

Abstract
Spinal muscular atrophy with respiratory distress type 1 (SMARD1) is a rare autosomal recessive disorder caused by a defect in the immunoglobulin mu binding protein 2 (IGHMBP2) gene, leading to motor neuron degeneration. We identified an infant with SMARD1 by targeted exome sequencing from a consanguineous Syrian family having a history of recurrent infant deaths. The patient initially presented intrauterine growth retardation, poor sucking, failure to thrive, and respiratory failure at the age of two months, and an inborn error of metabolism was suspected at first. Over a period of one month, the infant showed rapid progression of distal muscular weakness with hand and foot contractures, which were suggestive of neuromuscular disease. Using targeted exome sequencing, the mutation in IGHMBP2 was confirmed, although the first report was normal. Targeted exome sequencing enabled identification of the genetic cause of recurrent mysterious deaths in the consanguineous family. Additionally, it is suggested that a detailed phenotypic description and communication between bioinformaticians and clinicians is important to reduce false negative results in exome sequencing.
Keywords: Exome Sequencing, Infant Death, Spinal Muscular Atrophy with Respiratory Distress Type 1
Graphical Abstract
INTRODUCTION
Sudden infant death syndrome is caused by multifactorial risks such as developmental, environmental, and biological problems.1 Specific genetic alterations may trigger unexpected deaths in infancy, including inborn errors of metabolism such as fatty acid oxidation disorders, inherited arrhythmia, mitochondrial disorders, or neuromuscular disease.1 Especially in a case of multiple inexplicable deaths in a single consanguineous family, it is important to investigate mutations causing lethal autosomal recessive disorders. Spinal muscular atrophy with respiratory distress type 1 (SMARD1, OMIM #604320), also known as distal spinal muscular atrophy-1 or distal hereditary motor neuronopathy type VI, is a rare autosomal recessive neuromuscular disorder resulting from an immunoglobulin mu binding protein 2 (IGHMBP2) mutation, which could lead to sudden unexpected infant death.2 Here, we report a case of SMARD1 in a consanguineous family with a history of recurrent infant deaths, which was identified by targeted exome sequencing.
CASE DESCRIPTION
A 2-month-old girl was referred to our hospital due to poor oral intake and respiratory failure in April 2016. She was born at 40 weeks of gestation with a 2-kg birth weight by planned repeat caesarean section. She reportedly had intrauterine growth retardation despite the absence of records of height and head circumference at birth. Until the first month of age, the patient gained weight properly with exclusive breastfeeding; gradually, her sucking weakened and weight gain stopped.
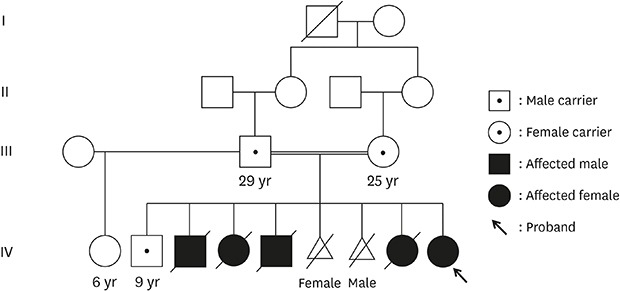
The patient had healthy consanguineous parents from Syria. Her four siblings suddenly died of unknown causes within 10 weeks of life, and two siblings were artificially aborted due to suspected fetal hydrops (Fig. 1). All deaths were preceded by a few days of feeding difficulties and sudden onset of cyanosis. The family had multiple consanguineous marriages, but there were no affected children other than the parents' children.
Fig. 1. Pedigree of the patient with spinal muscular atrophy with respiratory distress type 1. (A) Partial Sanger sequencing of IGHMBP2 (NM_002280.2) homozygous for c.1273C>T (p.Arg425Cys) in exon 9 in the patient (IV-9) and each mutation inherited from the patient's parent (III-2 and III-3). (B) Sequencing of her surviving brother (IV-2) as a heterozygous carrier of the mutation. A written consent was obtained for publication of patient and family information.

IGHMBP2 = immunoglobulin mu binding protein 2.
At presentation, the patient's height, weight, and head circumference was 49 cm, 3.2 kg, and 39 cm, respectively, and all were under the 3rd percentile. Dysmorphic features were not observed. On neurological examination, she was found to be hypotonic; she showed grade-V muscle power in all extremities with positive palmar and plantar grasp reflexes, and a normal deep tendon reflex. Her sucking power was weak despite the normal coordination of swallowing; hence, she needed to feed through the nasogastric tube. She also required mechanical ventilation owing to shallow respiration with decreased breathing sounds by pulmonary auscultation, and respiratory acidosis. Laboratory findings showed mildly elevated levels of liver enzymes, including aspartate aminotransferase of 137 IU/L (reference range [RR], 22–63), alanine aminotransferase of 110 IU/L (RR, 12–45), and gamma-glutamyl transferase of 101 IU/L (RR, 8–90); other values were unremarkable. Chest radiographs, abdominal ultrasound, and brain magnetic resonance imaging demonstrated no abnormalities.
Initially, considering her family history suggesting an autosomal recessive disorder and mildly elevated levels of liver enzymes with feeding intolerance, an inborn error of metabolism was suspected. The results of investigations for inborn errors of metabolism, including tandem mass spectrometry, serum amino acid, and urine organic acid analysis, demonstrated no significant abnormalities. Enzyme assays for lysosomal storage disorders, such as Gaucher disease, Niemann-Pick disease types A and B, and lipoprotein lipase deficiency, were performed and the results were normal. The targeted exome sequencing using the Illumina MiSeq platform and a TruSight One Panel (Illumina Inc., San Diego, CA, USA) including 125,395 probes targeting a 12-Mb region spanning 4,813 genes was performed in this subject. The mean coverage of the target region was greater than 80×, and more than 97% of the target regions were covered with more than 10 reads. However, the initial result was reported as negative when the annotated variants were screened by gene lists for the inborn error metabolism.
Several attempts to wean the patient from the mechanical ventilator failed because of hypoventilation and respiratory acidosis. The patient's effort for respiration, including weak chest wall movement, was inspected; however, she seemed unable to produce adequate negative pressure or inspiratory flow that could be detectable by a mechanical ventilator as the patient's spontaneous breathing. Therefore, she received tracheostomy at 3 months of age; at that time, the patient was re-evaluated. She showed an increase in weight (3.8 kg) and height (58 cm) owing to formula feeding through the nasogastric tube. She was found to be hypertonic and needed muscle relaxants because of the frequent rigidity of both extremities. Her shoulder and hip movements were remarkably weak, and no movements were noticed in any of her distal extremities. Both wrist and ankle joint contractures, and fatty pads on palms and soles were definite (Fig. 2). All tendon reflexes were absent. Unexplained tachycardia and excessive sweating were also detected. Laboratory findings, including liver enzyme levels, showed no abnormalities. X-ray of the lower limbs showed hyperextension of both knees and atrophy of the calf muscle with interstitial fatty hyperplasia (Fig. 2). On electrophysiological examination, motor and sensory potentials were found to be absent in all extremities and fingers, and needle electromyography in the distal muscle showed abnormal spontaneous activities, which corresponded to severe sensorimotor polyneuropathy.
Fig. 2. Clinical features and radiographs of the lower limbs of the patient. (A) Clinical photographs showing distal lower limb atrophy, and hand and ankle contracture at 3 months of age. (B) Both lateral knee radiographs showing atrophy of the calf muscle and interstitial fatty hyperplasia.

Regarding her clinical course, data from exome sequencing were reviewed again as the initial diagnostic algorithm was focused on genes for inborn error of metabolism. Homozygotic mutations of c.1273C>T (p.Arg425Cys) in IGHMBP2 (NM_002280.2) were identified and confirmed by Sanger sequencing (Fig. 1). Each mutation was inherited from the parents, and her surviving brother was a heterozygous carrier of the mutation. Although p.Arg425Cys was previously reported in a patient with SMARD1 showing compound heterozygous variants, the result of segregation analysis was not demonstrated and no functional test was performed in the previous report.3 The patient was discharged with a home mechanical ventilator and a nasogastric feeding tube at 6 months of age; she died of accidental airway obstruction 5 months later, at home. The patient's parents provided written informed consent for publication of patient and family information.
DISCUSSION
We present a case of SMARD1, confirmed by exome sequencing, in a consanguineous Syrian family with a history of recurrent infant deaths. At first, owing to the family history of sudden and inexplicable deaths of previous siblings of both sexes and consanguinity in the parents, an autosomal recessive genetic disorder was suspected. Additionally, two aborted fetuses with fetal hydrops and feeding intolerance of the patient with mildly elevated liver enzyme levels yielded a false impression of an inborn error of metabolism. However, over a period of one month, we observed rapid progression of the weakness of distal muscles, which led to a suspicion of neuromuscular disorder. Using targeted exome sequencing, she was eventually diagnosed with SMARD1, which was molecularly confirmed by the presence of an IGHMBP2 mutation.
SMARD1 is characterized by diaphragmatic paralysis and respiratory failure.2 The affected patients demonstrate progressive distal muscle weakness resulting in foot deformities and contractures and fatal respiratory distress within the first few weeks of life.4 Most patients with SMARD1 die within the first year of life because of respiratory failure, and thus far, the condition has no effective treatment other than symptomatic care.2
Our patient had a unique family history of unknown infantile deaths at 10–12 weeks after birth.
Mutations in the gene encoding the IGHMBP2, mapped on chromosome 11q13, are known to cause SMARD1 or Charcot-Marie-Tooth disease type 2S.5,6 Mutations in IGHMBP2 result in a broad clinical spectrum of axonal neuropathies due to degeneration of α-motor neurons in the brain stem and anterior horns of the spinal cord, which is suggested to result in impaired maturation of mRNAs in neurons.7 Charcot-Marie-Tooth disease type 2S involves slowly progressive sensorimotor axonal polyneuropathy, but no significant respiratory compromise, with a longer survival compared to SMARD1.7,8 In addition, spinal muscular atrophy type 1 and SMARD1 have similar clinical manifestations, including fetal respiratory symptoms in the first few weeks of life; however, they are recognized as separate clinical entities of neuromuscular disease caused by the involvement of different genes.2,4,9 Compared to those with SMARD1, patients with SMA1 caused by SMN1 mutation present predominant proximal muscle weakness without diaphragm involvement and a bell-shaped thorax due to intercostal muscle weakness.9
This patient presented intrauterine growth retardation and autonomic dysregulation, such as unexplained tachycardia or excessive sweating, atrophy of the calf muscle replaced by fat mass causing wrist and ankle joint contractures, as previously reported,10-12 as well as poor sucking and respiratory failure. However, diaphragmatic paralysis was not observed until 8 months of age. Respiratory failure in our patient was expected to be mainly due to respiratory muscle weakness rather than diaphragmatic paralysis, although eventuation of the diaphragm is highly suggestive of SMARD1, among neuromuscular disorders. Several cases of atypical phenotype with SMARD1, such as those with no severe signs of respiratory involvement despite an early onset diaphragmatic paralysis, have been found in the literature.10,12,13 Moreover, particularly in this patient, we observed muscle tone changes, as a result of which she was initially hypotonic and then became hypertonic. It was noticed that both knee joints were fixed in hyperextension, as shown in Fig. 2. This might be due to the compensational responsiveness of the limb girdle muscles against distal neuromuscular paralysis, as reported in a similar case report of a patient with SMARD1 presenting initially with hypertonia.12 Only one SMARD1 patient carrying p.Arg425Cys of IGHMBP2 has been reported in literature, and this male patient had another heterozygous truncated mutation with c.2611+1G>T IGHMBP2. Compared to our patient with homozygous mutations of c.1273C>T (p.Arg425Cys) IGHMBP2, he presented early symptoms of bilateral diaphragmatic paralysis at the age of 1 month and died at 5 months of age, despite diaphragmatic plication and mechanical ventilator support.3 Additionally, since the author did not state the nationality of the parents and the result of segregation analysis in their patient, we could not assess that the p.Arg425Cys in French report was also inherited from a Syrian.
Furthermore, in SMARD1, no clear phenotype-genotype correlations have been reported even in siblings with the same mutations.4,14 Because of the heterogeneity of phenotypes, targeted exome sequencing may be useful for early diagnosis in rare fatal cases.
In conclusion, we present a Syrian consanguineous family with SMARD1, confirmed by targeted exome sequencing. SMARD1 should be considered in cases with early respiratory insufficiency or near-miss sudden infant death syndrome. Targeted exome sequencing can be useful to identify the genetic cause in cases of consanguinity and recurrent mysterious deaths. In addition, it is suggested that a detailed phenotypic description and communication between bioinformaticians and clinicians is important to reduce false negative results in exome sequencing.
ACKNOWLEDGMENTS
We thank the patient and her family for their participation in this study. We wish to thank Dr. Ja-Hyun Jang at Green Cross genome for providing genetic result and careful review of the article.
Footnotes
Disclosure: The authors have no potential conflicts of interest to disclose.
- Conceptualization: Kim YM.
- Writing - original draft: Kim YA, Kim YM.
- Writing - review & editing: Kim YA, Jin HY.
References
- 1.Opdal SH, Rognum TO. The sudden infant death syndrome gene: does it exist? Pediatrics. 2004;114(4):e506–e512. doi: 10.1542/peds.2004-0683. [DOI] [PubMed] [Google Scholar]
- 2.Eckart M, Guenther UP, Idkowiak J, Varon R, Grolle B, Boffi P, et al. The natural course of infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1) Pediatrics. 2012;129(1):e148–56. doi: 10.1542/peds.2011-0544. [DOI] [PubMed] [Google Scholar]
- 3.Gitiaux C, Bergounioux J, Magen M, Quijano-Roy S, Blanc T, Bonnefont JP, et al. Diaphragmatic weakness with progressive sensory and motor polyneuropathy: case report of a neonatal IGHMBP2-related neuropathy. J Child Neurol. 2013;28(6):787–790. doi: 10.1177/0883073812450209. [DOI] [PubMed] [Google Scholar]
- 4.Porro F, Rinchetti P, Magri F, Riboldi G, Nizzardo M, Simone C, et al. The wide spectrum of clinical phenotypes of spinal muscular atrophy with respiratory distress type 1: a systematic review. J Neurol Sci. 2014;346(1-2):35–42. doi: 10.1016/j.jns.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 5.Grohmann K, Wienker TF, Saar K, Rudnik-Schöneborn S, Stoltenburg-Didinger G, Rossi R, et al. Diaphragmatic spinal muscular atrophy with respiratory distress is heterogeneous, and one form Is linked to chromosome 11q13-q21. Am J Hum Genet. 1999;65(5):1459–1462. doi: 10.1086/302636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grohmann K, Schuelke M, Diers A, Hoffmann K, Lucke B, Adams C, et al. Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat Genet. 2001;29(1):75–77. doi: 10.1038/ng703. [DOI] [PubMed] [Google Scholar]
- 7.Cottenie E, Kochanski A, Jordanova A, Bansagi B, Zimon M, Horga A, et al. Truncating and missense mutations in IGHMBP2 cause Charcot-Marie Tooth disease type 2. Am J Hum Genet. 2014;95(5):590–601. doi: 10.1016/j.ajhg.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schottmann G, Jungbluth H, Schara U, Knierim E, Morales Gonzalez S, Gill E, et al. Recessive truncating IGHMBP2 mutations presenting as axonal sensorimotor neuropathy. Neurology. 2015;84(5):523–531. doi: 10.1212/WNL.0000000000001220. [DOI] [PubMed] [Google Scholar]
- 9.Rudnik-Schöneborn S, Forkert R, Hahnen E, Wirth B, Zerres K. Clinical spectrum and diagnostic criteria of infantile spinal muscular atrophy: further delineation on the basis of SMN gene deletion findings. Neuropediatrics. 1996;27(1):8–15. doi: 10.1055/s-2007-973741. [DOI] [PubMed] [Google Scholar]
- 10.Messina MF, Messina S, Gaeta M, Rodolico C, Salpietro Damiano AM, Lombardo F, et al. Infantile spinal muscular atrophy with respiratory distress type I (SMARD 1): an atypical phenotype and review of the literature. Eur J Paediatr Neurol. 2012;16(1):90–94. doi: 10.1016/j.ejpn.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 11.San Millan B, Fernandez JM, Navarro C, Reparaz A, Teijeira S. Spinal muscular atrophy with respiratory distress type 1 (SMARD1) report of a Spanish case with extended clinicopathological follow-up. Clin Neuropathol. 2016;35(2):58–65. doi: 10.5414/NP300902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han C, Mai J, Tian T, He Y, Liao J, Wen F, et al. Patient with spinal muscular atrophy with respiratory distress type 1 presenting initially with hypertonia. Brain Dev. 2015;37(5):542–545. doi: 10.1016/j.braindev.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 13.Stalpers XL, Verrips A, Poll-The BT, Cobben JM, Snoeck IN, de Coo IF, et al. Clinical and mutational characteristics of spinal muscular atrophy with respiratory distress type 1 in The Netherlands. Neuromuscul Disord. 2013;23(6):461–468. doi: 10.1016/j.nmd.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 14.Joseph S, Robb SA, Mohammed S, Lillis S, Simonds A, Manzur AY, et al. Interfamilial phenotypic heterogeneity in SMARD1. Neuromuscul Disord. 2009;19(3):193–195. doi: 10.1016/j.nmd.2008.11.013. [DOI] [PubMed] [Google Scholar]