

Abstract
Oncolytic virotherapies have emerged as new modalities for cancer treatment. We previously reported that coxsackievirus B3 (CVB3) is a novel oncolytic virus (OV) with a strong ability to lyse human non-small cell lung cancer cells; however, its non-specific toxicity against normal cells remains to be resolved. To improve its safety profile, microRNA target sequences complementary to miR-34a/c, which is expressed preferentially in normal cells, were inserted into the 5′ UTR or 3′ UTR of the CVB3 genome. In the presence of miR-34a/c, the gene-modified CVB3 could not replicate in normal cells. We also found that the pathogenicity of CVB3 was reduced to a greater extent by targeting miR-34a than miR-34c; in addition, it was more effective to insert the target sequences into the 3′ UTR rather than the 5′ UTR of the viral genome. Ultimately, we developed a double-miR-34a targeting virus (53a-CVB) by inserting miR-34a targets in both the 5′ UTR and 3′ UTR of the virus. 53a-CVB was minimally toxic to cells in normal tissue, but maintained nearly its full oncolytic activity in mice xenografted with human lung cancer. 53a-CVB is the first miR-34-regulated OV and represents a promising platform for the development of safe and effective anti-cancer therapies.
Keywords: oncolytic virus, coxsackievirus B3, miRNA
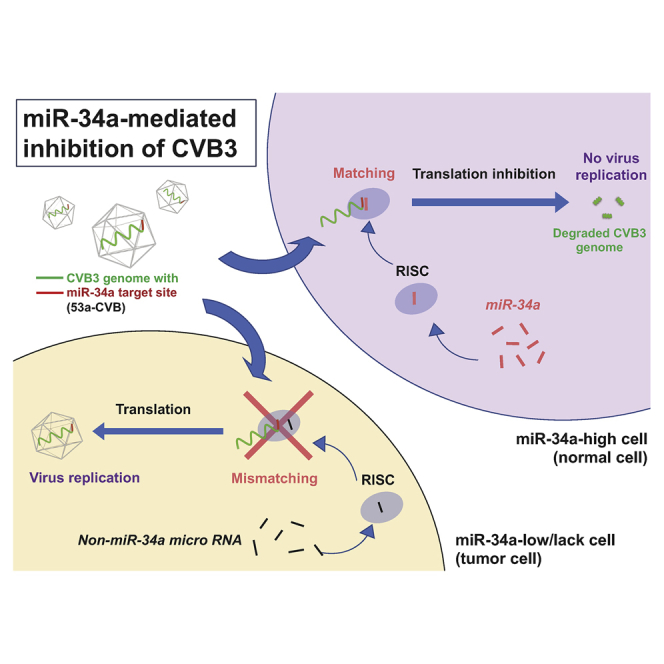
Graphical Abstract
Introduction
Lung cancer is the second most common cancer in both men and women in the United States, and it is by far the leading cause of cancer death over the past three decades.1 Five-year survival of lung cancer patients is still only 18.6% for all stages, despite the use of intensive combined therapies and recent advances in molecular targeting therapies. To improve this poor prognosis, new therapeutic modalities are urgently required.
In the past decade, oncolytic viruses (OVs) were developed as a new class of therapeutic agents for cancer treatment. These agents function by promoting tumor cell lysis via preferential replication of OVs in tumor cells, followed by activation of the host’s antitumor immunity.2, 3, 4 Very recently, talimogene laherparepvec (T-Vec), an oncolytic herpes simplex virus type 1 engineered to contain granulocyte-macrophage colony-stimulating factor (GM-CSF) cDNA, was approved by the Food and Drug Administration of the United States as the first OV immunotherapy for advanced melanoma.5 Using other types of viruses, such as adenoviruses, reoviruses, measles viruses, and Newcastle disease viruses, many other OVs have been developed and are currently undergoing pre-clinical and clinical studies for many types of cancers.6 However, tumor cells often become resistant to OVs because of downregulation of their receptors, leading to insufficient proliferation and transmission of viruses. Therefore, novel OVs targeting different tumor-expressing receptors with strong proliferative ability would be useful in the clinical setting.
Coxsackievirus is a member of genus Enterovirus, family Picornaviridae, and is further divided into two groups: coxsackie A (CVA) and coxsackie B (CVB). It is a non-enveloped virus that is icosahedral in structure, and the capsid encloses a single strand of positive-sense RNA genome, approximately 7.4 kb in length.7, 8 A strain of CVA, CVA21, that uses intracellular adhesion molecule-I and decay-accelerating factor (DAF) as its receptors was developed as an OV.9 Various clinical trials of CVA21 targeting non-small cell lung cancer (NSCLC), melanoma, breast cancers, prostate cancers, or head and neck cancers have been carried out since 2005.9 To obtain a novel potent OV, we previously screened 28 enterovirus strains and found that coxsackievirus B3 (CVB3), which uses coxsackievirus and adenovirus receptor (CAR) and DAF as its receptors, was a highly potent OV capable of targeting a wide range of human tumors, including NSCLC.10, 11 Despite these promising results, safety issues persist: due to its broad tropism, CVB3 can cause myocarditis, pancreatitis, and aseptic meningitis.12, 13, 14 Accordingly, elimination of these virulent features of CVB3 is essential for developing CVB3-based OVs in the clinical setting.
To inhibit virus replication in normal organs, we took advantage of the ability of microRNAs (miRNAs) to regulate gene expression. Several reports have shown that the off-target toxicity of OVs can be reduced by inserting miRNA target sequences into the viral genomes.15, 16, 17, 18 miRNAs mainly cause degradation or translational inhibition of mRNAs by guiding the RNA-induced silencing complex (RISC) to mRNA targets, which contain sequences that are imprecisely complementary to miRNAs.19, 20 In addition, the expression profiles of many miRNAs can be clearly distinguished between normal and cancerous cells.21 miRNA-34 (miR-34) is a family of tumor suppressor miRNAs, transcriptionally activated by TP53, that comprises three members: miR-34a, miR-34b, and miR-34c. miR-34a is expressed in most normal tissues, but not in many cancer cells; miR-34b and miR-34c share a common precursor transcript and are expressed predominantly in normal lung and brain.22, 23, 24
Therefore, we hypothesized that insertion of target sequences for miR-34a (miR-34aT) and miR-34c (miR-34cT) in the CVB3 genome would reduce pathogenesis of CVB3. We demonstrated that CVB3 harboring miR-34 target sequences could be useful for cancer treatment. This is the first report to demonstrate the effectiveness of miR-34-controlled gene regulation in an OV.
Results
Expression of miR-34a and miR-34c in Cancer Cells and Normal Tissues
To determine the expression levels of miR-34a and miR-34c in cancer and normal cells, we first examined an NSCLC H1299 and normal bronchial epithelium BEAS-2B cells for the expression of both miR-34a and miR-34c, using qRT-PCR (Figure 1A). H1299 cells expressed much lower levels of miR-34 than BEAS-2B cells. We also examined miR-34a expression in normal organs of mice and found that it was expressed at higher levels in all organs, particularly in brain, pancreas, heart, and lung, than in H1299 cells. Brain and lung also expressed miR-34c abundantly (Figure 1B).
Figure 1.

Insertion of miR-34 Target Sequences in CVB3 Efficiently Decreases Cytotoxicity in miR-34-Expressing Cells
Expression levels of miR-34a and miR-34c in the human NSCLC cell line H1299 and the normal bronchus epithelium cell line BEAS-2B (A), and in C57BL/6 mouse tissues (B). Values were measured by qRT-PCR and normalized against endogenous 18S rRNA. Data represent means ± SD of triplicate assays. (C) Schematic diagram of WT-CVB genome and insertion site of miR-34aTs (34aT) in 5-CVBs (5a-CVB and 5c-CVB) and miR-34cTs (34cT) in 3-CVBs (3a-CVB and 3c-CVB). (D) Representative images of H1299 cells transfected with miR-34a mimics (miR-34a-H1299) and miR-34c mimics (miR-34c-H1299), followed by inoculation with indicated viruses at an MOI of 10−4 to 1. Cytotoxicity of each virus was determined 72 h post-infection by crystal violet staining. All experiments were repeated at least three times. (E) Expression levels of miR-34a and miR-34c in a human cancer cell lines: NSCLC, A549; cervical cancer, HeLa; and pancreatic cancer, AsPC. Data represent means (values above each bar) ± SD of relative level of H1299 cells from triplicate assays, and dotted lines indicate average level of BEAS-2B. (F) Cell viability of cancer cell lines and BEAS-2B was determined by MTS assay 72 h after inoculation with the indicated viruses. Data represent means ± SD of relative viable cell number versus un-infected cell number from triplicate assays. **p < 0.01; ***p < 0.001 versus WT-CVB (Tukey’s test); †p < 0.05 5a-CVB versus 5c-CVB (Tukey’s test).
Insertion of miRNA Target Sequences in 5′ UTR or 3′ UTR of CVB3 Genome
These results suggested that insertion of miR-34aT or miR-34cT into the CVB3 genome would suppress viral translation and proliferation in normal cells expressing these miRNAs. Based on previous reports about insertion-tolerant sites of picornaviruses,25, 26 we predicted that miRNA target sequences (miRTs) could be inserted in the 5′ region immediately upstream of the start codon, as well as in the 3′ region just downstream of the stop codon. In addition, insertion of four copies of miRT in viral vector genomes can be efficiently controlled by complementary miRNAs.27 Therefore, we engineered miRNA-regulated CVB3s (miRT-CVBs) by inserting four tandem miR-34aTs or miR-34cTs into the 5′ UTR (nt 743) (5-CVBs: 5a-CVB and 5c-CVB) or 3′ UTR (nt 7,305) (3-CVBs: 3a-CVB and 3c-CVB) of the CVB3 genome (Figure 1C). We also constructed control CVB3s (Ctrl-CVB) by inserting an miRNA target gene corresponding to C. elegans miR-39, which does not exist in mammalian cells, in the 3′ UTR as 3-CVB.
Improved Tumor Specificity of CVB3 by Inserting miRTs in UTRs
To examine the effect of miRT insertion in the CVB3 genome on cytotoxicity, we transfected synthetic miR-34a or miR-34c mimics to H1299 cells. After confirming successful transfection of both miRNA mimics at almost the same level, the cells were inoculated with miRT-CVBs (Figure S1A). Seventy-two hours later, in untransfected H1299 cells, all miRT-CVBs induced massive cell lysis, as did wild-type CVB3 (WT-CVB) and Ctrl-CVB (Figure 1D, left panel). By contrast, H1299 cells transfected with miR-34a or miR-34c exhibited much less cell lysis when infected with miRT-CVBs harboring complementary miRTs. 3-CVBs exhibited less cytotoxicity than 5-CVBs, and miRT-CVBs with miR-34aT exhibited less cytotoxicity than miRT-CVBs with miR-34cT (Figure 1D, middle and right panels). These results indicated that insertion of miRTs made CVB3 less toxic only in cells expressing miR-34a or miR-34c.
To further examine the effect of miRT-CVBs on tumor and normal cells, we inoculated WT-CVB or miRT-CVBs into several tumor cell lines, including H1299, A549, HeLa, and AsPC, as well as BEAS-2B. All tumor cells expressed less miR-34c than BEAS-2B cells, but A549 and HeLa cells expressed higher levels of miR-34a than BEAS-2B cells (Figure 1E). As expected, 5c-CVB and 3c-CVB exhibited strong cytotoxicity, comparable with that of WT-CVB in all tumor cells, even at an MOI of 0.001 (Figure 1F; Figure S1B). Moreover, 5a-CVB and 3a-CVB unexpectedly induced strong cytotoxicity in miR-34a-high A549 and HeLa cells, as well as in miR-34a-low H1299 and AsPC cells (Figure 1F; Figures S1B and S1C). Normal bronchus epithelium BEAS-2B cells were much more resistant to WT-CVB than tumor cells, but at a 100-fold higher titer (MOI of 0.1), only 30% of cells survived (Figure 1F; Figures S1B and S1C). Importantly, in contrast with the results obtained with tumor cells, the majority of miRT-CVBs exhibited reduced cytotoxicity in BEAS-2B cells (Figure 1F; Figures S1B and S1C). 5a-CVB resulted in 60% viability at an MOI of 0.1, whereas the cytopathic effect of 5c-CVB was almost the same as that of WT-CVB. In addition, more than 80% of cells survived when inoculated with 3-CVBs. These findings suggest that insertion of miR-34aT or miR-34cT into the 3′ UTR of CVB3 genome is an effective strategy for reducing cytotoxicity in normal cells without losing antitumor activity.
Antitumor Activity of miRT-CVBs in Mouse Tumor Models
To investigate the antitumor activity of miRT-CVBs in vivo, we injected miRT-CVBs into tumors derived from H1299 cells transplanted into BALB/c nude mice. After subcutaneously transplanted tumors reached 0.5 cm in diameter on day 2, we injected miRT-CVBs or WT-CVBs intratumorally (i.t.) at 1 × 106 median tissue culture infectious dose (TCID50) on days 2, 4, 6, 8, and 10. Control mice exhibited continuous tumor growth, whereas all virus-treated groups exhibited complete tumor regression (Figures 2A and 2B) with no death during the observation period, indicating that miRT-CVBs were preserving antitumor activities as effectively as the original WT-CVB (Figures 2C and 2D). Importantly, although transient weight loss was observed in WT-CVB-treated mice and 5-CVBs after the injections started, no weight loss was observed in 3-CVB-treated mice, suggesting that the toxicity of 3-CVBs was reduced in non-tumor tissues (Figures 2E and 2F).
Figure 2.

Effect of miRT-CVBs on Tumors and Hosts in Mouse Tumor Models
BALB/c nude mice received s.c. transplantation of 5 × 106 H1299 cells. Arrows indicate the timing of five doses (5 × 106 TCID50) of i.t. injection of indicated viruses or vehicle control. 5-CVBs (A, C, and E), 3-CVBs (B, D, and F), WT-CVB, and vehicle control were injected intratumorally. (A and B) Tumor volume was measured every 2 days. Data represent means ± SD of each group. (C and D) Overall survival of transplanted mice. Kaplan-Meier survival curve is shown, and the statistical difference between the control group and each virus-treated group was evaluated by log rank test. (E and F) Body weight was measured every 2 days. Data represent means ± SD. Each group consists of four or five mice.
Reduced Normal Organ Injury by miRT-CVB Treatment
To determine whether insertion of miRTs in 5′ UTR or 3′ UTR could alleviate WT-CVB-associated pathogenicity, we performed blood biochemistry tests and pathological examination of our xenograft mouse models. Nude mice received a single i.t. injection of viruses (1 × 106 TCID50) 2 days after inoculation of H1299 cells, and blood samples and mouse organs were collected 2 days after virus injection. Moreover, there were no significant changes in serum blood urea nitrogen (BUN) or total bilirubin (T-bil) levels in mice treated with any types of CVBs (Figures S2A and S2B). In WT-CVB-treated mice, serum aspartate aminotransferase (AST), alanine aminotransferase (ALT), lactate dehydrogenase (LDH), and amylase levels were significantly increased (Figures 3A and 3B), and histological signs of pancreatic injury, such as destruction of acinar cells with inflammation, were observed (Figures 3C and 3D). Although 5-CVBs failed to reduce the WT-CVB-associated pathogenicity (Figures 3A and 3C), 3-CVBs did not significantly increase the level of any enzyme except ALT, and they also induced less pancreatic injury (Figures 3B and 3D). There was no significant difference in serum enzyme levels between 3a-CVB- and 3c-CVB-treated mice. Because elevated LDH reflects damage to many organs, the reduced LDH elevation in 3-CVB-treated mice could have resulted from restriction of virus replication in a wide range of organs. Although CVB3 has been reported to cause some pathological changes in mouse heart tissue,26, 28, 29 we did not observe any significant pathological findings in the heart, such as myocardial necrosis or inflammation, in mice treated with any type of virus, including WT-CVB (Figures S3A and S3B). In addition, we did not observe any pathological findings in the livers of any mice (Figures S4A and S4B). These results indicated that 3-CVBs are safer than 5-CVBs and WT-CVB, and that in particular 3a-CVB was the best miRT-CVB in terms of antitumor effect and safety, although this virus still induced mild ALT elevation.
Figure 3.

Blood Biochemistry Test and Pathological Examination of Mice Xenografted with Human Tumors after Treatment with miRT-CVBs
H1299 cells (5 × 106) were transplanted s.c. into BALB/c nude mice; then vehicle control or indicated viruses were inoculated at 5 × 106 TCID50 2 days after transplantation. 5-CVBs (A and C), 3-CVBs (B and D), WT-CVB, or vehicle (A and B) was inoculated. Blood samples were collected at 2 days post-inoculation, and serum ALT, AST, LDH, and amylase levels were measured. Data represent means ± SD of eight or nine mice in each group. *p < 0.05; **p < 0.01; ***p < 0.001 versus control group (Tukey’s test); ††p < 0.01 5a-CVB versus 5c-CVB group (Tukey’s test). (C and D) Representative pathological images of pancreas. Magnification: 10× + 2× zoom. Scale bars, 100 μM. All experiments were repeated twice.
Double Insertion of miR-34aTs Improves the Safety of CVBs without Diminishing Their Antitumor Effect
To further improve the safety of 3a-CVB, we modified CVB3 to be more downregulated by miR-34a. For this purpose, we inserted four tandem miR-34aTs not only in the 3′ UTR, but also in the 5′ UTR of the CVB3 genome (53a-CVB), to increase the number of miR-34a-binding sites (Figure 4A). We first compared the cytotoxicity of 53a-CVB in H1299 cells with that of 5a-CVB, 3a-CVB3, and WT-CVB. 53a-CVB exerted strong cytotoxicity in H1299 cells, like other miRT-CVBs, but less cytotoxicity than 5a-CVB and 3a-CVB in miR-34a-transfected H1299 cells (Figure 4B). Although viral replication of miRT-CVBs was slower than that of WT-CVB, virus production of miRT-CVBs reached almost the same level as that of WT-CVB at 12 h or later in H1299 cells, and decreased virus production was found in cells with a complementary combination of miRNAs and miRT-CVBs (Figure 4C). 53a-CVB also killed other cell lines very efficiently, although HeLa cells were slightly more resistant to 53a-CVB than to WT-CVB (Figure 4D; Figures S1D and S1E). 53a-CVB also exerted significantly lower cytotoxicity than 5a-CVB, 3a-CVB, and WT-CVB in BEAS-2B cells because of a significant reduction in virus replication (Figure 4D; Figure S1F). Furthermore, we observed an antitumor effect of 53a-CVB using H1299 cell-injected mice, and found that 53a-CVB induced complete tumor regression, similar to 5a-CVB, 3a-CVB, and WT-CVB, resulting in no tumor-associated death (Figures 4E and 4F). In contrast with WT-CVB- and 5a-CVB-treated mice, neither 3a-CVB- nor 53a-CVB-treated mice lost weight after starting the treatment (Figure 4G).
Figure 4.

Antitumor Effect of a Double-miR-34aT-Inserted CVB3, 53a-CVB, In Vitro and In Vivo
(A) Schematic diagram of 53a-CVB, showing insertion sites of miR-34aT constructs in the 5′ UTR and 3′ UTR of the CVB3 genome. (B) Representative images of H1299 cells transfected with miR-34a mimics, followed by inoculation with indicated viruses at an MOI of 10−4 to 1. The cytotoxicity of each virus was determined by crystal violet staining at 72 h post-infection. All experiments were repeated at least three times. (C) Replication kinetics of miRT-CVBs were evaluated using single-step growth curve analysis (MOI = 3) in H1299 cells transfected with miR-34a mimics (miR-34a-H1299) or miR-34c mimics (miR-34c-H1299). Data are represented as mean virus titer ± SD. (D) The cell viability of cancer cell lines and BEAS-2B was determined by MTS assay 72 h after inoculation with the indicated viruses. Data represent means ± SD of relative viable cell number versus un-infected cell number from triplicate assays. *p < 0.05; ***p < 0.001 versus CVB3-WT (Tukey’s test); †p < 0.05 CVB3-53A versus CVB3-3A (Tukey’s test). For in vivo studies, BALB/c nude mice received s.c. transplantation of 5 × 106 H1299 cells. Arrows indicate timing of five doses (5 × 106 TCID50) of i.t. injection of indicated viruses or vehicle control. Tumor volume (E) and body weight (G) were monitored every 2 days. (F) Survival curves of mice treated with indicated viruses. Differences between control group and each virus-treated group were statistically evaluated by log rank test. Data represent means ± SD. Each group consists of five mice. (H) A549 cells (miR-34a-high) were treated with 10 μM LY294022, 10 μM PD0335901, or DMSO for 1 h, followed by inoculation with indicated CVBs. Sixteen hours later, living cell numbers were determined by MTS assay. *p < 0.05; **p < 0.01 versus WT-CVB (Dunnett’s test).
Aberrant Activation of Oncogenic Pathways May Defeat miRT-Mediated Inhibition of CVB3 Replication
As shown in Figure 4D, although miR-34a was expressed at higher levels in A549 and HeLa cells than in normal bronchial BEAS-2B cells, 53a-CVB exerted stronger cytotoxicity in these tumor cells as 5a-CVB or 3a-CVB at much lower titers than in non-tumor cells. Because CVB3 replication is dependent on signaling pathways such as phosphatidylinositol 3-kinase (PI3K)/Akt and mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway, which are aberrantly activated in tumor cells,12, 30 we hypothesized that those activated pathways would override downregulation of CVB3 replication by miR-34a in tumor cells. To demonstrate this hypothesis, we examined the inhibited effect of those pathways on the cytotoxicity of miRT-CVBs in cancer cell lines. While a PI3K inhibitor LY294002 or MEK inhibitor PD0325901 decreased the cytotoxicity of WT-CVB to some extent in A549 cells, these inhibitors further decreased the cytotoxicity of miR-34aT-CVBs, particularly 53a-CVB (Figure 4H; Figure S5), which supports our hypothesis. In contrast, a reduction in cytotoxicity by those inhibitors was independent of the type of CVBs in AsPC cells expressing a very low level of miR-34a. This result further suggests that miR-34a-induced inhibition of miR-34aT-CVB replication could be overridden by aberrantly activated PI3K/Akt and/or MAP/ERK/MEK pathways in tumor cells.
Extremely Low Toxicity of 53a-CVB in Tumor Mouse Models
To confirm the safety of 53a-CVB in vivo, we evaluated organ injury by performing blood biochemistry tests and pathological examination in mice injected with H1299 cells. As in previous experiments (Figure 3A), although no CVBs increased serum BUN or T-bil levels (Figure S2C), WT-CVB and 5a-CVB significantly increased serum AST, ALT, LDH, and amylase, whereas 3a-CVB induced mild elevation of ALT (Figure 5A). By contrast, there was no significant elevation of these enzymes, including ALT, in mice treated with 53a-CVB, suggesting almost complete elimination of CVB-induced organ toxicity (Figure 5A). Moreover, a pathological study revealed that the 53a-CVB-treated mice did not have pancreatitis, in contrast with mice treated with the other miRT-CVBs (Figure 5B). Moreover, there were no pathological changes in the heart or liver in mice treated with 53a-CVB, as in mice treated with other CVBs (Figures S3C and S4C).
Figure 5.

Extremely Low Pathogenesis of 53a-CVB in Mice Transplanted with H1299 Cells
H1299 cells (5 × 106) were transplanted s.c. into BALB/c nude mice; then the indicated viruses or vehicle controls were inoculated with 5 × 106 TCID50 2 days after transplantation. (A) Mouse blood samples were collected 2 days post-infection, followed by immediate analysis of serum ALT, AST, LDH, and amylase. Data represent means ± SD (n = 8/group). ***p < 0.001 versus control mice (Tukey’s test); †p < 0.05, 53a-CVB group versus 3a-CVB3 group (Tukey’s test). (B) Representative pathological findings of pancreas. All experiments were repeated twice. (C) Viral load in mouse organs and serum. Copy numbers of the virus genome in each organ or serum were quantified by qRT-PCR. The limit of detection is indicated by the dashed line (2 × 104 copies of virus RNA per ng). Data represent means ± SD of triplicated experiments. (D) Immunohistochemistry of mouse heart. Expression of CVB3 VP1 in myocardial cells treated with three doses of virus injections (brown signals). (B and D) Magnification, 10× + 2× zoom. Scale bars, 100 μM.
Biodistribution of CVBs in Tumor-Harboring Mice
To determine the proliferation site of CVBs in mice transplanted with human lung cancer, we examined copy numbers of the CVB genome in each mouse organ. In mice treated with WT-CVB, the pancreas contained the highest number of viral copies besides tumors, followed by the spleen, heart, and lungs. Of note is that in 53a-CVB-treated mice, the pancreas showed about 1/100 of the viral copy numbers detected in WT-CVB-treated mice, and the other organs did not show any detectable viruses, even though the tumors of both mice had almost the same viral load (Figure 5C, left). To address why there was a low but detectable level of viral load in all organs except the pancreas of WT-CVB-treated, but not 53a-CVB-treated, mice despite having no obvious signs of tissue damage in these organs, we examined the serum viral load and found a high viral load in WT-CVB-treated mice (Figure 5C, right). These results suggested that the pancreas was a viral reservoir providing viruses to circulating blood, and the high circulating viral load resulted in the detection of viruses in uninfected organs of WT-CVB-treated mice. These data were consistent with the maintained antitumor effect and very low toxicity of 53a-CVB, indicating that 53a-CVB is an excellent OV with the highest efficacy and safety.
Reduced Toxicity of 53a-CVB in a Myocarditis Mouse Model
Because CVB3 is known to induce myocarditis, we further evaluated the cardiac toxicity of 53a-CVB by using a mouse model. In mice receiving three doses of WT-CVB injected in the tumors, there were signs of myocarditis such as mononuclear cell infiltration in heart cells and expression of CVB VP1 in myocardial cells (Figure 5D; Figure S6). In contrast, 53a-CVB-treated mice did not show any evidence of myocarditis, further suggesting that 53a-CVB is safe.
No Obvious Toxicity of 53a-CVB in an Immunocompetent Mouse Bearing Mouse Lung Cancer
To address whether any immunogenic response against CVBs could induce an adverse effect in vivo, we evaluated the toxicity of CVBs in syngeneic tumor transplantation models. C57BL/6J mice bearing subcutaneous tumors of mouse lung cancer TC-1 cells, which are susceptible to CVB3 (Figure 6A), received an i.t. injection of WT-CVB, 5a-CVB, 3a-CVB, or 53a-CVB. After 48 h, blood and organs were collected from those mice, and there were very similar findings to that of nude mouse xenograft models. This result further indicated that 53a-CVB also has extremely low toxicity in immunocompetent mice (Figures 6B and 6C).
Figure 6.

Low Toxicity of 53a-CVB Treatment in an Immunocompetent Mouse with Lung Cancer
(A) Representative images of mouse lung cancer TC-1 cells inoculated with indicated viruses at an MOI of 10−2 to 1. The cytotoxicity of each virus was determined at 72 h post-infection. (B) Blood biochemistry test of an immunocompetent mouse bearing TC-1 cells. C57BL/6J mice were injected with indicated CVBs or vehicle controls 2 days after syngeneic TC-1 cell transplantation. Mouse serum samples were measured for ALT, AST, LDH, and amylase. Data represent means ± SD (n = 4/group). **p < 0.005; ***p < 0.001 versus control group (Tukey’s test). (C) Pathological images of the pancreas of mice injected with vehicle control, WT-CVB, 5a-CVB, 3a-CVB, or 53a-CVB. Original magnification ×20. Scale bars, 100 μM.
Discussion
OVs are considered to be promising tools for cancer treatment, even for patients with disease that is resistant or refractory to chemotherapies or radiotherapies.3 Although many OVs have been developed over the past two decades, the number of successful examples remains limited because of insufficient tumoricidal effects caused by low receptor expression or insufficient viral amplification and transmission, off-target toxicity to normal tissues, and unwelcome induction of neutralizing antibodies. These problems must be resolved in order to develop novel OVs with improved outcomes. In a previous study, we showed that CVB3 exerts a strong antitumor effect in a wide variety of tumors because of the broad expression of its receptors and its rapid replication in tumor cells.10 However, we observed non-negligible toxicities, particularly in pancreases, during the further characterization of WT-CVB for the purpose of clinical translation. Although WT-CVB did not cause death in NSCLC xenograft mice, those toxicities must be eliminated in order for CVB3-based OV to be used in the clinic. To this end, we adopted a gene expression regulation method using miRNA.
To date, the target sequences of let-7 miRNA family members, which are widely expressed in normal tissues, have been used to reduce toxicity of OVs in normal tissues. Insertion of the let-7 targets in OVs, however, often decreases not only normal tissue toxicity but also antitumor activity because of the extensive overlap in target sequences among its various family members.31 Therefore, we tried to find other miRNA-regulating systems with stronger on-target effects and milder off-target effects. miR-34a, which is downregulated in various cancers, was reported to inhibit the tumor growth of many types of cancers including NSCLC in vitro and in vivo.32, 33, 34, 35, 36, 37, 38 Consistent with a previous report,24 we observed generally higher expression of miR-34a and miR-34c in normal tissues than in cancer cells (Figures 1A and 1B). Conveniently, in contrast with let-7, the miR-34 family consists of only three members, suggesting less interference among the family members. These findings encouraged us to develop safer and novel CVB3-based OVs by inserting miR-34 target sequences into the viral genome. As expected, the insertion of miR-34aT and miR-34cT in CVB3s specifically reduced the cytotoxicity in miR-34a- and miR-34c-transfected H1299 cells, respectively (Figure 1D), and the miRT-CVBs showed very low toxicity in normal organs without losing the antitumor effect in mice harboring H1299 xenografts. Interestingly, despite the high level of expression of miR-34a, A549 and HeLa cells were killed by miR-34aT-CVBs much more efficiently than normal bronchial BEAS-2B cells. The miR-34a-independent replication in cancer cells was suggested to be at least partially dependent on the PI3K/Akt and/or MAP/ERK/MEK pathways, which are aberrantly activated in many tumor cells (Figure 4H; Figure S5). Therefore, miR-34aT-CVBs, particularly 53a-CVB, are thought to have antitumor effects in a wide variety of tumors, with low toxicity. Additionally, the slower amplification of miRT-CVBs (Figure 4C) did not affect the antitumor effect in vitro and in vivo (Figures 1F, 2, 4D, and 4E), but could contribute to reducing the circulating virus (Figure S7), resulting in less toxicity in normal organs.
When inserted into CVBs, miR-34aT was more effective than miR-34cT at reducing toxicities in normal tissues, possibly because all organs (including heart and pancreas, which are susceptible to CVB3) express miR-34a, whereas miR-34c is expressed only in lung and brain. Another possible reason for the difference in efficacy is the difference in miRNA target sites. Although miR-34a and miR-34c belong to the same seed family and share the same seed region (5′-GGCAGTG-3′),22 the complementary sequences of the seed region and adjacent base of miR-34a are an 8-mer-type site, whereas that of miR-34c is a 7-mer-m8-type site (Figure S8). Although controversy persists on this issue, the 8-mer-type site is thought to induce miRNA inhibition much more strongly than other types of sites, including 7-mer-m8.39, 40, 41, 42 Therefore, the toxicities of 5a-CVB and 3a-CVB should be lower in miR-34a-expressing cells than those of 5c-CVB and 3c-CVB in miR-34c-expressing cells (Figure 1D).
On the other hand, the insertion site of miRT is another important factor to consider when seeking to suppress viral replication while maintaining oncolytic activity. Previously, the 5′ UTR26, 43, 44 and 3′ UTR15, 17 of CVB3 tolerate sequence insertion, although the core sequence of the internal ribosome entry site (IRES) was located at nt 432–639 of CVB3 genome (5′ UTR),45 and in the 3′ UTR, sites at nt 7,387 and 7,359–7,360 do not tolerate sequence insertion.26 Thus, we avoided those sensitive sites for the purpose of miR-34aT and miR-34cT insertion. As expected, we could successfully insert miRTs into the 5′ UTR, immediately upstream of the start codon, and into the 3′ UTR at nt 7,305, without losing the antitumor effect of the original CVB3. We also found that 3-CVBs exhibited significantly lower pathogenicity than 5-CVBs (Figure 3). Although we have not yet determined the reason, it seems that the effects of miRNAs are dependent on the locations of their target sequences. Several experimental40, 46, 47, 48 and computational49, 50 studies reported that an miRNA can inhibit an mRNA more efficiently when the target sequence is in the 3′ UTR than in the 5′ UTR or open reading frame. Although the mechanism underlying the reduced efficacy of miRNA in the 5′ UTR remains unknown, it may involve detachment of RISC from the mRNA: RISC could easily be detached from miRTs when ribosomes bind to the 5′ UTR at the beginning of translation, resulting in failure of miRNA regulation.20
Although miRNA-mediated regulation is sometimes saturable and cannot always control target expression,51 8× miRT was reported to induce miRNA regulation more strongly than 4× in the single location.52 Ruiz et al.17 reported that when miRNA-targeted viruses were serially passaged in vitro in the presence of the cognate miRNAs, large deletions within the insertion were observed. Multiple insertions of miRTs seemed to be a practical and easy strategy for improving the safety of 3a-CVB and avoiding the emergence of total deletion of an miRT insertion. Therefore, we engineered 53a-CVB by inserting 4× miR-34aT sequences in both the 5′ and 3′ UTRs. Even if deletion of miR-34aT occurred in one of the two miR-34aT sites in the 5′ and 3′ UTRs, miR-34a was still accessible to the other miR-34aT site in the CVB genome, and virus replication would still be controllable by miR-34a. As expected, 53a-CVB exhibited an additive reduction in toxicity relative to 5a-CVB and 3a-CVB, with little toxicity in WT-CVB-sensitive organs such as the liver and pancreas in mouse tumor models (Figure 5). Moreover, 53a-CVB did not lose any of the antitumor activity of the original WT-CVB in vitro or in vivo (Figures 4B–4E), and could thus induce complete regression of tumors in all mice without obvious signs of adverse effects.
In conclusion, we successfully developed a potent and safe OV, 53a-CVB, by inserting miR-34aTs in both the 5′ UTR and 3′ UTR of WT-CVB. The effects on antitumor immunity, an important mechanism of tumor inhibition by WT-CVB, and on other types of tumors remain to be addressed in future studies. Nonetheless, our findings indicate that 53a-CVB is a promising OV that could be useful in the clinical setting because of its minimal toxicity and strong oncolytic effects.
Materials and Methods
Mice
Four- to five-week-old female BALB/c nude mice and C57BL/6J mice were purchased from Oriental Yeast (Tokyo, Japan). We have received the approval for the animal experiments from the University of Tokyo and Japanese government. All animal experiments were carried out under the Guidelines for Animal Experiments of The University of Tokyo and Law 105 Notification 6 of the Japanese Government.
Cell Lines
Human NSCLC (NCI-H1299), pancreatic cancer (AsPC-1), cervical adenocarcinoma (HeLa), and human normal lung bronchial epithelium (BEAS-2B) cell lines were purchased from the American Type Culture Collection (Manassas, VA, USA). A human lung carcinoma (A549) cell line was purchased from Riken Cell Bank (Wako, Japan).
Construction of miR-34a/cT-Containing CVB Plasmids
As a plasmid coding for full-length CVB3 (Nancy strain) infectious clone cDNA, pBluescript II KS-CVB3 was used as the starting material for genetic manipulations. To insert miR-34a/cT in CVB cDNA, pBluescript II KS-CVB3 was first linearized by PCR (94°C for 2 min; 30 cycles of 98°C for 10 s, 60°C for 30 s, and 68°C for 6 min; and finally 72°C for 7 min), then treated with DpnI (TOYOBO, Osaka, Japan) for 1 h at 37°C to digest any residual template plasmid. To prepare the insert fragments of miR-34a/cT or miR-39T (control), we used synthetic DNA oligomers (Table 1) as templates for PCR. Following the PCRs, the vector was purified by gel extraction (approximately 10 kb), and the linearized vectors and inserts were ligated using the In-Fusion HD Cloning Kit (Takara Bio, Kusatsu, Japan). The closed circular plasmid clones were obtained by transformation with DH5alpha (TOYOBO). The insertion of miR-34a/cT was confirmed by sequence analysis.
Table 1.
Sequences of Inserted miRNA Targets for Novel Recombinant CVB3 Viruses
| miRT | Inserted Sequence (5′–3′) |
|---|---|
| miR-34aT × 4 | ACAACCAGCTAAGACACTGCCAcgat ACAACCAGCTAAGACACTGCCAaccggt ACAACCAGCTAAGACACTGCCAtcac ACAACCAGCTAAGACACTGCCA |
| miR-34cT × 4 | GCAATCAGCTAACTACACTGCCTcgat GCAATCAGCTAACTACACTGCCTaccggt GCAATCAGCTAACTACACTGCCTtcac GCAATCAGCTAACTACACTGCCT |
| miR-39T × 4 (control) | CAAGCTGATTTACACCCGGTGAcgat CAAGCTGATTTACACCCGGTGAaccggt CAAGCTGATTTACACCCGGTGAtcac CAAGCTGATTTACACCCGGTGA |
Uppercase letters represent the miRNA target sequences, and lowercase letters represent spacer elements.
Production of Recombinant Viruses
Plasmids containing the full-length miRT-CVB cDNA were linearized with SalI-HF (NEB, Ipswich, MA, USA), and the reaction was terminated by adding 1/10th volume of ammonium acetate solution (5 M) and two volumes of ethanol. Transcription reaction was performed using the MEGAscript T7 Transcription kit (Thermo Fisher Scientific, Waltham, MA, USA) and purified by phenol-chloroform extraction and ethanol precipitation. H1299 cells were transfected with 50 μg RNA of each virus using Lipofectamine 3000 (Thermo Fisher Scientific). At 24–30 h after transfection, the culture medium was collected and used to inoculate H1299 cells. After 6–10 h, the cells were collected with a cell scraper followed by three freeze-thaw cycles; then the cell lysate was centrifuged to separate cell debris. The supernatants were collected as virus stocks, and the aliquots were stored at −80°C.
Virus Titration and Single-Step Growth Curve Analysis
H1299 cells (5 × 103 cells/well) were seeded into 96-well plates and incubated at 37°C for 8 h. Ten-fold serial dilutions of each virus stock were prepared, and 50 μL of each dilution was added to each of eight replicate wells. After 5 days of culture, cell lysis in each well was evaluated, and the titer was calculated by TCID50 as previously described.53 To determine the replication of CVBs in H1299 with or without miR-34a or miR-34c transfection, or BEAS-2B cells, the cells were subjected to single-step growth curve analysis. In brief, the cells were inoculated with viruses at an MOI of 3 for 1 h, after which the medium was replaced. At 6, 12, 24, and 48 h post-infection, cells and supernatants were harvested and stored at −80°C. Then all samples were subjected to three freeze-thaw cycles, and cell debris was removed by centrifugation. The supernatant was used for titration with H1299 cells, as described above.
qRT-PCR of miRNA
Total miRNA was extracted from cells or tissues using the mirVana miRNA Isolation Kit (Thermo Fisher Scientific). For miRNA qPCR, cDNA was synthesized from total miRNA using the TaqMan MicroRNA Reverse Transcription Kit (Thermo Fisher Scientific). Subsequently, real-time PCR was carried out using TaqMan Universal PCR Master Mix (Thermo Fisher Scientific) to detect levels of mature miR-34a/c. U6 small nuclear RNA (snRNA) was used as the endogenous control. Fold changes were calculated by relative quantification (2−ΔΔCt method).
Crystal Violet Staining
Cells were infected with viruses at appropriate MOI for 1 h; then the viral supernatant was replaced with fresh media. After 72 h, the cells were stained with crystal violet as previously described.54
miRNA Mimics Transfection Experiment
miRNA oligonucleotide mimics were purchased from Bioneer Corporation (Bioneer, Daejeon, Korea). miRNA mimics were transfected into H1299 cells using Lipofectamine RNAiMAX (Thermo Fisher Scientific) at a concentration of 10 μM. Twenty-four hours later, the copy numbers of miR-34a/c were determined by qRT-PCR, as described above. The transfected cells were inoculated with viruses at an MOI of 10−4 to 1, and after 72 h, the cells were subjected to crystal violet staining, as described above.
MTS Assay for Cell Viability
In vitro cell viability was measured using cells (1 × 106) infected with miRT-CVBs by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) assay using CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA). To determine the effect of cellular signaling pathway inhibitors on the cytotoxicity of miRT-CVBs, we pretreated cells with 2% fetal bovine serum (FBS) media containing 10 μM MEK1/MEK2 inhibitor PD0325901 (Wako Pure Chemical, Osaka, Japan) or 10 μM PI3K inhibitor LY294002 (Santa Cruz Biotechnology, Dallas, TX, USA) for 1 h and then inoculated with miRT-CVBs at an MOI of 10. Sixteen hours after infection, an MTS assay was performed, as described above.
Quantification of CVB3 Copy Numbers in Mouse Organs
To quantify the miRT-CVB genome in mouse organs, we collected tissues for RNA extraction by using an RNeasy Plus Mini Kit (QIAGEN, Germany). To obtain cDNA, we performed reverse transcription for 20 ng of the total RNA with ReverTra Ace qPCR Master Mix (TOYOBO). Subsequently, 2/100 volume of cDNA was subjected to real-time PCR by using PrimeTime Gene Expression Master Mix (Integrated DNA Technologies, USA) and CVB genome-specific primers (forward: 5′-GTGCAAGGCCCTGCCTTT-3′; reverse: 5′-AACGGCCCACCTGTCATAGA-3′) according to the manufacturer’s protocol. pBluescript II KS-CVB3 was used to create the standard curve for the calculation of the viral copy numbers. Because the molecular weight of the plasmid is 6.8 × 106 (10,318 bp × 660) Da, 1 ng of the plasmid contains 8.85 × 107 (1 × 10−9 × 6.023 × 1023/6.8 × 106) copies of the virus genome.
In Vivo Therapeutic Studies
To establish xenograft mouse models, 5 × 106 H1299 cells were injected subcutaneously (s.c.) into the right flank of BALB/c nude mice. To establish immunocompetent mouse tumor models, 1 × 105 TC-1 cells were injected s.c. into C57BL/6J mice. After 2 days, when the tumor size reached 0.5 cm in diameter, 5 × 106 TCID50 WT-CVB, miRT-CVBs, or vehicle (Opti-MEM; Thermo Fisher Scientific) was injected into the tumors. For multiple injection studies, miRT-CVBs or vehicle was injected i.t. on days 2, 4, 6, 8, and 10 for the total of five times. Tumor size and body weight were measured every other day for 60 days after tumor transplantation, and tumor volume was calculated as (length × width2)/2. Mice were euthanized when the diameter of tumors exceeded 1.0 cm or signs of skin ulceration were evident. To address safety issues, the mice were euthanized 2 days after virus injection, and whole blood, heart, liver, and pancreas were collected for biochemical and histopathological analysis. In the myocarditis model, the virus was injected three times 2 days before euthanasia; then the heart was collected for immunohistochemistry (IHC).
Biochemical Analyses
Serum levels of BUN, AST, ALT, T-bil, Amy, and LDH were measured using an auto-analyzer (SPOTCHEM EZ-SP-4430; Arkray, Kyoto, Japan).
Histopathological Examination
Mouse organs were fixed in formalin for 24 h, washed five times with PBS, and then dehydrated in 75% alcohol. The organs were embedded in paraffin and processed for sectioning and H&E staining by the Pathology Core Laboratory of the Institute of Medical Science, The University of Tokyo (IMSUT). For IHC, heart sections were incubated with mouse monoclonal anti-enterovirus PV1 antibody (1:400, clone 5-D8/1; Dako, Jena, Germany) for 1 h at room temperature. Then the sections were incubated with goat anti-mouse immunoglobulin by using the EnVision+ System, HRP (horseradish peroxidase; Dako) followed by diaminobenzidine reaction (brown signals in Figure 5D) (Cell Signaling Technology, USA). The prepared sections were observed and diagnosed by a pathologist at IMSUT Hospital using a BZ-9000 fluorescence microscope (KEYENCE, Osaka, Japan).
Statistical Analysis
All statistical analyses and graphical representations were performed using GraphPad Prism software, version 5.0a (GraphPad Prism, USA). Survival curves were plotted according to the Kaplan-Meier method, and statistical differences were evaluated by log rank test. MTS and biochemical analyses were compared by one-way ANOVA (Tukey’s multiple comparison test and Dunnett’s multiple comparison test). All differences were considered statistically significant at p < 0.05.
Author Contributions
Y.J. conducted the experiments, prepared figures, and participated in study design and writing the manuscript. S.M. designed the study and provided expertise and advice. Y.S. and K.T. provided expertise and advice, and contributed to writing and revision of the manuscript. Y.T., M.S., J.L., L.H., Y.H., Y.M., K.H., and A.I. supported the experiments. Y.O. contributed to histopathological examination.
Conflicts of Interest
Project Division of ALA Advanced Medical Research is receiving research funding from Shinnihonseiyaku Co., NeoPharma Japan Co., and Takara Bio, Inc.
Acknowledgments
This work was supported by research funding from Shinnihonseiyaku Co., Ltd. and NeoPharma Japan Co., Ltd. We thank Tomoko Ando, Tomohiro Kumaki, and Ai Sugawara (IMSUT) for technical assistance, and Michiko Ushijima (IMSUT) for administrative assistance.
Footnotes
Supplemental Information includes eight figures and can be found with this article online at https://doi.org/10.1016/j.omto.2019.01.003.
Supplemental Information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2018. CA Cancer J. Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Ries S.J., Brandts C.H. Oncolytic viruses for the treatment of cancer: current strategies and clinical trials. Drug Discov. Today. 2004;9:759–768. doi: 10.1016/S1359-6446(04)03221-0. [DOI] [PubMed] [Google Scholar]
- 3.Kaufman H.L., Kohlhapp F.J., Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015;14:642–662. doi: 10.1038/nrd4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Russell S.J., Peng K.W. Oncolytic virotherapy: a contest between apples and oranges. Mol. Ther. 2017;25:1107–1116. doi: 10.1016/j.ymthe.2017.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andtbacka R.H.I., Kaufman H.L., Collichio F., Amatruda T., Senzer N., Chesney J., Delman K.A., Spitler L.E., Puzanov I., Agarwala S.S. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 6.Twumasi-Boateng K., Pettigrew J.L., Kwok Y.Y.E., Bell J.C., Nelson B.H. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer. 2018;18:419–432. doi: 10.1038/s41568-018-0009-4. [DOI] [PubMed] [Google Scholar]
- 7.Tuthill T.J., Groppelli E., Hogle J.M., Rowlands D.J. Picornaviruses. Curr. Top. Microbiol. Immunol. 2010;343:43–89. doi: 10.1007/82_2010_37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Linden L., Wolthers K.C., van Kuppeveld F.J.M. Replication and inhibitors of enteroviruses and parechoviruses. Viruses. 2015;7:4529–4562. doi: 10.3390/v7082832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fountzilas C., Patel S., Mahalingam D. Review: oncolytic virotherapy, updates and future directions. Oncotarget. 2017;8:102617–102639. doi: 10.18632/oncotarget.18309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyamoto S., Inoue H., Nakamura T., Yamada M., Sakamoto C., Urata Y., Okazaki T., Marumoto T., Takahashi A., Takayama K. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res. 2012;72:2609–2621. doi: 10.1158/0008-5472.CAN-11-3185. [DOI] [PubMed] [Google Scholar]
- 11.Shafren D.R., Williams D.T., Barry R.D. A decay-accelerating factor-binding strain of coxsackievirus B3 requires the coxsackievirus-adenovirus receptor protein to mediate lytic infection of rhabdomyosarcoma cells. J. Virol. 1997;71:9844–9848. doi: 10.1128/jvi.71.12.9844-9848.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garmaroudi F.S., Marchant D., Hendry R., Luo H., Yang D., Ye X., Shi J., McManus B.M. Coxsackievirus B3 replication and pathogenesis. Future Microbiol. 2015;10:629–653. doi: 10.2217/fmb.15.5. [DOI] [PubMed] [Google Scholar]
- 13.Rhoades R.E., Tabor-Godwin J.M., Tsueng G., Feuer R. Enterovirus infections of the central nervous system. Virology. 2011;411:288–305. doi: 10.1016/j.virol.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spindler K.R., Hsu T.H. Viral disruption of the blood-brain barrier. Trends Microbiol. 2012;20:282–290. doi: 10.1016/j.tim.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelly E.J., Hadac E.M., Greiner S., Russell S.J. Engineering microRNA responsiveness to decrease virus pathogenicity. Nat. Med. 2008;14:1278–1283. doi: 10.1038/nm.1776. [DOI] [PubMed] [Google Scholar]
- 16.Leber M.F., Bossow S., Leonard V.H.J., Zaoui K., Grossardt C., Frenzke M., Miest T., Sawall S., Cattaneo R., von Kalle C., Ungerechts G. MicroRNA-sensitive oncolytic measles viruses for cancer-specific vector tropism. Mol. Ther. 2011;19:1097–1106. doi: 10.1038/mt.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruiz A.J., Hadac E.M., Nace R.A., Russell S.J. MicroRNA-detargeted mengovirus for oncolytic virotherapy. J. Virol. 2016;90:4078–4092. doi: 10.1128/JVI.02810-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shayestehpour M., Moghim S., Salimi V., Jalilvand S., Yavarian J., Romani B., Mokhtari-Azad T. Targeting human breast cancer cells by an oncolytic adenovirus using microRNA-targeting strategy. Virus Res. 2017;240:207–214. doi: 10.1016/j.virusres.2017.08.016. [DOI] [PubMed] [Google Scholar]
- 19.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 20.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 21.Iorio M.V., Croce C.M. microRNA involvement in human cancer. Carcinogenesis. 2012;33:1126–1133. doi: 10.1093/carcin/bgs140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rokavec M., Li H., Jiang L., Hermeking H. The p53/miR-34 axis in development and disease. J. Mol. Cell Biol. 2014;6:214–230. doi: 10.1093/jmcb/mju003. [DOI] [PubMed] [Google Scholar]
- 23.Zhang D.G., Zheng J.N., Pei D.S. P53/microRNA-34-induced metabolic regulation: new opportunities in anticancer therapy. Mol. Cancer. 2014;13:115. doi: 10.1186/1476-4598-13-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010;17:193–199. doi: 10.1038/cdd.2009.56. [DOI] [PubMed] [Google Scholar]
- 25.Rohll J.B., Moon D.H., Evans D.J., Almond J.W. The 3′ untranslated region of picornavirus RNA: features required for efficient genome replication. J. Virol. 1995;69:7835–7844. doi: 10.1128/jvi.69.12.7835-7844.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He F., Yao H., Wang J., Xiao Z., Xin L., Liu Z., Ma X., Sun J., Jin Q., Liu Z. Coxsackievirus B3 engineered to contain microRNA targets for muscle-specific microRNAs displays attenuated cardiotropic virulence in mice. J. Virol. 2015;89:908–916. doi: 10.1128/JVI.02933-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown B.D., Gentner B., Cantore A., Colleoni S., Amendola M., Zingale A., Baccarini A., Lazzari G., Galli C., Naldini L. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat. Biotechnol. 2007;25:1457–1467. doi: 10.1038/nbt1372. [DOI] [PubMed] [Google Scholar]
- 28.Fuse K., Chan G., Liu Y., Gudgeon P., Husain M., Chen M., Yeh W.C., Akira S., Liu P.P. Myeloid differentiation factor-88 plays a crucial role in the pathogenesis of Coxsackievirus B3-induced myocarditis and influences type I interferon production. Circulation. 2005;112:2276–2285. doi: 10.1161/CIRCULATIONAHA.105.536433. [DOI] [PubMed] [Google Scholar]
- 29.Pinkert S., Klingel K., Lindig V., Dörner A., Zeichhardt H., Spiller O.B., Fechner H. Virus-host coevolution in a persistently coxsackievirus B3-infected cardiomyocyte cell line. J. Virol. 2011;85:13409–13419. doi: 10.1128/JVI.00621-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheung P.K.M., Yuan J., Zhang H.M., Chau D., Yanagawa B., Suarez A., McManus B., Yang D. Specific interactions of mouse organ proteins with the 5'untranslated region of coxsackievirus B3: potential determinants of viral tissue tropism. J. Med. Virol. 2005;77:414–424. doi: 10.1002/jmv.20470. [DOI] [PubMed] [Google Scholar]
- 31.Ruiz A.J., Russell S.J. MicroRNAs and oncolytic viruses. Curr. Opin. Virol. 2015;13:40–48. doi: 10.1016/j.coviro.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 32.Ma Z.L., Hou P.P., Li Y.L., Wang D.T., Yuan T.W., Wei J.L., Zhao B.T., Lou J.T., Zhao X.T., Jin Y., Jin Y.X. MicroRNA-34a inhibits the proliferation and promotes the apoptosis of non-small cell lung cancer H1299 cell line by targeting TGFβR2. Tumour Biol. 2015;36:2481–2490. doi: 10.1007/s13277-014-2861-5. [DOI] [PubMed] [Google Scholar]
- 33.Sun H.B., Tian J., Xian W.H., Xie T.T., Yang X.D. miR-34a inhibits proliferation and invasion of bladder cancer cells by targeting orphan nuclear receptor HNF4G. Dis. Markers. 2015;2015:879254. doi: 10.1155/2015/879254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim Y., Park E.Y., Chang E., Kang H.G., Koo Y., Lee E.J., Ko J.Y., Kong H.K., Chun K.H., Park J.H. A novel miR-34a target, protein kinase D1, stimulates cancer stemness and drug resistance through GSK3/β-catenin signaling in breast cancer. Oncotarget. 2016;7:14791–14802. doi: 10.18632/oncotarget.7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang J., Li Y., Daniels G., Sfanos K., De Marzo A., Wei J., Li X., Chen W., Wang J., Zhong X. LEF1 targeting EMT in prostate cancer invasion is regulated by miR-34a. Mol. Cancer Res. 2015;13:681–688. doi: 10.1158/1541-7786.MCR-14-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gallardo E., Navarro A., Viñolas N., Marrades R.M., Diaz T., Gel B., Quera A., Bandres E., Garcia-Foncillas J., Ramirez J., Monzo M. miR-34a as a prognostic marker of relapse in surgically resected non-small-cell lung cancer. Carcinogenesis. 2009;30:1903–1909. doi: 10.1093/carcin/bgp219. [DOI] [PubMed] [Google Scholar]
- 37.Wiggins J.F., Ruffino L., Kelnar K., Omotola M., Patrawala L., Brown D., Bader A.G. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. 2010;70:5923–5930. doi: 10.1158/0008-5472.CAN-10-0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song C., Lu P., Sun G., Yang L., Wang Z., Wang Z. miR-34a sensitizes lung cancer cells to cisplatin via p53/miR-34a/MYCN axis. Biochem. Biophys. Res. Commun. 2017;482:22–27. doi: 10.1016/j.bbrc.2016.11.037. [DOI] [PubMed] [Google Scholar]
- 39.Bartel D.P. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lewis B.P., Burge C.B., Bartel D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 41.Agarwal V., Bell G.W., Nam J.W., Bartel D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife. 2015;4:e05005. doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu W., San Lucas A., Wang Z., Liu Y. Identifying microRNA targets in different gene regions. BMC Bioinformatics. 2014;15(Suppl 7):S4. doi: 10.1186/1471-2105-15-S7-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barnes D., Kunitomi M., Vignuzzi M., Saksela K., Andino R. Harnessing endogenous miRNAs to control virus tissue tropism as a strategy for developing attenuated virus vaccines. Cell Host Microbe. 2008;4:239–248. doi: 10.1016/j.chom.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jopling C.L., Yi M., Lancaster A.M., Lemon S.M., Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 45.Liu Z., Carthy C.M., Cheung P., Bohunek L., Wilson J.E., McManus B.M., Yang D. Structural and functional analysis of the 5′ untranslated region of coxsackievirus B3 RNA: in vivo translational and infectivity studies of full-length mutants. Virology. 1999;265:206–217. doi: 10.1006/viro.1999.0048. [DOI] [PubMed] [Google Scholar]
- 46.Grimson A., Farh K.K.H., Johnston W.K., Garrett-Engele P., Lim L.P., Bartel D.P. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Farh K.K.H., Grimson A., Jan C., Lewis B.P., Johnston W.K., Lim L.P., Burge C.B., Bartel D.P. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science. 2005;310:1817–1821. doi: 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- 48.Lim L.P., Lau N.C., Garrett-Engele P., Grimson A., Schelter J.M., Castle J., Bartel D.P., Linsley P.S., Johnson J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 49.Hamzeiy H., Allmer J., Yousef M. Computational methods for microRNA target prediction. Methods Mol. Biol. 2014;1107:207–221. doi: 10.1007/978-1-62703-748-8_12. [DOI] [PubMed] [Google Scholar]
- 50.Martin H.C., Wani S., Steptoe A.L., Krishnan K., Nones K., Nourbakhsh E., Vlassov A., Grimmond S.M., Cloonan N. Imperfect centered miRNA binding sites are common and can mediate repression of target mRNAs. Genome Biol. 2014;15:R51. doi: 10.1186/gb-2014-15-3-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Doench J.G., Petersen C.P., Sharp P.A. siRNAs can function as miRNAs. Genes Dev. 2003;17:438–442. doi: 10.1101/gad.1064703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bofill-De Ros X., Gironella M., Fillat C. miR-148a- and miR-216a-regulated oncolytic adenoviruses targeting pancreatic tumors attenuate tissue damage without perturbation of miRNA activity. Mol. Ther. 2014;22:1665–1677. doi: 10.1038/mt.2014.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Darling A.J., Boose J.A., Spaltro J. Virus assay methods: accuracy and validation. Biologicals. 1998;26:105–110. doi: 10.1006/biol.1998.0134. [DOI] [PubMed] [Google Scholar]
- 54.Meng X., Nakamura T., Okazaki T., Inoue H., Takahashi A., Miyamoto S., Sakaguchi G., Eto M., Naito S., Takeda M. Enhanced antitumor effects of an engineered measles virus Edmonston strain expressing the wild-type N, P, L genes on human renal cell carcinoma. Mol. Ther. 2010;18:544–551. doi: 10.1038/mt.2009.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.