as means ± S.E.M. of the fractional release of cellular [3H]DA per 5 min in WT and mutants. (C,D) kinetic analysis of [3H]DA uptake in WT hDAT and Y88F/H547A in the presence or absence of AMPH (1 µM). Data were presented as means ± S.E.M. (n = 5/group). *p < 0.05 compared to their respective controls (paired t test) #p < 0.05 compared to WT hDAT value (unpaired Student’s t test).
Table 3.
Kinetic analysis of AMPH-stimulated [3H]DA efflux in WT hDAT and mutants.
| WT hDAT | Y88F | Y88A | H547A | Y88F/H547A | |
|---|---|---|---|---|---|
| V max | 0.42 ± 0.03 | 0.28 ± 0.06 | 0.29 ± 0.01 | 0.22 ± 0.04** | 0.20 ± 0.03** |
| K m | 10.2 ± 3.2 | 7.61 ± 2.00 | 9.80 ± 5.11 | 9.49 ± 6.52 | 9.31 ± 4.7 |
Data are presented as mean ± S.E.M. of the Vmax and Km values for AMPH-stimulated [3H]DA in WT hDAT and its mutants from 5–13 independent experiments. *p < 0.05 compared to WT hDAT values (Bonferroni’s multiple comparison test).
Double mutation of Tyr88 and His547 could induce DAT conformational change by altering AMPH interaction with DAT. We first examined the affinity for AMPH to inhibit [3H]DA uptake in WT and Y88F/H547A (Table 1), however, no significant difference in the IC50 value was found between WT (926 ± 167.4 nM) and Y88F/H547A [1279 ± 200.5 nM, t(8) = 1.32, p = 0.22]. Next, we performed the kinetic analysis of [3H]DA uptake in WT and Y88F/H547A in presence of AMPH (1 µM, final concentration). As shown in Fig. 6C, two-way ANOVA analysis on the Vmax of [3H]DA uptake reveled significant main effects of mutation (F(1, 16) = 9.50, p < 0.01) and AMPH (F(1, 16) = 11.99, p < 0.01). No significant interaction of mutation × AMPH treatment was found (F(1, 16) = 1.61, p = 0.22). Post hoc analyses show that the Vmax value in Y88F/H547A (18.9 ± 2.81 pmol/min/105 cells) was higher than that in WT hDAT (11.5 ± 0.94 pmol/min/105 cells, t(8) = 2.49, p < 0.05, n = 5/group) in the absence of AMPH. AMPH decreased the Vmax in both WT hDAT (32%, 7.80 ± 0.97 pmol/min/105 cells, t(8) = 2.76, p < 0.01) and Y88F/H547A (42%, 10.9 ± 1.33 pmol/min/105 cells, t(8) = 2.58, p < 0.05) compared with their respective controls. There was a significant difference in the magnitude of the AMPH-induced decrease in Vmax values between WT hDAT and Y88F/H547A (t(8) = 1.86, p < 0.05). Furthermore, two-way ANOVA analysis on Km values revealed significant main effects of mutation [F(1, 16) = 21.06, p < 0.001] and AMPH [F(1, 16) = 10.17, p < 0.01],whereas no significant interaction of mutation × AMPH treatment was found [F(1, 16) = 0.13, p = 0.73]. In the absence of AMPH, the Km values of [3H]DA uptake was greater in Y88F/H547A (5.12 ± 0.76 µM) than that in WT hDAT [1.43 ± 0.15 µM, t(8) = 4.79, p < 0.01). Compared to the controls, AMPH significantly increased the Km value in WT hDAT [4.08 ± 0.56 µM; t(8) = 4.63, p < 0.01], which was attenuated in Y88F/H547A (7.24 ± 1.15 µM).
Discussion
Our recent studies demonstrated that single hDAT mutants, Y88F, K92M, and H547A differentially regulate DAT reuptake but attenuate Tat’s inhibitory effect on DAT function. This study sought to determine whether multiple mutations on these hDAT residues (Y88F/H547A) and (Y88F/K92M/H547A) influence the direct interactions between Tat and DAT. There were three major findings. First, compared with WT hDAT, Y88F/H547A enhances Vmax of DA uptake, whereas Y88F/K92M/H547A dramatically reduces the Vmax. Second, consistent with the computational modeling prediction, Y88F/H547A attenuates Tat-induced inhibition of DA uptake observed in WT hDAT. The third finding is that Y88F/H547A displays an attenuation of zinc ion modulation of [3H]WIN35,428 binding, increased basal DAT-mediated DA efflux, and reduced AMPH-stimulated DA efflux. Collectively, these findings suggest that targeting DAT residues Tyr88 and His547 may have potential therapeutic benefits on Tat-induced dysregulation of dopaminergic system seen in patients with HAND.
According to our computationally modeled structures, Y548-Y470-Y551 is a stable motif (denoted as the YYY motif for convenience) essential for the conformational conversion of hDAT required for the DA transport process35. This YYY motif has hydrophobic contact with a typical U-turn loop associated with extracellular loop 6 (EL6) and transmembrane 10 (TM10a). Any structural changes affecting the stability of the YYY motif could destabilize the R85-D476 salt bridge of hDAT, and the salt bridge is key for the conformational conversion36–39. H547 is expected to significantly affect the hDAT stability because H547 is adjacent to Y548 of the YYY motif35, which is consistent with the observed increase in DA uptake by the H547A mutation18. On the other hand, the side chain of K92 residue (TM1b) forms a favorable salt bridge with the side chain of D313 (TM6a) during the molecular dynamics simulation on the hDAT-DA binding complex, and the bridge is also essential for the conformational conversion of hDAT required for the DA uptake process40. For this reason, disrupting the K92-D313 salt bridge will likely impair or slow down the DA uptake process, which has been validated pharmacologically by mutations on K92 (K92M)17 and D313 (D313N)41. In addition, the aromatic ring on the side chain of Y88 is sandwiched by TM1b and extracellular loop 4 (EL4) in a hydrophobic region of hDAT. Therefore, mutation of Y88 to phenylalanine retains the normal reuptake function of hDAT and demonstrated no difference compared to WT hDAT17. However, Y88F increased the Km of DA binding by about 50%, suggesting that Y88F may slightly alter the local structure around Y8817. In the current study, the double mutant Y88F/H547A retains the increased Vmax observed in H547A, suggesting a critical role of the mutated H547 residue in regulating transporter activity. In contrast, the combination of the mutated residues Y88, K92, and H547 does not follow a simple math addition, because compared to WT hDAT, a 96% reduction of Vmax was observed in Y88F/K92M/H547A, whereas the Vmax was decreased by 71% in K92M17. These results suggest that the inclusion of the K92 mutation in the K92-D313 salt bridge is a critical factor in the differential DAT kinetics in the triple mutation of hDAT. As Y88 is spatially adjacent to R85 and K92 in helix TM1b, the double mutations on both Y88F and K92M are expected to induce more structural disturbance to the R85-D476 and K92-D313 salt brides, thus further decreasing Vmax of the transporter. Therefore, triple mutant Y88F/K92M/H547A has a lower Vmax compared to, single mutant K92M. Although including K92M mutation significantly decreases the Vmax for DA uptake in the triple mutant Y88F/K92M/H547A compared to Y88F/H547A, no significant change in the Bmax of [3H]WIN 35,428 binding is found in the double or triple mutants, suggesting that the multiple mutations may alter DAT re-uptake function rather than the substrate binding sites on DAT. This conclusion is also supported by the increased Km in Y88F/H547A and no change in Km in Y88F/K92M/H547A. However, single point mutations of Y88, K92, and H547 differentially affect the Vmax with no changes in the Km17,18. One possible explanation is that allosteric modulation of the dynamic DA transport process may be responsible for the enhanced Vmax and its associated DA uptake potency.
We further determined the effect of double mutant Y88F/H547A on the basal DA uptake via hDAT and binding potency for DA, GBR12909, and cocaine. Y88F/H547A had decreased the potency for [3H]DA to inhibit DA uptake relative to WT hDAT, which is similar to that in Y88F and H547A16,18. However, Y88F/H547A increased the DA uptake potency for cocaine and GBR12909, which is consistent with the single mutation of Y88F relative to WT hDAT17. Similar to Y88F17, Y88F/H547A displayed decreased potency for DA to inhibit [3H]WIN35,428 binding. However, the inhibition potency of [3H]WIN35,428 binding for cocaine was decreased in Y88F/H547A but increased in Y88F17. Y88F/H547A, similar to Y88F17, retained the [3H]WIN35,428 binding potency for GBR12909. GBR12909 is a selective inhibitor of DA uptake, binding to the piperazine acceptor site without influencing DA release42; while cocaine is a competitive inhibitor for DAT and prefers to an outward-facing transport state, leading to a decrease in DA transport43,44. According to our computational modeling prediction, both hDAT residues Y88 and H547 are the critical binding sites on DAT for Tat20. Therefore, our findings demonstrate that through allosteric modulation, mutation of Y88 and H547 alters the inhibition potency for DA uptake and DAT binding site for substrate and DAT inhibitors40.
Our previous study has demonstrated that mutant H547A displays increased sensitivity to PKC-mediated downregulation of DAT function by PMA compared to WT hDAT18. Interestingly, the current study shows no difference in PMA-mediated reduction of DA uptake between Y88F/H547A and WT hDAT, indicating that Y88F/H547A-induced increase in Vmax is independent of PKC activity. It has been shown that PKC-mediated phosphorylation of DAT downregulates DA uptake velocity45–47. Through allosteric modulatory effect, single mutation of H547 may alter the levels of PKC-induced phosphorylation of DAT activity, thereby causing the increased Vmax. However, double mutation of Y88 and H547 may attenuate the intermolecular structural interaction across PKC phosphorylation sites. Indeed, compared with WT hDAT, H547A, and Y88F/H547A enhances the Vmax values by 196%18 and 96%, respectively. In addition to PKC-mediated regulation of DAT function, activation of palmitoyl acyltransferase, a group of enzymes that transfer palmityl group to -SH group on cysteine on a protein, enhances DA uptake via DAT palmitoylation48. Therefore, we will investigate whether Y88F/H547A-induced increased DA uptake is dependent on palmitoylation activity.
A major finding is that the double mutant Y88F/H547A completely attenuates the Tat-mediated inhibition of DAT reuptake function, which is consistent with our previous reports on Y88F- or H547A-induced attenuation of the Tat’s effect17,18. According to our computational modeling prediction, either the Y88 or H547 residue could form hydrogen bonds with Tat residue K19 or R49 independently, suggesting that the double mutation of Y88 and H547 eliminated Tat-DAT binding. Hence, our pharmacological study further validates the computational prediction for the complementary hydrophobic interactions between Tat and hDAT. Furthermore, since we have demonstrated that Tat interacts with DAT allosterically19,22, the current findings also suggest that double mutation of Y88 and H547 attenuates Tat effect through a conformational mechanism. First, Y88F/H547A attenuates zinc-mediated increase in [3H]WIN35,428 binding relative to WT hDAT. Second, Y88F/H547A augments basal DA efflux when compared to WT hDAT, which opposes our previous reports showing that neither Y88F nor H547A affected the basal efflux17,18. The elevated basal DA efflux in Y88F/H547A may be attributed to its elevated Vmax of DA uptake and DA accumulation. Taken together, Y88F/H547A, via an alteration of DAT conformational transitions, may influence the binding structure of hDAT-Tat complex, thereby disrupting Tat-induced inhibition of DA uptake. Studying the multiple mutation of DAT residues in Tat-DAT intermolecular interaction will greatly contribute to our ongoing proof of concept studies using novel allosteric modulators to establish their potential for therapeutic application in HAND.
AMPH, a substrate of DAT, competitively inhibits the reuptake of DA and elicits DAT-mediated DA efflux by reversal of the transporter49. Our computational modeling has demonstrated that the dynamic DA uptake process includes outward-open, outward-occluded, and inward-open states21, which can be reversed by DA substrate and DAT mutation49–51. The current results indicate that the potency for AMPH inhibition of [3H]DA uptake was not altered in Y88F/H547A compared with WT hDAT, suggesting that the mutation of Tyr88 and His547 does not alter the interaction of AMPH with hDAT. However, AMPH-mediated DA efflux was reduced in H547A and Y88F/H547A, suggesting that mutations of Tyr88 and His547 residues alter the conformational equilibrium of the DAT transport transitions. Furthermore, AMPH decreases the Vmax of DA uptake in both WT hDAT and Y88F/H547A, which is consistent with the previous report51, however, the magnitude of the AMPH-mediated reduction of the Vmax in Y88F/H547A was greater than that in WT hDAT. Interestingly, the AMPH-mediated reduction of the Vmax was accompanied by increased Km values in WT hDAT but not Y88F/H547A. Given that DAT-mediated DA transport is accompanied by reuptake of two Na+ and Cl− ions, AMPH influx would transport Na+ through DAT and enhance its levels at the inner face of the transporter52. Therefore, AMPH decreases DA transport but increases Km values in WT hDAT, whereas altered transporter conformation by Y88F/H547A may attenuate the increased Km. Taken together, the results along with attenuated zinc-regulated [3H] WIN35,428 binding and increased basal DA efflux in Y88F/H547A further support that mutations of Tyr88 and His547 cause an alteration in conformational transitions, thereby disrupting the inhibitory effect of Tat on DAT reuptake function.
In conclusion, our data further demonstrate DAT Tyr88 and His547 as important residues for DAT function and the Tat-DAT interaction. Double mutation of these residues preserves single mutant H547A-mediated enhancement of DA uptake and diminishes Tat-induced inhibition of DA transport18. An allosteric modulatory mechanism may contribute to Y88F/H547A-mediated attenuation of Tat-induced inhibition of DA transport. Understanding the multiple mutation effects of the identified DAT residues might provide a basis for a novel approach for developing compounds to attenuate Tat binding to DAT by an allosteric mechanism in HIV-1-infected individuals.
Materials and Methods
Prediction of Tat-DAT binding model
We performed computational modeling and simulations to model the binding structure of hDAT with HIV-1 clade B type Tat based on the nuclear magnetic resonance (NMR) structures of Tat53 and the constructed structure of hDAT-DA complex21. Protein docking and molecular dynamics simulations were employed to identify the conformation of hDAT-Tat complex. The energy-minimized complex structure used in this work were extracted from long-time equilibrated molecular dynamics simulation trajectories in our previous work17,18,20,35.
Construction of plasmids
We selected the mutations of Y88, H547, Y88/H547 and Y88/K92/H547 based on predictions of computational modeling and simulations (Fig. 1) and our previous studies17,18. Single mutants, Y88F, K92M, and H547A, are expected to abolish hydrogen bonds between hDAT and Tat. Double (H547A/Y88F) and triple (H547A/Y88F/K92M) mutations were generated based on the wild type hDAT (WT hDAT) sequence (NCBI, cDNA clone MGC: 164608 IMAGE: 40146999) by site-directed mutagenesis. We then used the synthetic cDNA encoding hDAT subcloned into pcDNA3.1 + as a template to generate mutants using QuikChange™ site-directed mutagenesis Kit (Agilent Tech, Santa Clara CA). We performed the DAT sequence for confirming the sequence of the mutant construct at University of South Carolina EnGenCore facility. Plasmids DNA were propagated and purified using a plasmid isolation kit (Qiagen, Valencia, CA, USA).
Cell culture and DNA transfection
In the current study, we used rat pheochromocytoma cells (PC12 cells, CRL-1721, American Type Culture Collection (ATCC), Manassas, VA), which were maintained in Dulbecco’s modified eagle medium (DMEM, Life Technologies, Carlsbad, CA) supplemented with 15% horse serum, 2.5% bovine calf serum, 2 mM glutamine and antibiotics, 100 U/mL penicillin and 100 µg/mL streptomycin. Cells were cultured at 37 °C in a 5% CO2 incubator. Once cells were growing to 100% confluence on plates, cells were seeded at a density of 1 × 105 cells/cm2 in 24-well plates and transfected with WT hDAT or mutants by Lipofectamine 2000 (Life Technologies). Twenty-four hours after transfection, intact cells or cell suspensions were used for experiments.
[3H]DA uptake assay
In order to determine whether DAT mutants alter DAT function, we evaluated the maximal velocity (Vmax) or Michaelis-Menten constant (Km) of [3H]DA uptake in intact PC12 cells transfected with WT hDAT or mutants as reported previously16,17. Twenty-four hours after transfection, cells in 24-well plates were washed twice with Krebs-Ringer-HEPES (KRH) buffer (final concentration in mM: 125 NaCl, 5 KCl, 1.5 MgSO4, 1.25 CaCl2, 1.5 KH2PO4, 10 D-glucose, 25 HEPES, 0.1 EDTA, 0.1 pargyline, and 0.1 L-ascorbic acid; pH 7.4) and incubated with nomifensine (10 µM, final concentration) as nonspecific uptake for 10 min at room temperature. A fixed concentration of [3H]DA (500,000 dpm/well, specific activity, 21.2 Ci/mM; PerkinElmer Life and Analytical Sciences, Boston, MA) mixed with one of six concentrations of unlabeled DA (final DA concentrations, 0.03–5 µM) were applied to cells at room temperature for additional 8 min. The specific DAT-mediated DA uptake was calculated by subtracting non-specific uptake from total uptake (in the absence of nomifensine). The reaction was terminated by removal of solution from wells and quickly washed three times with ice cold KRH buffer. Cells were then lysed in 500 μL of 1% SDS for an hour and radioactivity was measured using a liquid scintillation counter (Tri-Carb 2900TR; PerkinElmer Life and Analytical Sciences, Waltham, MA).
To confirm whether Y88F/H547A-induced increase in Vmax was dependent on PKC regulatory mechanism, the Vmax of [3H]DA uptake was measured in the presence or absence of 1 µM PMA, a PKC activator (Tocris, Bristol, UK) as the previous reports29,30. Intact PC12 cells transfected with WT or Y88F/H547A were preincubated with or without PMA for 30 min at 37 °C followed by additional 8 min incubation with six concentrations of mixed [3H]DA as described above. To determine the effect of amphetamine (AMPH) on Y88F/H547A-mediated transporter conformational transitions, Vmax or Km of [3H]DA uptake in WT and Y88F/H547A was determined in the presence or absence of 1 µM AMPH (Sigma-Aldrich, St. Louis, MO) as reported previously51.
To determine whether these mutations alter the affinity of DAT substrate or inhibitors as well as Zn2+-regulation of DA transport, we performed the competitive inhibition of DA uptake in intact PC12 cells transfected with WT or mutant, which were preincubated with a series of final concentrations of DA (1 nM-1 mM), GBR12909 (1 nM-10 µM), cocaine (1 nM-1 mM), AMPH (1 nM-1 mM) or ZnCl2 (1, 10, and 100 μM) at room temperature for 10 min followed by additional 8 min incubation with a fixed concentration of [3H]DA (0.05 µM, final concentration).
In order to determine whether double mutation of Y88 and H547 disrupts Tat and DAT interaction, we performed the specific [3H]DA uptake in PC12 cells transfected with WT or mutant in the presence or absence of Tat protein. In brief, cells were dissociated with trypsin/EDTA (0.25%/0.1%, 1 mL for one 10 cm dish) and resuspended in culture medium and incubated at room temperature for 10 min. The dissociated cells were collected by centrifugation at 400 × g for 5 min at 4 °C and washed once with phosphate-buffered saline followed by additional 5 min centrifugation (400 × g, 4 °C). Finally, the resulted cell pellets were resuspended in KRH assay buffer. The cell suspensions from WT hDAT or Y88F/H547A mutant were then preincubated with or without recombinant Tat1–86 (rTat1–86, 140 nM, final concentration, Diatheva, Fano, Italy) at room temperature for 20 min followed by additional 8 min incubation with a mixed [3H]DA uptake (0.05 µM, final concentration). Non-specific [3H]DA uptake was determined in the presence of 10 µM nomifensine. The update reaction was terminated by immediate filtration through Whatman GF/B glass filters (presoaked with 1 mM pyrocatechol for 3 h) followed by three washes with 3 mL of ice-cold KRH buffer containing pyrocatechol using a Brandel cell harvester (model M-48; Brandel Inc., Gaithersburg, MD). Radioactivity was determined as described above.
[3H]WIN 35,428 Binding Assay
Binding assays were used to determine the kinetic parameters (Bmax or Kd) of [3H]WIN 35,428 binding in intact PC12 cells transfected with WT hDAT or mutant. Cells were washed with sucrose-phosphate buffer twice (final concentration in mM: 2.1 NaH2PO4, 7.3 Na2HPO47H2O, and 320 sucrose, pH 7.4) and then incubated with one of the six concentrations of [3H]WIN 35,428 (84 Ci/mmol, PerkinElmer, 0.5–30 nM final concentrations) in a final volume of 500 μL on ice for 2 h. Nonspecific binding at each concentration of [3H]WIN 35,428 was determined by adding in the presence of 30 µM cocaine (final concentration) and subtracted from total binding to obtain the specific binding. For competitive inhibition experiments, assays were performed in a final volume of 250 μL. Intact PC12 cells transfected with WT or mutated hDATs were incubated in buffer containing 25 μL of [3H]WIN 35,428 (final concentration, 5 nM) and one of ten concentrations of DA (1 nM- 100 μM), cocaine (1 nM- 100 μM), GBR12909 (0.01 nM- 1 μM) or ZnCl2 (1, 10, 100 μM) on ice for 2 h. Assays were terminated by removing reaction reagents in each well followed by two washes with ice-cold assay buffer. Cells were lysed with 1% SDS for an hour and subjected to the radioactivity measurement as described above.
[3H]DA efflux assay
Basal DA efflux was performed at room temperature as described previously16–18. Intact PC12 cells transfected with WT hDAT or its mutant were preloaded with 0.05 μM [3H]DA for 20 min and then washed 3 times with KRH buffer prior to collecting fractional efflux samples. To obtain an estimate of the total amount of [3H]DA in the cells at the zero time point, cells from a set of wells (four wells/sample) were lysed rapidly in 1% SDS after preloading with [3H]DA. To collect factional efflux samples, buffer (500 μL) was added into a separate set of cell wells and transferred to scintillation vials after 1 min as an initial fractional efflux, and another 500 μl buffer was added to the same wells and collected after 10 min as second fractional efflux. Additional fractional efflux at 20, 30, 40, 50 min, respectively, was repeated under the same procedure. After last fractional efflux, cells were lysed and counted as total amount of [3H]DA remaining in the cells from each well.
To further determine DAT mutation-mediated transporter confirmation transitions, basal efflux and AMPH-stimulated DA efflux in WT hDAT and its mutants were measured as reported previously51. PC12 cells transfected with WT hDAT and its mutants growing in 24-well plates were preloaded with [3H]DA (50 nM, final concentration) for 20 min at room temperature. Subsequently, cells in all plate wells were washed with KRH for three times, cells were then incubated with KRH buffer containing AMPH (5–250 µM) or vehicle (KRH buffer alone, as baseline control) for 15 min at room temperature. After incubation, the buffer solution incubated with AMPH or vehicle was collected as AMPH-stimulated efflux or baseline efflux. Cells were lysed in 1% SDS and subject to count for obtaining the initial amount of DA (total DA) in the cells.
Data analysis
In the current study, all data are presented as mean ± SEM with n as the number of independent experiments for each experiment group. Pharmacological parameter values (Vmax, Km, Bmax, and Kd) and IC50 from [3H]DA uptake and [3H]WIN 35,428 binding were calculated by nonlinear regression analysis using a one-site model with variable slope. To determine the significant difference in the pharmacological parameters between unpaired sample groups, such as WT hDAT and its mutants, unpaired Student’s t test was used for the statistical comparisons. Significant differences between sample groups were analyzed with separate ANOVAs followed by post-hoc, Bonferroni multiple comparison test as indicated in the results Section of each experiment. All statistical analyses were performed using IBM SPSS Statistics version 25, and differences were considered significant at p < 0.05.
Acknowledgements
Research reported in this publication was supported by National Institutes of Health grants to JZ (R01DA035714 and R21DA041932).
Author Contributions
W.L.S., Y.Y. and J.Z. contributed to the original writing and editing the manuscript. All authors provided critical manuscript revision for important intellectual content. All authors have read and approved the final manuscript version. C.G.Z. and J.Z. were responsible for study concept and design. P.Q., R.M., M.S., Y.Y. and C.G.Z. contributed to the data acquisition.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Heaton RK, et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology. 2010;75:2087–2096. doi: 10.1212/WNL.0b013e318200d727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.King JE, Eugenin EA, Buckner CM, Berman JW. HIV tat and neurotoxicity. Microbes and infection / Institut Pasteur. 2006;8:1347–1357. doi: 10.1016/j.micinf.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 3.Clifford DB, Ances BM. HIV-associated neurocognitive disorder. The Lancet infectious diseases. 2013;13:976–986. doi: 10.1016/S1473-3099(13)70269-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brack-Werner R. Astrocytes: HIV cellular reservoirs and important participants in neuropathogenesis. AIDS. 1999;13:1–22. doi: 10.1097/00002030-199901140-00003. [DOI] [PubMed] [Google Scholar]
- 5.Frankel AD, Young JA. HIV-1: fifteen proteins and an RNA. Annu Rev Biochem. 1998;67:1–25. doi: 10.1146/annurev.biochem.67.1.1. [DOI] [PubMed] [Google Scholar]
- 6.Johnston JB, et al. HIV-1 Tat neurotoxicity is prevented by matrix metalloproteinase inhibitors. Ann Neurol. 2001;49:230–241. doi: 10.1002/1531-8249(20010201)49:2<230::AID-ANA43>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 7.Power C, et al. Neuronal death induced by brain-derived human immunodeficiency virus type 1 envelope genes differs between demented and nondemented AIDS patients. J Virol. 1998;72:9045–9053. doi: 10.1128/jvi.72.11.9045-9053.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rappaport J, et al. Molecular pathway involved in HIV-1-induced CNS pathology: role of viral regulatory protein, Tat. Journal of leukocyte biology. 1999;65:458–465. doi: 10.1002/jlb.65.4.458. [DOI] [PubMed] [Google Scholar]
- 9.Zhu J, Ananthan S, Zhan CG. The role of human dopamine transporter in NeuroAIDS. Pharmacol Ther. 2018;183:78–89. doi: 10.1016/j.pharmthera.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nieoullon A. Dopamine and the regulation of cognition and attention. Prog Neurobiol. 2002;67:53–83. doi: 10.1016/S0301-0082(02)00011-4. [DOI] [PubMed] [Google Scholar]
- 11.Cools R. Dopaminergic modulation of cognitive function-implications for L-DOPA treatment in Parkinson’s disease. Neurosci Biobehav Rev. 2006;30:1–23. doi: 10.1016/j.neubiorev.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 12.Tye KM, et al. Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature. 2013;493:537–541. doi: 10.1038/nature11740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lammel S, et al. Input-specific control of reward and aversion in the ventral tegmental area. Nature. 2012;491:212–217. doi: 10.1038/nature11527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Purohit V, Rapaka R, Shurtleff D. Drugs of abuse, dopamine, and HIV-associated neurocognitive disorders/HIV-associated dementia. Molecular neurobiology. 2011;44:102–110. doi: 10.1007/s12035-011-8195-z. [DOI] [PubMed] [Google Scholar]
- 15.Berger JR, Arendt G. HIV dementia: the role of the basal ganglia and dopaminergic systems. Journal of psychopharmacology. 2000;14:214–221. doi: 10.1177/026988110001400304. [DOI] [PubMed] [Google Scholar]
- 16.Midde NM, et al. Mutation of tyrosine 470 of human dopamine transporter is critical for HIV-1 Tat-induced inhibition of dopamine transport and transporter conformational transitions. J Neuroimmune Pharmacol. 2013;8:975–987. doi: 10.1007/s11481-013-9464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Midde NM, et al. Mutations at tyrosine 88, lysine 92 and tyrosine 470 of human dopamine transporter result in an attenuation of HIV-1 Tat-induced inhibition of dopamine transport. J Neuroimmune Pharmacol. 2015;10:122–135. doi: 10.1007/s11481-015-9583-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quizon PM, et al. Molecular mechanism: the human dopamine transporter histidine 547 regulates basal and HIV-1 Tat protein-inhibited dopamine transport. Scientific reports. 2016;6:39048. doi: 10.1038/srep39048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu J, Mactutus CF, Wallace DR, Booze RM. HIV-1 Tat protein-induced rapid and reversible decrease in [3H]dopamine uptake: dissociation of [3H]dopamine uptake and [3H]2beta-carbomethoxy-3-beta-(4-fluorophenyl)tropane (WIN 35,428) binding in rat striatal synaptosomes. J Pharmacol Exp Ther. 2009;329:1071–1083. doi: 10.1124/jpet.108.150144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yuan Y, et al. Molecular mechanism of HIV-1 Tat interacting with human dopamine transporter. ACS chemical neuroscience. 2015;6:658–665. doi: 10.1021/acschemneuro.5b00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuan Y, Huang X, Zhu J, Zhan CG. Computational modeling of human dopamine transporter structures, mechanism and its interaction with HIV-1 transactivator of transcription. Future medicinal chemistry. 2016;8:2077–2089. doi: 10.4155/fmc-2016-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu J, Ananthan S, Mactutus CF, Booze RM. Recombinant human immunodeficiency virus-1 transactivator of transcription1-86 allosterically modulates dopamine transporter activity. Synapse. 2011;65:1251–1254. doi: 10.1002/syn.20949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pariser JJ, Partilla JS, Dersch CM, Ananthan S, Rothman RB. Studies of the biogenic amine transporters. 12. Identification of novel partial inhibitors of amphetamine-induced dopamine release. J Pharmacol Exp Ther. 2008;326:286–295. doi: 10.1124/jpet.108.139675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rothman RB, et al. Studies of the biogenic amine transporters 15. Identification of novel allosteric dopamine transporter ligands with nanomolar potency. J Pharmacol Exp Ther. 2015;353:529–538. doi: 10.1124/jpet.114.222299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rothman RB, Dersch CM, Ananthan S, Partilla JS. Studies of the biogenic amine transporters. 13. Identification of “agonist” and “antagonist” allosteric modulators of amphetamine-induced dopamine release. J Pharmacol Exp Ther. 2009;329:718–7288. doi: 10.1124/jpet.108.149088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun WL, et al. Allosteric modulatory effects of SRI-20041 and SRI-30827 on cocaine and HIV-1 Tat protein binding to human dopamine transporter. Scientific reports. 2017;7:3694. doi: 10.1038/s41598-017-03771-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pristupa ZB, Wilson JM, Hoffman BJ, Kish SJ, Niznik HB. Pharmacological heterogeneity of the cloned and native human dopamine transporter: disassociation of [3H]WIN 35,428 and [3H]GBR 12,935 binding. Mol Pharmacol. 1994;45:125–135. [PubMed] [Google Scholar]
- 28.Reith ME, Coffey LL. [3H]WIN 35,428 binding to the dopamine uptake carrier. II. Effect of membrane fractionation procedure and freezing. J Neurosci Methods. 1994;51:31–38. doi: 10.1016/0165-0270(94)90023-X. [DOI] [PubMed] [Google Scholar]
- 29.Navaroli DM, et al. The plasma membrane-associated GTPase Rin interacts with the dopamine transporter and is required for protein kinase C-regulated dopamine transporter trafficking. J Neurosci. 2011;31:13758–13770. doi: 10.1523/JNEUROSCI.2649-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Melikian HE, Buckley KM. Membrane trafficking regulates the activity of the human dopamine transporter. J Neurosci. 1999;19:7699–7710. doi: 10.1523/JNEUROSCI.19-18-07699.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loland CJ, Norgaard-Nielsen K, Gether U. Probing dopamine transporter structure and function by Zn2 + -site engineering. Eur J Pharmacol. 2003;479:187–197. doi: 10.1016/j.ejphar.2003.08.068. [DOI] [PubMed] [Google Scholar]
- 32.Norregaard L, Frederiksen D, Nielsen EO, Gether U. Delineation of an endogenous zinc-binding site in the human dopamine transporter. EMBO J. 1998;17:4266–4273. doi: 10.1093/emboj/17.15.4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gnegy ME. The effect of phosphorylation on amphetamine-mediated outward transport. Eur J Pharmacol. 2003;479:83–91. doi: 10.1016/j.ejphar.2003.08.059. [DOI] [PubMed] [Google Scholar]
- 34.Robertson SD, Matthies HJ, Galli A. A closer look at amphetamine-induced reverse transport and trafficking of the dopamine and norepinephrine transporters. Molecular neurobiology. 2009;39:73–80. doi: 10.1007/s12035-009-8053-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan Y, et al. Role of Histidine 547 of Human Dopamine Transporter in Molecular Interaction with HIV-1 Tat and Dopamine Uptake. Scientific reports. 2016;6:27314. doi: 10.1038/srep27314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang X, Zhan CG. How dopamine transporter interacts with dopamine: insights from molecular modeling and simulation. Biophysical journal. 2007;93:3627–3639. doi: 10.1529/biophysj.107.110924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gedeon PC, Indarte M, Surratt CK, Madura JD. Molecular dynamics of leucine and dopamine transporter proteins in a model cell membrane lipid bilayer. Proteins. 2010;78:797–811. doi: 10.1002/prot.22601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmitt KC, Reith ME. The atypical stimulant and nootropic modafinil interacts with the dopamine transporter in a different manner than classical cocaine-like inhibitors. PLoS One. 2011;6:e25790. doi: 10.1371/journal.pone.0025790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manepalli S, Surratt CK, Madura JD, Nolan TL. Monoamine transporter structure, function, dynamics, and drug discovery: a computational perspective. AAPS J. 2012;14:820–831. doi: 10.1208/s12248-012-9391-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu J, Ananthan S, Zhan CG. The role of human dopamine transporter in NeuroAIDS. Pharmacol Ther. 2018;183:78–89. doi: 10.1016/j.pharmthera.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen N, Vaughan RA, Reith ME. The role of conserved tryptophan and acidic residues in the human dopamine transporter as characterized by site-directed mutagenesis. J Neurochem. 2001;77:1116–1127. doi: 10.1046/j.1471-4159.2001.00312.x. [DOI] [PubMed] [Google Scholar]
- 42.Andersen PH, Jansen JA, Nielsen EB. [3H]GBR 12935 binding in vivo in mouse brain: labelling of a piperazine acceptor site. Eur J Pharmacol. 1987;144:1–6. doi: 10.1016/0014-2999(87)90002-1. [DOI] [PubMed] [Google Scholar]
- 43.Reith ME, Berfield JL, Wang LC, Ferrer JV, Javitch JA. The uptake inhibitors cocaine and benztropine differentially alter the conformation of the human dopamine transporter. J Biol Chem. 2001;276:29012–29018. doi: 10.1074/jbc.M011785200. [DOI] [PubMed] [Google Scholar]
- 44.Loland CJ, Norregaard L, Litman T, Gether U. Generation of an activating Zn(2 + ) switch in the dopamine transporter: mutation of an intracellular tyrosine constitutively alters the conformational equilibrium of the transport cycle. Proc Natl Acad Sci USA. 2002;99:1683–1688. doi: 10.1073/pnas.032386299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huff RA, Vaughan RA, Kuhar MJ, Uhl GR. Phorbol esters increase dopamine transporter phosphorylation and decrease transport Vmax. J Neurochem. 1997;68:225–232. doi: 10.1046/j.1471-4159.1997.68010225.x. [DOI] [PubMed] [Google Scholar]
- 46.Vaughan RA, Huff RA, Uhl GR, Kuhar MJ. Protein kinase C-mediated phosphorylation and functional regulation of dopamine transporters in striatal synaptosomes. J Biol Chem. 1997;272:15541–15546. doi: 10.1074/jbc.272.24.15541. [DOI] [PubMed] [Google Scholar]
- 47.Zhu SJ, Kavanaugh MP, Sonders MS, Amara SG, Zahniser NR. Activation of protein kinase C inhibits uptake, currents and binding associated with the human dopamine transporter expressed in Xenopus oocytes. J Pharmacol Exp Ther. 1997;282:1358–1365. [PubMed] [Google Scholar]
- 48.Moritz AE, et al. Reciprocal Phosphorylation and Palmitoylation Control Dopamine Transporter Kinetics. J Biol Chem. 2015;290:29095–29105. doi: 10.1074/jbc.M115.667055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol. 2005;75:406–433. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 50.Khoshbouei H, et al. N-terminal phosphorylation of the dopamine transporter is required for amphetamine-induced efflux. PLoS Biol. 2004;2:E78. doi: 10.1371/journal.pbio.0020078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guptaroy B, et al. A juxtamembrane mutation in the N terminus of the dopamine transporter induces preference for an inward-facing conformation. Mol Pharmacol. 2009;75:514–524. doi: 10.1124/mol.108.048744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khoshbouei H, Wang H, Lechleiter JD, Javitch JA, Galli A. Amphetamine-induced dopamine efflux. A voltage-sensitive and intracellular Na + -dependent mechanism. J Biol Chem. 2003;278:12070–12077. doi: 10.1074/jbc.M212815200. [DOI] [PubMed] [Google Scholar]
- 53.Peloponese JM, Jr., et al. 1H-13C nuclear magnetic resonance assignment and structural characterization of HIV-1 Tat protein. C R Acad Sci III. 2000;323:883–894. doi: 10.1016/S0764-4469(00)01228-2. [DOI] [PubMed] [Google Scholar]