

Abstract
The human kinome comprises more than 50 pseudo-kinases with unclear biological function due to the absence of apparent catalytic activity, and therefore, with presumably little interest for cancer drug discovery. However, it is now acknowledged that several of them, such as Pragmin family members, play roles as important as those of active kinases in human cancer. How these pseudo-kinases promote tumor formation is largely unknown. Recently, independent structural analyses of three Pragmin pseudo-kinases (Pragmin, SGK223, and SGK269/PEAK1) revealed a split helical dimerization (SHED)-based mechanism of action. Additional sequence-structure analysis identified C19orf35 as a new member of the Pragmin family. Based on the results of these molecular studies, we present a unified model on how Pragmin pseudo-kinases may regulate oncogenic signaling, and suggest potential therapeutic strategies to block their tumor activity.
Keywords: Pseudo-kinase, cell signaling, cancer, phosphorylation, structure, cell migration, cell growth, therapeutic target
The Pragmin subfamily of pseudo-kinases comprises Pragmin, the human ortholog SGK223 and the structural homologs SGK269/PEAK1 as well as a new incomer C19orf35, also named PEAK3. Pragmin was originally discovered as a signaling protein and effector of the small GTPase Rnd2 from a rat expression library to induce cell contraction [1]. Later, Pragmin was identified as a key component of a nuclear Notch transcriptional complex containing the Notch intracellular domain, NICD, and the co-activator Maml-1 to regulate Notch-dependent transcription, consistent with additional nuclear functions [2]. SGK269/PEAK1 was isolated as a pseudopodium-enriched atypical kinase 1 from a proteomic study (hence PEAK1) to regulate cell adhesion [3]. C19orf35 was identified lately as an additional member of the subfamily from an in silico sequence-structure analysis and has yet unknown function [4]. Consistent with signaling functions, Pragmin/SGK223 and PEAK1/SGK269 regulate cell growth and adhesion induced by various extracellular cues (EGF, VEGF, Notch, TGFbeta, collagen and fibronectin) [2,3,5-8]. Mechanistically, they undergo tyrosine phosphorylation allowing the recruitment of important effectors, such as Grb2 and Shc, for intracellular signaling [3,5,9]. Compelling evidence now support an essential role for these pseudo-kinases in human cancer. For instance, Pragmin/SGK223 and PEAK1/SGK269 were found overexpressed in various adenocarcinoma cells from the colon, the lung and the pancreas [3,10-13]. Additionally, their tumor expression levels were associated with the aggressiveness of the disease in patients with colorectal cancer [13] (unpublished observations); and PEAK1/SGK269 was reported as also an early biomarker for pancreatic development and progression [10]. They exhibit oncogenic activity as their overexpression enhances tumor cell growth and migration in vitro and promotes tumorigenesis in vivo, while their downregulation reduce these cell neoplastic transforming properties [2,3,6-11,14]. Proteomic studies also identified these pseudo-kinases as novel substrates of oncogenic tyrosine kinases (Src, Lyn, HER2, DDR1) and critical components of tumor signaling driven by various oncogenes (KRAS, Notch and TGFbeta) [2,6-10,14-17] (Figure 1). Therefore, targeting their tumor activity may be of therapeutic interest in these human adenocarcinomas.
Figure 1.
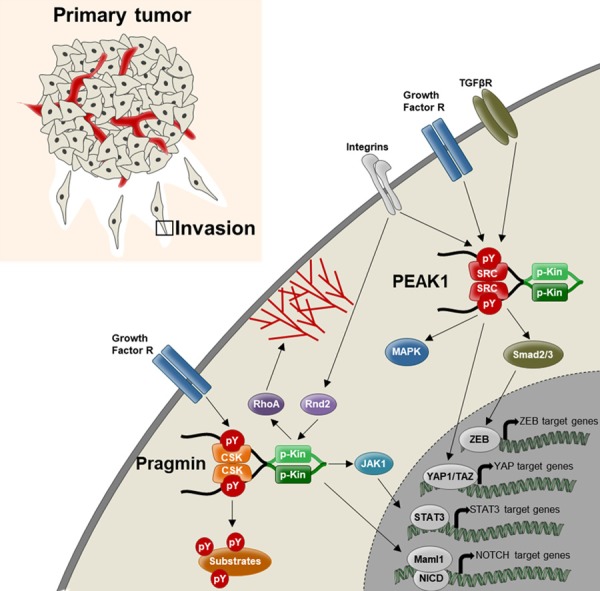
Proposed tumor activity of Pragmin pseudo-kinases. Integrins and growth factor receptors induce pseudo-kinase activation by a double-act mechanism (see Figure 3) enhancing MAPKs, Jak/Stat3, Notch, ZEB1, YAP/TAZ and Rho-pathways to induce gene expression and cytoskeleton rearrangement necessary for tumor cell growth and migration.
How these pseudo-kinases induce malignant cell transformation is largely unclear. Due to the presence of a kinase domain, Pragmin oncogenic functions have been linked to protein phosphorylation. For instance, SGK223 was shown to regulate protein tyrosine phosphorylation in cancer cells and both Pragmin and PEAK1 were found associated with protein tyrosine kinase activity in vitro [3,4,14]. Furthermore, an intact kinase domain was found necessary for PEAK1 to induce tumor cell growth [10] and for SGK223 to activate Notch-dependent transcription [18]. However, the exact role of these kinase domains in protein tyrosine phosphorylation and malignant cell transformation is unclear. Indeed, these signaling proteins were scored as pseudo-kinases due to the absence of conserved sequences needed for catalytic reaction including the the Gly-rich loop and the so-called DFG [19].
Recently, three structural analyses brought important molecular insight on their mechanism of action [4,20,21]. First of all, the three independent crystal structures confirmed that these proteins share a protein-kinase fold. They also revealed a rearranged catalytic site with both significant sequence variations compared to catalytically active protein-kinase and also a tightly closed conformation. Indeed, these three structures showed a conserved inhibitory triad (D978, Y981 and Q1021 in Pragmin) surrounding the catalytic lysine. These features suggested that no ATP binding can occur and no ATP binding by either the crystallized C-terminal of Pragmin in vitro or the full-length protein in cellulo, was detected [4]. Furthermore, any attempt to resuscitate ATP binding by mutating the inhibitory triad were unsuccessful [4]. This data confirms the kinase inactive nature of these signaling proteins, which raises the mechanism by which they may induce protein tyrosine phosphorylation. By proteomics, it was then discovered that Pragmin uses the tyrosine kinase CSK to induce protein tyrosine phosphorylation in human cells [4], which was primed by a pTyr-SH2 dependent mechanism upon Pragmin phosphorylation on its major tyrosine phosphorylation site present in the EPIYA sequence [22]. Although not experimentally verified, PEAK1 may use a similar mechanism through an interaction with a tyrosine kinase of the Src family (SFKs) upon phosphorylation on Tyr 665 [23] and an equivalent scenario might be expected for C19orf35 (Figure 2).
Figure 2.
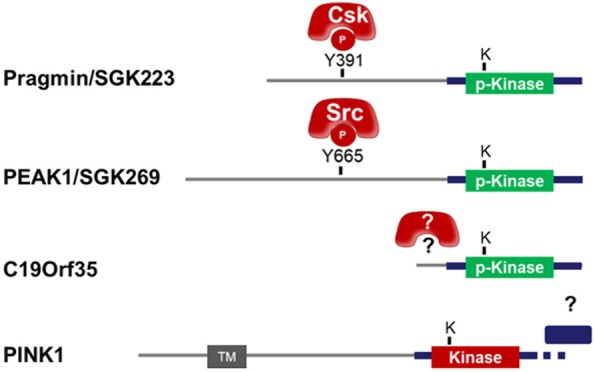
The growing family of Pragmin pseudo-kinases. Modular structure of Pragmin pseudo-kinases with the associated activated protein kinase shown in red. The modular structure of the closest active protein kinase homolog PINK1 is also shown. Predicted additional regulatory mechanisms are highlighted by question marks.
Interestingly, these three crystal structures revealed an original dimerization domain, named split helical dimerization (SHED) in PEAK1, comprising a long helix N-terminal to the pseudo-kinase domain and four additional helices from the C-terminal extension [4,20,21]. The sequence motifs conserved in those extensions are found surrounding the pseudo-kinase domain of C19orf35, accordingly a new member of the family [4]. Finally, by taking advantage of the crystal structures to design mutations, it was shown that protein tyrosine phosphorylation activation and signaling by Pragmin/SGK223 relies on its conserved dimerization [4,20]. Remarkably, Pragmin dimerization dramatically enhances the CSK capacity to induce protein tyrosine phosphorylation in human cells independently from SFKs [4]. It will thus be interesting to test whether this dimerization mechanism also operates in PEAK1 for SFKs activation. This mechanism however differs from the previously described mode of action where a pseudo-kinase heterodimerizes back-to-back to an active kinase for regulating its activity [24]. Here, the homodimerization would bring in proximity their natively disordered N-terminal extension (from 100 up to 900 residues long) and their attached partners thanks to various motifs such as the phosphorylated EPIYA segment. Surprisingly, the most closely related and active protein-kinase, PINK1, is a serine/threonine kinase whose recently solved crystal structure highlighted the presence of a similar domain in its C-terminus, and in similar position relative to the protein-kinase domain but lacking the long N-terminal helix [25]. It is therefore postulated that PINK1 activity may be regulated by dimerization as soon as its N-terminus or a helical partner complete its partial SHED module (Figure 2). In this view, we might wonder whether a partner of Pragmin or PEAK1, can bring a long helix to break down their dimer to regulate their activity or cellular localization.
From these structural analyses, we propose a conserved “double-act” mechanism by which Pragmin pseudo-kinases induces protein phosphorylation and signaling [26] (Figure 3). In this model, the SHED dimerization domain flanking its pseudo-kinase domain is important for Pragmin-mediated activation of the targeted non-receptor tyrosine kinase to induce phosphorylation of specific oncogenic substrates. In agreement, PEAK1 was shown to use Src activity to promote cell transformation in the pancreas [10] and Pragmin may use CSK to enhance cancer properties of gastric or colorectal adenocarcinoma cells [14,15], where CSK was found dramatically upregulated [27,28]. From this model, one can propose these pseudo-kinases as novel candidates for the development of new therapeutics. For instance, Pragmin pseudo-kinases inhibition can rely on the targeting of their activated protein tyrosine kinase. Accordingly, one might predict the utility of clinically available Src inhibitors in tumors overexpressing PEAK1 and possibly Pragmin since they also efficiently target CSK activity [29]. Pseudo-kinase inhibition may also involve drugs that destabilize their dimeric conformation with the guidance of the structure, although sufficient evidence for the dimeric role of these oncogenic pseudo-kinases are still lacking. Finally, since Pragmin tumor activities are linked to their upregulation in human cancer, any drug targeting their expression may display anti-oncogenic effect. Consistently, this strategy was recently validated with eIF5A-dependent translation inhibitors to target PEAK1-dependent growth properties in pancreatic cancer cells [30]. Whether this strategy may apply to additional oncogenic pseudo-kinases is however currently unknown.
Figure 3.
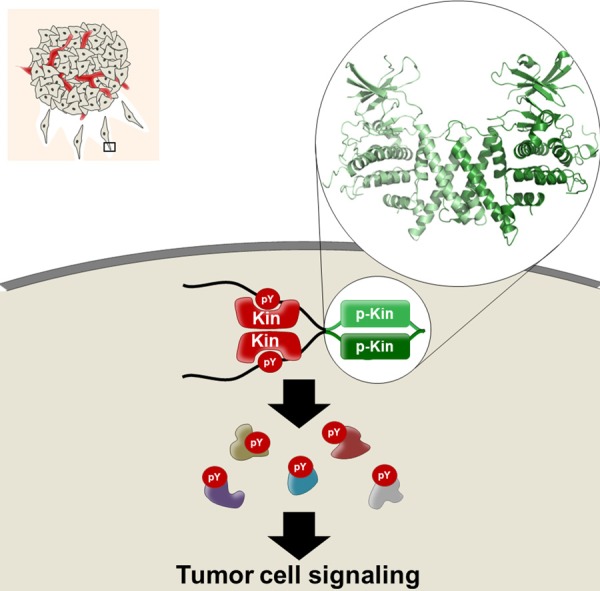
A double-act mechanism for Pragmin pseudo-kinase signaling in human cancer. SHED-mediated pseudo-kinase dimerization activates the associated protein tyrosine kinase upon tyrosine phosphorylation by upstream signals to phosphorylate specific cancer substrates.
Finally, these structural analyses raise a number of important questions on how Pragmin pseudo-kinases exert their tumor activities. For instance, an important nuclear role for Pragmin was reported in tumor cells [2], which may be linked to its adhesive function, possibly to coordinate cell adhesion and transcription for cell fate decision. It will thus be important to address how these functions are coordinated to promote tumor formation and whether our described kinase-dependent dimeric mechanism is involved in this process. Another important question comes from the strong similarities shared by the SHED domains, which predicts a combination of heterodimers within the family [21], as recently reported for SGK223/PEAK1 complexes [31]. How these heterodimers influence Pragmin pseudo-kinases signaling is another important question that needs to be addressed in the future. Finally, these structural studies predict a novel dimeric-based mechanism of tyrosine kinase activation. Deciphering such regulatory mechanism may bring additional important insight into pseudo-kinase and tyrosine kinase regulation.
Acknowledgements
This work was supported by INCA (PLBIO-2011-150), La Ligue Contre le Cancer (Equipe Labellisée LIGUE 2017), Montpellier SIRIC Grant (INCa-DGOS-Inserm 6045), CNRS and the University of Montpellier. SR is an INSERM investigator.
Disclosure of conflict of interest
None.
Abbreviations
- SHED
split helical dimerization
- NICD
Notch intracellular domain
- Maml-1
Mastermind-like protein 1
- PEAK1
pseudopodium-enriched atypical kinase 1
- EGF
Epidermal Growth Factor
- VEGF
Vascular Endothelial Growth Factor
- TGF beta
Transforming Growth Factor beta
- HER2
Human Epidermal Growth Factor Receptor-2
- DDR1
Discoidin Domain Receptor 1
- CSK
C-terminal Src kinase
- SFK
Src Family Kinase
- PINK1
PTEN-induced putative kinase 1
References
- 1.Tanaka H, Katoh H, Negishi M. Pragmin, a novel effector of Rnd2 GTPase, stimulates RhoA activity. J Biol Chem. 2006;281:10355–10364. doi: 10.1074/jbc.M511314200. [DOI] [PubMed] [Google Scholar]
- 2.Weaver KL, Alves-Guerra MC, Jin K, Wang Z, Han X, Ranganathan P, Zhu X, DaSilva T, Liu W, Ratti F, Demarest RM, Tzimas C, Rice M, Vasquez-Del Carpio R, Dahmane N, Robbins DJ, Capobianco AJ. NACK is an integral component of the notch transcriptional activation complex and is critical for development and tumorigenesis. Cancer Res. 2014;74:4741–4751. doi: 10.1158/0008-5472.CAN-14-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y, Kelber JA, Tran Cao HS, Cantin GT, Lin R, Wang W, Kaushal S, Bristow JM, Edgington TS, Hoffman RM, Bouvet M, Yates JR 3rd, Klemke RL. Pseudopodium-enriched atypical kinase 1 regulates the cytoskeleton and cancer progression. Proc Natl Acad Sci U S A. 2010;107:10920–10925. doi: 10.1073/pnas.0914776107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lecointre C, Simon V, Kerneur C, Allemand F, Fournet A, Montarras I, Pons JL, Gelin M, Brignatz C, Urbach S, Labesse G, Roche S. Dimerization of the Pragmin pseudo-kinase regulates protein tyrosine phosphorylation. Structure. 2018;26:545–554. e544. doi: 10.1016/j.str.2018.01.017. [DOI] [PubMed] [Google Scholar]
- 5.Zheng Y, Zhang C, Croucher DR, Soliman MA, St-Denis N, Pasculescu A, Taylor L, Tate SA, Hardy WR, Colwill K, Dai AY, Bagshaw R, Dennis JW, Gingras AC, Daly RJ, Pawson T. Temporal regulation of EGF signalling networks by the scaffold protein Shc1. Nature. 2013;499:166–171. doi: 10.1038/nature12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agajanian M, Runa F, Kelber JA. Identification of a PEAK1/ZEB1 signaling axis during TGFbeta/fibronectin-induced EMT in breast cancer. Biochem Biophys Res Commun. 2015;465:606–612. doi: 10.1016/j.bbrc.2015.08.071. [DOI] [PubMed] [Google Scholar]
- 7.Fujimura K, Wang H, Watson F, Klemke RL. KRAS oncoprotein expression is regulated by a self-governing eIF5A-PEAK1 feed-forward regulatory loop. Cancer Res. 2018;78:1444–1456. doi: 10.1158/0008-5472.CAN-17-2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeitany M, Leroy C, Tosti P, Lafitte M, Le Guet J, Simon V, Bonenfant D, Robert B, Grillet F, Mollevi C, El Messaoudi S, Otandault A, Canterel-Thouennon L, Busson M, Thierry AR, Martineau P, Pannequin J, Roche S, Sirvent A. Inhibition of DDR1-BCR signalling by nilotinib as a new therapeutic strategy for metastatic colorectal cancer. EMBO Mol Med. 2018;10:e7918. doi: 10.15252/emmm.201707918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Croucher DR, Hochgrafe F, Zhang L, Liu L, Lyons RJ, Rickwood D, Tactacan CM, Browne BC, Ali N, Chan H, Shearer R, Gallego-Ortega D, Saunders DN, Swarbrick A, Daly RJ. Involvement of Lyn and the atypical kinase SgK269/PEAK1 in a basal breast cancer signaling pathway. Cancer Res. 2013;73:1969–1980. doi: 10.1158/0008-5472.CAN-12-1472. [DOI] [PubMed] [Google Scholar]
- 10.Kelber JA, Reno T, Kaushal S, Metildi C, Wright T, Stoletov K, Weems JM, Park FD, Mose E, Wang Y, Hoffman RM, Lowy AM, Bouvet M, Klemke RL. KRas induces a Src/PEAK1/ErbB2 kinase amplification loop that drives metastatic growth and therapy resistance in pancreatic cancer. Cancer Res. 2012;72:2554–2564. doi: 10.1158/0008-5472.CAN-11-3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tactacan CM, Phua YW, Liu L, Zhang L, Humphrey ES, Cowley M, Pinese M, Biankin AV, Daly RJ. The pseudokinase SgK223 promotes invasion of pancreatic ductal epithelial cells through JAK1/Stat3 signaling. Mol Cancer. 2015;14:139. doi: 10.1186/s12943-015-0412-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kong R, Feng J, Ma Y, Zhou B, Li S, Zhang W, Jiang J, Zhang J, Qiao Z, Zhang T, Zang Q, He X. Silencing NACK by siRNA inhibits tumorigenesis in non-small cell lung cancer via targeting Notch1 signaling pathway. Oncol Rep. 2016;35:2306–2314. doi: 10.3892/or.2016.4552. [DOI] [PubMed] [Google Scholar]
- 13.Huang L, Wen C, Yang X, Lou Q, Wang X, Che J, Chen J, Yang Z, Wu X, Huang M, Lan P, Wang L, Iwamoto A, Wang J, Liu H. PEAK1, acting as a tumor promoter in colorectal cancer, is regulated by the EGFR/KRas signaling axis and miR-181d. Cell Death Dis. 2018;9:271. doi: 10.1038/s41419-018-0320-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leroy C, Fialin C, Sirvent A, Simon V, Urbach S, Poncet J, Robert B, Jouin P, Roche S. Quantitative phosphoproteomics reveals a cluster of tyrosine kinases that mediates SRC invasive activity in advanced colon carcinoma cells. Cancer Res. 2009;69:2279–2286. doi: 10.1158/0008-5472.CAN-08-2354. [DOI] [PubMed] [Google Scholar]
- 15.Senda Y, Murata-Kamiya N, Hatakeyama M. C-terminal Src kinase-mediated EPIYA phosphorylation of Pragmin creates a feed-forward C-terminal Src kinase activation loop that promotes cell motility. Cancer Sci. 2016;107:972–980. doi: 10.1111/cas.12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang H, Lapek J, Fujimura K, Strnadel J, Liu B, Gonzalez DJ, Zhang W, Watson F, Yu V, Liu C, Melo CM, Miller YI, Elliott KC, Cheresh DA, Klemke RL. Pseudopodium-enriched atypical kinase 1 mediates angiogenesis by modulating GATA2-dependent VEGFR2 transcription. Cell Discov. 2018;4:26. doi: 10.1038/s41421-018-0024-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strnadel J, Choi S, Fujimura K, Wang H, Zhang W, Wyse M, Wright T, Gross E, Peinado C, Park HW, Bui J, Kelber J, Bouvet M, Guan KL, Klemke RL. eIF5A-PEAK1 signaling regulates YAP1/TAZ protein expression and pancreatic cancer cell growth. Cancer Res. 2017;77:1997–2007. doi: 10.1158/0008-5472.CAN-16-2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin K, Zhou W, Han X, Wang Z, Li B, Jeffries S, Tao W, Robbins DJ, Capobianco AJ. Acetylation of mastermind-like 1 by p300 drives the recruitment of NACK to initiate notch-dependent transcription. Cancer Res. 2017;77:4228–4237. doi: 10.1158/0008-5472.CAN-16-3156. [DOI] [PubMed] [Google Scholar]
- 19.Eyers PA, Murphy JM. Dawn of the dead: protein pseudokinases signal new adventures in cell biology. Biochem Soc Trans. 2013;41:969–974. doi: 10.1042/BST20130115. [DOI] [PubMed] [Google Scholar]
- 20.Patel O, Griffin MDW, Panjikar S, Dai W, Ma X, Chan H, Zheng C, Kropp A, Murphy JM, Daly RJ, Lucet IS. Structure of SgK223 pseudokinase reveals novel mechanisms of homotypic and heterotypic association. Nat Commun. 2017;8:1157. doi: 10.1038/s41467-017-01279-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ha BH, Boggon TJ. The crystal structure of pseudokinase PEAK1 (Sugen kinase 269) reveals an unusual catalytic cleft and a novel mode of kinase fold dimerization. J Biol Chem. 2018;293:1642–1650. doi: 10.1074/jbc.RA117.000751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Safari F, Murata-Kamiya N, Saito Y, Hatakeyama M. Mammalian Pragmin regulates Src family kinases via the Glu-Pro-Ile-Tyr-Ala (EPIYA) motif that is exploited by bacterial effectors. Proc Natl Acad Sci U S A. 2011;108:14938–14943. doi: 10.1073/pnas.1107740108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bristow JM, Reno TA, Jo M, Gonias SL, Klemke RL. Dynamic phosphorylation of tyrosine 665 in pseudopodium-enriched atypical kinase 1 (PEAK1) is essential for the regulation of cell migration and focal adhesion turnover. J Biol Chem. 2013;288:123–131. doi: 10.1074/jbc.M112.410910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacobsen AV, Murphy JM. The secret life of kinases: insights into non-catalytic signalling functions from pseudokinases. Biochem Soc Trans. 2017;45:665–681. doi: 10.1042/BST20160331. [DOI] [PubMed] [Google Scholar]
- 25.Schubert AF, Gladkova C, Pardon E, Wagstaff JL, Freund SMV, Steyaert J, Maslen SL, Komander D. Structure of PINK1 in complex with its substrate ubiquitin. Nature. 2017;552:51–56. doi: 10.1038/nature24645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Preuss F, Mathea S, Knapp S. A pseudo-kinase double act. Structure. 2018;26:527–528. doi: 10.1016/j.str.2018.03.008. [DOI] [PubMed] [Google Scholar]
- 27.Benistant C, Bourgaux JF, Chapuis H, Mottet N, Roche S, Bali JP. The COOH-terminal Src kinase Csk is a tumor antigen in human carcinoma. Cancer Res. 2001;61:1415–1420. [PubMed] [Google Scholar]
- 28.Sirvent A, Benistant C, Pannequin J, Veracini L, Simon V, Bourgaux JF, Hollande F, Cruzalegui F, Roche S. Src family tyrosine kinases-driven colon cancer cell invasion is induced by Csk membrane delocalization. Oncogene. 2010;29:1303–1315. doi: 10.1038/onc.2009.450. [DOI] [PubMed] [Google Scholar]
- 29.Sirvent A, Benistant C, Roche S. Oncogenic signaling by tyrosine kinases of the SRC family in advanced colorectal cancer. Am J Cancer Res. 2012;2:357–371. [PMC free article] [PubMed] [Google Scholar]
- 30.Fujimura K, Wright T, Strnadel J, Kaushal S, Metildi C, Lowy AM, Bouvet M, Kelber JA, Klemke RL. A hypusine-eIF5A-PEAK1 switch regulates the pathogenesis of pancreatic cancer. Cancer Res. 2014;74:6671–6681. doi: 10.1158/0008-5472.CAN-14-1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu L, Phua YW, Lee RS, Ma X, Jenkins Y, Novy K, Humphrey ES, Chan H, Shearer R, Ong PC, Dai W, Saunders DN, Lucet IS, Daly RJ. Homo- and heterotypic association regulates signaling by the SgK269/PEAK1 and SgK223 pseudokinases. J Biol Chem. 2016;291:21571–21583. doi: 10.1074/jbc.M116.748897. [DOI] [PMC free article] [PubMed] [Google Scholar]