

Abstract
The historical relationship between cancer and inflammation has long been evaluated, and dates back to the early work of Virchow (1863), where he hypothesised that chronic inflammation as a direct cause of tissue injury and infection, could actually promote tissue proliferation. At that period in time however, the exact mechanisms that mediated this relationship were little understood. Subsequent studies have since then demonstrated that chronic inflammation plays significant roles in microenvironments, mostly in the progression of tumours, probably, through over-secretion of proinflammatory cytokines and other immune-killing apparatus such as reactive oxygen species (ROS) which cause damage to normal cells leading to DNA damage and increased cellular mutation rates. Recently, the identification of DNA lesion 5-chlorocytosine (5-CIC) created by hypochlorous acid (HOCl) secreted to nullify or kill infectious agents and toll-like receptor 4 (TLR4)-mediated chronic inflammation in the human gut, has become the latest evidence linking inflammation directly to cancer. The key to cellular survival and adaptation under unfavourable or pathological conditions is the expression of heat shock proteins (HSPs) also called molecular chaperones. These proteins play essential roles in DNA repair processes by maintaining membrane integrity, orderliness and stability of client proteins that play prominent roles in DNA repair mechanisms. More so, HSPs have also been shown to modulate the effects of pro-inflammatory/apoptotic cytokines through the inhibition of cascades leading to the generation of ROS-mediated DNA damage, while promoting the DNA repair mechanism, thus playing prominent roles in various stages of DNA repair and cancer progression. Hence, studies targeting HSPs and their inhibitors in inflammation, DNA damage, and repair, could improve current cancer therapeutic efficiency. Here the focus will be on the relationship between HSPs, inflammation and cancer, as well as roles of HSPs in DNA damage response (DDR).
Keywords: Heat shock proteins, inflammation, cancer, DNA damage response, 5-chlorocytosine, hypochlorous acid
Heat shock proteins
Due to continuous exposure to cellular stressors (e.g. heat, heavy metal stress, viral and bacterial infections, nutrient deficiency, exercise, oxidative stress, cold, acute or chronic inflammatory diseases, ischemia injury, drought, UV-light), cells frequently produce a family of evolutionary conserved proteins termed heat shock proteins (HSPs) or molecular chaperones [1,2]. These proteins enable cell survival under pathological conditions, metabolic changes and environmental adaptation, which may lead to protein aggregation and denaturation thus resulting in several neurodenerative disorders such as motor neuron, frontal temporal lobar degeneration, Alzheimer’s, Huntington’s and Parkinson’s diseases, as well as aging and cancer [3-5]. Although most of these polypeptides are generally expressed upon thermal induction and stress, some that are involved in the folding of nascent proteins, signal transduction and translocation of organelles across cellular membranes, are constitutively expressed under physiological states and are termed heat shock cognate (Hsc) proteins [6-8]. These constitutively expressed HSPs perform important ‘housekeeping’ activities, which are critical in maintaining cellular homeostasis [9].
HSPs were first identified in the salivary glands of Drosophila larvae by Ritossa in the early 1960s and are broadly categorized into two main groups: high molecular weight and small heat shock proteins. High molecular weight HSPs (hHSPs) range from 40 to 110 kDa, (including HSPs 110, 100, 90, 70 and 60), and are ATP-dependent. They bind nascent and non-native proteins, and actively refold them into their 3-D structure. They also direct irreparable proteins to proteasomal degradation [9]. Small molecular weight HSPs (sHSPs) on the other hand, range from 8 to 30 kDa (including HSPs 40, 27, 15, 10 and ubiquitin), and are ATP-independent. These proteins, especially those which are encoded by HSPB genes, are structurally classified by flanking C-and N-terminal residues characterized by the existence of well-preserved α-crystallin and β-sandwich domains [10]. They are involved in the refolding of misfolded peptides, degradation of irreversible proteins and prevention of stress labile polypeptide aggregations [11]. More so, these proteins have been shown to be involved in complex immune responses to diseases such as cancer, as well as participate in the modulation of several inflammatory cascades, especially in the microenvironment [10].
Inflammation
Inflammation, forms part of the non-specific immune system and is the first line of defense in response to all forms of cellular injuries (e.g. viral or bacterial infections, toxins and chemicals) and is identified normally by heat, pain, redness, and swelling in the inflamed area [12]. Inflammatory response clears cellular damage caused by cellular injuries and initiates cellular repair and healing. It can be broadly classified into two: acute and chronic inflammation. Acute inflammation is a short-term and a self-limiting response that occurs under physiological conditions. It is thought to be beneficial to the cellular system when short-lived, but if long-termed and uncontrolled, it could degenerate into chronic inflammation characterized by increased inflammatory mediators, acute-phase reactants, inflammatory signalling pathway activation and increased production of abnormal cytokines [13]. Chronic inflammation leads to the destruction of several healthy cells and tissues, and in most cases, degenerates into organ failure and mortality, as observed in several human inflammatory diseases (HIDs) and cancer [14]. The major causes of chronic inflammation are currently still a topic of debate. Recent studies, however, have shown that inflammatory instigators such as microbial infections or bodily injuries are not the only causes. Hormonal imbalance and malfunctioned tissues caused by complications from other human diseases such as aging, HIDs and cancer also play a major role [15,16].
Inflammatory response is normally mediated by proinflammatory signalling molecules as well as the secretion of proteins called cytokines. Cytokines are a large group of molecular proteins that are secreted normally by the Helper T-cells and macrophages. These molecules mediate specific interactions and communications between immune cells thus, stimulating inflammatory reactions and immune responses [17]. Generally, cytokines are named according to their mode of action and/or site of production; examples include interleukins produced by leukocytes, lymphokines produced by lymphocytes, chemokines produced to play chemotactic roles, monokines produced by monocytes and interferons [17,18]. Interleukins (ILs) are the minute component of cytokines immunomodulatory proteins secreted by one leukocyte to act on neighbouring leukocytes, therefore acting as “second molecular messengers” which activate these cells during inflammation and immune response [19]. Unlike other amino acids and steroidal-derived hormones which act on the endocrine system, interleukins act in an autocrine or paracrine manner due to their effects on neighbouring cells. ILs can also act as both pro- and anti-inflammatory stimulators and have been reported to modulate immune cell activation, growth, and differentiation during inflammatory reactions, infection and regulation of neuronal proteins, cell injury and invasion [20]. Those ILs which can stimulate inflammation and immune response under both physiological and pathological conditions are grouped into classes (IL1-IL17) according to their common structural elements and peptide sequence conservation. This makes them ideal therapeutic targets in several human diseases [18].
Cancer
Cancer is a group of diseases caused by uncontrolled cell growth, proliferation and invasion, and has the ability to metastasize to other parts of the body through the bloodstream and lymphatic nervous system [21]. Despite improvements in treatment modalities, this disease persists as one of the leading global health threats in both developed and developing countries. In the United States alone, cancer accounts for higher mortality rate more than heart diseases in individuals below the age of 85 years [22]. Furthermore, statistical and epidemiological studies compiled by the National Centre for Health Statistics (NCHS), National Cancer Institute (NCI), North American Association of Central Cancer Registries (NAACCR) and Centres for Disease Control and Prevention (CDC) have shown that 1 in every 4 deaths was as a result of cancer and 1,529,560 new cancer cases and 569,490 deaths were predicted to occur in 2010 [22].
Cancer can be broadly classified according to the tumour cells’ originality; these include carcinoma, leukaemia, sarcoma, blastoma and germ cell tumour [23]. Carcinomas originate from epithelial cells such as colon, lung, breast, pancreas and prostate, and are most common in adults [24]. Leukaemia develops from the cell lining of the blood vessels, and they account for almost 30% of cancer cases in young children [25]. Sarcoma develops from connecting tissues outside the bone such as cartilage, nerve, fat and bone. Blastoma originates from immature cells or embryos and is more common in children than in adults [26]. Germ cell tumours originate from the gonads, testes and the ovaries [27].
Cancer and inflammation
Genetic mutations, environmental factors (e.g. ultraviolet radiation, air pollution, ionizing radiation, cancer-causing chemicals and occupational exposures), lifestyle (e.g. smoking, lack of exercise, imbalanced diet and excess alcohol consumption) and hormonal imbalance, have emerged as the major sources of cancer morbidity and mortality worldwide [28,29]. Although bacterial and viral infections such as human papillomavirus (HPV) are now gaining momentum, persistence chronic inflammation resulting from long-term invasion of infectious (e.g. Helicobacter pylori) and non-infectious (e.g. chronic pancreatitis, esophagitis, Barrett’s and metaplasia) agents contribute immensely towards the development of several cancer types [30]. In fact, it is estimated that nearly 25% of all cancer cases are connected to chronic inflammation due to infectious or physico-chemical agents, with persistence H. pylori and hepatitis A and C promoting the risk of stomach and hepatocellular carcinoma, respectively [31]. This could be due to improper stimulation and regulation of inflammatory cytokines, as well as immune cells leading to DNA damage and mutation of oncogenes, which in turn leads to tumour development. An adequate stimulation of inflammatory response can lead to quick clearance of the immune cell apparatus and inflammatory cytokines (e.g. chemokines, interleukins) via phagocytosis and apoptosis, immediately after acute inflammation [32]. The phagocytosis of apoptotic factors by phagocytes further supports anti-apoptotic action by promoting the production of anti-apoptotic transforming growth factor-β (TGF-β). Transforming growth factor-β in complex with a serine/threonine kinase binds to TGF-β receptors to trigger signalling cascades that regulate chemotaxis, cell differentiation, proliferation and several immune cell activators resulting in the inhibition of cancer progression [33,34]. However, inadequate regulation of inflammatory response results in chronic inflammation characterized by massive cellular and tissue destruction and neoplastic transformation, which may lead to cancer progression and other inflammatory diseases [35,36].
Link between HSPs, inflammation and cancer: a potential cancer therapeutic target
The link between chronic inflammation and cancer has long been an area of debate. Nevertheless, the precise mechanisms by which persistence inflammation leads to cancer are now beginning to unfold. A DNA lesion or site of structural damage in the DNA called 5-chlorocytosine (5-CIC) created by inflammatory cells during the inflammatory response, has been identified as a major DNA mutant that links chronic inflammation to cancer. In response to infectious agents, biological systems stimulate the synthesis of the immune apparatus referred to as “battery of chemical warfare” with the aim of killing infectious agents and initiating cellular repair. These immune apparatus includes T-and B-lymphocytes, leukocytes, interleukins, cytokines, chemokines, reactive nitrogen species (RNS) (e.g. hydrogen peroxide, hypochlorous acid, and nitric oxide) and ROS. The reactive oxygen molecules designed to nullify the effects of invading pathogens can also in the process cause collateral damage to normal healthy cells resulting in irreparable DNA damage and mutation [37].
For instance, hypochlorous acid (HOCl) generated from the oxidative reaction between H2O2 and chloride ions (Cl-) catalysed by the heme enzyme myeloperoxidase, plays major roles in the killing of foreign pathogens [38]. However, when in excess and under acidic conditions, this molecule is capable of chlorinating cytosine on the DNA strand to form 5-chlorocytosine, resulting in a lesion seen at the inflammatory site and present during inflammatory-related diseases such as atherosclerosis [39,40]. Furthermore, because 5-chlorocytosine is relatively unstable, its conversion to 5-chlorouracil either spontaneously or through enzymatic deamination, causes irreparable damages and mutations in DNA which can lead to cancer [41,42].
The other mechanism directly linking inflammation to cancer is the incidence of toll-like receptor 4 (TLR4) expressions in colon cancer [43]. TLR4 plays a significant role in activating the immune system, inflammatory response, as well as clearance of invading pathogens; however, this receptor can also sustain chronic inflammation, for example, the TLR4 response to bacteria found in the human gut, due to frequent food intake. These bacteria constantly gets replenished, therefore, cannot be completely cleared by the immune response. In this way, TLR4 can sustain persistent chronic inflammation, which is not only involved in cancer formation but also promotes different phases of cancer progression [44].
Moreover, one of the key factors of cellular survival under stressful conditions is the expression of highly regulated and conserved proteins termed HSPs. These proteins are upregulated in response to stress signals such as inflammation and the onset of tumours. Therefore, it is not surprising that these proteins are elevated in every facet of cancer development and inflammatory reactions. According to recent studies, HSPs form part of an immune-inflammatory complex that responds to infectious agents, cellular damage as well as DNA breaks [16]. The DNA is frequently subjected to lesion-inducing agents, it exogenous (genotoxic chemicals, UV or ionizing radiation) or endogenous (reactive oxidants), which subsequently leads to the generation of several DNA lesions including 5-CIC, double-stranded breaks, single-stranded breaks, and single base modifications. In the case where these errors are not properly repaired, they may lead to genome instability which increases the risk of oncogenesis, as a result, multi-dangerous gene mutations [45]. The evidence of heat shock proteins in reducing mutation frequency in Saccharomyces cerevisiae responding to elevated temperature, as well as the elevation of HSP70 in glioblastoma cell lines induced by UV or γ-rays interaction with p53, may however suggest their functions in apoptosis, cell cycle control and DNA repair processes [46,47]. Furthermore, HSP70 and HSP27 in particularly have demonstrated association with base excision repair (BER) enzymes such as human AP endonuclease (APE1) and uracil DNA glycosylase (UDG). This observation suggests the significant roles of HSPs in DNA repair mechanisms, as well as presenting HSPs as modulators of certain DNA repair systems; and since most therapies targeting cancer exert their action by disrupting the normal integrity, structure, and functions of DNA, proper DNA repair mechanisms mediated by HSPs are therefore crucial in maintaining genome stability, integrity and orderliness, which may increase cancer therapeutics efficiency [48,49].
Interestingly, scientific evidence exist showing that HSPs interact with and stabilize key client proteins that play crucial roles in DNA damage response (DDR) mechanisms and cancer progression. DDR mechanisms refer to pathways that are involved in cell-cycle checkpoints, activation of signalling networks, detection of DNA damage, and induction of cell death or DNA repair [50]. The mechanisms that are involved in the DNA repair processes include base excision repair (BER), direct DNA damage reversal and nuclear excision repair (NER). The ability of DDR to respond to DNA damage is controlled by the balancing act between client protein synthesis and their degradation mediated by HSPs. HSP90 in particular binds to DDR proteins and prevent them from E3 ubiquitin-CHIP (chaperoning) mediated ubiquitination and degradation [49]. These proteins include checkpoint kinase 1 (CHKI), O6-meG-DNA methyltransferase (MGMT), MLH1 and MLH2, and APE1, among others.
Emerging evidence has shown that cells inability to repair DNA damage caused by stress has been associated with the loss of genomic integrity, organisation, structure, and function, which may increase the rate of tumour development [51]. These findings reveal the significant roles of HSPs in genetic stability, DDR and tumour progression [52]. Moreover, the roles of HSPs in inflammation cascades have been well elucidated. According to Ikwegbue and co-workers, HSPs exert cytoprotection against inflammatory reactions through the modulation of inflammation cascades that lead to the activation of pro-inflammatory cytokines such as TNF-α, thereby attenuating chronic inflammation [16]. So far there is no study linking HSPs directly to 5-CIC lesion and DDRs, but it can be hypothesised that a similar mechanism observed in other DNA damages and DDRs is followed. Based on these findings, it is tempting to propose that any study that is targeting HSPs (especially HSP90) and their inhibitors in membrane and genetic stability, as well as inflammation, could give new mechanistic insight and may serve as a promising target to improve cancer therapeutics efficacy as proposed in the model (Figure 1).
Figure 1.
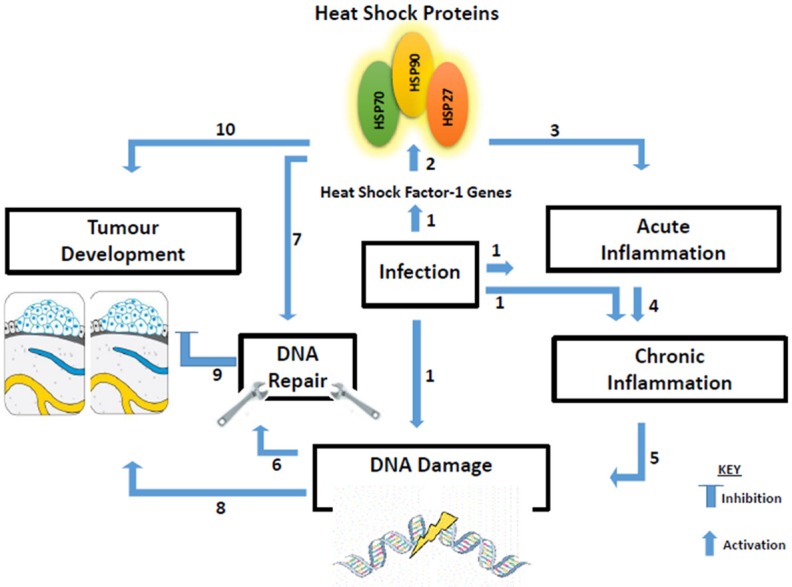
Proposed model showing biological cooperativity between heat shock proteins and inflammation as a promising target towards improved cancer therapeutics. (1) The presence of infection causes the transcription of the heat factor-1 (HSF) gene, which enhances the synthesis of heat shock proteins (HSPs). Infection could also activate acute inflammation, and sometimes initiate chronic inflammatory response and DNA damage directly; (2) Synthesis and activation of HSPs (70, 90, and 27); (3) HSPs together with several immune cells including macrophages, neutrophils, interleukins, and several cytokines as well as ROS (HOCl), form part of acute inflammation aimed at eliminating infectious agents and initiating cellular repair; (4) Nevertheless, if the immune cell apparatus, especially ROS (HOCl in particular) are not properly stimulated, inflammation could persist and degenerates to chronic inflammation. (5) Chronic inflammation is characterized by massive cells and tissue destruction, resulting in DNA damage; (6) DNA damage activates several DDR pathways which can repair DNA damage and prevent accumulation of damaged proteins and tumour development; (7) Central to DNA repair mechanism is the synthesis of heat shock proteins, which maintain and stabilize the DDR proteins and prevent peptides aggregation and cancer; (8) DNA damage and lesions created by chronic inflammation can directly lead to tumour development; (9) If the DDR mechanisms fail due to the presence of HSPs inhibitors, the result could be malignant cells progression; (10) Improper stimulation of HSPs during tumour formation can lead to tumour invasion, progression, and metastasis.
Conclusion
Tremendous efforts have been made over the years in devising strategies to prevent, manage, cure and even vaccinate against cancer, yet with several unsatisfactory results. More so, recent studies have focussed on testing therapeutic efficacy of HSPs and HSP inhibitors in cells and animal models, while other efforts are focussed on conducting clinical trials using HSPs in different stages of cancer development. However, any study aimed at understanding the functions of excess HOCl during inflammatory response, and the roles 5-CIC lesion (created by HOCl) which links chronic inflammation to DNA damage, and crucial roles of HSPs in maintaining the integrity and stability of the proteins that are involved in DNA repair processes, could be promising targets in improving current cancer therapies. Based on this promising aspect of HSPs and its inhibitors in inflammation, DNA repair, and cancer, one can therefore be optimistic that improved therapies and a cure for cancer is to be expected soon.
Acknowledgements
Abidemi Paul Kappo is sincerely grateful to the National Research Foundation (NRF), South Africa for a Thuthuka Research Grant and to the University of Zululand Research Committee for funding (Grant No: C387). Paul Ikwegbue and Priscilla Masamba are equally thankful to the National Research Foundation (NRF), South Africa for postgraduate bursaries towards their studies. Londiwe Mbatha acknowledges the University of Zululand Research Committee for a Postdoctoral Research Fellowship, while Babatunji Oyinloye is grateful to Afe Babalola University for an educational advancement scholarship.
Disclosure of conflict of interest
None.
References
- 1.Laplante AF, Moulin V, Auger FA, Landry J, Li H, Morrow G, Tanguay RM, Germain L. Expression of heat shock proteins in mouse skin during wound healing. J Histochem Cytoch. 1998;46:1291–1301. doi: 10.1177/002215549804601109. [DOI] [PubMed] [Google Scholar]
- 2.Al-Whaibi MH. Plant heat-shock proteins: a mini review. J KSUS. 2011;23:139–150. [Google Scholar]
- 3.Ou JR, Tan MS, Xie AM, Yu JT, Tan L. Heat shock protein 90 in Alzheimer’s disease. BioMed Res Int. 2014;2014:1–7. doi: 10.1155/2014/796869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang X, Shi J, Tian J, Robinson AC, Davidson YS, Mann DM. Expression of one important chaperone protein, heat shock protein 27, in neurodegenerative diseases. Alzheimer’s Res Ther. 2014;6:78. doi: 10.1186/s13195-014-0078-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leak RK. Heat shock proteins in neurodegenerative disorders and aging. J Cell Commun Signal. 2014;8:293–310. doi: 10.1007/s12079-014-0243-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D’Souza SM, Brown IR. Constitutive expression of heat shock proteins Hsp90, Hsc70, Hsp70 and Hsp60 in neural and non-neural tissues of the rat during postnatal development. Cell Stress Chaperon. 1998;3:188. doi: 10.1379/1466-1268(1998)003<0188:ceohsp>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Somji S, Todd JH, Sens MA, Garrett SH, Sens DA. Expression of the constitutive and inducible forms of heat shock protein 70 in human proximal tubule cells exposed to heat, sodium arsenite, and CdCl (2) Environ Health Perspect. 1999;107:887. doi: 10.1289/ehp.99107887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Place SP, Hofmann GE. Constitutive expression of a stress-inducible heat shock protein gene, hsp70, in phylogenetically distant Antarctic fish. Pol Biol. 2005;28:261–267. [Google Scholar]
- 9.Edkins AL, Boshoff A. Edited by Heat Shock Proteins of Malaria. Springer; 2014. General structural and functional features of molecular chaperones; pp. 5–45. [DOI] [PubMed] [Google Scholar]
- 10.Chatterjee S, Burns TF. Targeting heat shock proteins in cancer: a promising therapeutic approach. Int J Mol Sci. 2017;18:1978. doi: 10.3390/ijms18091978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finka A, Goloubinoff P. Proteomic data from human cell cultures refine mechanisms of chaperone-mediated protein homeostasis. Cell Stress Chaperon. 2013;18:591–605. doi: 10.1007/s12192-013-0413-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferrero-Miliani L, Nielsen O, Andersen P, Girardin S. Chronic inflammation: importance of NOD2 and NALP3 in interleukin-1β generation. Clin Exp Immunol. 2007;147:227–235. doi: 10.1111/j.1365-2249.2006.03261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 14.Oyinloye BE, Adenowo AF, Kappo AP. Reactive oxygen species, apoptosis, antimicrobial peptides and human inflammatory diseases. Pharm. 2015;8:151–175. doi: 10.3390/ph8020151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 16.Ikwegbue PC, Masamba P, Oyinloye BE, Kappo AP. Roles of heat shock proteins in apoptosis, oxidative stress, human inflammatory diseases, and cancer. Pharm. 2017;11:2. doi: 10.3390/ph11010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang JM, An J. Cytokines, inflammation and pain. Int Anesthesiol Clin. 2007;45:27. doi: 10.1097/AIA.0b013e318034194e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feghali CA, Wright TM. Cytokines in acute and chronic inflammation. Front Biosci. 1997;2:d12–d26. doi: 10.2741/a171. [DOI] [PubMed] [Google Scholar]
- 19.Brocker C, Thompson D, Matsumoto A, Nebert DW, Vasiliou V. Evolutionary divergence and functions of the human interleukin (IL) gene family. Hum Genomics. 2010;5:30. doi: 10.1186/1479-7364-5-1-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Commins SP, Borish L, Steinke JW. Immunologic messenger molecules: cytokines, interferons, and chemokines. J Allergy Clin Immunol. 2010;125:S53–S72. doi: 10.1016/j.jaci.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 21.Adekiya TA, Aruleba RT, Khanyile S, Masamba P, Oyinloye BE, Kappo AP. Structural analysis and epitope prediction of MHC class-1-chain related protein-a for cancer vaccine development. Vaccines. 2017;6:1. doi: 10.3390/vaccines6010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA: Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 23.Young JL Jr, Miller RW. Incidence of malignant tumors in US children. J Pediatr. 1975;86:254–258. doi: 10.1016/s0022-3476(75)80484-7. [DOI] [PubMed] [Google Scholar]
- 24.Morris J. Information: theoretical and applied. Prog Pathol. 2001;5:165–183. [Google Scholar]
- 25.Varricchio CG. A cancer source book for nurses. United Kingdom: Edited by Jones and Bartlett Publishers; 2004. pp. 13–30. [Google Scholar]
- 26.Scotting PJ, Walker DA, Perilongo G. Childhood solid tumours: a developmental disorder. Nat Rev Cancer. 2005;5:481. doi: 10.1038/nrc1633. [DOI] [PubMed] [Google Scholar]
- 27.Oosterhuis JW, Looijenga LH. Testicular germ-cell tumours in a broader perspective. Nat Rev Cancer. 2005;5:210. doi: 10.1038/nrc1568. [DOI] [PubMed] [Google Scholar]
- 28.Czene K, Lichtenstein P, Hemminki K. Environmental and heritable causes of cancer among 9.6 million individuals in the Swedish family-cancer database. Int J Cancer. 2002;99:260–266. doi: 10.1002/ijc.10332. [DOI] [PubMed] [Google Scholar]
- 29.Danaei G, Vander Hoorn S, Lopez AD, Murray CJ, Ezzati M Comparative Risk Assessment collaborating group (Cancers) Causes of cancer in the world: comparative risk assessment of nine behavioural and environmental risk factors. Lancet. 2005;366:1784–1793. doi: 10.1016/S0140-6736(05)67725-2. [DOI] [PubMed] [Google Scholar]
- 30.Shacter E, Weitzman SA. Chronic inflammation and cancer. Oncol. 2002;16:217–229. [PubMed] [Google Scholar]
- 31.Korniluk A, Koper O, Kemona H, Dymicka-Piekarska V. From inflammation to cancer. Ir J Med Sci. 2017;186:57–62. doi: 10.1007/s11845-016-1464-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vallespí MG, García I. Heat-shock proteins in inflammation and cancer. Biotecnol Apl. 2008;25:208–215. [Google Scholar]
- 33.Nakao A, Afrakhte M, Morn A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH. Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature. 1997;389:631. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- 34.Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Bio. 2012;13:616. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu H, Ouyang W, Huang C. Inflammation, a key event in cancer development. Mol Cancer Res. 2006;4:221–233. doi: 10.1158/1541-7786.MCR-05-0261. [DOI] [PubMed] [Google Scholar]
- 36.Salem ML, Attia ZI, Galal SM. Acute inflammation induces immunomodulatory effects on myeloid cells associated with anti-tumor responses in a tumor mouse model. J Adv Res. 2016;7:243–253. doi: 10.1016/j.jare.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fedeles BI, Freudenthal BD, Yau E, Singh V, Chang SC, Li D, Delaney JC, Wilson SH, Essigmann JM. Intrinsic mutagenic properties of 5-chlorocytosine: a mechanistic connection between chronic inflammation and cancer. Proc Natl Acad Sci U S A. 2015;112:E4571–E4580. doi: 10.1073/pnas.1507709112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khor HK, Fisher M, Schoneich C. Potential role of methionine sulfoxide in the inactivation of the chaperone GroEL by hypochlorous acid (HOCl) and peroxynitrite (ONOO-) J Biol Chem. 2004;279:19486–93. doi: 10.1074/jbc.M310045200. [DOI] [PubMed] [Google Scholar]
- 39.Hazen SL, Heinecke JW. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J Clin Invest. 1997;99:2075–2081. doi: 10.1172/JCI119379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang Q, Blount B, Ames BN. 5-chlorouracil, a marker of DNA damage from hypochlorous acid during inflmmation: a GC-MS assay. J Biol Chem. 2003;278:32834–40. doi: 10.1074/jbc.M304021200. [DOI] [PubMed] [Google Scholar]
- 41.Chen HJ, Row SW, Hong CL. Detection and quantification of 5-chlorocytosine in DNA by stable isotope dilution and gas chromatography/negative ion chemical ionization/mass spectrometry. Chem Res Toxicol. 2002;15:262–268. doi: 10.1021/tx015578g. [DOI] [PubMed] [Google Scholar]
- 42.Hale JT, Bigelow JC, Mathews LA, McCormack JJ. Analytical and pharmacokinetic studies with 5-chloro-2’-deoxycytidine. Biochem Pharmacol. 2002;64:1493–1502. doi: 10.1016/s0006-2952(02)01413-2. [DOI] [PubMed] [Google Scholar]
- 43.Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S, Hsu D, Xu R, Harpaz N, Dannenberg AJ. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterol. 2007;133:1869–81. doi: 10.1053/j.gastro.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hunter P. The inflammation theory of disease: The growing realization that chronic inflammation is crucial in many diseases opens new avenues for treatment. EMBO Rep. 2012;13:968–970. doi: 10.1038/embor.2012.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pennisi R, Ascenzi P, di Masi A. Hsp90: a new player in DNA repair? Biomolecules. 2015;5:2589–2618. doi: 10.3390/biom5042589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nunes E, Candreva EC, Keszenman D, Salvo VA. The mutagenic effect of elevated temperatures in yeast is blocked by a previous heat shock. Mutat Res Fund Mol Mech Mut. 1993;289:165–170. doi: 10.1016/0027-5107(93)90066-o. [DOI] [PubMed] [Google Scholar]
- 47.Matsumoto H, Wang X, Ohnishi T. Binding between wild-type p53 and hsp72 accumulated after UV and γ-ray irradiation. Cancer Lett. 1995;92:127–133. doi: 10.1016/0304-3835(95)03769-s. [DOI] [PubMed] [Google Scholar]
- 48.Mendez F, Sandigursky M, Franklin WA, Kenny MK, Kureekattil R, Bases R. Heat-shock proteins associated with base excision repair enzymes in HeLa cells. Radiat Res. 2000;153:186–195. doi: 10.1667/0033-7587(2000)153[0186:hspawb]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 49.Sottile ML, Nadin SB. Heat shock proteins and DNA repair mechanisms: an updated overview. Cell Stress Chaperon. 2018;23:303–315. doi: 10.1007/s12192-017-0843-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bridge G, Rashid S, Martin SA. DNA mismatch repair and oxidative DNA damage: implications for cancer biology and treatment. Cancers. 2014;6:1597–1614. doi: 10.3390/cancers6031597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]