

Abstract
Background
Kufor‐Rakeb syndrome (KRS) is a rare autosomal recessive neurologic disease with diverse phenotypic features. Herein we report an Iranian KRS family with seizure and action myoclonus in addition to other typical manifestations of this syndrome.
Method
All family members underwent careful neurologic examination. Exome sequencing was performed and ATP13A2 variation genotyped in all family members.
Results
Cognitive deficits, hypokinesia, rigidity, spasticity, brisk deep tendon reflexes, upward gaze palsy, tremor, and facial‐faucial‐finger mini‐myoclonus were the common manifestations of all affected siblings. Two cases had seizure and the most severely affected sibling demonstrated severe action myoclonus. Exome sequencing identified a homozygous nonsense mutation c.2455C>T;p.Arg819* in ATP13A2 gene.
Conclusions
We reported five KRS affected siblings who manifested myoclonus and seizure. The most severely affected one demonstrated action myoclonus, which has not been reported so far.
Keywords: action myoclonus, ATP13A2 mutation, Kufor‐Rakeb syndrome, neurodegeneration with brain iron accumulation, seizure
Kufor‐Rakeb syndrome (KRS), which is a rare neurologic disease with autosomal recessive inheritance, was first described in 1994.1, 2 ATP13A2 that encodes a member of subfamily P5 of the P‐type superfamily of ion pumps was identified as cause of KRS in 2006.3 Until very recently, 13 disease‐causing mutations were identified in patients from various populations.4 Review of phenotypic features of the patients harboring the mutations revealed intra‐familial and inter‐familial heterogeneity. Bradykinesia, rigidity, initial favorable response to L‐dopa, slow saccades, and supranuclear vertical gaze palsy were almost always reported. Pyramidal involvement, including spasticity, increased deep tendon reflexes (DTRs), and Babinski sign were also common presentations.5, 6 Cognitive decline was found in most of the patients, but its severity was variable. Tremor, dystonia, and bulbar dysfunction were variable manifestations. Iron accumulation in the basal ganglia was reported once.7 Herein, we report an Iranian KRS family with seizure and action myoclonus in addition to other manifestations of this syndrome.
Subjects and Methods
Subjects
This research was performed in accordance with the Declaration of Helsinki and with approval of the ethics board of the University of Tehran. The KRS103 pedigree studied here was a large pedigree with a neurologic disorder, including five affected siblings. The siblings were born to consanguineous unaffected parents (Fig. S1) suggesting an autosomal recessive inheritance.
Exome Sequencing
Exome sequencing, Illumina HiSeq®2000 system (Illumina) was performed on the DNAs of two affected individuals, KRS103‐IV‐2 and KRS103‐IV‐10. Sequence alignment and variant calling were performed against human reference genome UCSC NCBI37/hg19. Preliminary filtering was done to identify all homozygous variations. Subsequently, SNPs with a reported minimal allele frequency of > 0.01 in the public and local databases were removed. Finally, variants that were not present in both siblings and that did not affect amino acid change or splicing were filtered out.
Screening of c.2455C>T in ATP13A2
Exon 22 of ATP13A2 and adjacent intronic sequences that contain nucleotide c.2455C>T were PCR amplified from the DNAs of all available KRS103 family members and subsequently Sanger sequenced. Sequences were analyzed with the Sequencher 5.2 software. ATP13A2 reference sequences used were NC_000001.11, NM_022089.3, and NP_071372.1.
Results
Clinical Findings
Mean age at disease onset was 11.0 ± 1.0 years (range: 10–12). The presenting symptoms in all patients were slowness of movements, clumsiness, loss of dexterity, disequilibrium, and speech disturbance. All patients had cognitive deficits, slow saccades, and vertical gaze limitation or palsy. They showed signs of extrapyramidal system involvement (i.e., hypokinesia and rigidity, partially levodopa‐responsive, especially at onset). Patients also had spasticity and brisk DTRs and three of them (IV‐2, IV‐9 and IV‐13) had Babinski sign. A common finding in all affected siblings was presence of jerky movements in the perioral area, tongue and fingers compatible with facial‐faucial‐finger mini‐myoclonus. Subject IV‐2 showed severe action myoclonus of arms, head, and neck. Case IV‐9 had severe postural instability and needed assistance for walking; subject IV‐2 (the oldest) was bedridden due to severe ataxia. A more detailed report on their clinical features is following (Table 1).
Table 1.
Phenotypic Features of Cases With ATP13A2 Mutation
| KRS‐103‐IV‐2 | KRS‐103‐IV‐9 | KRS‐103‐IV‐10 | KRS‐103‐IV‐13 | KRS‐103‐IV‐14 | |
|---|---|---|---|---|---|
| Gender | F | F | F | M | F |
| Age at onset (y) | 10 | 10 | 12 | 11 | 12 |
| Age at exam (y) | 45 | 33 | 31 | 26 | 24 |
| Presenting symptoms | Slowness of movements | Social isolation, seizure | Speech disorder, disequilibrium, Seizure | Slowness of movements, hand clumsiness | Speech disorder, slowness of movements, disequilibrium |
| Cognitive deficit | Severe | Prominent | Mild | Mild | Mild |
| Psychiatric problems | Visual hallucination, depression | Visual hallucination, depression | Depression, anxiety | Visual hallucination, depression | depression |
| Hypokinesia | + | + | + | + | + |
| Rigidity | + | + | + | + | + |
| Dystonia | ‐ | ‐ | ‐ | ‐ | ‐ |
| Tremor | + | + | + | + | + |
| Postural instability | + | + | ‐ | ‐ | ‐ |
| Spasticity | + | + | + | + | + |
| Increased deep tendon reflexes | + | + | + | + | + |
| Babinski sign | + | + | ‐ | + | ‐ |
| Pes cavus | + | + | + | + | + |
| Slow saccades | + | + | + | + | + |
| Vertical gaze palsy | + | + | + | + | + |
| Dysarthria | + | + | + | + | + |
| Bladder/bowel dysfunction | + | + | ‐ | ‐ | ‐ |
| FFF Myoclonus | + | + | + | + | + |
| Seizure | ‐ | + | + | ‐ | ‐ |
| Ataxia | Severe | Mild | Mild | Mild | Absent |
| Brain MRI | Diffuse brain atrophy more prominent in fronto‐temporal region | Not done | Normal | Normal | Not done |
| Other features | Action myoclonus, pseudobulbar affect | Freezing of gait | ‐ | Drooling | Drooling |
Abbreviations: F, Female; M, Male; FFF, facial‐faucial finger.
Patients
Krs103‐iv‐2
The patient's problems started with slowing of movements and gait disturbances when she was 10 years. After her third pregnancy, she had aggravation of gait dysfunction and became bedridden with bowel and bladder incontinence. Subsequently, the clinical picture was enriched by severe jerks in arms and neck. She had severe cognitive deterioration and pseudobulbar affect. Neurologic examination showed scanning speech, vertical gaze palsy, and severe myoclonic jerks of head, neck, and upper limbs. She had action myoclonus, which got worse on approaching the target. There was tongue and perioral muscle twitches. She was unable to walk. She had pes cavus, tendon reflexes were brisk, and plantar reflexes were up going bilaterally (Video S1). Brain MRI showed severe diffuse atrophy more prominent in frontotemporal areas (Fig. 1A).
Figure 1.
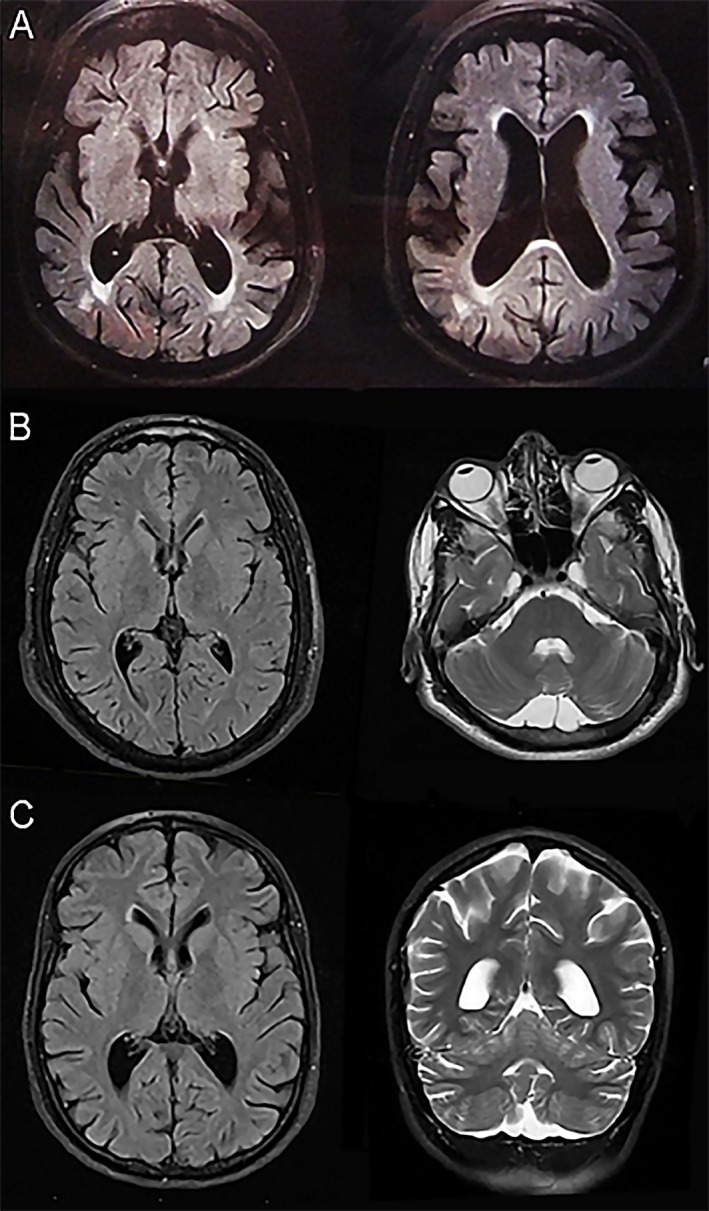
Brain MRI of case IV‐2 demonstrates cerebral volume loss and normal basal ganglia (A) Brain MRI of case IV‐10 (B) and case IV‐13 (C) illustrate normal basal ganglia and cerebellum.
Krs103‐iv‐9
This patient had problems in primary school (around the age 10), was socially isolated, and had two generalized tonic‐clonic seizures. She had progressive slowness of movement and difficulty walking. Prominent pathologic findings on her examination were facial hypomimia, slurred and low tone speech, vertical gaze palsy, tongue and perioral jerks, severe hypokinesia of limbs, finger mini‐myoclonus, pes cavus, brisk DTRs, and bilateral extensor plantar reflexes. Gait was slow, spastic, and wide‐based. She also had postural instability (Video S2).
Krs103‐iv‐10
This 33‐year‐old woman with normal birth history and psychomotor development presented balance problems and difficulties walking and speaking at age 12. She also had two episodes of generalized tonic‐clonic seizures. On examination her speech was slurred. There was vertical gaze palsy, and tremors of the tongue and perioral muscles. There was mild hypokinesia and rigidity of extremities. DTRs were increased without Babinski sign. Gait was mildly spastic with impaired tandem gait (Video S3). Brain MRI was normal (Fig. 1B).
Krs103‐iv‐13
This patient's problem started at the age of 11, when he noticed clumsiness of his hands and difficulty in doing fine motor tasks. He was slow in all his movements, which improved to some extent with levodopa. On examination, he had facial hypomimia, slow horizontal saccades, vertical gaze palsy (upward), and tremor‐like movements of the tongue and perioral area. There was hypokinesia of limbs more on the right side; DTRs were brisk with sustained ankle clonus and bilateral Babinski signs. He had mild dysdiadochokinesia. Gait was mildly spastic and wide‐based with decreased right arm swing (Video S4). His brain MRI was same as the case 103‐IV‐10 (Fig. 1C).
Krs103‐iv‐14
This patient's problems started with movement's slowness, dysarthria, and disequilibrium at 12 years of age. At the time of our visit, the main complaints were slowness of all movements, tremor of the tongue, and drooling, which responded partially to levodopa. Examination revealed masked face, slurred speech, vertical gaze palsy, hypokinesia, pes cavus, and brisk DRTs. There was mild mini‐myoclonus in fingers, tongue and perioral region (Video S5).
Genetic Analysis
The filtering criteria used for analysis of the exome data identified a novel candidate variation, c.2455C>T in ATP13A2 that causes p.Arg819* (Fig. S2). This variation segregated with disease status in the pedigree. It was assessed that this variant in the homozygous state was the likely cause of the neurologic disorder in this family. The affected codon encodes an amino acid in the intracellular loop between membrane spanning regions six and seven of the protein and the mutation is predicted to cause deletion of 358 amino acids from its C‐terminus. Previously, a homozygous mutation c.2572C>T within this region (p.Gln858*) had been reported in other Iranian families with presentations similar to our cases, but without action myoclonus and seizure.4
Discussion
Affected individuals in the first pedigree diagnosed with KRS in 1994, showed a combination of parkinsonism with supranuclear gaze palsy, facial‐faucial‐finger mini‐myoclonus, early dementia, visual hallucinations, and age at onset of 12–15 years. These patients had a good response to levodopa, which caused subsequent dyskinesias. MRI showed diffuse cerebral and cerebellar atrophy, without signal changes in basal ganglia.1 Quinn et al. reported a similar disease in two Pakistani cousins with akinetic rigid syndrome since the age of 16 years with mild response to levodopa with hand and neck dyskinesia. They showed pyramidal signs and dystonia, saccadic abnormalities, and cognitive decline. These patients were first reported as affected with a form of pallido‐pyramidal syndrome, but subsequently diagnosed as KRS.5 In one case, Schneider et al. reported iron deposition in the basal ganglia, suggesting that KRS should be considered among the neurodegeneration with brain iron accumulation (NBIA) syndromes.7 Other reports of KRS were added to the literature, mostly demonstrating the aforementioned clinical features.3, 4, 8, 9 Nevertheless, KRS is a disease with multiple presentations: parkinsonism seems to be the most prominent feature of this syndrome and most reported cases suffer from hypokinesia and rigidity, with various degrees of response to levodopa and occasional levodopa‐induced dyskinesia.2, 4, 5, 6
The unique feature of the family presented here is severe action myoclonus of arms and head in the most severely affected subject, which has not been reported previously. While facial‐faucial‐finger mimi‐myoclonus is a common finding in KRS, severe action myoclonus of arms and head is unusual for KRS patients and to the best of our knowledge, has not been previously reported. Also seizure at early stage of disease is a rare feature. Seizure was only once previously reported at end‐stage of disease in KRS patients.10
Although electrophysiology was not done to characterize the myoclonus in our patient with action myoclonus, according to clinical characteristics, action induced and involvement of head and hand, it seems to be cortical in origin. Moreover, the presence of generalized tonic‐clonic seizures point toward cortical involvement in KRS. Thus, these features, as well as pyramidal signs seem to be due to cortical pathology as opposed to parkinsonian features that are due to basal ganglia involvement in KRS.
In summary, we reported a KRS family with certain clinical manifestations that have not been previously reported. These features included severe action myoclonus and early onset seizure supporting cortical involvement in KRS. Genetic analysis of the patients revealed they carry a novel homozygous mutation in ATP13A2 gene.
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
M.R.:1A, 1B, 1C, 3A
F.S.:1B, 1C
A.E.L.:1A, 3B
E.E.:2A, 3B
A.F.:2A, 3B
J.B.:2B, 3B
J.H.:2A
A.A.:1C, 2A, 2B, 3B
Disclosures
Ethical Compliance Statement: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflict of Interest: All authors assert absence of conflicts of interests and absence of financial interests.
Financial Disclosures from previous 12 months: The authors report no financial disclosure.
Supporting information
Additional Supporting Information may be found online in the supporting information tab for this article:
Figure S1. Iranian KRS pedigree with p.Arg819* mutation in ATP13A2. Present age (years) of some individuals is indicated under their ID numbers. KRS genotypes of individuals tested are presented. Note: filled circles and squares = affected; unfilled circles and squares = not affected. Abbreviations: KRS, Kufor‐Rakeb syndrome; M, mutated ATP13A2 allele; N, wild‐type ATP13A2 allele.
Figure S2. DNA sequence chromatogram showing the homozygous mutation c.2572C>T in ATP13A2 and the wild type sequence.
Video S1. Case KRS103‐IV‐2: There is vertical gaze palsy, tongue, and perioral twitches, coarse myoclonus of upper limbs and action myoclonus, getting worse on approaching the target.
Video S2. Case KRS103‐IV‐9: This video demonstrates vertical gaze palsy, hypokinesia of upper limbs, increased DTRs, positive Babinski sign, slow and spastic gait.
Video S3. Case KRS103‐IV‐10: The video shows perioral and tongue twitches, limitation of vertical gaze, hypokinesia in hands, increased muscle stretch reflexes, pes cavus and flexor plantar reflexes.
Video S4. Case KRS103‐IV‐13: The patient shows hypokinesia more on the right limbs, hyperreflexia, ankle clonus, Babinski sign, mild spastic gait and up gaze palsy.
Video S5. Case KRS103‐IV‐14: This video depicts mild hypokinesia, mild spastic gait, vertical gaze palsy and tongue twitches
Acknowledgements
The authors acknowledge the patients and their family members for consenting to participate in this study. The authors confirm that the approval of an institutional review board was not required for this work.
Relevant disclosures and conflicts of interest are listed at the end of this article.
Supporting information may be found in the online version of this article.
References
- 1. Najim al‐Din AS, Wriekat A, Mubaidin A, Dasouki M, Hiari M. Pallido‐pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor‐Rakeb syndrome. Acta Neurol Scand 1994;89:347–352. [DOI] [PubMed] [Google Scholar]
- 2. Williams DR, Hadeed A, Al‐Din ASN, Wreikat A‐L, Lees AJ. Kufor Rakeb disease:autosomal recessive, levodopa‐responsive parkinsonism with pyramidal degeneration, supranuclear gaze palsy, and dementia. Mov Disord 2005;20:1264–1271. [DOI] [PubMed] [Google Scholar]
- 3. Ramirez A, Heimbach A, Gründemann J, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P‐type ATPase. Nat Genet 2006;38:1184–1191. [DOI] [PubMed] [Google Scholar]
- 4. Malakouti‐Nejad M, Shahidi GA, Rohani M, et al. Identification of p.Gln858* in ATP13A2 in two EOPD patients and presentation of their clinical features. Neurosci Lett 2014;577:106–111. [DOI] [PubMed] [Google Scholar]
- 5. Quinn NP, Goadsby PJ, Lees AJ. Hereditary juvenile parkinsonism with pyramidal signs and mental retardation. Eur J Neurol 1995;2:23–26. [DOI] [PubMed] [Google Scholar]
- 6. Tranchant C, Koob M, Anheim M. Parkinsonian‐Pyramidal syndromes: a systematic review. Parkinsonism Relat Disord 2017;39:4–16. [DOI] [PubMed] [Google Scholar]
- 7. Schneider SA, Paisan‐Ruiz C, Quinn NP, et al. ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov Disord 2010;25:979–984. [DOI] [PubMed] [Google Scholar]
- 8. Bras J, Verloes A, Schneider SA, Mole SE, Guerreiro RJ. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid‐lipofuscinosis. Hum Mol Genet 2012;21:2646–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crosiers D, Ceulemans B, Meeus B, et al. Juvenile dystonia‐parkinsonism and dementia caused by a novel ATP13A2 frameshift mutation. Parkinsonism Relat Disord 2011;17:135–138. [DOI] [PubMed] [Google Scholar]
- 10. Behrens MI, Brüggemann N, Chana P, et al. Clinical spectrum of Kufor‐Rakeb syndrome in the Chilean kindred with ATP13A2 mutations. Mov Disord 2010;25:1929–1937. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found online in the supporting information tab for this article:
Figure S1. Iranian KRS pedigree with p.Arg819* mutation in ATP13A2. Present age (years) of some individuals is indicated under their ID numbers. KRS genotypes of individuals tested are presented. Note: filled circles and squares = affected; unfilled circles and squares = not affected. Abbreviations: KRS, Kufor‐Rakeb syndrome; M, mutated ATP13A2 allele; N, wild‐type ATP13A2 allele.
Figure S2. DNA sequence chromatogram showing the homozygous mutation c.2572C>T in ATP13A2 and the wild type sequence.
Video S1. Case KRS103‐IV‐2: There is vertical gaze palsy, tongue, and perioral twitches, coarse myoclonus of upper limbs and action myoclonus, getting worse on approaching the target.
Video S2. Case KRS103‐IV‐9: This video demonstrates vertical gaze palsy, hypokinesia of upper limbs, increased DTRs, positive Babinski sign, slow and spastic gait.
Video S3. Case KRS103‐IV‐10: The video shows perioral and tongue twitches, limitation of vertical gaze, hypokinesia in hands, increased muscle stretch reflexes, pes cavus and flexor plantar reflexes.
Video S4. Case KRS103‐IV‐13: The patient shows hypokinesia more on the right limbs, hyperreflexia, ankle clonus, Babinski sign, mild spastic gait and up gaze palsy.
Video S5. Case KRS103‐IV‐14: This video depicts mild hypokinesia, mild spastic gait, vertical gaze palsy and tongue twitches