

Abstract
Neuroblastoma is one of the common solid tumors of childhood. Nearly half of neuroblastoma patients are classified into the high-risk group, and their 5-year event-free survival (EFS) rates remain unsatisfactory in the range of 30-40%. High-risk neuroblastoma is characterized by amplification of the MYCN gene and excessive expression of its protein product, N-Myc. Because N-Myc is a transcription factor for various pro-proliferative proteins, the excessive expression causes aberrant or blocked neuronal differentiation during development of sympathetic nervous system, which is a central aspect of neuroblastoma genesis. The current main treatment for high-risk neuroblastoma is intensive chemotherapy using anti-cancer drugs that induce apoptosis in tumor cells, but intensive chemotherapy has another serious risk of long-lasting side effects, so-called “late effects”, that occur many years after chemotherapy has ended. As a solution for such situation, differentiation therapy has been expected as a mild chemotherapy with a low risk of late effects, and an application of retinoic acid (RA) and its derivatives as treatment for high-risk neuroblastoma has long been attempted. However, the clinical outcome has not been sufficient with the use of retinoids, including all-trans retinoic acid (ATRA), mainly because of the inhibition of differentiation caused by N-Myc. In the present study, we succeeded in synergistically accelerating the ATRA-induced neuronal differentiation of MYCN-amplified neuroblastoma cells by combining a peptide derived from tenascin-C, termed TNIIIA2, which has a potent ability to activate β1-integrins. Accelerated differentiation was caused by a decrease in N-Myc protein level in neuroblastoma cells after the combined treatment of TNIIIA2 with ATRA. That is, combination treatment using ATRA with TNIIIA2 induced proteasomal degradation in the N-Myc oncoprotein of neuroblastoma cells with MYCN gene amplification, and this caused acceleration of neuronal differentiation and attenuation of malignant properties. Furthermore, an in vivo experiment using a xenograft mouse model showed a therapeutic potential of the combination administration of ATRA and TNIIIA2 for high-risk neuroblastoma. These results provide a new insight into differentiation therapy for high-risk neuroblastoma based on N-Myc protein degradation.
Keywords: Neuroblastoma, proto-oncogene, mycn, N-Myc, differentiation therapy, retinoic acid, ATRA, tenascin-C, integrin, ubiquitin-proteasome system
Introduction
Neuroblastoma is one of the most common solid tumors in childhood and appears mainly in the sympathetic nervous system [1]. Approximately 40% of patients are classified into the high-risk group, in which the 5-year EFS rate is below 50% [2]. Most high-risk patients characteristically have tumors with extra copies of the MYCN gene encoding the N-Myc oncoprotein [2]. The MYCN gene amplification is closely correlated with poor prognosis and the 5-year EFS rates of patients with this gene amplification remain below 30% [2]. Because N-Myc is a transcription factor for various proteins associated with cell cycle progression, the excessive expression of N-Myc causes aberrant or blocked sympathetic neuronal differentiation during development, which is a central aspect of neuroblastoma genesis [3]. Thus, excessively expressed N-Myc has been considered the most important target for the treatment of high-risk neuroblastoma [4]. However, there is no effective therapeutic agent targeting N-Myc due to its lack of a specific catalytic site, which could be targeted by small molecule inhibitors [5].
The current main treatment for high-risk neuroblastoma patients is intensive chemotherapy using anti-cancer drugs that induce apoptosis in tumor cells [6], but the survival rates have remained unsatisfied during the past 30 years [7]. Additionally, intensive chemotherapy in pediatric cancer patients has another serious risk of long-lasting side effects, so-called “late effects”, that occur many years after chemotherapy has ended [8]. Therefore, there is an urgent need for alternative therapeutic approaches without the risk of the late effects. Differentiation therapy using RA has been investigated as a mild chemotherapy, which does not depend on killing cancer cells [9]. In fact, ATRA has been used for the treatment of acute promyelocytic leukemia (APL) [10]. However, differentiation therapy using RA is extremely limited and has never provided sufficient therapeutic effects on the other malignancies, including solid cancers [11]. Among various RA derivatives, 13-cis RA is currently served as a maintenance treatment after remission of high-risk neuroblastoma, but the clinical benefit in 5-year overall survival rate is not proven [12-14]. Further improvement of differentiation therapy is required to improve the current outcome for high-risk neuroblastoma patients.
Cell adhesion to the extracellular matrix (ECM) via integrins plays a key role in cell regulation such as survival, proliferation and even differentiation [15,16]. We previously found that a 22-mer peptide derived from tenascin-C, TNIIIA2, has potent and sustained ability to promote cell adhesion to the ECM by activating β1-integrins [17]. Our previous studies indicated that a variety of cellular processes can be regulated through β1-integrin activation by peptide TNIIIA2 [18-20]. Notably, the present study demonstrated that combination treatment of ATRA with TNIIIA2 induced proteasomal degradation of N-Myc in neuroblastoma cells with MYCN amplification. This N-Myc protein degradation was accompanied by a remarkable induction of neuronal differentiation in neuroblastoma cells, resulting in a marked decrease in malignant properties, such as anchorage-independent proliferation and tumorigenicity. Moreover, an in vivo experiment using a neuroblastoma xenograft mouse model showed that combination treatment of ATRA with TNIIIA2 successfully prevented tumor growth and was accompanied by a clear decrease in N-Myc protein level in the tumors. These results provide an important basis to develop a strategy for high-risk neuroblastoma treatment based on differentiation therapy.
Materials and methods
Cells
The human neuroblastoma cell line IMR-32 was obtained from Riken Cell Bank. MEM (Gibco) with 10% FBS, 2.2 g/L NaHCO3, 2 mM L-glutamine, and penicillin-streptomycin solution (FUJIFILM Wako) was used for IMR-32 cell culture. The human neuroblastoma cell line Kelly was obtained from ATCC. RPMI1640 medium (Nissui) supplemented with 10% FBS, 2.2 g/L NaHCO3, 2 mM L-glutamine, and penicillin-streptomycin solution was used for Kelly cell culture. Cells were incubated in a 5% CO2 incubator at 37°C.
Reagents
The synthetic TNIIIA2 peptide (RSTDLPGLKAATHYTITIRGVTC) was purchased from Eurofins genomics (Whitefield, India). ATRA was purchased from FUJIFILM Wako (Osaka, Japan). CS-1 peptide (LHPGEILDVPST) was obtained from Eurofins genomics. GRGDSP peptide was purchased from Calbiochem. MG-132 (Carbobenzoxy-L-leucyl-L-leucyl-L-leucinal) was obtained from Merck Millipore Ltd. (Tokyo, Japan). Anti-β1-integrin-activating monoclonal antibody (mAb), HUTS-4, was purchased from Millipore.
Cell adhesion assay
IMR-32 cells were harvested and suspended (1 × 104 cells/well) in serum-free medium with TNIIIA2 (1.5, 3, 50 µg/mL). They were incubated in a 96-well plate coated with fibronectin (2 µg/mL) in a 5% CO2 incubator at 37°C for 45 minutes. Adhered cells were fixed with 4% formalin and 5% glycerol. Fixed cells were stained with crystal violet and the number of spread and attached cells in 4 fields of each well were counted.
Flow cytometric analysis
Active-β1-integrins on the cells were evaluated by flow cytometric analysis using anti-β1-integrin antibody (Clone: AG89) conjugated with phycoerythrin (Medical & Biological Laboratories Co., Ltd.), which recognizes the active conformation-specific epitope of β1-integrin, and BD FACS Aria (BD Bioscience) as previously described [17].
Differentiation and measurement of axon-like neurites
IMR-32 cells were incubated with MEM including 1% FBS, ITS Mix (5 µg/mL insulin, 5 µg/mL transferrin and 5 ng/mL selenium) and reagents or inhibitors. After incubation, photographs were taken using microscope and Motic Image Plus 2.2S or 2.3S (Shimadzu), the number of differentiated cells was counted, and the length of axon-like neurites was measured using curvimeter. Cells were fixed and stained using Diff-Quick stain kit as needed (Sysmex).
Semi quantitative RT-PCR
RNA was extracted and purified by RNeasy Plus Mini Kit (QIAGEN) or Mammalian RNA purified Kit (Sigma). The isolated RNA was then transcribed into cDNA using QuantiTect® Reverse Transcription Kit (QIAGEN) according to the manufacturer’s instructions. Samples were run for RT-PCR on TaKaRa Thermal Cycler Dice mini (Takara Bio Inc.). Following primers were used: Actin forward; 5’-TGAAGTACCCCATTGAACACG-3’, Actin reverse; 5’-GTGCTAGGAGCCAGGGCAGT-3’, MYCN forward; 5’-GACCACAAGGCCCTCAGTAC-3’, MYCN reverse; 5’-GTGGATGGGAAGGCATCGTT-3’, GAPDH forward; 5’-CCCATGTTCGTCATGGGTGT-3’, GAPDH reverse; 5’-TGGTCATGAGTCCTTCCACGATA-3’, GAP43 forward; 5’-GGAGAAGGCACCACTACTGC-3’, GAP43 reverse; 5’-GGCGAGTTATCAGTGGAAGC-3’, Neurofilament light chain (NF-L) forward; 5’-ACCAAGACCTCCTCAACGTG-3’, NF-L reverse; 5’-TCAGCCTTGGCAGCCTCAAT-3’, AURKA forward; 5’-GCAGATTTTGGGTGGTCAGT-3’, and AURKA reverse; 5’-CAAAAGGAGGCTTCCCAACT-3’. 1 μM of each pair of the indicated primers, and 10 ng cDNA were mixed and PCR was performed in the following conditions: Actin; a 30 seconds initial denaturation at 95°C, followed by 35 cycles of 30 seconds at 94°C, 15 seconds at 55°C, and 50 seconds at 68°C, and 5 minutes at 68°C, MYCN (Figure 3Aa) and GAPDH (Figure 2D); a 30 seconds initial denaturation at 94°C, followed by 30 or 33 cycles of 30 seconds at 94°C, 30 seconds at 57°C, and 30 seconds at 68°C, and 7 minutes at 68°C, NF-L; a 60 seconds initial denaturation at 95°C, followed by 35 cycles of 30 seconds at 95°C, 30 seconds at 55°C, and 30 seconds at 72°C, and 7 minutes at 72°C, GAP43; a 2 minutes initial denaturation at 95°C, followed by 33 cycles of 30 seconds at 95°C, 30 seconds at 55°C, and 30 seconds at 72°C, and 7 minutes at 72°C, and MYCN (Figures 3B and 7E), GAPDH (Figures 3B, 6C and 7E) and AURKA; a 2 minutes initial denaturation at 95°C, followed by 27 cycles of 30 seconds at 95°C, 30 seconds at the appropriate anneal temperatures (MYCN; 58°C, GAPDH and AURKA; 55°C), and 30 seconds at 72°C, and 7 minutes at 72°C. Products were run in agarose gel electrophoresis and the band intensity was scanned by Printgraph (ATTO).
Figure 3.
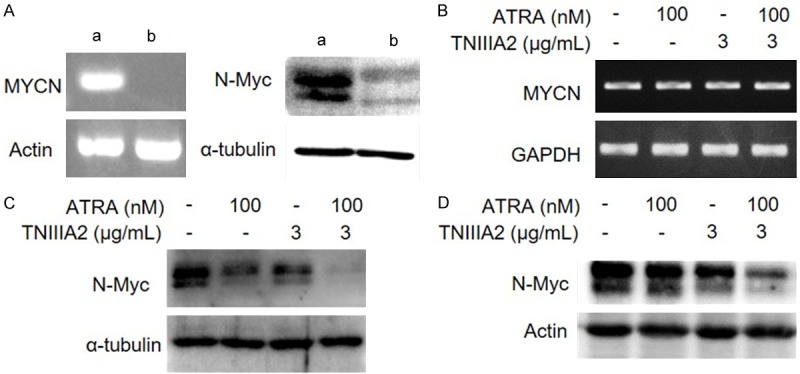
The combination of ATRA and TNIIIA2 strongly decreases the protein expression of N-Myc in IMR-32 and Kelly cells. (A) Left panel: MYCN gene expression of IMR-32 (a) and SK-N-SH (b) cells evaluated by RT-PCR. Right panel: N-Myc protein expression of IMR-32 (a) and SK-N-SH (b) cells detected by western blotting. (B and C) IMR-32 cells were cultured in the presence or absence of ATRA and TNIIIA2 as indicated. After culturing for 6 days, expressions of MYCN gene (B) and N-Myc protein (C) were evaluated by RT-PCR and western blotting, respectively. (D) Kelly cells, another MYCN amplified human neuroblastoma cell line, were cultured under the same conditions as performed in IMR-32 cells. N-Myc protein expression was detected by western blotting.
Figure 2.
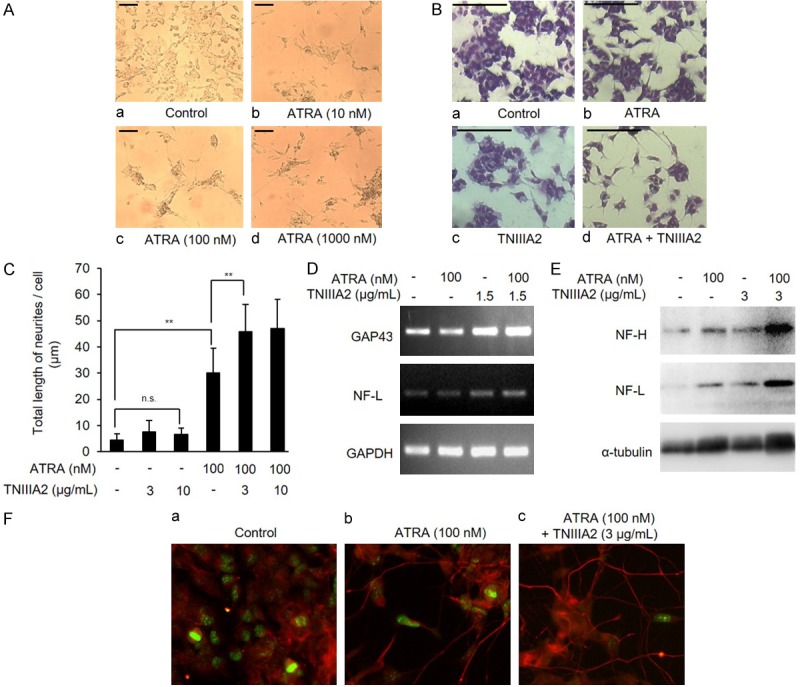
TNIIIA2 synergistically promotes ATRA-induced neuronal differentiation of IMR-32. (A) Dose-dependency of ATRA in induction of neuronal-like morphological changes. IMR-32 cells were cultured in the medium with ATRA (10, 100, 1000 nM) for 6 days. Microscopic observation was performed (scale bars: 100 µm). (B) IMR-32 cells were cultured in the medium containing ATRA (100 nM) with or without TNIIIA2 (3 µg/mL) as indicated. After culturing for 6 days, cells were fixed and stained using Diff-Quick stain kit. Microscopic observation was performed (scale bars: 100 µm). (C) IMR-32 cells cultured under the conditions as described above were subjected to measurement of the length of axon-like protrusions using a curvimeter. Data are shown as the means ± SD (n=4). Statistical analysis was performed by ANOVA and post hoc test. **P<0.01. (D and E) IMR-32 cells cultured as above were subjected to detection of neuronal differentiation markers. Gene expression of GAP43, NF-L or GAPDH as evaluated by RT-PCR (D) and protein expression of NF-H, NF-L and α-tubulin as detected by western blotting (E). (F) IMR-32 cells cultured in a glass bottom dish under the same conditions as above were fixed, permeabilized with Triton-X, and incubated with anti-Ki-67 or NF-H antibody, followed by incubation with FITC-conjugated and Alexa 633-conjugated secondary antibodies, respectively. Immunofluorescence localization of Ki-67 (green) and NF-H (red) was visualized by using immunofluorescence microscopy.
Figure 7.
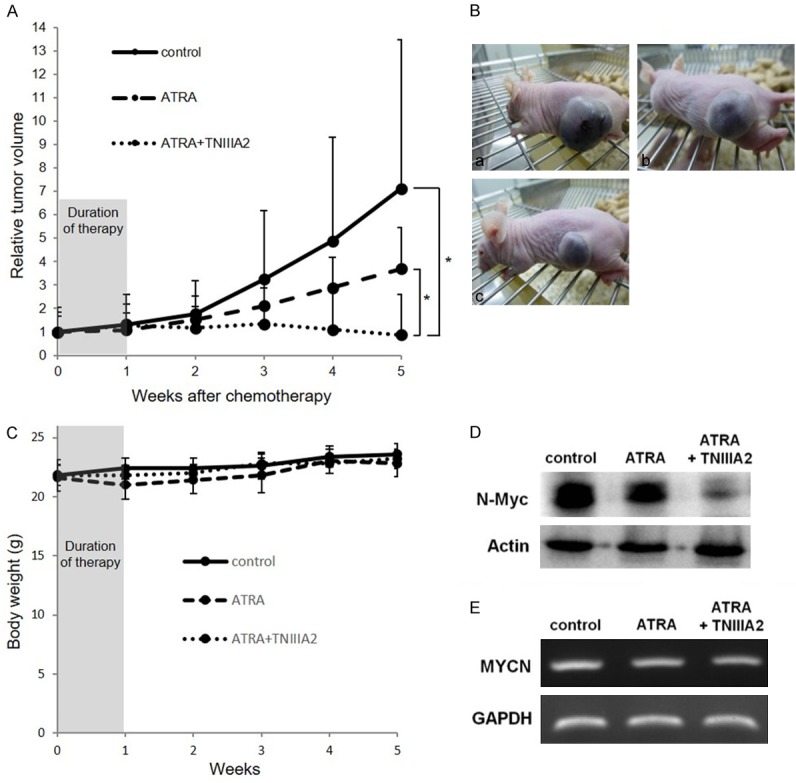
The combination of ATRA with TNIIIA2 decreased tumor growth in parallel with N-myc protein decrease in tumor. (A) IMR-32 cells were injected into the left back side of nude mice. After 8 weeks of injections, the nude mice were divided into 3 groups and subjected to daily chemotherapy with vehicle, ATRA (200 μg i.p./head), or the ATRA and TNIIIA2 (alternately 500 μg i.p./head and 250 μg i.v./head) combination for initial 1 week. Data are shown as the means ± SD (n=4). Statistical analysis was performed by Mann-Whitney U test. *P<0.05. (B) Representative photographs of mice were taken at 5 weeks: (a) control, (b) ATRA, (c) ATRA + TNIIIA2. (C) Body weight of each mouse was measured in every week. (D and E) Protein expression of N-Myc and gene expression of MYCN in tumors of each group at 5 weeks were evaluated by western blotting (D) and RT-PCR (E), respectively.
Figure 6.
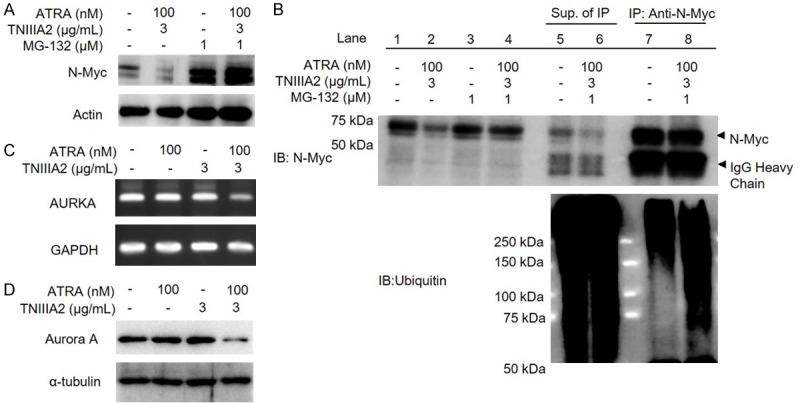
Decrease in N-myc protein level induced by the combination of ATRA with TNIIIA2 is caused by proteasomal degradation. (A and B) IMR-32 cells were cultured in the medium with ATRA (100 nM) and TNIIIA2 (3 µg/mL) in the presence or absence of MG-132, a proteasome inhibitor. A part of cell lysates was subjected to western blotting for N-Myc protein (A). Another part of cell lysates was subjected to immunoprecipitation using anti-N-Myc antibody (B). Ubiquitinated proteins were detected by western blotting using anti-ubiquitin antibody. (C and D) To evaluate the association of Aurora A in the decreased protein level of N-Myc, IMR-32 cells cultured with ATRA in the presence or absence of TNIIIA2 were subjected to RT-PCR and western blotting to detect gene expression of AURKA (C) and protein expression of Aurora A (D), respectively.
Western blotting
Cells were dissolved with the sodium dodecyl sulfate (SDS) sample buffer and subjected to SDS-PAGE and immunoblot analysis using anti-neurofilament heavy chain (NF-H) antibody (Abcam), anti-NF-L antibody (Sigma), anti-α-tubulin antibody (Santa Cruz Biotechnology), anti-N-Myc antibody (Calbiochem), anti-actin antibody (Sigma), anti-talin antibody (Biomol), anti-Aurora A antibody (Cell Signaling Technology) and anti-ubiquitin antibody (Millipore). Immunoblots were detected using Las 3000 mini (FUJIFILM).
Fluorescence microscope
IMR-32 cells were incubated in glass bottom dish (Matsunami) with the medium for differentiation induction for 6 days. Adhered cells were fixed with 10% formalin and 5% glycerol for 1 hour. Fixed cells were treated with 0.03% Triton-X for 5 minutes. After washing with phosphate buffered saline, cells were incubated with anti-Ki-67 (Abcam) and NF-H (Abcam) antibodies in a 5% CO2 incubator at 37°C for 1 hour. Then, cells were incubated with FITC-conjugated (Beckman Coulter) and Alexa 633-conjugated (Invitrogen) secondary antibodies in a 5% CO2 incubator at 37°C for 1 hour. Immunofluorescence localization of Ki-67 (green) and NF-H (red) was visualized by using immunofluorescence microscope, BIOREVO BZ-9000 (KEYENCE).
Colony formation assay
IMR-32 cells (1 × 105 cells/well) incubated with MEM containing ATRA (100 nM) in the presence or absence of TNIIIA2 (6 µg/mL) in a 6-well plate for 6 days. After removing medium, cells were treated with 0.05% collagenase (FUJIFILM Wako) and incubated until the intercellular adhesion got loose. Harvested cells (4 × 103 cells/well) were suspended in medium with 0.35% soft agar and 7.5% FBS, Bacto Agar (BD Bioscience), and the suspensions were laid on the solid layer of medium with 0.7% soft agar and 10% FBS and placed still at 4°C for 8 minutes. Finally, medium with 10% FBS was applied over the solid layers. After incubation in a 5% CO2 incubator at 37°C for 14 days, the number of colonies in 4 fields per well was counted and photographs were taken. 16 days after the cell count, photographs were taken again. The half volume of medium of the upper layer was replaced with fresh one every week during incubation.
Tumorigenicity
The animal procedure was approved by the Institutional Animal Care and Use Committee (IACUC) of Tokyo University of Science. IMR-32 cells were incubated with MEM containing ATRA (100 nM) in the presence or absence of TNIIIA2 (3 µg/mL). Cells were harvested and then injected into the back side of nude mice (Balb/c Slc-nu/nu, Sankyo Labo Service Corporation Inc.) with Matrigel® (BD Bioscience, 2 μg/mL). The volume of tumors was measured, and photographs were taken after 28 days. Tumor volumes were estimated (volume = (a2 × b)/2, a<b) using a caliper.
Knockdown of talin
200 μL of Opti-MEM (Invitrogen) including talin small interference RNA (siRNA) (Sigma) or siPerfect Negative Control (Sigma) and 2 μL of Lipofectamine RNAi Max (Invitrogen) were mixed and placed still for 20 minutes at room temperature. 1 mL of IMR-32 cell suspension (6 × 104 cells/mL) was added to the solution and the mixture was incubated in a 5% CO2 incubator at 37°C for 48 hours. Knockdown of talin was confirmed by western blotting.
Immunoprecipitation
IMR-32 cells were cultured with or without the combination of TNIIIA2 (3 µg/mL) and ATRA (100 nM) for 6 days. Cells were treated with or without MG-132 (1.0 µM) 24 hours before lysis. Cells were dissolved with the RIPA buffer (0.025 M Tris, 0.15 M NaCl, 0.001 M EDTA, 1% NP-40, 5% glycerol, pH 7.4) and prepared protein concentration for 1 mg/mL. Cell lysates were added 5 µg of anti-N-Myc antibody and rotated for 1 hour in 4°C. After rotated, cell lysates were mixed with 20 µL Protein G-Agarose and rotated for overnight in 4°C. After washed by RIPA buffer 3 times, cell lysates were subjected to immunoblot analysis using anti-N-Myc antibody and anti-ubiquitin antibody.
Neuroblastoma xenograft model
The animal procedure was approved by the IACUC of Tokyo University of Science. IMR-32 cell suspension (2.0 × 106 cells) containing Matrigel® (2 μg/mL) was injected into the back side of each Balb-c nude mouse (Balb/c Slc-nu/nu). After 8 weeks passed since the transplantation, these mice were allotted to 3 groups to set equally the average size of tumor in each group for evaluation of a therapeutic effect by agents. Each group was subjected to chemotherapy with vehicle, ATRA (200 μg i.p./head), or the ATRA and TNIIIA2 (alternately 500 μg i.p./head and 250 μg i.v./head) combination for initial 1 week. Tumor volumes were estimated (volume = (a2 × b)/2, a<b) using a caliper at the indicated time. After monitoring tumor size for 5 weeks, tumors were subjected to western blotting and RT-PCR analysis.
Database analysis
A publicly available RNA-sequencing data from a cohort of 498 neuroblastoma tumors (GSE62564) was analyzed using R2 program (Genomics Analysis and Visualization Platform, http://r2.amc.nl). The expression data of tenascin-C in low/high-risk group and MYCN non-amplification/amplification group were downloaded from R2 and analyzed by unpaired student’s t-test. A Kaplan-Meier survival curve was also generated using R2 and the cut-off level of tenascin-C for the curve was the median value of tenascin-C expression.
Results
Combination treatment of TNIIIA2 synergistically accelerated the ATRA-induced neuronal differentiation of human neuroblastoma cell line IMR-32 with MYCN amplification
IMR-32 cells were used in this study as a human neuroblastoma cell line with MYCN gene amplification. Consistent with our previous report using various cell lines [17], TNIIIA2 also enhanced adhesion of IMR-32 cells to the fibronectin substrate (Figure 1A), through activation of β1-integrins (Figure 1B). Prior to the effects of TNIIIA2 on neuronal differentiation, dose-dependency of neuronal differentiation induced by ATRA was examined. When IMR-32 cells were stimulated with ATRA at increased concentrations, extension of axon-like protrusions were observed in some cells at 100 nM (Figure 2Ac). Higher concentration (1000 nM) of ATRA caused no remarkable increase in the extension of axon-like protrusions (Figure 2Ad). When neuronal differentiation by ATRA was then stimulated in the presence of TNIIIA2, a marked synergistic increase in the length of axon-like neurites became evident, although TNIIIA2 alone showed no significant effect on the axon-like neurites (Figure 2B and 2C). Acceleration of ATRA-induced differentiation by TNIIIA2 was further proven by evaluation of neuronal marker expressions as follows. RT-PCR showed that the gene expression levels of NF-L and GAP43 were significantly enhanced by combination treatment of ATRA with TNIIIA2 (Figure 2D). Similarly, western blotting analysis showed that protein expressions of NF-H and NF-L in cells treated with ATRA were clearly increased by the addition of TNIIIA2 (Figure 2E). Stimulation with TNIIIA2 without ATRA was less effective on the expression of neuronal differentiation markers than the combination treatment (Figure 2D and 2E). Furthermore, Ki-67 staining showed a significant decrease in the number of proliferating cells following ATRA-TNIIIA2 combination treatment (Figure 2F), suggesting that the transformation of the cell phase from proliferation to differentiation was promoted by the combination treatment. These results showed that the addition of TNIIIA2 synergistically accelerated the ATRA-induced neuronal differentiation of IMR-32 cells with MYCN gene amplification.
Figure 1.

Promotion of IMR-32 cell adhesion to the fibronectin substrate through activation of β1-integrin. (A) IMR-32 cells were suspended in serum-free medium with TNIIIA2 (1.5, 3, 50 µg/mL) and then incubated in a 96-well plate coated with fibronectin (2 µg/mL). Adhered cells were stained with crystal violet and counted (■: % of cells spread, □: % of cells attached). Data are shown as the means ± SD (n=3). Statistical analysis was performed by ANOVA and post hoc test for the total percentages of spread and adhered cells. **P<0.01. (B) IMR-32 cells suspended in serum-free medium (5 × 105 cells/mL) were incubated with vehicle (a and b), MnCI2 (1 mM) (c) or TNIIIA2 (3 µg/mL) (d) for 45 minutes. Each suspension was further incubated with phycoerythrin-conjugated AG89, active-β1-integrin-recognizing mAb, for 1 hour (b-d). Active β1-integrins were detected by flow cytometry.
Combination treatment of ATRA with TNIIIA2 caused a decrease in intracellular level of N-Myc protein, resulting in attenuation of the malignant properties of IMR-32 cells
High expression of N-Myc is a major cause of aberrant neuronal differentiation in neuroblastoma [3]. Therefore, accelerated neuronal differentiation by a combination treatment of ATRA with TNIIIA2 might be caused by a decrease in the intracellular levels of N-Myc in IMR-32 cells. The MYCN gene and N-Myc protein were confirmed to be highly expressed in IMR-32 cells, but not in SK-N-SH neuroblastoma cells without MYCN gene amplification [21] (Figure 3A). This high expression of MYCN in IMR-32 cells was barely influenced by treatment with ATRA, TNIIIA2 or their combination (Figure 3B). In contrast, the intracellular level of N-Myc in IMR-32 cells was remarkably decreased by combination treatment of ATRA with TNIIIA2, but weakly by ATRA or TNIIIA2 alone (Figure 3C). A decrease in N-Myc protein level by ATRA-TNIIIA2 combination was also observed in another MYCN-amplified human neuroblastoma cell line, Kelly (Figure 3D).
High protein levels of N-Myc are thought to play important roles in tumor initiation and malignant progression [3,4]. Thus, as the representative cancer-associated malignant properties, the anchorage-independent growth in soft agarose and tumorigenicity in mice were examined. IMR-32 cells, which were pre-treated with ATRA, TNIIIA2 or their combination, were cultured in soft agarose or injected to mice for evaluating colony formation in vitro and tumor formation in vivo, respectively. The number of colonies formed in soft agarose was decreased in IMR-32 cells pre-treated with ATRA alone, but not in those treated with TNIIIA2 alone (Figure 4A). The decrease in the number of colonies became highly significant by pre-treating the cells additionally with TNIIIA2 (Figure 4A). Injections of IMR-32 cells pre-treated with TNIIIA2 or ATRA alone formed tumors in all mice (Figure 4Ba-d), while the average tumor size in mice injected with cells pre-treated with ATRA was smaller than that of vehicle control (Figure 4Bf). In contrast, importantly, no definite tumor formation was detected in all mice injected with IMR-32 cells pre-treated with the ATRA-TNIIIA2 combination (Figure 4Ba, 4Be and 4Bf). From these observations, a decrease in the intracellular level of N-Myc induced by ATRA-TNIIIA2 combination treatment seemed to cause a remarkable attenuation of malignant properties, concomitant with neuronal differentiation.
Figure 4.
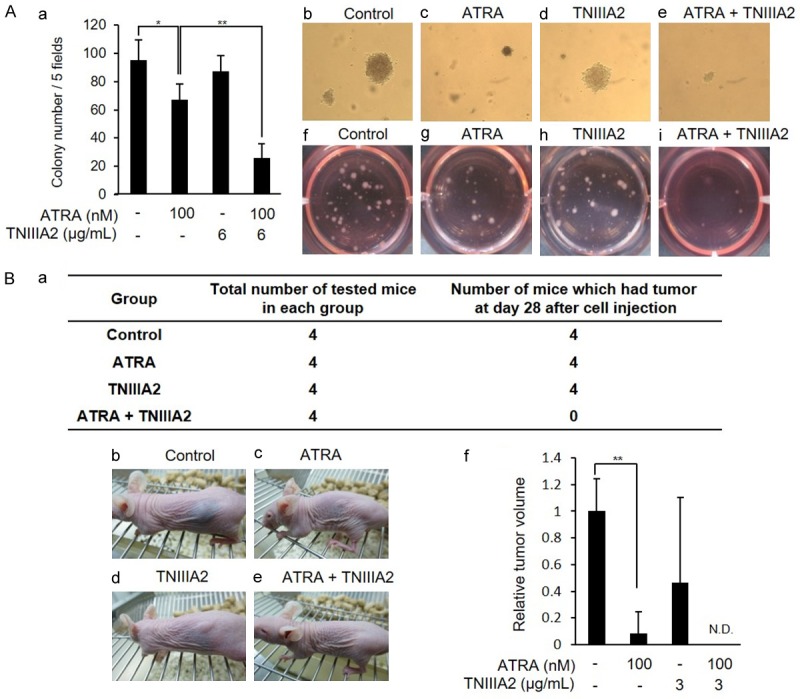
The combination of ATRA with TNIIIA2 strongly suppresses tumorigenic potential of IMR-32 cells. (A) IMR-32 cells were cultured with ATRA (100 nM) in the presence or absence of TNIIIA2 (6 µg/mL) for 6 days. Cells were then harvested and mixed with 0.35% soft agar containing ATRA (100 nM), TNIIIA2 (6 µg/mL) or their combination and subjected to colony formation assay, as described in Materials and methods. (a) The number of colonies was counted after 14 days. Data are shown as the means ± SD (n=3). Statistical analysis was performed by ANOVA and post hoc test. *P<0.05, **P<0.01. Photographs were taken after 14 days (b-e) and 30 days (f-i). (B) IMR-32 cells were cultured with ATRA (100 nM) in the presence or absence of TNIIIA2 (3 µg/mL) for 6 days. Cells were harvested and then injected into the back side of nude mice with Matrigel® (2 μg/mL). The incidence of tumor formation during bleeding for 28 days is summarized in (a). Representative photographs of tumor formation after 28 days are in (b-e), and tumors formed in each group of mice are represented as a relative value to the control in (f). Data are shown as the means ± SD (n=3). Statistical analysis was performed by ANOVA and post hoc test. **P<0.01.
Molecular basis for a decrease in intracellular level of N-Myc by ATRA-TNIIIA2 combination treatment
To determine whether decreased N-Myc could be coupled to β1-integrin activation by TNIIIA2, HUTS-4, an anti-β1-integrin-activating mAb [22], was used instead of TNIIIA2. As shown in Figure 5A, when treating IMR-32 cells with ATRA in the presence of HUTS-4, a significant decrease in N-Myc protein level was observed, but only a weak decrease was evident with HUTS-4 alone. The involvement of β1-integrin activation was also demonstrated by knockdown of talin that functions as an important molecule to keep the β1-integrin conformation active [23]. That is, down-regulation of talin expression by siRNA in IMR-32 cells (Figure 5Ba) completely abrogated the decrease in N-Myc protein level induced by ATRA-TNIIIA2 combination treatment (Figure 5Bb). Additionally, the decrease of N-Myc was also blocked by peptide CS-1, a specific antagonist for integrin α4β1, but not by GRGDSP, an integrin α5β1 antagonist (Figure 5C). Peptide CS-1 had little effect on the N-Myc protein expression level (Figure 5Ca). These results thus suggested that activation of β1-integrin, especially α4β1, played a critical role in the decreased expression of N-Myc in neuroblastoma cells, which was induced by ATRA-TNIIIA2 combination treatment.
Figure 5.
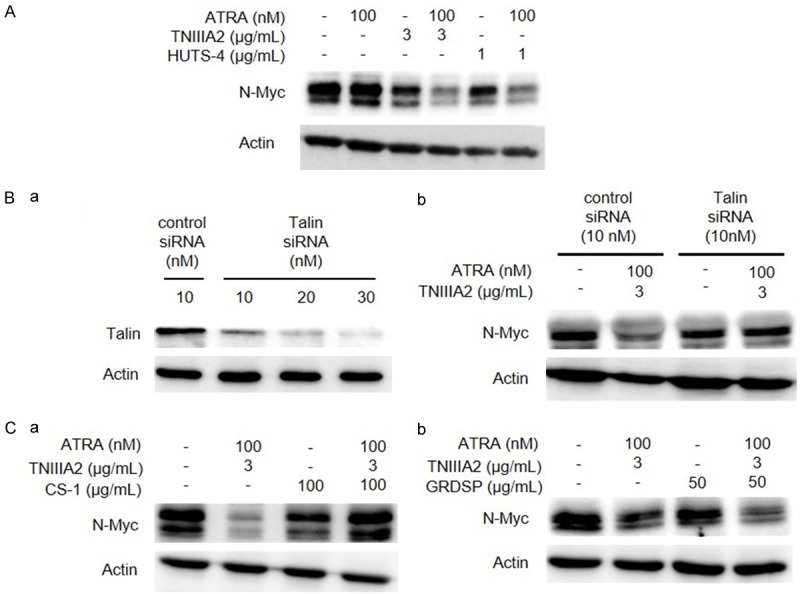
Activation of Integrin α4β1 is responsible for N-Myc protein degradation by the combination of ATRA and TNIIIA2. (A) IMR-32 cells were cultured with or without ATRA in the presence or absence of HUTS-4, instead of TNIIIA2, as indicated. After culturing for 6 days, N-Myc protein expression was detected by western blotting. (B) IMR-32 cells were transfected with control siRNA or talin siRNA as described in Materials and methods. (a) Down-regulation of talin was verified by western blotting. (b) IMR-32 cells treated with control siRNA or talin siRNA, were cultured in the medium with ATRA (100 nM) and TNIIIA2 (3 µg/mL), and expression of N-Myc protein was detected by western blotting. (C) IMR-32 cells were cultured in the medium with ATRA and TNIIIA2 in the presence or absence of integrin antagonistic peptides: CS-1 (a) for Integrin α4β1 and GRGDSP (b) for α5β1. N-Myc protein expression was detected by western blotting.
In general, the balance of de novo protein synthesis and its degradation contributes on keeping certain expression levels of intracellular proteins [24]. Since the decreased N-Myc protein expression by ATRA-TNIIIA2 combination treatment was shown to occur at the post-transcriptional level (see Figure 3), the implication of proteasomal degradation was examined. When IMR-32 cells were treated with ATRA-TNIIIA2 combination in the presence of MG-132, a proteasome inhibitor, the ATRA-TNIIIA2 co-treatment-induced decrease of N-Myc was completely canceled (Figure 6A).
We next investigated the molecular processes coupled to the decreased expression of N-Myc protein, which is induced by ATRA-TNIIIA2 combination treatment. After IMR-32 cells were treated with ATRA-TNIIIA2 combination in the presence of MG-132, cell lysates were subjected to immunoprecipitation with anti-N-Myc antibody, and ubiquitinated N-Myc was then detected by anti-ubiquitin antibody. As shown in Figure 6B (lane 7 and 8 in lower panel), immunoblots larger than 75 kDa in lane 8 became more significant than those in lane 7, indicating that ubiquitination of N-Myc was promoted by ATRA-TNIIIA2 combination treatment.
We next examined the impact of Aurora A which binds to ubiquitinated N-Myc and prevents its proteasomal degradation [25]. Both the gene expression of AURKA coding Aurora A and protein expression of Aurora A in IMR-32 cells were decreased by ATRA-TNIIIA2 combination, but not changed by ATRA or TNIIIA2 alone (Figure 6C and 6D). Thus, ATRA-TNIIIA2 combination treatment was shown to promote the ubiquitination of N-Myc protein in IMR-32 cells, which might have led to N-Myc protein degradation.
Therapeutic potential of the ATRA-TNIIIA2 co-administration in neuroblastoma
To evaluate the therapeutic effect of the ATRA-TNIIIA2 combination administration, we used a neuroblastoma xenograft model. IMR-32 cells were injected subcutaneously in the left flank of nude mice. After 8 weeks of injections, mice were divided into 3 groups based on average tumor size. For chemotherapy, vehicle, ATRA, or ATRA-TNIIIA2 combination was injected into the mice every day for 1 week. After monitoring tumor sizes for 5 weeks, the expressions of the MYCN gene and N-Myc protein in the tumors were measured by RT-PCR and western blot analysis.
As shown in Figure 7A and 7Ba, conspicuous tumor growth was observed in the vehicle control group. The administration of ATRA suppressed tumor growth, while the difference of tumor sizes between control and ATRA group was not statistically significant (Figure 7A and 7Bb). ATRA-TNIIIA2 co-administration suppressed tumor growth significantly and completely even though duration of treatment was only 1 week, and average tumor size was reduced (Figure 7A and 7Bc). There was no significant change in body weight in each group (Figure 7C). The prevention of tumor growth by ATRA-TNIIIA2 co-administration was in parallel with a clear decrease in N-Myc protein level in tumors (Figure 7D), which closely coincided with the in vitro data. The MYCN gene expression level was not influenced by co-administration (Figure 7E).
Discussion
Treating cancer by inducing normal differentiation of tumor cells is an attractive strategy, but the clinical application of differentiation therapy for cancer treatment has been limited to date, especially for solid cancers [11]. Differentiation therapy using RA derivatives, including ATRA, has long been investigated for high-risk neuroblastoma. However, the clinical outcome has not been sufficient via RA derivatives [12-14,26], probably because of the inhibition of differentiation caused by N-Myc which is highly expressed in high-risk neuroblastoma. Besides the low therapeutic efficacy, an additional problem recently became apparent in pediatric cancer treatment. Since most pediatric cancers often occur in children younger than 4 years of age, it is difficult to find chemotherapeutic side effects in these young patients. Intensive chemotherapy in pediatric cancer patients, which is often applied to high-risk neuroblastoma, causes serious health problems months or years after treatments have ended [8]. Therefore, alternative therapeutic approaches have been urgently needed to develop chemotherapy that has high therapeutic efficacy with a low risk of late effects.
Here, we showed that the treatment of neuroblastoma cells with ATRA in combination with TNIIIA2 enabled stimulation of proteasomal degradation of N-Myc (Figure 6A). High expression of N-Myc is a cause of not only aberrant neuronal differentiation but also the malignant features of neuroblastoma. Thus, a significant decrease in N-Myc might be responsible for the efficient induction of neuronal differentiation and attenuation of malignant properties in neuroblastoma cells. Furthermore, an in vivo experiment using a neuroblastoma xenograft model showed that ATRA-TNIIIA2 combination treatment had potent ability to inhibit tumor growth (Figure 7A), and N-Myc protein level was significantly decreased in tumor tissues, in agreement with in vitro results (Figure 7D). In contrast to conventional anti-cancer drugs, the anti-tumor effect of ATRA-TNIIIA2 combination treatment does not depend on cancer cell death but is based on accelerated neuronal differentiation in response to N-Myc protein degradation. The proteasomal degradation of N-Myc stimulated here is a physiological process functioning as a turnover system for this oncoprotein. Thus, ATRA-TNIIIA2 combination treatment might provide a new strategy for differentiation therapy with a low risk of late effects for high-risk neuroblastoma.
Tenascin-C, the parent molecule of TNIIIA2, is transiently and highly expressed in malignant tumors, such as glioblastoma, lung cancer and breast cancer, and its expression level correlates with poor prognoses for these cancers [27-29]. Therefore, tenascin-C has been involved in tumor initiation and progression, while its substantial role in cancer pathogenesis has been unclear. Recently, we showed that tenascin-C and TNIIIA2 could be involved in the pathogenesis of colon cancer and its lung metastasis [18]. This result seems to be contrary to the present results, which showed the beneficial effects of TNIIIA2 on neuroblastoma treatment. Tenascin-C is also highly expressed in neuroblastoma [30], while retrospective database analysis showed that the expression level of tenascin-C in low-risk/MYCN non-amplified neuroblastoma patients is higher than that in high-risk/MYCN-amplified patients (Figure 8A and 8B). Additionally, high tenascin-C expression was correlated with favorable outcome of neuroblastoma patients in a Kaplan-Meier analysis where the median value of tenascin-C expression was used as a cutoff (Figure 8C). It might be possible that at least in high-risk neuroblastoma with MYCN amplification, TNIIIA2-relating peptides endogenously released from tenascin-C molecule function repressively in the pathogenesis of neuroblastoma. Besides the correlation between tenascin-C level and prognosis, further statistical analysis to obtain information on N-Myc and TNIIIA2 expression in neuroblastoma patients would be required.
Figure 8.
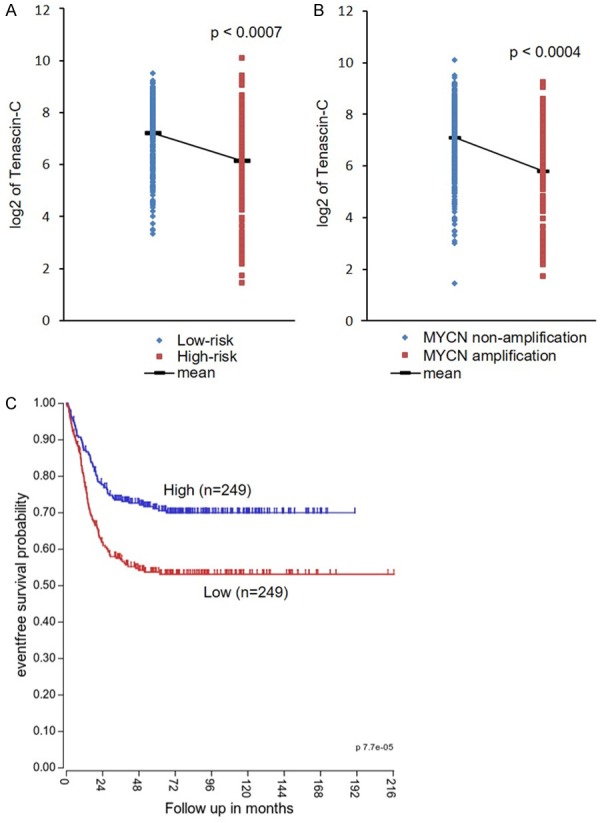
Correlation between tenascin-C expression and prognosis in neuroblastoma patients. A and B: Tenascin-C expressions of low/high risk neuroblastoma patients and MYCN non-amplification/amplification neuroblastoma patients were analyzed by R2 program and unpaired student’s t-test. C: A Kaplan-Meier event free survival curve of neuroblastoma patients who have low/high level of tenascin-C expression was generated by R2.
Combination treatment of ATRA with TNIIIA2 stimulated the proteasomal degradation of N-Myc protein, while TNIIIA2 or ATRA alone had little effect (Figure 3C). This N-Myc protein degradation by ATRA-TNIIIA2 combination treatment was accompanied by a remarkable decrease in the expression level of Aurora A (Figure 6D). Aurora A is known to be able to inhibit the proteasomal degradation of N-Myc [25]. This decrease in the level of Aurora A was also caused by ATRA-TNIIIA2 combination treatment, but not by TNIIIA2 or ATRA alone (Figure 6D). These results indicate that suppression of Aurora A protein expression requires functional coordination between RA and β1-integrin signals. In our study, activation of integrin α4β1 was associated with N-Myc protein degradation in IMR-32 cells adhering to the fibronectin substrate that is an abundant and ubiquitous ECM scaffold for mammalian cells (Figure 5Ca). In contrast, some reports show that neuronal differentiation of neuroblastoma cells by RA is accompanied by an increase in integrin α1β1 expression [30,31]. It is presumed that the activation of β1 subfamily of integrins, independent of α integrin partner, may be functional in N-Myc protein degradation.
On the other hand, it was previously reported that PP2A, a protein phosphatase, is associated with N-Myc protein degradation [32]. PP2A, which is upregulated by ATRA [33], also has an ability to decrease the intracellular level of Aurora A via proteasomal degradation [34]. Additionally, β1-integrin is able to interact with PP2A and influence the phosphatase activity [35-37]. PP2A might be implicated in the N-Myc protein degradation induced by ATRA-TNIIIA2 combination treatment in our study, in which the combination of TNIIIA2 with ATRA was essential. Further investigation should be performed to define whether the combination treatment of ATRA with TNIIIA2 induces N-Myc protein degradation through PP2A signal cascade. Precise understanding of the signaling crosstalk between integrin and ATRA could make the differentiation therapy more effective.
Acknowledgements
We acknowledge the support of Takeda Science Foundation and thank Dr. Akimune Asanuma, Dr. Masanao Watanabe, and Naoto Ishibashi of KOWA COMPANY, LTD. for their kind help in this study.
Disclosure of conflict of interest
None.
References
- 1.Cheung NK, Dyer MA. Neuroblastoma: developmental biology, cancer genomics, and immunotherapy. Nat Rev Cancer. 2013;13:397–411. doi: 10.1038/nrc3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, Faldum A, Hero B, Iehara T, Machin D, Mosseri V, Simon T, Garaventa A, Castel V, Matthay KK. The International neuroblastoma risk group (INRG) classification system: an INRG task force report. J. Clin. Oncol. 2009;27:289–297. doi: 10.1200/JCO.2008.16.6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakagawara A, Li YY, Izumi H, Muramori K, Inada H, Nishi M. Neuroblastoma. Jpn J Clin Oncol. 2018;48:214–241. doi: 10.1093/jjco/hyx176. [DOI] [PubMed] [Google Scholar]
- 4.Huang M, Weiss WA. Neuroblastoma and Mycn. Cold Spring Harb Perspect Med. 2013;3:22. doi: 10.1101/cshperspect.a014415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen H, Liu H, Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther. 2018;3:5. doi: 10.1038/s41392-018-0008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foster J, Smith V. High-risk neuroblastoma treatment review. Children. 2018;5:114. doi: 10.3390/children5090114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berthold F, Spix C, Kaatsch P, Lampert F. Incidence, survival, and treatment of localized and metastatic neuroblastoma in Germany 1979-2015. Pediatr Drugs. 2017;19:577–593. doi: 10.1007/s40272-017-0251-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedman DN, Henderson TO. Late effects and survivorship issues in patients with neuroblastoma. Children. 2018;5:107. doi: 10.3390/children5080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leszczyniecka M, Roberts T, Dent P, Grant S, Fisher PB. Differentiation therapy of human cancer: basic science and clinical applications. Pharmacol Ther. 2001;90:105–156. doi: 10.1016/s0163-7258(01)00132-2. [DOI] [PubMed] [Google Scholar]
- 10.Degos L, Dombret H, Chomienne C, Daniel MT, Miclea JM, Chastang C, Castaigne S, Fenaux P. All-trans-retinoic acid as a differentiating agent in the treatment of acute promyelocytic leukemia. Blood. 1995;85:2643–2653. [PubMed] [Google Scholar]
- 11.Xu WP, Zhang X, Xie WF. Differentiation therapy for solid tumors. J Dig Dis. 2014;15:159–165. doi: 10.1111/1751-2980.12122. [DOI] [PubMed] [Google Scholar]
- 12.Matthay KK, Villablanca JG, Seeger RC, Stram DO, Harris RE, Ramsay NK, Swift P, Shimada H, Black CT, Brodeur GM, Gerbing RB, Reynolds CP. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. N Engl J Med. 1999;341:1165–1173. doi: 10.1056/NEJM199910143411601. [DOI] [PubMed] [Google Scholar]
- 13.Matthay KK, Reynolds CP, Seeger RC, Shimada H, Adkins ES, Haas-Kogan D, Gerbing RB, London WB, Villablanca JG. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children’s oncology group study. J. Clin. Oncol. 2009;27:1007–1013. doi: 10.1200/JCO.2007.13.8925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matthay KK. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children’s oncology group study (vol 27, pg 1007, 2009) J. Clin. Oncol. 2014;32:1862–1863. doi: 10.1200/JCO.2007.13.8925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giancotti FG, Ruoslahti E. Transduction - integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- 16.Myers JP, Santiago-Medina M, Gomez TM. Regulation of axonal outgrowth and pathfinding by integrin-ECM interactions. Dev Neurobiol. 2011;71:901–923. doi: 10.1002/dneu.20931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saito Y, Imazeki H, Miura S, Yoshimura T, Okutsu H, Harada Y, Ohwaki T, Nagao O, Kamiya S, Hayashi R, Kodama H, Handa H, Yoshida T, Fukai F. A peptide derived from tenascin-C induces β1 integrin activation through syndecan-4. J Biol Chem. 2007;282:34929–34937. doi: 10.1074/jbc.M705608200. [DOI] [PubMed] [Google Scholar]
- 18.Suzuki H, Sasada M, Kamiya S, Ito Y, Watanabe H, Okada Y, Ishibashi K, Iyoda T, Yanaka A, Fukai F. The promoting effect of the extracellular matrix peptide TNIIIA2 derived from tenascin-c in colon cancer cell infiltration. Int J Mol Sci. 2017;18:181. doi: 10.3390/ijms18010181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanaka R, Seki Y, Saito Y, Kamiya S, Fujita M, Okutsu H, Iyoda T, Takai T, Owaki T, Yajima H, Taira J, Hayashi R, Kodama H, Matsunaga T, Fukai F. Tenascin-C-derived peptide TNIIIA2 highly enhances cell survival and platelet-derived growth factor (PDGF)-dependent cell proliferation through potentiated and sustained activation of integrin α5β1. J Biol Chem. 2014;289:17699–17708. doi: 10.1074/jbc.M113.546622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanaka R, Owaki T, Kamiya S, Matsunaga T, Shimoda K, Kodama H, Hayashi R, Abe T, Harada YP, Shimonaka M, Yajima H, Terada H, Fukai F. VLA-5-mediated adhesion to fibronectin accelerates hemin-stimulated erythroid differentiation of K562 cells through induction of VLA-4 expression. J Biol Chem. 2009;284:19817–19825. doi: 10.1074/jbc.M109.009860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang JH, Rychahou PG, Ishola TA, Qiao JB, Evers BM, Chung DH. Mycn silencing induces differentiation and apoptosis in human neuroblastoma cells. Biochem Biophys Res Commun. 2006;351:192–197. doi: 10.1016/j.bbrc.2006.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luque A, Gomez M, Puzon W, Takada Y, Sanchez-Madrid F, Cabanas C. Activated conformations of very late activation integrins detected by a group of antibodies (huts) specific for a novel regulatory region (355-425) of the common beta 1 chain. J Biol Chem. 1996;271:11067–11075. doi: 10.1074/jbc.271.19.11067. [DOI] [PubMed] [Google Scholar]
- 23.Wegener KL, Partridge AW, Han J, Pickford AR, Liddington RC, Ginsberg MH, Campbell ID. Structural basis of integrin activation by talin. Cell. 2007;128:171–182. doi: 10.1016/j.cell.2006.10.048. [DOI] [PubMed] [Google Scholar]
- 24.Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–774. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- 25.Otto T, Horn S, Brockmann M, Eilers U, Schuttrumpf L, Popov N, Kenney AM, Schulte JH, Beijersbergen R, Christiansen H, Berwanger B, Eilers M. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell. 2009;15:67–78. doi: 10.1016/j.ccr.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 26.Villablanca JG, London WB, Naranjo A, McGrady P, Ames MM, Reid JM, McGovern RM, Buhrow SA, Jackson H, Stranzinger E, Kitchen BJ, Sondel PM, Parisi MT, Shulkin B, Yanik GA, Cohn SL, Reynolds CP. Phase II study of oral capsular 4-hydroxyphenylretinamide (4-HPR/fenretinide) in pediatric patients with refractory or recurrent neuroblastoma: a report from the children’s oncology group. Clin Cancer Res. 2011;17:6858–6866. doi: 10.1158/1078-0432.CCR-11-0995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herold-Mende C, Mueller MM, Bonsanto MM, Schmitt HP, Kunze S, Steiner HH. Clinical impact and functional aspects of tenascin-C expression during glioma progression. Int J Cancer. 2002;98:362–369. doi: 10.1002/ijc.10233. [DOI] [PubMed] [Google Scholar]
- 28.Gocheva V, Naba A, Bhutkar A, Guardia T, Miller KM, Li CM, Dayton TL, Sanchez-Rivera FJ, Kim-Kiselak C, Jailkhani N, Winslow MM, Del Rosario A, Hynes RO, Jacks T. Quantitative proteomics identify tenascin-C as a promoter of lung cancer progression and contributor to a signature prognostic of patient survival. Proc Natl Acad Sci U S A. 2017;114:E5625–E5634. doi: 10.1073/pnas.1707054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Helleman J, Jansen M, Ruigrok-Ritstier K, van Staveren IL, Look MP, Gelder ME, Sieuwerts AM, Klijn JG, Sleijfer S, Foekens JA, Berns E. Association of an extracellular matrix gene cluster with breast cancer prognosis and endocrine therapy response. Clin Cancer Res. 2008;14:5555–5564. doi: 10.1158/1078-0432.CCR-08-0555. [DOI] [PubMed] [Google Scholar]
- 30.Linnala A, Lehto VP, Virtanen I. Neuronal differentiation in SH-SY5Y human neuroblastoma cells induces synthesis and secretion of tenascin and upregulation of alpha(v) integrin receptors. J Neurosci Res. 1997;49:53–63. doi: 10.1002/(sici)1097-4547(19970701)49:1<53::aid-jnr6>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 31.Rozzo C, Chiesa V, Ponzoni M. Integrin up-regulation as marker of neuroblastoma cell differentiation: correlation with neurite extension. Cell Death Differ. 1997;4:713–724. doi: 10.1038/sj.cdd.4400304. [DOI] [PubMed] [Google Scholar]
- 32.Gustafson WC, Weiss WA. Myc proteins as therapeutic targets. Oncogene. 2010;29:1249–1259. doi: 10.1038/onc.2009.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vuocolo S, Purev E, Zhang DM, Bartek J, Hansen K, Soprano DR, Soprano KJ. Protein phosphatase 2A associates with Rb2/p130 and mediates retinoic acid-induced growth suppression of ovarian carcinoma cells. J Biol Chem. 2003;278:41881–41889. doi: 10.1074/jbc.M302715200. [DOI] [PubMed] [Google Scholar]
- 34.Horn V, Thelu J, Garcia A, Albiges-Rizo C, Block MR, Viallet L. Functional interaction of Aurora-A and PP2A during mitosis. Mol Biol Cell. 2007;18:1233–1241. doi: 10.1091/mbc.E06-12-1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pankov R, Cukierman E, Clark K, Matsumoto K, Hahn C, Poulin B, Yamada KM. Specific β1 integrin site selectively regulates Akt/Protein kinase B signaling via local activation of Protein phosphatase 2A. J Biol Chem. 2003;278:18671–18681. doi: 10.1074/jbc.M300879200. [DOI] [PubMed] [Google Scholar]
- 36.Ivaska J, Nissinen L, Immonen N, Eriksson JE, Kahari VM, Heino J. Integrin α2β1 promotes activation of Protein phosphatase 2A and dephosphorylation of Akt and Glycogen synthase kinase 3β. Mol Cell Biol. 2002;22:1352–1359. doi: 10.1128/mcb.22.5.1352-1359.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xia H, Nho R, Kleidon J, Kahm J, Henke CA. Polymerized collagen inhibits fibroblast proliferation via a mechanism involving the formation of a β1 integrin/PP2A/TSC2 complex that suppresses S6K1 activity. J Biol Chem. 2008;283:20350–20360. doi: 10.1074/jbc.M707489200. [DOI] [PubMed] [Google Scholar]