

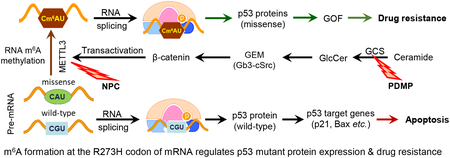
Abstract
Mutant p53 proteins that promote cancer cell invasive growth, metastasis and drug resistance emerge as therapeutic targets. Previously, we reported that suppression of ceramide glycosylation restored wild-type p53 protein and tumor suppressing function in cancer cells heterozygously carrying p53 R273H, a hot-spot missense mutation; however, the mechanisms underlying the control of mutant protein expression remain elusive. Herein, we report that an N6-methyladenosine (m6A) at the point-mutated codon 273 (G>A) of p53 pre-mRNA determines the mutant protein expression. Methylation of the transited adenosine was catalyzed by methyltransferase like 3 (METTL3), and this m6A-RNA promoted a preferential pre-mRNA splicing; consequently, the produced p53 R273H mutant protein resulted in acquired multidrug resistance in colon cancer cells. Furthermore, glycosphingolipids (particularly globotriaosylceramide) generated from serial ceramide glycosylation were seen to activate cSrc and β-catenin signaling so as to upregulate METTL3 expression, in turn promoting expression of p53 R273H mutant protein, with consequent drug resistance. Conversely, either silencing METTL3 expression by using small interfering RNA (siRNA) or inhibiting RNA methylation with neplanocin A suppressed m6A formation in p53 premRNA, and substantially increased the level of phosphorylated p53 protein (Ser15) and its function in cells heterozygously carrying the R273H mutation, thereby re-sensitizing these cells to anticancer drugs. Concordantly, suppression of ceramide glycosylation repressed METTL3 expression and m6A formation in p53 pre-mRNA, thus sensitizing cells carrying R273H to anticancer drugs. This study uncovers a novel function of pre-mRNA m6A as a determinant of mutant protein expression in cancer cells heterozygously carrying the TP53 R273H mutation. Suppressing both RNA methylation and ceramide glycosylation might constitute an efficacious and specific approach for targeting TP53 missense mutations coding for a G>A transition, thereby improving cancer treatments.
Keywords: tumor suppressor p53, missense mutation, RNA methylation, N6-methyladenosine, drug resistance, glucosylceramide synthase
Graphical Abstract
1. Introduction
The p53 protein, produced from mRNA encoded by the TP53 gene, plays a perhaps more crucial role than any other tumor suppressor in preventing tumorigenesis and tumor progression. Moreover, p53 is an essential and powerful transcription factor, acting through the binding of its DNA binding domain (DBD) to corresponding promoter elements, whereby it can transactivate the expression of p53 target genes, approximately 246 human genes, including p21, Bax and PUMA [1, 2]. In response to DNA damage stress and other oncogenic stresses, cells highly express p53 protein, which, with concomitant upregulation of the expression of p53 target genes, normally enables p53 to trigger cell-cycle arrest, senescence, and cell death by apoptosis or ferroptosis [2–4]. It has been found that TP53 mutation is one of the most common genetic alterations in cancers, appearing in approximately 42% of cases across 12 tumor types carrying mutant TP53, the presence of which correlates to poor prognosis [5, 6]. Among all such TP53 mutations, more than 75% are missense point-mutations extant in the region encoding the DBD, and these produce full-length, missense proteins that function aberrantly with respect to their transactivation of p53 target genes [7, 8]. The transition from guanine to adenine (G>A) at codons 175, 248, and 273 accounts for 11.2% of all TP53 mutations in cancers appearing in the colon and lungs [9–11] (http://p53.free.fr/Database/p53_cancer/all_cancer.html). p53 missense proteins that lack the tumor suppression activity of wild-type p53 (wt p53) instead often exhibit oncogenic gain-of-function (GOF) [12]. Knock-in mouse models that express hot-spot mutant alleles R172H or R270H (R175H or R273H in human) manifest GOF by conferring a broader tumor spectrum and more tumor metastases, as compared with wt p53-expressing mice [13]. TP53 mutants appear with increased frequency in tumors diagnosed at advanced stages, or with more metastases, and in recurrences of cancers in colon, ovaries and breasts [14, 15]. Missense p53 mutants thus deserve strong attention with respect to therapeutic targeting aimed at improving cancer treatments.
Under normal conditions, p53 protein levels are low, owing to feedback regulation by p53-activated MDM2-mediated degradation. In cancer cells, wt p53 can be activated by stress conditions, including oncogenic activation (oncogenic stress) and DNA damage [16]. Missense p53 mutants are expressed at high levels in cancer cells, in part owing to failure of mutant proteins to induce expression of MDM2 [10]. The small molecules PRIMA-1 and APR-246 promote refolding of p53 mutant proteins (R273H, R175H) by the binding of the reactive methylene quinuclidinone (MQ) moiety to cysteine, thereby enabling mutant protein to activate p53 target genes, including p21, Bax and PUMA, in tumor cells [17, 18]. As an augmentation to effecting the refolding of missense proteins for reactivating p53 function, our recent work indicates that it is possible to eliminate mutant protein while restoring wt p53 expression in cancer cells. Inhibition of glucosylceramide synthase (GCS) restores wt p53 protein levels, and abolishes oncogenic GOF, in cells heterozygously carrying a TP53 R273H mutation [19].
Unearthing how cells select pre-mRNA molecules to generate mRNA transcripts coding protein for wt vs. mutant can be expected to reveal therapeutic intervention opportunities for restoring p53 in cells carrying TP53 missense mutations. DNA sequences determine the sequences of pre-mRNA; however, further RNA processing, including pre-mRNA splicing and RNA methylation, contributes to the posttranscriptional regulation of protein expression [20]. During pre-mRNA splicing, a dynamically assembled spliceosome of nuclear ribonucleoprotein (snRNP) complexes recognizes splice sites in pre-mRNA and catalyzes two transesterification reactions, so as to excise introns and splice exons together to form a mature and functional mRNA for translation to produce proteins [20, 21]. Alternative splicing, in which extrinsic and non-spliceosomal RNA-binding proteins (classical/canonical hnRNPs, SR proteins, tissue-specific RNA-binding proteins) are involved in recognizing introns in pre-mRNA, allowing generation of more than one unique mRNA species from a single gene [20, 21]. Alterative splicing can generate mRNAs that differ in their untranslated regions or coding sequences through mechanisms that include exon-skipping, a choice between exons, the use of alternative splice sites, or intron retention [20]. Aberrant RNA splicing has been found, in a growing number of instances, to underlie human diseases, including cancers [22]. Upregulation of epithelial-restricted splicing proteins (ERSP1, ERSP2) and SR proteins (SRSF1, SRSF3) in cancer cells contributes to cancer progression [23, 24].
Modulation of alternative pre-mRNA splicing with ceramide allows cancer cells to express pro-apoptotic isoforms of BCL-x and caspase-9 [25, 26], and even to restore wt p53 levels and functions in cancer cells carrying a p53-deletion mutation [27]. In pre-mRNA, adenosine can be methylated by a methyltransferase complex containing methyltransferase-like 3 (METTL3) to form N6-methyladenosine (m6A) [28, 29]. Emerging evidence implicates that the m6A reader protein YTHDF1 selectively binds m6A-modified pre-mRNAs, with higher affinity, and that this can in turn cause alternative splicing, thereby altering protein translation and levels thereof, thus bringing about m6A-dependent deleterious changes in protein expression in cancer cells [28, 30]. Herein, we report our study results of the methylation of adenosine to generate m6A in R273H missense mutant of p53 pre-mRNA transcripts, and consequent preferential effects on mutant protein expression and oncogenic GOF in association with cancer drug resistance.
2. Materials and methods
2.1. Cell lines and cell culture
Human colon cancer SW48 cells, and also cells of its corresponding heterozygous TP53 missense mutant line (SW48/TP53 (R273H/+), introduced by a CRISPR/Cas9 editing system), were purchased from Horizon Discovery (HD 103–008, Waterbeach, Cambridge, UK) [31]. SW48-Dox and TP53-Dox sublines of SW48 and SW48/TP53 (R273H/+) cells were generated by culture in 10% FBS RPMI-1640 medium including 2 mM L-glutamine and 25 mM sodium bicarbonate, supplemented with 10% FBS and 800 μg/mL geneticin (G418), with the addition of 25 nM doxorubicin (Dox) for 16 weeks (~26 passages) [19]. SW48–5FU and TP53–5FU sublines of SW48 and SW48/TP53 (R273H/+), respectively, were cultured in medium including 1 μM 5-fluorouracil (5-FU) for 16 weeks (~27 passages). Human WiDr (homozygous R273H p53) colon cancer cells were purchased from ATCC (Manassas, VA, USA). Cells were cultured in RPMI-1640 or ATCC-formulated EMEM containing 10% fetal bovine serum (FBS), 100 units/mL penicillin, 100 μg/mL streptomycin and 584 mg/L l-glutamine, and maintained in an incubator humidified with 95% air and 5% CO2 at 37°C. Cell culture medium and supplementary materials were purchased from Thermo Fisher Scientific (Waltham, MA, USA) unless otherwise stated. The cell lines were analyzed by short tandem repeat (STR) profiling (Gene Resources Core Facility, Johns Hopkins University) and compared against publically available databases (DSMZ and ATCC) for authentication.
2.2. Cell viability assay
Cell viability was determined by quantitation of ATP, an indicator of live cells, using the CellTiter-Glo luminescent cell viability assay kit (Promega, Madison, WI, USA), as described previously [19, 32]. Briefly, cells (4000 cells/well) were grown in 96-well plates with 10% FBS supplemented RPMI-1640 medium. Cells were treated with indicated agents in 5% FBS medium for 72 hours. For combination treatments, cells (3×106/100-mm dish; 4000 cells/well in 96-well plates) were pretreated with d-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP; 5 μM) or neplanocin A (NPC, 20 nM), fumonisin B1 (FB1, 25 μM), Shiga toxin 1B-subunit (STxB, 100 nM), PP2 (500 nM) and FH535 (5 μM) in 5% FBS medium for 48 hr. ATP quantitation for cell viability assessment was accomplished with a Synergy HT microplate reader (BioTek, Winnooski, VT, USA), following incubation with CellTiter-Glo reagent.
2.3. Induction and detection of apoptosis
Apoptosis and cell cycle analysis were carried out using an APO-DIRECT kit (BD Biosciences, San Jose, CA, USA) or propidium iodide (PI), as described previously [32, 33]. Briefly, cells were pretreated with NPC (20 nM) or vehicle in 5% FBS medium for 3 days, and then co-treated with NPC and Dox (100 nM) for an additional 48 hr to induce apoptosis. After fixing with paraformaldehyde (1% w/v) and 70% ethanol, cells (1 × 106 cells/ml) were incubated with FITC-labeled Br-dUTP (5 μg/100 μl) and then PI (5 μg/100 μl) in staining buffer containing RNase for apoptosis detection. For cell cycle analysis, fixed cells (1 × 106 cells/ml) were incubated with PI (5 μg/100 μl) in staining buffer containing RNase. Cells were then analyzed using an imaging flow cytometry system, namely an ImageStreamX Mark II with high-resolution microscope (Amnis, Seattle, WA, USA). For each sample, 5000 events were counted in triplicate. Data were analyzed with the IDEAS® Software package (Amnis).
2.4. N6-Methyladenosine RNA immunoprecipitation (MeRIP) and RT-qPCR
After treatments, total RNA was extracted from cells of SW48-Dox, TP53-Dox and WiDr cell lines using SV total RNA isolation kits (Promega). MeRIP was performed using the Magna MeRIP m6A kit (Cat. # 17–10499, EMD Millipore, Billerica, MA, USA), as described in previous studies [28, 34] and per kit instructions. Briefly, equal amounts of total RNA (300 μg in 1 μg/μl aliquots) were fragmented with heating in a thermocycler, extracted, and validated with 1.5% agarose gel electrophoresis, and then quantitated via NanoDrop spectrometry. Fragmented RNA samples (~100 nt, 280 μg) were incubated with anti-m6A antibody (clone 17-3-4-1; 10 μg) bound-protein A/G magnetic beads in IP buffer for 2 hours at 4°C, eluted with elution buffer, and purified in RNeasy mini-columns. For control experiments, MeRIP was carried out with mouse IgG replacing m6A antibody, under the same conditions.
Equal amounts RNA input (fragmented RNA; 100 ng) and MeRIP RNA (equal to 100 ng input) from cell extract samples were amplified by reverse transcription quantitative polymerase chain reaction (RT-qPCR) with the QuantiTect SYBR Green RT-PCR kit (Qiagen, Germantown, MD, USA), coupling to the MyiQ Real-Time PCR detection system, as described previously [35]. To detect the m6A at codon 273 of p53, a 95-bp fragment in the region of human TP53 mRNA (codons 249–280; accession number BC003596.1) was generated by using the upstream primer (5´-AGGCCCATCCTCACCATCAT-3´) and downstream primer (5´-CTCCCAGGACAGGCACAAAC-3´). This 95-bp product that includes the point-mutation site (codon 273) would be transited and methylated in p53 R273H mutants. MeRIP-positive control RNA was generated using MeRIP primers human EEF1A1 (accession# NM_001402) positive: forward primer (5´-CGGTCTCAGAACTGTTTGTTTC-3´) and reverse primer (5´-AAACCAAAGTGGTCCACAAA-3´) to amplify the methylated region (stop codon); MeRIP-negative control RNA was generated using MeRIP primers human EEF1A1 negative: forward primer (5´-GGATGGAAAGTCACCCGTAAG-3´) and reverse primer (5´-TTGTCAGTTGGACGAGTTGG-3´) to amplify the unmethylated region (exon 5). A standard curve was generated using threshold cycle (Ct) values of serially diluted input RNA (fragmented) from SW48-Dox cells, calculating the fold enrichment as the ratio of Cm6A to Cnegativecontrol. During RT-PCR reactions, cDNA synthesis was conducted at 50°C for 10 min; polymerase was then inactivated at 95°C for 1 min, and 40 cycles were run from denature (95°C, 10 sec) to anneal (60°C, 30 sec). All samples were analyzed in triplicate, repeated at least two times.
2.5. Gene Silencing of METTL3 and Gb3 synthase
Silencing of METTL3 and globotriaosylceramide (Gb3) synthase was accomplished as described previously [27, 32, 36]. siRNA-METTL3 (siMETTL3, 100 nM) or siRNA-Gb3 synthase (siGb3S) and siRNA-scrambled control (siRNA-SC; 100 nM) were introduced into TP53-Dox cells (3 × 106/100-mm dish; 4000 cells/well in 96-well plates) after overnight growth, facilitating with Lipofectamine 2000 in Opti-MEM reduced-serum medium (Thermo Fisher Scientific) for 4 h. The cells continuously grew in 5% FBS medium for an additional 48 h. In combination treatment groups, cells were then treated with Dox (5 μM) in 5% FBS medium for an additional 48 h.
2.6. Immunocytochemistry
Under the various treatment conditions, cells (50,000 cells/chamber) were grown in 4-chamber slides for 48 hr. After methanol fixation, cells were blocked with 5% goat serum in phosphate-buffered saline (PBS), and incubated with antibodies against METTL3 (1:2000 dilution) and phosphorylated p53 Ser15 (1:200 dilution) in blocking solution at 4°C, overnight. Primary antibodies were further recognized by Qdot 605- or Alexa Fluor 488-conjugated goat IgG (1:50 or 1:2000, respectively). Cell nuclei were counterstained with DAPI (4´,6-diamidino-2-phenylindole) in mounting solution (Vector Laboratories, Burlingame, CA, USA). Images (100 × magnification) were captured using an Olympus BX63 automated fluorescence microscope with monochrome CCD camera (Olympus, Tokyo, Japan). Alexa Fluor 488-conjugated goat anti-mouse IgG (H+L) and Qdot 605-conjugated goat anti-rabbit IgG (H+L) secondary antibodies were purchased from Thermo Fisher Scientific.
2.7. Western blotting analysis
Western blotting was carried out as described previously [27, 37]. Briefly, cells or tissue homogenates were lysed in NP40 cell lysis buffer (Biosource, Camarillo, CA, USA) to extract total cellular proteins once a given treatment was finished. Detergent-soluble proteins were measured by using a bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL, USA). Equal amounts of proteins (50 or 100 μg/lane) were resolved by using 4─20% gradient SDS-PAGE (Thermo Fisher Scientific). After transferring, blots of nitrocellulose membrane were blocked in 5% fat-free milk in 0.05% Tween-20, 20 mM phosphate-buffered saline, pH 7.4 (PBST), and then incubated separately with each one of the primary antibodies (1:500 or 1:2000 dilution), at 4°C overnight. After PBS washing, these blots were incubated with corresponding horseradish peroxidase conjugated secondary antibodies (1:5000 dilutions) and developed using SuperSignal West Femto substrate (Thermo Fisher Scientific). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control for cellular protein. Relative protein levels present were calculated from the optical density (OD) values of bands in triplicated blotting, normalized against corresponding GAPDH OD values in the same membranes. Antibodies against human p53 phosphorylated at Ser15 or for poly(ADP-ribose) polymerase (PARP; Cat. # 9542S) were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibody for methyltransferase-like 3 (METTL3) was purchased from Thermo Fisher Scientific. Antibodies for PUMA, p21, Bax, p53, β-catenin, phosphorylated cSrc and GAPDH were obtained from Santa Cruz Biotechnology (Dallas, TX, USA).
2.8. Immunoprecipitation and protein identification
After treatments, p53 proteins were precipitated with anti-p53 or anti-phosphorylated-p53 antibodies and identified by mass spectrometry (MS), accomplished as described previously [38]. In-gel trypsin digestion and tandem mass spectrometry (MS/MS) analysis of SDS-PAGE protein bands was conducted at the Proteomics Core at University of Arkansas for Medical Sciences. The Mascot database was drawn on for protein identification, which was accomplished using Scaffold software (v. 4.8, Proteome Software, Portland, OR, USA).
2.9. Chemicals and statistical analysis
All chemicals used in this study were obtained from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise stated. PDMP was purchased from Matreya (State College, PA, USA). (─)-Neplanocin A (NPC), which was demonstrated to block m6A formation by acting as a selective inhibitor of S-adenosylhomocysteine (SAH) hydrolase and thereby act as an indirect inhibitor of METTL3 activity (due to excess SAH accumulation) [39], was purchased from Cayman Chemical (Ann Arbor, MI, USA). Fumonisin B1 (FB1), a ceramide synthase inhibitor [27], was purchased from Biomol (Plymouth Meeting, PA, USA). A cSrc kinase inhibitor, 4-amino-5-(4-chlorophenyl)-7-(t-butyl)-pyrazolo[3,4-d]pyrimidine (PP2) [40] was purchased from Enzo Life Sciences (Plymouth Meeting, PA, USA). An effective β-catenin/Tcf inhibitor, FH535 [41], was purchased from Sigma-Aldrich. Shiga toxin 1B-subunit (STxB) was kindly provided by Dr. Anne V. Lane (Division of Geographic Medicine and Infectious Diseases, Tufts Medical Center, and Tufts University School of Medicine, Boston, MA) [42]. siRNAs targeting human METTL3 (sc-92172) [29], Gb3 synthase (sc-72831) [36] and the scrambled control (siRNA-SC, sc-37007) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
All experiments were repeated at least 2 times. All data were analyzed using the GraphPad Prism software (GraphPad, San Diego, CA, USA). Statistical analyses, which included the two-tailed Student’s t-test or ANOVA, were used to compare the continuous variables between groups. Statistical significance was considered for p<0.05.
3. Results
3.1. p53 R273H with RNA m6A Methylation Correlates to Cancer Drug Resistance
The R273H mutation of TP53 is the most common one detected, accounting for approximately 3.1% of p53 mutant-associated cancer cases [11]. p53 R273H protein is encoded from mutant mRNA, in which CAU replaces CGU at codon 273 (Fig. 1A). Colon cancer TP53-Dox or TP53–5FU cells, which heterozygously carried R273H mutant p53 (mut p53, introduced by a CRISPR/Cas9 system in SW48 cells) and were long-term exposed to low-dose doxorubicin (Dox) or 5-flurouracil (5-FU), displayed multidrug resistance, as compared with SW48-Dox or SW48–5FU cells, which were cells that were comparably long-term exposed to low concentration Dox or 5-FU, but which carry only wt p53. To wit, in TP53-Dox cells, the IC50 values for Dox increased by ten-fold, and for oxaliplatin (Oxa) or 5-FU, increased by two-fold (Figs 1B, 1C, Table 1), respectively, over the IC50 values observed for SW48-Dox cells that were subjected to the same treatments. In subline TP53–5FU, the IC50 values for 5-FU increased by 2-fold, for Oxa increased by 12-fold, and for Dox by 5-fold, compared to SW48–5FU cells, respectively (Table 1). Consistently, colon cancer WiDr cells that carry homozygous R273H presented substantial resistance to these drugs, as compared either to SW48-Dox and TP53-Dox, or to SW48–5FU and TP53–5FU cells (Table 1).
Fig. 1. m6A RNA methylation correlated with doxorubicin resistance in colon cancer cells harboring p53 R273H mutations.

A, R273H mutation in p53. Codon 273 of p53 RNA was CAU, rather CGU, in TP53-Dox cells. B, Cell responses to doxorubicin. After pretreatments with siMETTL3 or NPC for 3 days, SW48-Dox (wild-type p53) and TP53-Dox (p53/R273H+) cells were treated with doxorubicin (Dox) combined with these agents for 72 hr. siMETTL3, 100 nM siRNA against human METTL3; siRNA-SC, 100 nM siRNA scrambled control; NPC, 20 nM neplanocin A. *, p<0.001, compared to SW48-Dox cells; ** or #, p<0.001 compared to TP53-Dox cells treated with vehicle or siRNA-SC (not depicted in this graph). C, IC50 values for Dox in cells treated with combinations of NPC, siMETTL3 or PDMP. *, p<0.001, compared to SW48-Dox cells; ** or #, p<0.001 compared to TP53-Dox cells treated with vehicle or siRNA-SC.
Table 1. IC50 values for doxorubicin and other drugs in cancer cells.
Cells were pretreated with METTL3 siRNA (100 nM), neplanocin A (NPC, 20 nM) or PDMP (5 μM). #, WiDr cells were pretreated with 0.5 μM NPC or 10 μM PDMP. *p<0.001 to SW48 cells; **, p<0.001 compared to vehicle control. Data presented as mean ± SD.
| Cell Lines and Pretreatments | Doxorubicin (nM) | Oxaliplatin (μM) | 5-Fluorouracil (μM) |
|---|---|---|---|
| SW48-Dox | 45.60 ± 8.02 | 2.21 ± 0.11 | 6.11 ± 0.79 |
| TP53-Dox | 599.9 ± 9.87* | 5.30 ± 0.83* | 12.5 ± 1.07* |
| SW48/5-FU | 83.60 ± 5.12* | 0.95 ± 0.04 | 7.6 ± 0.85 |
| TP53/5-FU | 413 ± 20.8* | 11.8 ± 4.39* | 17.05 ± 0.92* |
| WiDr | 740.88 ± 23.96* | 17.8 ± 0.98* | 19.11 ± 0.77* |
| METTL3 siRNA | |||
| SW48-Dox | 47.03 ± 3.07 | ||
| TP53-Dox | 55.27 ± 5.13** | ||
| Neplanocin A | |||
| SW48-Dox | 35.6 ± 6.1 | 1.09 ± 0.1 | 2.35 ± 0.16 |
| TP53-Dox | 51.17 ± 6.21** | 2.02 ± 0.37** | 7.30 ± 1.02** |
| WiDr# | 259.79 ± 35.81** | 9.07 ± 1.57** | 7.52 ± 0.57** |
| PDMP | |||
| SW48-Dox | 43.27 ± 3.74 | 2.05 ± 0.09 | 5.53 ± 0.65 |
| TP53-Dox | 49.42 ± 3.84** | 2.04 ± 0.31** | 4.59 ± 0.15** |
| WiDr# | 484.60 ± 11.23** | 12.05 ± 1.68** | 13.97 ± 2.73** |
Further, we employed small interfering RNA (siRNA) to silence METTL3 expression (siMETTL3), and found that siMETTL3 treatments (100 nM) substantially sensitized the response of drug-resistant TP53-Dox cells to anticancer drugs. To wit, the IC50 values for Dox decreased by 7-fold in TP53-Dox cells treated with siMETTL3 in combination with Dox (Figs 1b, 1c, Table 1). Also, based on reports that neplanocin A (NPC) could decrease METTL3 activity indirectly in m6A modification by inhibition of S-adenosylhomocysteine (SAH) hydrolase [39], we examined the responsiveness of TP53-Dox cells to drugs after NPC treatments (20 nM). We found that NPC, like siMETTL3, could also sensitize TP53-Dox cells to anticancer drugs, decreasing the IC50 values for Dox by 12-fold, for Oxa by 2-fold and for 5-FU by 2-fold, respectively, comparing to vehicle control (Figs 1B, 1C, Table 1).
In parallel experiments, treatments with the GCS inhibitor PDMP that sufficiently inhibited GCS, and consequently decreased glycosphingolipid production, significantly sensitized the responsiveness of TP53-Dox cells, but not SW48-Dox cells, to these same three drugs (Fig. 1C, Table 1). To wit, the IC50 values decreased, respectively, for Dox by 12-fold (Fig. 1C), and for Oxa or 5-FU, by 2-fold, in TP53-Dox cells treated with PDMP in individual combination with each of these three anticancer drugs (Table 1).
We further examined the effects of p53 R273H on apoptosis. Cells were pretreated with NPC (20 nM) or vehicle, and then exposed to Dox (100 nM, 48 hr) to induce apoptosis. Flow cytometry assay with a FITC BrdU kit showed that Dox induced apoptosis in SW48-Dox cells, whereas TP53-Dox cells displayed resistance to the apoptosis, as the apoptotic cells were approximately 4-fold fewer than those seen in SW48-Dox (5.3% vs. 25.6% of total cells) (Figs 2A, 2B). NPC treatments significantly increased Dox efficacy in inducing apoptosis in TP53-Dox cells, but notin SW48-Dox cells. In TP53-Dox cells treated with NPC, apoptotic cells significantly increased, by approximately 5-fold (25% vs. 5% of total cells), comparing to vehicle treatments (Figs 2A, 2B). Further, Western blotting analysis indicated that cleaved PARP, a DNA nick sensor that was cleaved from PARP by activated caspase-3 or caspase-7 in apoptosis, was significantly increased, by approximately 8-fold in TP53-Dox cells treated with NPC and Dox in combination (Fig. 2C). These results indicate that drug resistance of cancer cells heterozygously carrying R273H mutant TP53 highly correlates to activity of METTL3, an enzyme responsible for m6A RNA methylation.
Fig. 2. Inhibition of m6A methylation increased Dox-induced apoptosis in TP53-Dox cancer cells.

SW48-Dox and TP53-Dox cells were treated with neplanocin A (NPC, 20 nM) or vehicle for 3 days, and then exposed to Dox (100 nM, for 48 h) with these treatments to induce apoptosis. Imaging flow cytometry was accomplished with an ImageStream X Mark II system, and the data analyzed with IDEAS® Software. A, Apoptosis assay with imaging flow cytometry. BrdU, bromodeoxyuridine-FITC; BF, bright field; PI, propidium iodide. Top-panel, images of apoptotic and nonapoptotic cells of TP53-Dox cells identified using imaging flow cytometry for analysis. Apoptotic cells labelled with BrdU-FITC (green) are presented as percentages of total cells detected in each sample. B, NPC effects on apoptosis. **, p<0.001 compared to SW48-Dox cells; *, p<0.001 compared to cells treated with vehicle. C, Representative Western blot of PARP protein. After above treatments, equal amounts of detergent-soluble proteins extracted (50 μg/lane) were resolved using 4–20% gradient SDS-PAGE, and then immunoblotted with PARP antibody. Cleaved-P, cleaved PARP; GAPDH served as internal control. Protein levels (bottom panel) are represented as means of their optical densities normalized against GAPDH from three settings of blots. *, p<0.01 compared to corresponding cells treated with vehicle.
3.2. m6A Modification in pre-mRNA Transited Codon 273 Determines the Mutant Protein Expression in Cells Heterozygously Carrying TP53 R273H Mutation
We next wished to identify the roles of m6A modification in regulating protein expression of the R273H p53 mutant. TP53-Dox cells (heterozygous for the missense codon for R273) expressed mutant p53 protein, which could not be phosphorylated, with consequent loss of transactivation of p53 target genes or p53-responsive genes involved in cell proliferation arrest and apoptosis [19, 31]. The levels of phosphorylated p53 (pp53), as well as levels of p21, Bax and PUMA, in TP53-Dox cells, particularly those exposed to Dox (100 nM for last 48 h), were significantly lower than in SW48-Dox cells under the same conditions, due to the fact that in the latter cells, DNA damage stress could upregulate wt p53 expression (Figs. 3A, 3B). Interestingly, NPC treatments (20 nM) significantly increased the levels of pp53, by more than 7-fold (0.52 vs 0.07), and of p21, PUMA and Bax, by approximately 3-fold, in TP53-Dox cells, as compared to vehicle, respectively (Figs 3A). Treatments with PDMP, which inhibits GCS activity and the downstream synthesis of GSLs including globotriaosylceramide (Gb3) [43], decreased METTL3 protein levels by approximately 3-fold (0.31 vs 0.81), increased the pp53 levels by 6-fold, and increased p21 or PUMA and Bax levels by 3-fold, compared to vehicle treatments in TP53-Dox cells. The increases in protein levels of pp53, p21, Bax and PUMA brought about by NPC and PDMP were also observed for the treated TP53-Dox cells absent Dox exposure (Fig. 3B), although these differences were not significant (Fig. 3B), and overall the levels of these proteins in these cells were lower than in those subjected to Dox exposure that cause DNA damage stress (Figs. 3A, 3B). Importantly, treatments with NPC or PDMP were without significant effect on levels of pp53, or of p21, Bax or PUMA, in the wt p53 SW48-Dox cells (Figs 3A, 3B).
Fig. 3. m6A methylation determined missense protein expression in cells carrying p53 R273H mutation.


A, Protein expression of p53 and p53 target genes in cells exposed to Dox. Cells were treated with NPC (20 nM), PDMP (5 μM) or vehicle for 5 days, with exposed to Dox (100 nM) during the last 48 h of treatments. Equal amounts of detergent-soluble proteins extracted (50 μg/lane) were resolved using 4–20% gradient SDS-PAGE, and then immunoblotted with corresponding antibodies. Top panel, representative Western blots. METTL3, methyltransferase like 3; pp53, phosphorylated p53 (Ser15). Protein levels (bottom panel) are represented as mean ± SD of their optical densities normalized against GAPDH from three settings of blots. *, p<0.001 compared to SW48-Dox cells; #, p<0.001 compared to TP53-Dox cells treated with vehicle. B, Protein Expression of p53 and p53 target genes in cells exposed to vehicle. Cells were treated with NPC (20 nM), PDMP (5 μM) or vehicle for 5 days, with exposed to vehicle during the last 48 h of treatments. C, Immunostaining for METTL3 and pp53. Scale bar represents 25 μM in photomicrographs (100× magnification). Red, METTL3-Qdot 605; green, pp53-Alexa Fluor 488. D, Western blotting of p53 and pp53 after immunoprecipitation (IP). Cells were treated with PDMP (5 μM, 6 days) or vehicle, with exposed to Dox (100 nM) during the last 48 h of treatments. Equal amounts of detergent-soluble proteins (500 μg) were precipitated with indicated antibody and then immunoblotted after non-denaturing 12% PAGE resolution. Relative levels of p53 or pp53 are represented as means of their optical densities normalized against SW48-Dox cells with vehicle. *, p<0.001 compared to SW48-Dox with vehicle; **, p<0.001 compared to TP53-Dox with vehicle. E, MS/MS proteomics analysis of p53. Equal amounts of proteins were precipitated with pp53 antibody, resolved and assessed via mass spectroscopic (MS/MS) analysis.
In corroboration with the above-described body of results, an inverse correlation of pp53 expression to m6A modification with METTL3 was comparably observed in TP53-Dox cells upon immunofluorescence staining. Increased levels of pp53 displayed prominently in cell nuclei (green, pp53-Alexa Fluor 488) of TP53-Dox cells treated with PDMP (GCS inhibition), with decreased METTL3 protein levels (red, METTL3-Qdot 605), and likewise when these cells were alternatively treated with NPC (inhibition m6A methylation), in both cases comparing to TP53-Dox cells treated with vehicle (Fig. 3C). Furthermore, we identified p53 protein after immunoprecipitation (IP) with anti-p53 or anti-pp53 antibodies. Western blotting after IP showed that levels of wt p53 (pp53) were suppressed in TP53-Dox cells as compared to SW48-Dox, and PDMP treatments restored the pp53 levels in TP53-Dox cells (Fig. 3D). Based on two peptides (amino acids 102–110: TYQGSYGFR; amino acids 307–319: ALPNNTSSPQPK), tandem mass spectrometry (MS/MS) analysis of proteins resolved by SDS-PAGE identified p53 protein expressed in SW48-Dox and TP53-Dox cells treated with PDMP, but not TP53-Dox in the absence of PDMP treatment (Fig 3D, 3E).
In human cells, METTL3 is an essential methyltransferase for the methylation of adenosines to generate m6As in RNA [29, 30]. As anticipated, siRNA silencing of METTL3 expression reduced METTL3 protein levels to 20% vs. vehicle treatment in TP53-Dox cells, which, interestingly, concomitantly enhanced the pp53 levels 5-fold over the levels seen for vehicle control treatment (Fig. 4A). These results clearly implicate m6A methylation catalyzed by METTL3 (Fig. 4B) as being involved in regulating the expression of mut-p53 vs. wt-p53 protein in cells heterozygously carrying the p53 R273H mutation. Perhaps more noteworthy, however, was that PDMP treatments also reduced METTL3 protein levels, to 20% vs. vehicle, once again concomitantly enhancing pp53 protein levels by more than 5-fold (Fig 4A).
Fig. 4. m6A methylation in the mRNA of p53 R273H.

A, Silencing METTL3 enhanced pp53 levels in TP53-Dox cells. TP53-Dox cells were treated with siRNA against METTL3 (100 nM, 6 days) and PDMP (5 μM, 6 days). Equal amounts of detergent-soluble proteins extracted (50 μg/lane) were resolved using 4–20% gradient SDS-PAGE, and then immunoblotted with corresponding antibodies. pp53, phosphorylated p53 (Ser15); METTL3, methyltransferase-like 3. Protein levels are represented below bands, as their OD values normalized against GAPDH from three settings of blots and compared to vehicle treatment. *, p<0.001 compared to vehicle. B, Methylation to m6A catalyzed by METTL3. C, RNA m6A methylation in cells. Equal amounts of total RNA extracted from cells of SW48-Dox (wt p53), TP53-Dox (TP53; R273H/+), WiDr (R273H+/+) and TP53-Dox treated with neplanocin A (NPC, 20 nM, 6 days) or PDMP (5 μM, 6 days) were assessed with MeRIP RT-qPCR. *, p<0.001, compared to SW48-Dox cells; **, p<0.001, compared to vehicle control in TP53-Dox cells. D, Real-time PCR chart, as described above.
To directly identify the presence of m6A in R273H pre-mRNA, we precipitated RNA transcripts with anti-m6A antibody, and then analyzed a region of p53 mRNA that included the segment coding for R273 (p53-c273, 95 bp, codons 249–280) using quantitative RT-PCR (MeRIP qRT-PCR). The levels of m6A p53-c273 in TP53-Dox and in WiDr cells were 16-fold (330 vs. 20 pg/μg RNA) and 19-fold (376 vs. 20 pg/μg RNA) higher, respectively, than in SW48-Dox cells (Fig 4C, 4D). These results directly evidenced that the adenosine transited from guanosine at codon 273 of the mutant pre-mRNA (-CAU-), due to DNA point mutation (Fig. 1A), was extensively converted to m6A by METTL3 (Fig. 4B). NPC treatments significantly decreased m6A p53-c273 levels, by 2-fold (140 vs. 329 pg/μg RNA), in TP53-Dox cells, as compared to vehicle control (Fig. 4C, 4D). Perhaps more interestingly, PDMP treatments also decreased m6A p53-c273 levels, and to a greater degree (by 9-fold: 36 vs. 329 pg/μg RNA), in TP53-Dox cells, as compared to vehicle. Altogether, current work directly pinpoints that m6A production, which only appears in the adenosine-transited codon of the R273H-mutant p53 pre-mRNA, positively regulates missense protein expression in cancer cells carrying p53 R273H mutations. This result also implies that inhibition of METTL3-catalyzed RNA methylation of R273H-mutant might be an effective approach for restoring wt p53 expression in cells heterozygously carrying R273H.
3.3. Glycosphingolipid Gb3 Mediates Signaling of cSrc and β-catenin to Increase METTL3 Expression and RNA m6A Methylation
To explore how GSLs modulate m6A methylation and protein expression of the p53 R273H mutant, we assessed and characterized the effects of ceramide, Gb3, cSrc and β-catenin on METTL3 expression in TP53-Dox cells. GCS, a rate-limiting enzyme in GSL synthesis, converts ceramide to glucosylceramide (GlcCer), which serves as a precursor for production of further-elaborated GSLs, including globo-series GSLs and gangliosides, upon serial glycosylation in cells. Previous works indicate that certain globo-series GSLs (in particular, globotriaosylceramide Gb3) play important roles in modulating transcription of gene expression via cSrc and β-catenin signaling pathways [36, 37]. It has been elucidated that the active proto-oncogene protein cSrc elevates the levels of β-catenin, which in turn upregulates the expression of cyclin D1, c-Myc, MDR1 and fibroblast growth factor 2 (FGF2) [36, 37, 44]. PDMP treatment, which inhibits GCS activity and ceramide glycosylation [19], reduced levels of phosphorylated cSrc (pcSrc), β-catenin and METTL3, and thence restored pp53 levels (Fig. 5A, 5B); however, fumonisin B1 (FB1), an inhibitor of ceramide synthase [27, 45], could not affect the levels of these proteins (inclusive of pp53) in TP53-Dox cells (Fig. 5A, 5B). These results indicate that GSLs, rather than ceramides, modulate METTL3 expression.
Fig. 5. Globotriaosylceramide modulates METTL3 and m6A methylation on p53 R273H mutant.

A, Representative Western blot. TP53-Dox cells were treated with PDMP (5 μM), FB1 (fumonisin B1, 25 μM), siGb3S (siRNA against Gb3 synthase, 100 nM), STxB (Shiga toxin 1 B subunit, 100 nM), PP2 (Src kinase inhibitor, 500 nM) or FH535 (β-catenin/Tcf inhibitor, 5 μM) for 6 days, with exposure to doxorubicin (100 nM) during the last 48 h of treatments. Equal amounts of detergent-soluble proteins (50 μg/lane) were resolved and then immunoblotted with corresponding antibodies. B, Relative protein levels in TP53-Dox cells with treatments. Protein levels are represented as mean ± SD of their OD values normalized against GAPDH from three settings of blots. *, p<0.001 compared to vehicle. #, p<0.001 compared to PDMP treatment. C, IC50 values for Dox in cells after treatments. TP53-Dox cells were pretreated with PDMP (5 μM), FB1 (25 μM), siGb3S (100 nM), STxB (100 nM), PP2 (10 μM) or FH535 (5 μM) for 48 h, and then co-treated with these agents and various concentrations of Dox for an additional 72 h. *, p<0.001 compared to vehicle control. #, p<0.001 compared to PDMP treatments.
GSLs are tightly associated with membrane proteins and signal transducers in GSL-enriched microdomains (GEMs, also named lipid rafts), and the downstream functional effects of cSrc kinase correlate with Gb3 alterations [46, 47]. Silencing the expression of Gb3 synthase with siRNA (siGb3S) to decrease Gb3 synthesis, or disrupting the interaction of GSLs (Gb3) with other proteins in GEMs with Shiga toxin 1B subunit (STxB, a specific ligand for Gb3) [48], significantly suppressed the levels of pcSrc, β-catenin and METTL3, while increasing pp53 levels, in TP53-Dox cells (Fig. 5A, 5B). Furthermore, inhibition of cSrc with PP2 (4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine, a selective Src kinase inhibitor) significantly decreased the levels of cSrc, β-catenin and METTL3 while restoring pp53 levels in TP53-Dox cells. Suppression of the transactivation effects of β-catenin/Tcf with FH535 ((N-(2-methyl-4-nitro)-2,4-dichlorosulfonamide, a β-catenin/Tcf inhibitor) significantly decreased the protein levels of β-catenin and METTL3, and then increased the levels of pp53, in TP53-Dox cells (Fig. 5A, 5B). Furthermore, consistent with the significantly increased levels of pp53, TP53-Dox cells were sensitive to Dox after inhibiting ceramide glycosylation with PDMP (Fig. S2), Gb3 synthesis with siGb3S, GEM interactions with STxB, cSrc kinase activity with PP2, and β-catenin transactivation with FH535, but not upon inhibiting ceramide synthesis with FB1 (Fig. 5C). The IC50 values for Dox in TP53-Dox cells significantly decreased, by more than 12-fold, upon treatment with PDMP, and by approximately 5-fold upon treatments with siGb3S, STxB, PP2 or FH535, as compared to vehicle treatments (Fig. 5C).
4. Discussion
Our present study elucidated that methylation of adenosine in TP53 codon R273H RNA transcripts promotes the selection of m6A-R273H pre-mRNA for splicing and mutant protein expression, which in turn results in drug resistance in cancer cells heterozygously carrying TP53 R273H mutation. Both accumulation of genetic mutations and dysregulated RNA processing occur in cancer cells, and it has become increasingly clear that the latter contributes key components of cancerous behaviors and tumor progression. Recent studies showed that RNA methylation converting adenosine into m6A, and preferential pre-mRNA splicing brought about as a consequence of enhanced pre-mRNA recognition and processing by YTHDF1, YTHDF2 and other m6A-readers, can globally either upregulate or down-regulate expression of genes, including NANOG, POU5F1, KLF4, SOX2, c-MYC, EGFR, ASB2 and RARA, in ways that enrich cancer stem cells and promote tumor progression [49, 50]. Our present study, for the first time, indicates that methylation of a single transited adenosine in a mutant codon of pre-mRNA can determine mutant protein expression from heterozygous TP53 R273H mutation. To wit, selective expression of the R273H mutant protein in cancer cells relies on the m6A at codon 273, and relevant premRNA structure, which together confer preference for binding to spliceosome and splicing [51]. G-to-A transition is the most common base substitution in gene mutation, and has been reported in other codons (R175H, R248Q) of TP53 missense mutations, as well as in many other genes [7, 9, 52]. Whether or not m6A production in these cases also leads to preferential splicing to regulate mutant protein expression needs to be investigated in further studies.
TP53 missense mutation is an attractive specific target for improving cancer treatments, and abolishing preferential p53 mutant protein expression might prove to be more an efficacious strategy thereto. DNA damage stress in cancer cells exposed to anticancer drugs can upregulate expression of mutant proteins, such as R273H, when mutated genes coding for them are present, which at minimum invariably results in drug resistance, thereby compromising the efficacy of treatments [53] (Fig. 1, Table 1). We hereby advance suppression of m6A levels as a possible approach for quashing mutant p53 protein expression while restoring wt p53 protein expression and function, so as to efficaciously sensitize cancer cells that carry heterozygous R273H mutation (Fig. 1, Table 1).
Our current studies also revealed that glycosphingolipids, in particular Gb3, can upregulate the expression of METTL3 via promotion of cSrc/β-catenin signaling, thereby enhancing RNA methylation so as to facilitate or favor mutant protein expression and consequent drug resistance. Devising suitable ways of targeting these pivotal pathways, perhaps most profitably in combination, should provide the basis for clinically viable and valuable therapeutic approaches that will improve treatments of cancers found to carry comparably activing mutations of tumor suppressor genes.
Altogether, our study uncovers a novel function of a pre-mRNA m6A in determining mutant protein expression, in this case for the TP53 R273H mutation. Moreover, we found that ceramide glycosylation in cross-talk with RNA methylation contributes to mutant protein expression and cancer drug resistance in cancer cells carrying this missense mutation. Therefore, suppressing m6A-RNA methylation or/and ceramide glycosylation might constitute a specific and highly efficacious approach for targeting R273H TP53 missense mutation.
Acknowledgements
This work was supported by National Institutes of Health Grants (R15CA167476) from the National Cancer Institute and (P20GM103424–11) from the National Institute of General Medical Sciences (to Y.Y.L.). This work was also partially supported by National Institutes of Health Grant (P20GM121293) from the National Institute of General Medical Sciences (to A.J.T.).
Abbreviations:
- FB1
fumonisin B1
- Gb3
obotriaosylceramide
- GCS
glucosylceramide synthase
- GOF
gain-of-function; G>A, guanine transited adenosine
- m6A
N6-methyladenosine
- MeRIP
N6-methyladenosine RNA immunoprecipitation
- METTL3
methyltransferase like 3
- NPC
neplanocin A
- Oxa
oxaliplatin
- PARP
poly(ADP-ribose) polymerase (PARP
- PDMP
d-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol
- pp53
phosphorylated p53 (Ser15)
- siRNA
silencing RNA
- SRSF
serine/arginine-rich splicing factor
- STxB
Shiga toxin 1 B-subunit
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author disclose statement
Authors claim no competing financial interests exist.
References
- [1].Fischer M, Census and evaluation of p53 target genes, Oncogene 36(28) (2017) 3943–3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lane DP, Cancer. p53, guardian of the genome, Nature 358(6381) (1992) 15–6. [DOI] [PubMed] [Google Scholar]
- [3].Harris SL, Levine AJ, The p53 pathway: positive and negative feedback loops, Oncogene 24(17) (2005) 2899–908. [DOI] [PubMed] [Google Scholar]
- [4].Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W, Ferroptosis as a p53-mediated activity during tumour suppression, Nature 520(7545) (2015) 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Soussi T, Wiman KG, TP53: an oncogene in disguise, Cell Death Differ 22(8) (2015) 1239–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MD, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, Wilson RK, Raphael BJ, Ding L, Mutational landscape and significance across 12 major cancer types, Nature 502(7471) (2013) 333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, Olivier M, TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data, Hum Mutat 37(9) (2016) 865–76. [DOI] [PubMed] [Google Scholar]
- [8].Sabapathy K, Lane DP, Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others, Nat Rev Clin Oncol 15(1) (2018) 13–30. [DOI] [PubMed] [Google Scholar]
- [9].Leroy B, Anderson M, Soussi T, TP53 mutations in human cancer: database reassessment and prospects for the next decade, Hum Mutat 35(6) (2014) 672–88. [DOI] [PubMed] [Google Scholar]
- [10].Muller PA, Vousden KH, p53 mutations in cancer, Nat Cell Biol 15(1) (2013) 2–8. [DOI] [PubMed] [Google Scholar]
- [11].Freed-Pastor WA, Prives C, Mutant p53: one name, many proteins, Genes Dev 26(12) (2012) 1268–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nigro JM, Baker SJ, Preisinger AC, Jessup JM, Hostetter R, Cleary K, Bigner SH, Davidson N, Baylin S, Devilee P, et al. , Mutations in the p53 gene occur in diverse human tumour types, Nature 342(6250) (1989) 705–8. [DOI] [PubMed] [Google Scholar]
- [13].Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T, Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome, Cell 119(6) (2004) 847–60. [DOI] [PubMed] [Google Scholar]
- [14].Iacopetta B, Russo A, Bazan V, Dardanoni G, Gebbia N, Soussi T, Kerr D, Elsaleh H, Soong R, Kandioler D, Janschek E, Kappel S, Lung M, Leung CS, Ko JM, Yuen S, Ho J, Leung SY, Crapez E, Duffour J, Ychou M, Leahy DT, O’Donoghue DP, Agnese V, Cascio S, Di Fede G, Chieco-Bianchi L, Bertorelle R, Belluco C, Giaretti W, Castagnola P, Ricevuto E, Ficorella C, Bosari S, Arizzi CD, Miyaki M, Onda M, Kampman E, Diergaarde B, Royds J, Lothe RA, Diep CB, Meling GI, Ostrowski J, Trzeciak L, Guzinska-Ustymowicz K, Zalewski B, Capella GM, Moreno V, Peinado MA, Lonnroth C, Lundholm K, Sun XF, Jansson A, Bouzourene H, Hsieh LL, Tang R, Smith DR, Allen-Mersh TG, Khan ZA, Shorthouse AJ, Silverman ML, Kato S, Ishioka C, Functional categories of TP53 mutation in colorectal cancer: results of an International Collaborative Study, Ann Oncol 17(5) (2006) 842–7. [DOI] [PubMed] [Google Scholar]
- [15].Russo A, Bazan V, Iacopetta B, Kerr D, Soussi T, Gebbia N, The TP53 colorectal cancer international collaborative study on the prognostic and predictive significance of p53 mutation: influence of tumor site, type of mutation, and adjuvant treatment, J Clin Oncol 23(30) (2005) 7518–28. [DOI] [PubMed] [Google Scholar]
- [16].Lakin ND, Jackson SP, Regulation of p53 in response to DNA damage, Oncogene 18(53) (1999) 7644–55. [DOI] [PubMed] [Google Scholar]
- [17].Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, Bergman J, Wiman KG, Selivanova G, Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound, Nat Med 8(3) (2002) 282–8. [DOI] [PubMed] [Google Scholar]
- [18].Lambert JM, Gorzov P, Veprintsev DB, Soderqvist M, Segerback D, Bergman J, Fersht AR, Hainaut P, Wiman KG, Bykov VJ, PRIMA-1 reactivates mutant p53 by covalent binding to the core domain, Cancer Cell 15(5) (2009) 376–88. [DOI] [PubMed] [Google Scholar]
- [19].Hosain SB, Khiste SK, Uddin MB, Vorubindi V, Ingram C, Zhang S, Hill RA, Gu X, Liu YY, Inhibition of glucosylceramide synthase eliminates the oncogenic functionof p53 R273H mutant in the epithelial-mesenchymal transition and induced pluripotency of colon cancer cells, Oncotarget 7(37) (2016) 60575–60592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Baralle FE, Giudice J, Alternative splicing as a regulator of development and tissue identity, Nat Rev Mol Cell Biol 18(7) (2017) 437–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lee Y, Rio DC, Mechanisms and Regulation of Alternative Pre-mRNA Splicing, Annu Rev Biochem 84 (2015) 291–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Scotti MM, Swanson MS, RNA mis-splicing in disease, Nat Rev Genet 17(1) (2016) 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP, ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing, Mol Cell 33(5) (2009) 591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shilo A, Siegfried Z, Karni R, The role of splicing factors in deregulation of alternative splicing during oncogenesis and tumor progression, Mol Cell Oncol 2(1) (2015) e970955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chalfant CE, Rathman K, Pinkerman RL, Wood RE, Obeid LM, Ogretmen B, Hannun YA, De novo ceramide regulates the alternative splicing of caspase 9 and Bcl-x in A549 lung adenocarcinoma cells. Dependence on protein phosphatase-1, J Biol Chem 277(15) (2002) 12587–95. [DOI] [PubMed] [Google Scholar]
- [26].Massiello A, Chalfant CE, SRp30a (ASF/SF2) regulates the alternative splicing of caspase-9 pre-mRNA and is required for ceramide-responsiveness, J Lipid Res 47(5) (2006) 892–7. [DOI] [PubMed] [Google Scholar]
- [27].Patwardhan GA, Hosain SB, Liu DX, Khiste SK, Zhao Y, Bielawski J, Jazwinski SM, Liu YY, Ceramide modulates pre-mRNA splicing to restore the expression of wild-type tumor suppressor p53 in deletion-mutant cancer cells, Biochim Biophys Acta 1841(11) (2014) 1571–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, Sorek R, Rechavi G, Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq, Nature 485(7397) (2012) 201–6. [DOI] [PubMed] [Google Scholar]
- [29].Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, Dai Q, Chen W, He C, A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation, Nat Chem Biol 10(2) (2014) 93–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lin S, Choe J, Du P, Triboulet R, Gregory RI, The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells, Mol Cell 62(3) (2016) 335–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA Jr., Kinzler KW, Vogelstein B, Papadopoulos N, A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53, Proceedings of the National Academy of Sciences of the United States of America 106(10) (2009) 3964–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Liu YY, Patwardhan GA, Bhinge K, Gupta V, Gu X, Jazwinski SM, Suppression of glucosylceramide synthase restores p53-dependent apoptosis in mutant p53 cancer cells, Cancer Res 71(6) (2011) 2276–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Henery S, George T, Hall B, Basiji D, Ortyn W, Morrissey P, Quantitative image based apoptotic index measurement using multispectral imaging flow cytometry: a comparison with standard photometric methods, Apoptosis 13(8) (2008) 1054–63. [DOI] [PubMed] [Google Scholar]
- [34].Xiang Y, Laurent B, Hsu CH, Nachtergaele S, Lu Z, Sheng W, Xu C, Chen H, Ouyang J, Wang S, Ling D, Hsu PH, Zou L, Jambhekar A, He C, Shi Y, RNA m6A methylation regulates the ultraviolet-induced DNA damage response, Nature 543(7646) (2017) 573–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liu YY, Yu JY, Yin D, Patwardhan GA, Gupta V, Hirabayashi Y, Holleran WM, Giuliano AE, Jazwinski SM, Gouaze-Andersson V, Consoli DP, Cabot MC, A role for ceramide in driving cancer cell resistance to doxorubicin, FASEB J 22(7) (2008) 2541–51. [DOI] [PubMed] [Google Scholar]
- [36].Liu YY, Gupta V, Patwardhan GA, Bhinge K, Zhao Y, Bao J, Mehendale H, Cabot MC, Li YT, Jazwinski SM, Glucosylceramide synthase upregulates MDR1 expression in the regulation of cancer drug resistance through cSrc and beta-catenin signaling, Mol Cancer 9 (2010) 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gupta V, Bhinge KN, Hosain SB, Xiong K, Gu X, Shi R, Ho MY, Khoo KH, Li SC, Li YT, Ambudkar SV, Jazwinski SM, Liu YY, Ceramide glycosylation by glucosylceramide synthase selectively maintains the properties of breast cancer stem cells, J Biol Chem 287(44) (2012) 37195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sengupta D, Byrum SD, Avaritt NL, Davis L, Shields B, Mahmoud F, Reynolds M, Orr LM, Mackintosh SG, Shalin SC, Tackett AJ, Quantitative Histone Mass Spectrometry Identifies Elevated Histone H3 Lysine 27 (Lys27) Trimethylation in Melanoma, Mol Cell Proteomics 15(3) (2016) 765–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Borcherding DR, Narayanan S, Hasobe M, McKee JG, Keller BT, Borchardt RT, Potential inhibitors of S-adenosylmethionine-dependent methyltransferases. 11. Molecular dissections of neplanocin A as potential inhibitors of S-adenosylhomocysteine hydrolase, J Med Chem 31(9) (1988) 1729–38. [DOI] [PubMed] [Google Scholar]
- [40].Chen JK, Capdevila J, Harris RC, Overexpression of C-terminal Src kinase blocks 14, 15-epoxyeicosatrienoic acid-induced tyrosine phosphorylation and mitogenesis, J Biol Chem 275(18) (2000) 13789–92. [DOI] [PubMed] [Google Scholar]
- [41].Handeli S, Simon JA, A small-molecule inhibitor of Tcf/beta-catenin signaling down-regulates PPARgamma and PPARdelta activities, Mol Cancer Ther 7(3) (2008) 521–9. [DOI] [PubMed] [Google Scholar]
- [42].Stone SM, Thorpe CM, Ahluwalia A, Rogers AB, Obata F, Vozenilek A, Kolling GL, Kane AV, Magun BE, Jandhyala DM, Shiga toxin 2-induced intestinal pathology in infant rabbits is A-subunit dependent and responsive to the tyrosine kinase and potential ZAK inhibitor imatinib, Front Cell Infect Microbiol 2 (2012) 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Abe A, Wild SR, Lee WL, Shayman JA, Agents for the treatment of glycosphingolipid storage disorders, Curr Drug Metab 2(3) (2001) 331–8. [DOI] [PubMed] [Google Scholar]
- [44].Chang YM, Bai L, Liu S, Yang JC, Kung HJ, Evans CP, Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530, Oncogene 27(49) (2008) 6365–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Meivar-Levy I, Futerman AH, Up-regulation of neutral glycosphingolipid synthesis upon long term inhibition of ceramide synthesis by fumonisin B1, J Biol Chem 274(8) (1999) 4607–12. [DOI] [PubMed] [Google Scholar]
- [46].Steelant WF, Kawakami Y, Ito A, Handa K, Bruyneel EA, Mareel M, Hakomori S, Monosialyl-Gb5 organized with cSrc and FAK in GEM of human breast carcinoma MCF-7 cells defines their invasive properties, FEBS Lett 531(1) (2002) 93–8. [DOI] [PubMed] [Google Scholar]
- [47].Kajiwara K, Yamada T, Bamba T, Fukusaki E, Imamoto F, Okada M, Oneyama C, c-Src-induced activation of ceramide metabolism impairs membrane microdomains and promotes malignant progression by facilitating the translocation of c-Src to focal adhesions, Biochem J 458(1) (2014) 81–93. [DOI] [PubMed] [Google Scholar]
- [48].Kovbasnjuk O, Mourtazina R, Baibakov B, Wang T, Elowsky C, Choti MA, Kane A, Donowitz M, The glycosphingolipid globotriaosylceramide in the metastatic transformation of colon cancer, Proceedings of the National Academy of Sciences of the United States of America 102(52) (2005) 19087–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Dai D, Wang H, Zhu L, Jin H, Wang X, N6-methyladenosine links RNA metabolism to cancer progression, Cell Death Dis 9(2) (2018) 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Jaffrey SR, Kharas MG, Emerging links between m6A and misregulated mRNA methylation in cancer, Genome Med 9(1) (2017) 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Xu C, Wang X, Liu K, Roundtree IA, Tempel W, Li Y, Lu Z, He C, Min J, Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain, Nat Chem Biol 10(11) (2014) 927–9. [DOI] [PubMed] [Google Scholar]
- [52].Gold B, Somatic mutations in cancer: Stochastic versus predictable, Mutat Res 814 (2017) 37–46. [DOI] [PubMed] [Google Scholar]
- [53].Brosh R, Rotter V, When mutants gain new powers: news from the mutant p53 field, Nature reviews 9(10) (2009) 701–13. [DOI] [PubMed] [Google Scholar]