

Abstract
The Plasmodium proteasome is an emerging antimalarial target due to its essential role in all the major life cycle stages of the parasite and its contribution to the establishment of resistance to artemisinin (ART)-based therapies. However, because of a similarly essential role for the host proteasome, the key property of any antiproteasome therapeutic is selectivity. Several parasite-specific proteasome inhibitors have recently been reported, however, their selectivity must be improved to enable clinical development. Here we describe screening of diverse libraries of non-natural synthetic fluorogenic substrates to identify determinants at multiple positions on the substrate that produce enhanced selectivity. We find that selection of an optimal electrophilic “warhead” is essential to enable high selectivity that is driven by the peptide binding elements on the inhibitor. We also find that host cell toxicity is dictated by the extent of coinhibition of the human β2 and β5 subunits. Using this information, we identify compounds with over 3 orders of magnitude selectivity for the parasite enzyme. Optimization of the pharmacological properties resulted in molecules that retained high potency and selectivity, were soluble, sufficiently metabolically stable and orally bioavailable. These molecules are highly synergistic with ART and can clear parasites in a mouse model of infection, making them promising leads as antimalarial drugs.
Graphical Abstract
INTRODUCTION
Plasmodium falciparum (Pf) is a protozoan parasite that causes the most severe form of malaria in humans. P. falciparum malaria remains a significant burden, leading to approximately 400 000 deaths annually with most being children under the age of five.1 Emerging resistance to the highly effective drug artemisinin (ART) and the partner drugs used in ART-based combination therapies (ACTs) is particularly worrisome as these drugs are used worldwide as first-line therapy. Many of the older drugs have already been compromised by the emergence and spread of resistance.2–6 Therefore, there is an urgent need for new drugs with novel mechanism of action and broad therapeutic potential, not only to widen the scope of treatment, but also to overcome existing multidrug resistance.
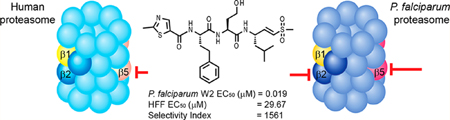
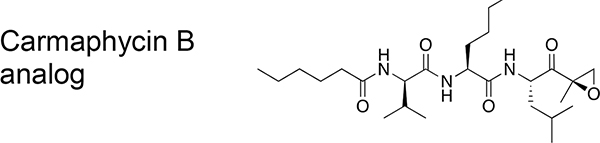
The Plasmodium proteasome has been validated as a promising target for antimalarial drugs because inhibitors of this essential enzyme attenuate parasite growth at all stages of its life cycle.7–12 More importantly, recent work has shown that proteasome inhibitors have a high degree of synergism when combined with ART.13,14 However, the main challenge for the development of proteasome inhibitors as antimalarial agents is optimization of selectivity for the parasite enzyme over its human counterpart. Understanding differences between the parasite and human proteasome ligand-binding preferences is therefore of paramount importance. We recently used a screen of natural peptide substrates15 coupled with high resolution cryo-electron microscopy (cryo-EM) and single particle analysis16 to identify a first generation of covalent irreversible inhibitors of P. falciparum proteasome that are over 100 times more potent for the parasite relative to the human proteasome.13 Our original lead vinyl sulfone molecule, WLL-vs (Figure 1) also showed 2 orders of magnitude selectivity for killing parasites compared to primary human cells, suggesting it has a relatively good “therapeutic window” where it could be dosed without toxicity to the host. In addition, a recent study has described a highly selective and potent inhibitor of the Plasmodium proteasome based on the epoxyketone-based carmaphycin B scaffold that was originally isolated from extracts derived from cyanobacterium Symploca sp.17 (Figure 1). This analog containing a D-amino acid at the P3 position shows similar potency and overall selectivity as the WLL vinyl sulfone but was not tested in mouse models of infection. However, while both classes of reported selective inhibitors effectively kill parasites at low nanomolar concentrations, both retain micromolar potency for the host proteasome, suggesting that additional optimization will be required to avoid long-term toxicity issues. In addition, due to the unique disease epidemiology of malaria, there is a need to optimize lead drug candidates for sufficient stability, blood exposure levels, and oral uptake. These are all criteria that have not been addressed for any of the currently published Plasmodium-specific proteasome inhibitors.
Figure 1.

Chemical structures of the Plasmodium-selective proteasome inhibitors WLL-vs and the recently reported Carmaphycin B analog. EC50 values and selectivity index values are shown for each compound. HFF = human foreskin fibroblasts, and EC50 = half maximal effective concentration.
This study reports our efforts to define the determinants of specificity of Plasmodium-specific proteasome inhibitors and use this information to further enhance the specificity of lead molecules and tune their pharmacological properties. We accomplished this goal by synthesizing and screening a library of fluorogenic peptide substrates that contain a range of diverse non-natural amino acids selected to be optimal for binding based on our prior high-resolution electron microscopy (EM) structure of the P. falciparum proteasome.16 Using this biased substrate library, we identified non-natural amino acid residues in multiple positions on the substrate that could be used to build a set of second-generation vinyl sulfone inhibitors with substantially improved selectivity over the original WLL-vs lead molecule. This selectivity for parasites was primarily derived from reduced activity against the human β2 subunit even after prolonged exposure times. We also examined the role of the reactive electrophile in controlling inhibitor specificity. These studies demonstrate that specificity gains resulting from selection of optimal peptide sequences in the backbone were lost when using the more reactive epoxyketone and boronate “warheads” which retained significant reactivity toward the human β2 subunit resulting in host cell toxicity. Finally, optimization of solubility and microsome stability resulted in a lead molecule with favorable in vivo properties and some oral bioavailability. This lead molecule showed strong synergy with ART and was able to clear parasites in a mouse model of infection, making it a promising lead for use in optimizing oral formulations and progressing to IND enabling studies.
RESULTS
Identification of Non-Natural Amino Acids That Enhance Selectivity for the Plasmodium Proteasome.
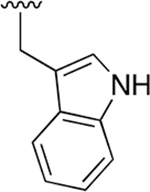
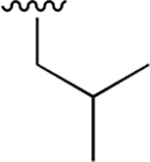
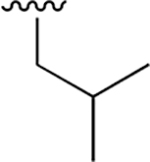
Our previous studies of the P. falciparum proteasome demonstrated that bulky aromatic residues in the P3 position of a substrate provide a key determinant for driving selectivity toward the parasite enzyme and away from the human enzyme.13 We therefore synthesized a set of 6 tripeptide vinyl sulfones that were identical in structure to the original WLL-vs inhibitor except for the residue in the P3 position, which was varied through a series of commercially available bulky aromatic non-natural amino acids (compounds 1–6, Table 1). To assess the potency and selectivity of the compounds, we treated ring stage P. falciparum W2 infected red blood cell cultures with compounds either for 1 h followed by washout (1 h EC50) or treated with no wash out, followed by measurement of the parasite viability 72 h later (72 h EC50). We also performed the same treatment on nonconfluent, replicating human foreskin fibroblast (HFF) cells to assess overall host cell toxicity. We chose HFF because they are primary cells that are likely to be more sensitive to toxicity of compounds compared to immortalized cell lines that are often used to assess host cell toxicity (i.e., HepG2 cells). We found that small substitutions on the para position of the aromatic ring of phenylalanine (methyl and fluoro) dramatically reduced potency and selectivity. However, a compound containing the large 1-naphthylalanine (compound 1) exhibited comparable activity to the parent WLL-vs (EC50 = 10 nM) and had slightly improved selectivity. Interestingly, the 2-naphthyl group caused a drop-in potency (compound 2; EC50 = 90 nM), which also translated into a lower overall selectivity. The homophenylalanine (Hfe) substitution (compound 3), however, resulted in substantially increased potency (EC50 = 3 nM) coupled with a 5-fold increase in selectivity (therapeutic index of 570) compared to WLL-vs and carmaphycin B analog (Table 1). We also made a version of compound 3 in which we replaced the P2 leucine with a methylserine (Ser(Me); compound 7) to enhance solubility and metabolic stability. This non-natural residue was chosen based on its use in orally available inhibitors of the human proteasome.18 Compound 7 showed slightly increased overall selectivity compared to 3 but it lost potency in parasite killing (Table 1). Finally, our prior EM structure suggested that the S1 pocket on the β2 and β5 subunits of the parasite proteasome could accommodate larger residues than the human enzyme. As a result, we also synthesized compounds 8–10 (Table 1) that contained a para-methylphenylalanine residue in the P1 position. These compounds, while highly potent for the parasite enzyme, also became more potent for the human enzyme resulting in an overall drop in selectivity. We therefore chose to focus on the scaffold from compound 7 for further optimization.
Table 1.
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. | Cap | R3 | R2 | R1 | 72 h EC50 (μM) | 1 h EC50 (μM) | ||||
| Pf | HFF | SI | Pf | HFF | SI | |||||
| WLL -vs |  |
 |
 |
 |
0.015 ± 0.005 | 1.52 ±0.41 | 101 | 0.147 ± 0.009 | 65.50 ± 15.2 | 446 |
| 1 | 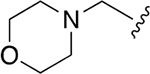 |
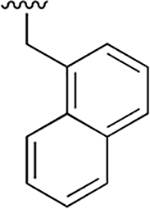 |
 |
 |
0.010 ± 0.002 | 1.80 ±0.39 | 178 | 0.061 ±0.011 | nd | nd |
| 2 | 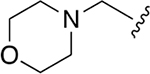 |
 |
 |
 |
0.090 ±0.016 | 1.67 ±0.26 | 19 | 0.495 ± 0.058 | nd | nd |
| 3 |  |
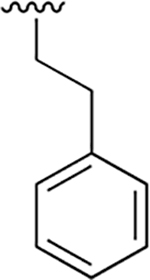 |
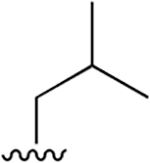 |
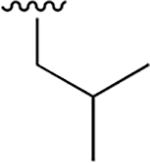 |
0.003 ± 0.0006 | 1.88 ±0.46 | 570 | 0.048 ± 0.006 | 75.65 ± 12.68 | 1576 |
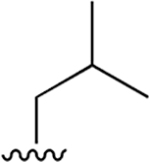
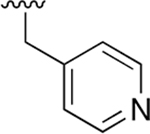
| 4 | 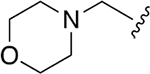 |
 |
 |
 |
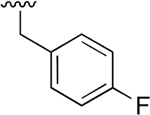
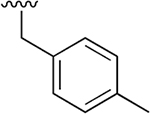
0.191 ± 0.036 | 1.67 ±0.32 | 9 | 6.27 ±2.64 | 135 ±42.2 | 21.5 |
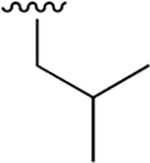
| 5 |  |
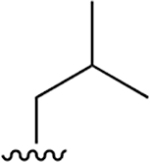
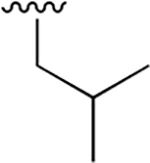
 |
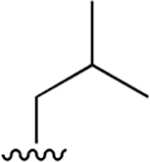 |
 |
0.062 ±0.013 | 1.01 ±0.16 | 16 | 2.03 ±0.34 | nd | nd |
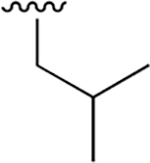
| 6 | 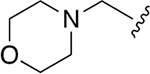 |
 |
 |
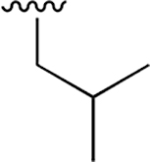 |
1.207 ± 0.093 | > 100 | nd | nd | nd | |
| 7 | 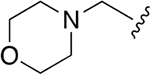 |
 |
 |
 |
0.022 ± 0.005 | 13.38 ±4.41 | 600 | 0.32 ± 0.035 | >250 | |
| 8 |  |
 |
 |
 |
0.015 ± 0.002 | 2.08 ±0.46 | 138 | 0.091 ± 0.028 | nd | nd |
| 9 |  |
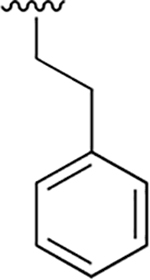 |
 |
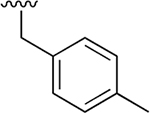 |
0.002 ± 0.0002 | 0.31 ± 0.098 | 172 | 0.008 ± 0.002 | 11.4 ±2.2 | 1443 |
| 10 | 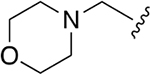 |
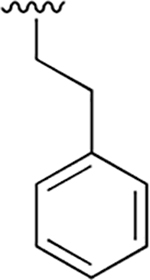 |
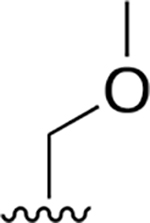 |
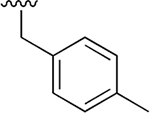 |
0.009 ± 0.002 | 4.36 ± 1.04 | 464 | 0.081 ±0.013 | 124 ±42.3 | 1536 |
 |
0.030 ± 0.008 | 2.75 ±0.50 | 92 | 0.342 ± 0.074 | >50 | |||||
EC50 values for each of the inhibitors 1–10 and the Carmaphycin B analog in 72 and 1 h treatment of P. falciparum W2 at ring stage parasites and non-confluent HFFs.
SI, selectivity index; nd, not determined.
One of the most commonly used tools to systematically define the specificity of proteases makes use of libraries of fluorogenic substrates.19,20 Screening can be accomplished using individual substrates assayed in parallel or through the use of positional scanning substrate combinatorial libraries (PS-SCLs) that contain a mixture of peptides where one or more of the positions are individually fixed while the remaining positions are randomized.21–23 These types of substrate libraries have also been used to identify optimal binding groups that can then be converted into inhibitors by replacing the reporter fluorophore with a reactive electrophile in an approach termed substrate activity screening (SAS).24–26 Furthermore, the use of non-natural amino acids in PS-SCLs has been shown to result in highly selective substrates and subsequent inhibitors of various protease targets.27–29 We therefore decided to use substrate libraries to more systematically screen for optimal residues that could be translated into inhibitors that have enhanced selectivity for the parasite proteasome. We synthesized a library of fluorogenic substrates with the 7-amino4-carbamoylmethylcoumarin (ACC) reporter group attached to a resin to facilitate rapid and highly parallel synthesis using standard solid-phase peptide synthesis methods30 (Figure 2A). As a test library, we synthesized Mor-Leu-Leu-P1-ACC (Mor = morpholinoacetyl) in which we scanned through the natural amino acids (minus methionine) at P1 position. Screening of this set of substrates using purified P. falciparum and human proteasomes confirmed our prior findings13 that the parasite proteasome has a preference for aromatic residues at P1 such as Phe, Tyr, Trp, and His (Figure 2B).
Figure 2.

Profiling the substrate specificity of the P. falciparum 20S proteasome using fluorogenic substrate libraries. (A) Schematic of coumarin-based fluorogenic substrate libraries containing non-natural amino acids at each of the P1, P2, and P3 positions showing processing by the protease to produce a fluorescent signal. (B) Plot of the slope over the linear range of the reaction (RFU/s) of each substrate containing the indicated P1 amino acid residue for the human proteasome (X-axis) and P. falciparum proteasome (Y-axis) normalized to the control substrate containing a P1 leucine (shown in blue). The points above the dashed line represent the P1 substrates with increased activity for the P. falciparum proteasome compared to the control substrate. The closer the points are to Y axis from the diagonal line, the more specific the substrate is for the P. falciparum proteasome over the human proteasome compared to the control substrate. (C–E) Plots as in (B) of the substrate processing rate for each of the P1, P2, and P3 positional scanning libraries containing non-natural amino acids compared to the template substrate Mor-Hfe-Ser(Me)-Leu-ACC (blue points). For each of the P1 (C), P2 (D), and P3 (E) positions, 45–50 non-natural amino acids were tested, while keeping the other positions fixed with the corresponding amino acids in the template sequence as indicated. The RFU/s values of each substrate for human and P. falciparum proteasome 20S were normalized to the values of a control Mor-Hfe-Ser(Me)-Leu-ACC. All experiments were performed at least in triplicate. (F) Structure of the optimal combination of residues found in the substrate profiling, Mor-Hfe-Ser(Me)-Thi-ACC. Mor = morpholinoacetyl, Hfe= homophenylalanine, Ser(Me) = methylserine, Thi = 2-thienylalanine, RFU = relative fluorescence unit.
We then synthesized a series of focused substrate libraries containing tripeptides based on the optimized template sequence in compound 7, Mor-Hfe-Ser(Me)-Leu (cap-P3P2-P1). For each library, we varied a single position (P1, P2, and P3) through 45–50 non-natural amino acids (Figure S2 of the Supporting Information, SI) while keeping the other positions fixed to the template sequence. We then measured processing of each substrate by purified P. falciparum and human proteasomes and normalized values to the control substrate, Mor-Hfe-Ser(Me)-Leu-ACC (Figure 2C–E). Scanning of the P1 library demonstrated that small aromatic and aliphatic residues (Thi, Fur, Pra, Nva, allylGly) have increased activity and selectivity relative to leucine. Among these improved residues, 2-thienylalanine (Thi) showed the strongest and most selective activity for the P. falciparum proteasome (Figure 2C). We also found that aliphatic side chains (propyl, butyl) are well tolerated in the P2 and this position could be used to tune the physicochemical properties. Screening of the P2 library of substrates showed that only the cyanobutyl (Cba) side chain displayed enhanced substrate specificity for the Pf proteasome over the human enzyme compared to the Ser(Me) in our template sequence (Figure 2D). The P3 library demonstrated, as expected, that the S3 pocket of the Plasmodium proteasome accommodates bulky and hydrophobic residues (Figure 2E). Interestingly, among the 47 amino acids tested at P3, the Hfe in the template molecule proved to be optimal in terms of overall activity and selectivity. Taken together, these substrate data identify multiple non-natural amino acids that could be used in each of the P1-P3 positions to optimize overall binding and selectivity for the parasite proteasome with the sequence Mor-Hfe-Ser(Me)-Thi being the most optimal combination of residues (Figure 2F).
Conversion of Optimal Substrates into Corresponding Vinyl Sulfone Inhibitors
We selected residues from the substrate screening that showed the most promising specificity and binding properties and used that information to build inhibitors by replacement of the ACC reporter with the vinyl sulfone electrophilic warhead (Compounds 11–17; Table 2). Encouragingly, the profiles found in the substrate screening translated into similar structure–activity relationships (SAR) for the vinyl sulfone inhibitors. Consistent with the fluorogenic substrate data, replacement of the P3 Hfe with a bulky but nonaromatic cyclohexylalanine (Cha; compound 11) reduced potency. Inhibitors with 2-aminobutyric acid (Abu; compound 12) and propargylglycine (Pra; compound 13) at the P1 position exhibited comparable activities (EC50 = 21 nM and 18 nM, respectively) and retained similar selectivity as the leucine P1. Notably, we observed a significant increase in potency and selectivity by incorporation of thiophene in the P1 position (compound 14). The compound with 2-furylalanine at the P2 position displayed similar potency (compound 16, EC50 = 7 nM) to the analog containing the methylserine. However, citrulline (Cit) substitution (compound 17) resulted in loss of activity probably due to poor permeability of the compound.
Table 2.
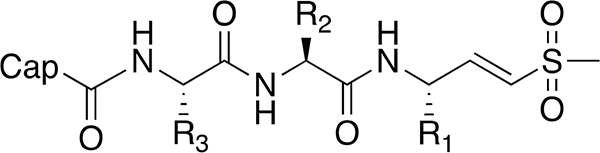 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. | Cap | R3 | R2 | R1 | 72 h EC50 (μM) | 1 h EC50 (μM) | ||||
| Pf | HFF | SI | Pf | HFF | SI | |||||
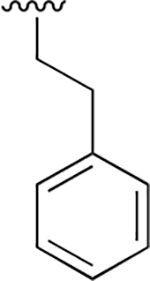
| 11 | 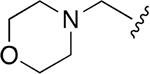 |
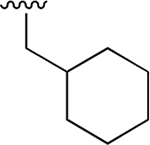 |
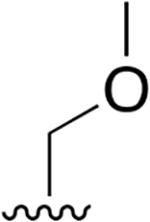 |
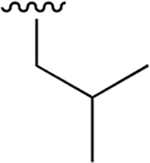 |
0.359 ± 0.042 | 78.41 ± 18.01 | 218 | nd | nd | nd |
| 12 |  |
 |
 |
 |
0.021 ±0.005 | 13.96 ±3.58 | 652 | 1.013 ±0.169 | >250 | |
| 13 |  |
 |
 |
 |
0.018 ±0.005 | 8.54 ±2.00 | 482 | 0.33 ± 0.047 | >250 | |
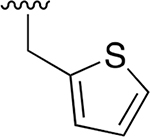
| 14 |  |
 |
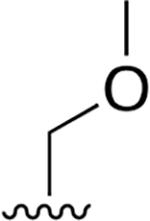 |
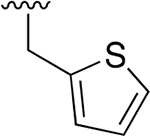 |
0.008 ± 0.002 | 9.80 ±2.96 | 1240 | 0.137 ± 0.095 | >250 | |
| 15 | 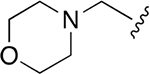 |
 |
 |
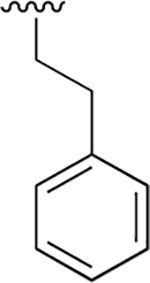 |
0.035 ±0.009 | 15.77 ±3.51 | 445 | 8.03 ±1.33 | >250 | |
| 16 | 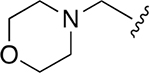 |
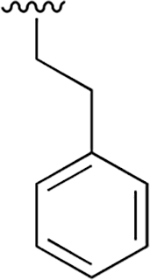 |
 |
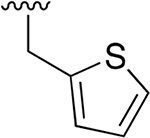 |
0.007 ±0.002 | 7.4 ±1.50 | 1138 | 0.051 ± 0.006 | 207 ±23.2 | 4043 |
| 17 | 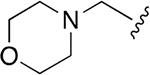 |
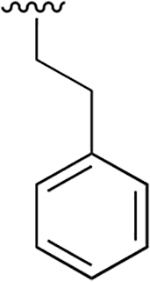 |
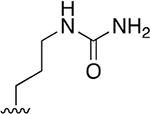 |
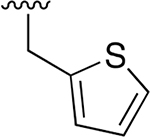 |
0.369 ±0.125 | 246 ±49.63 | 667 | >2.5 | >250 | |
EC50 values for inhibitors 11–17 in 72 and 1 h treatment of P. falciparum W2 at ring stage parasites and non-confluent HFFs.
SI, selectivity index; nd, not determined.
Assessment of Lead Compound Liabilities.
Given that epoxyketone-based inhibitors have been shown to have short half-lives in vivo due to hydrolysis of the reactive epoxide,31–34 we were concerned about the overall stability of the vinyl sulfone-based inhibitors. Therefore, we performed metabolite profiling studies on compound 7. Compound 7 exhibited good aqueous solubility (198 μM) and rat hepatocyte stability (t1/2; 75 min). Yet, it is predicted that the compound would have short half-life and be rapidly cleared in vivo based on its stability in human liver microsomes (Table S1). The formation of metabolites of compound 7 was liver microsome- and NADPH-dependent and the compound was stable to hydrolysis in human and mouse plasma (Figure S4). In fact, >85% of the compound remained stable in plasma after 6 h incubation. The tentative identity of the metabolites was deduced by comparison of the product ion spectra to that of the parent compound. The microsomal metabolites of inhibitor 7 were mainly derived from oxidative reactions on the morpholine cap and homophenylalanine side chain as well as some breakage of the morpholine ring and hydrolysis of the peptide backbone between Hfe and Ser(Me) (Figure S5–S6). These findings suggest that the vinyl sulfone and P1/P2 residues remain unmodified and that the most significant liabilities are related to the P3 Hfe and morpholino capping group.
Because a major metabolite of our lead molecule is a presumed hydroxylation on the Hfe side chain, we synthesized two vinyl sulfones containing P3 homotyrosine residues. These analogs inhibited the P. falciparum β2 and β5 subunits (Figure S7) but were found to be inactive at killing parasites in cellular assays suggesting a loss in cell permeability that resulted from addition of the hydroxyl group on the Hfe. Therefore, finding ways to block oxidative metabolism on Hfe may be important for retaining in vivo activity.
Optimization of Pharmacological Properties of Lead Molecules.
Given that major metabolism occurred on the morpholine ring of compound 7, we decided to make further modifications to replace the capping group with molecules that might improve stability and retain overall potency and selectivity of the lead molecules. We evaluated several five-membered aromatic or nonaromatic heterocycles as replacements for the morpholine cap based on prior use of these groups to generate Oprozomib (PR-047), a second-generation orally bioavailable human proteasome inhibitor.18 Within the same core Hfe-Ser(Me)-Leu-vs scaffold, compounds with 2-Me-5-thiazole, 5-Me-3-isoxazole, and 1-Et-3-Me-5-pyrazole rings showed potent activities in parasite killing (Table 3). The most selective of the three compounds was 18 but it was less selective than the original compound 7 with the morpholine cap. We then used these three selected capping groups on the optimized sequence, Hfe-Ser(Me)-Thi-vs Again, the 2-Me-5-thiazole cap showed a good selectivity profile (compound 23, SI= 716) and was the most potent compound yet described for killing P. falciparum (EC50= 3 nM).
Table 3.
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. | Cap | R3 | R2 | R1 | 72 h EC50 (μM) | 1 h EC50 (μM) | ||||
| Pf | HFF | SI | Pf | HFF | SI | |||||
| 18 | 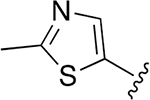 |
 |
 |
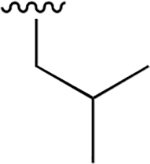 |
0.034 ± 0.006 | 13.63 ±2.63 | 402 | 0.384 ±0.071 | >250 | |
| 19 | 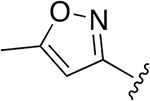 |
 |
 |
 |
0.009 ± 0.0059 | 1.7 ± 0.26 | 187 | 0.155 ± 0.060 | 54.3 ±11.2 | 349 |
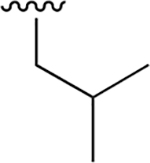
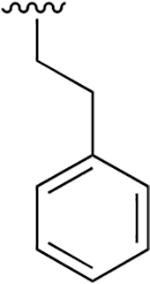
| 20 | 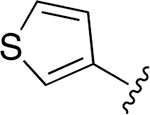 |
 |
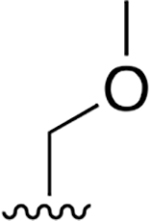 |
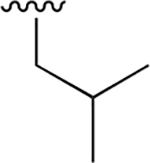 |
nd | nd | nd | nd | nd | nd |
| 21 | 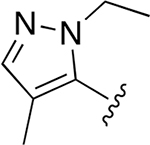 |
 |
 |
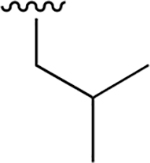 |
0.013 ± 0.004 | 2.75 ± 0.50 | 212 | 0.251 ± 0.027 | 95.3 ± 15.3 | 379 |
| 22 |  |
 |
 |
 |
nd | nd | nd | nd | nd | nd |
| 23 | 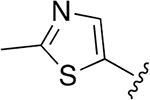 |
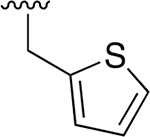
 |
 |
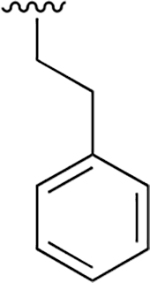
 |
0.003 ± 0.0007 | 2.22 ±0.077 | 716 | 0.048 ±0.009 | >250 | |
| 24 |  |
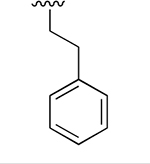
 |
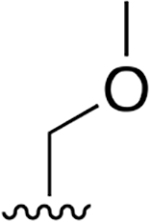 |
 |
0.002 ± 0.0004 | 0.727 ±0.092 | 346 | 0.012 ± 0.003 | 28.73 ±5.65 | 2355 |
| 25 | 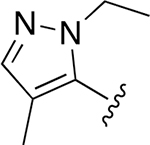 |
 |
 |
 |
0.004 ± 0.0007 | 0.985 ±0.093 | 281 | 0.021 ± 0.004 | 18.66 ±2.95 | 906 |
| 26 |  |
 |
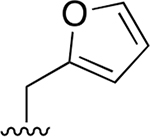 |
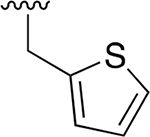 |
0.007 ± 0.002 | 3.45 ± 0.30 | 493 | 0.042 ±0.001 | 113 ±23.3 | 2690 |
| 27 | 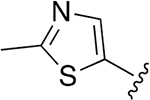 |
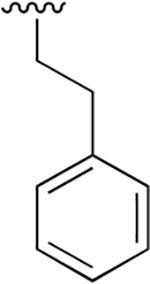 |
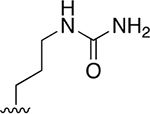 |
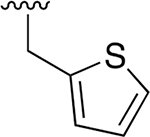 |
0.072 ± 0.008 | 73.39 ±7.21 | 1016 | >2.5 | >250 | |
| 28 | 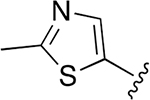 |
 |
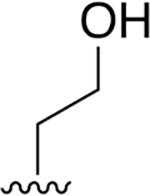 |
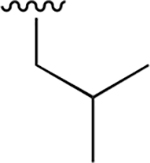 |
0.019 ± 0.003 | 29.67 ± 4.45 | 1561 | 0.648 ± 0.099 | >250 | |
| 29 | 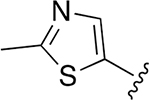 |
 |
 |
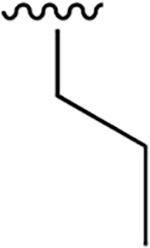 |
0.009 ±0.001 | 23.13 ± 3.53 | 2559 | 1.065 ± 0.242 | >250 | |
| 30 |  |
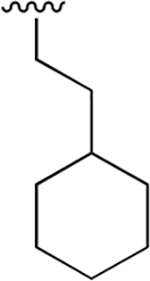 |
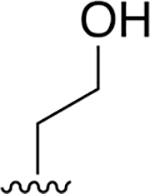 |
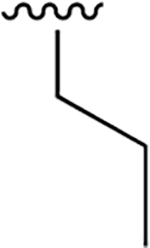 |
0.005 ±0.001 | 0.061 ±0.008 | 11.43 | 0.121 ± 0.032 | 42.45 ±7.7 | 351 |
EC50 values for inhibitors 18–30 in 72 and 1 h treatment of P. falciparum W2 at ring stage parasites and non-confluent HFFs.
SI, selectivity index; nd, not determined.
For further inhibitor design, we also considered physico-chemical properties such as molecular weight (MW), lipophilicity (cLogP), hydrogen bond donors/acceptors (HBDs/ HBAs), and topological polar surface area (tPSA) (Table S2). Inhibitor 23, although highly selective and exceptionally potent, has overall poor aqueous solubility (Table S1). Also, the P1 thiophene can be converted to reactive metabolites such as S-oxides and thiophene epoxides.35 Therefore, we synthesized two analogs with homoserine (Hse) at the P2 position to help enhance solubility and substituted the P1 thienylalanine by either leucine or norvaline. Compounds 28 and 29 both have low nanomolar activity at killing parasite in the 72 h treatment with exceptionally low toxicity against HFFs resulting in greatly enhanced selectivity ratios (Table 3). While compound 29 with norvaline at the P1 position decreased parasite survival after 72 h treatment, it was only active in the micromolar concentration range when used for a 1 h pulse treatment, suggesting that it may have reduced cellular uptake compared to compound 28. The selectivity profile of the most optimal inhibitors was also confirmed with a screen against P. falciparum Dd2 and HepG2 (Table S3). These data confirmed similar selectivity ratios as those observed for HFF cells but showed an even greater specificity of compound 28, which had no toxicity to HepG2 cells even after 72 h treatment with doses as high as 10 μM, supporting this as a promising lead molecule for further development.
Identification of Determinants That Drive Selectivity and Potency.
Having identified a number of lead molecules with enhanced parasite-specific killing, we wanted to better understand the determinants that drive the potency and selectivity. We therefore performed direct binding studies on purified proteasomes from both parasites and the human host using activity-based probes13,36 (Figure 3B). This approach allows direct assessment of compound binding to each of the active β subunits of the proteasome through the quantification of residual protein labeling after compound treatment. The resulting gel labeling data can then be used to calculate IC50 values for inhibitor binding at each subunit. Using this approach, we confirmed that potency for killing parasites tracked with the ability of the compounds to bind and inhibit both the β2 and β5 subunit of the parasite proteasome, consistent with our earlier studies.36 Thus, compounds 3, 23, 28, and 30 all were potent inhibitors of parasite growth (low nM EC50 values; Tables 1–3), which corresponded to low micromolar IC50 values in the competition labeling studies for both the β2 and β5 subunits. Compound 11, however, showed comparable β2 potency and reduced potency for the β5 subunit, with only partial inhibition of β5 even at 50 μM concentrations. This lack of β5 activity resulted in overall poor activity of the compound against parasite growth. Comparing the potency and selectivity index measured for each compound against parasite and human cell cultures (Figure 3C) to the competition labeling results for the human proteasome (Figure 3B), we found that even highly potent inhibition (nM) of the human β5 subunit did not result in host cell toxicity, consistent with reports that selective inhibition of host β5 activity is well tolerated in nontransformed mammalian cells.37,38 However, compounds that had potent inhibition of the β5 activity combined with even weak inhibition of the β2 subunit lost virtually all selectivity. This is most clearly illustrated when comparing compounds 28 and 30, for which both compounds had similar potencies for parasite killing, yet compound 30 was more than 100 times less selective than compound 28. The competition labeling results confirmed that both have similar potencies for the human β5 subunit yet compound 30 showed weak cross reactivity with the human β2 subunit when used at high dose. This apparently minor cross reactivity, therefore leads to a dramatic loss of selectivity due to the highly toxic effects of inhibiting both the β5 and β2 subunits in the human cells.
Figure 3.

Lead optimization of Plasmodium-selective proteasome inhibitors. (A) Structures of inhibitors designed based on the substrate screening data with enhanced selectivity and potency for the parasite proteasome. (B) Inhibition of purified P. falciparum and human 20S as assessed by competitive activity-based probe labeling. Purified 20S proteasomes were incubated with each inhibitor at the indicated final concentrations for 1 or 18 h followed by labeling of residual proteasome activity using the activity-based probes. The individual subunits of the proteasome (β1, β2 and β5) are indicated on the gel. The IC50 values were calculated for each inhibitor for the P. falciparum and human 20S proteasomes by quantification of labeled proteins using ImageJ and then normalizing to mock-treated control. (C) A stacked bar plot of EC50 values (logarithmic scale) of each inhibitor in 72 h treatment of P. falciparum W2 at ring stage parasites (black bar) and nonconfluent HFFs (white bar) with error bars, s.d. (upper panel), and selectivity index (comparing the P. falciparum activity versus HFF activity, HFF EC50/P. falciparum EC50, bottom panel) for selected inhibitors.
The immunoproteasome (i20S), which is a modified form of the constitutively active 20S proteasome core particle, has subtle differences in proteolytic activity and substrate preferences.39,40 Inhibition of immunoproteasomes can diminish immune responses against infection, therefore, we sought to profile our new inhibitors against human immunoproteasome as well. We used the broad-spectrum activity-based probe that specifically targets all active subunits of the constitutive proteasome and immunoproteasome.41 Although β1c, β1i, β5c, and β5i subunits cannot be clearly resolved on SDS-PAGE, we were still able to assess the inhibition of compounds for i20S and compare that to h20S and Pf20S inhibitions (Figure S8). Compounds 3, 11, and 30 that showed cross-reactivity toward the human β2c were also found to inhibit β2i at high concentrations (50 μM), while compounds 7, 14, 23, and 28 showed virtually no inhibition of this immunoproteasome subunit. To more effectively monitor inhibition of the β5i subunit, we used the β5i-selective fluorogenic substrate Ac-ANW-AMC.42 Compounds 23 and 30 exhibited submicromolar IC50 values, while the others showed only weak micromolar activity (Table S4). Overall, the selectivity of compounds was the same for β5i as β5c suggesting that enhanced cross-reactivity for the immunoproteasome is not likely to be a significant problem for our lead molecules.
Dominant Role for the Electrophile in the Selectivity of Lead Molecules.
The primary electrophiles used in the clinically approved human proteasome inhibitors bortezomib and carfilzomib are a boronic acid and an epoxyketone, respectively. Both of these electrophiles are substantially more reactive toward the active site threonine hydroxyl compared to the vinyl sulfone electrophile. This is because the vinyl sulfone is generally designed to target cysteine nucleophiles by way of a Michael addition reaction. Therefore, we reasoned that use of a highly reactive “warhead” on the inhibitor might reduce the overall selectivity of our lead molecules that derive selectivity through binding elements at the P1–P3 positions on the peptide scaffold. We replaced the vinyl sulfone group of the lead compound 28 with the epoxyketone and boronic acid electrophiles. Interestingly, the epoxyketone derivative 33 exhibited potent inhibition of β2 and β5 subunits of the P. falciparum proteasome, while the boronic acid derivative 34 inhibited all three catalytic subunits. In fact, the inhibitory activities of these analogs for the β2 subunit of the parasite proteasome were greater than that of the parent vinyl sulfone 28. However, analogs 33 and 34 also inhibited both β5 and β2 subunits of the human proteasome, which became more apparent when enzymes were treated with inhibitors for longer times (Figure 4A). This coinhibition of both subunits resulted in toxicity and a complete loss of selectivity of the compounds (Figure 4C and Table S5). The inhibitory activities of these analogs for human proteasome β2 and β5 subunits were also confirmed using fluorogenic substrate assays (Figure 4B). The boronate 34 showed potent inhibition of the human proteasome activity which was not reflected in the competition labeling experiments due to the fact that its reversible inhibition mechanism made it less effective at competing for the labeling by the irreversible activity-based probe. The loss of potency observed in parasite killing with the boronic acid derivative 34 despite strong inhibition of the P. falciparum proteasome in lysates also suggests poor cell permeability of the compound as another potential liability. Taken together, these results demonstrate that optimization of selectivity using peptide backbone changes requires the use of a suitable electrophile such as the vinyl sulfone to prevent potency being driven mainly by the reactivity of the electrophile.
Figure 4.

Choice of electrophile is important for obtaining optimal selectivity of proteasome inhibitors. (A) Structures of compounds 28, 33, and 34 and inhibition of purified P. falciparum and human proteasome 20S as assessed by competitive activity-based probe labeling. Purified 20S proteasomes were incubated with each inhibitor for indicated times followed by labeling of residual proteasome activity using the activity-based probes. The IC50 values were calculated for each inhibitor for the P. falciparum and human 20S proteasomes by quantifying labeled proteins using ImageJ and normalizing them to mock-treated control. (B) Inhibition of compounds 28, 33, and 34 for processing of the fluorogenic substrates, Suc-LLVY-AMC (β5) and Boc-LRR-AMC (β2) by human proteasome 20S. The inhibitors were incubated with h20S for 1 h followed by the addition of each fluorogenic substrate. The AMC molecules released by residual proteasomal activity were measured (excitation at 380 nm, emission at 460 nm) and the slope over the linear range of the reaction was used for the calculation of IC50 values using Prism. (C) A stacked bar plot of EC50 values (logarithmic scale) for the vinyl sulfone (VS)-, epoxyketone (EK)- and boronic acid (BA) derivatives 28, 33, and 34 in 72 h treatment of P. falciparum W2 at ring stage parasites (black bar) and nonconfluent HFFs (white bar) with error bars, s.d. (left) and selectivity index (comparing the P. falciparum activity to HFF activity, HFF EC50/P. falciparum EC50, right).
P. falciparum-Specific Proteasome Inhibitors Enhance Parasite Killing by ART in Both Sensitive and Resistant Parasites.
Recent data suggest that artemisinin-resistant (ART-R) parasites are vitally dependent on proteasomal degradation of ART-alkylated and poly ubiquitinated parasite proteins.14,43,44 In support of this finding, DHA and WLW-vs are highly synergistic in killing parasites in culture.13 Therefore, we tested our new P. falciparum-specific proteasome inhibitors 3, 7, 23, and 28 on artemisinin-sensitive (ART-S) parasites originated from Cambodia (Cam3.II) harboring a wild-type version of Kelch 13 (Cam3.II WT K13) as well parasites isogenic at the K13 locus (Cam3.II K13C580Y) that rendered them ART-R3. All of the tested inhibitors were potent on both ART-S and ART-R parasites, with killing activities in the nanomolar range. When parasites were exposed to combinations of the compounds and DHA (dihydroartemisinin) at the early ring stage of the intraerythrocytic cycle, we observed strong synergism in both ART-S and ART-R parasites (Figure 5A). In particular, the inhibitor 28 exhibited the strongest synergism among the compounds tested. These results confirm that Plasmodium-specific proteasome inhibitors potentiate ART action and can be used to eliminate ART-R parasites.
Figure 5.

The lead vinyl sulfone enhances parasite killing by ART and reduces parasite growth in vivo. (A) Isobolograms depicting P. falciparum in vitro culture-based fractional IC50 (FIC50) values in the presence of (a, b) dihydroartemisinin (DHA) and inhibitor 3, (c, d) DHA and 7, (e, f) DHA and 23, (g, h) DHA and 28 or (i, j) DHA and WLL-vs The dashed line represents additive interactions. Concave shapes (sum of FIC50 values <1) indicate synergistic effects between compounds. (B) Reduction of parasite burden after in vivo inhibitor treatment on day 4. CD1 mice were infected with P. berghei and treated with vehicle (n = 5) or 50 mg/kg compounds WLL-vs, 3 (n = 3), 7 or 28 (n = 5) once daily for 4 consecutive days starting 2 h after infection. Parasitemia was monitored daily by Giemsa-stained thin blood smears. (C) Plasma concentration–time profiles of inhibitor 28 after intravenous (50 mg/kg) and oral (100 mg/kg) administration to Sprague–Dawley rats (n = 6), error bars, s.d.
Lead Vinyl Sulfone Reduces Parasite Growth in Vivo without Host Toxicity.
Encouraged by the selective parasite killing and synergism with DHA in our culture model, we proceeded to test the efficacy of the new analogs compared to the original WLL-vs lead molecule in the Plasmodium berghei mouse model. In this model of human malaria infection, mice are infected and total parasite burden in blood is measured over time by simple blood smear analysis.45 We infected CD1 mice with P. berghei and treated with compounds at 50 mg/kg once daily for 4 consecutive days of intravenous injections. We observed almost complete reduction of parasite burden on day 4 (3: 99% and 28: 96% reduction of parasites, Figure 5B) suggesting that the new analogs are highly effective at killing parasites in vivo at levels comparable to WLL-vs (98% reduction). Given the overall high specificity ratio and low toxicity for compound 28, we selected it for further pharmacokinetic (PK) to evaluate its overall stability and oral uptake in rats. We therefore performed standard PK analysis following IV and PO administrations to Sprague–Dawley rats (Figure 5C) and found that after IV administration at 50 mg/kg, plasma concentrations of 28 reached a Cmax of 20 μM (12017 ng/mL) with a half-life (t1/2) of 0.49 h (Table S7). Following oral administration (100 mg/kg), the compound displayed some plasma exposure with a Cmax of 1 μM and an area under the curve (AUC) of 1294 h × ng/mL. The overall oral bioavailability (F) was 2.6%. While a promising sign that the compound could make it into blood upon oral dosing, this result suggests that additional formulation and compound optimization likely will be required to achieve therapeutically relevant doses of the drug by oral delivery.
DISCUSSION
The proteasome has proven to be a viable target in humans for the treatment of multiple myeloma.46–50 This multimeric complex has recently emerged as a viable target in Plasmodium falciparum, the causative agent of malaria, due to its central role in parasite growth, replication, and transmission.51 Its potential as an antimalarial target has recently been further strengthened by the fact that a functional proteasome is required for P. falciparum resistance to ART derivatives, which are the core components of global first-line ACTs. Therefore, inhibitors of the Plasmodium proteasome have the potential to block infection, replication, transmission, and possibly dissemination of ART resistance. However, to be viable as a therapy, compounds must be highly selective to avoid toxicity resulting from inhibition of the host proteasome. We recently reported on peptide vinyl sulfones that are capable of selective inhibition of the parasite enzyme. However, these lead molecules still suffer from some level of reactivity toward the human enzyme and also contain significant liabilities in terms of their pharmacological properties. Here we describe the use of a combination of substrate screening and rational design based on the prior high-resolution EM structure to optimize both the selectivity and physicochemical properties and provide a next generation of inhibitors of the Plasmodium proteasome. Using these new compounds, we were able to define the elements of specificity of compounds for the parasite enzyme over the human counterpart, which resulted in a lead molecule with high potency and exceptional selectivity that can clear parasites in mouse models of infection. Furthermore, the structural and SAR data presented here provide a roadmap for further optimization of compounds for advancement into clinical trials.
Most peptide-inspired proteasome inhibitors derive their subunit selectivity from the amino acid side chains that engage the S pockets of the enzyme active sites. Understanding the structural differences between the parasite and human proteasomes through high-resolution electron microscopy in our previous study facilitated the discovery of specific non-natural amino acids that could be used to greatly enhance the specificity of inhibitors for the parasite enzyme over the human host enzyme. Incorporation of non-natural amino acids into a substrate library design allowed us to generate 140 fluorogenic peptides (45–50 peptides/sublibrary), which were then assayed for specificity in a simple and rapid format. Coupled with an SAR investigation of the corresponding vinyl sulfone inhibitors, we identified bulky and hydrophobic P3 residues that are preferred by the parasite enzyme. Steric and conformational constraint appear to be key determinants of the specificity as narrower binding pockets of human β2 reduce the accessibility of ligands. Furthermore, our data show that toxicity of compounds toward host cells is associated specifically with an increase in potency and inhibition of the β2 subunit of the human enzyme. We also found that the P1 side chain can be exploited to enhance selectivity using small aliphatic and aromatic residues, exemplified by 2-thienylalanine. Our data further support the finding that the P1 residue directs the specificity for the β2 subunit of the parasite proteasome because only this subunit is compatible with bulky side chains such as Trp and Hfe at the P1 position. However, it is important to retain potency for the β5 subunit as β2-selective inhibitors are not effective at killing parasites on their own. Recently, Kirkman and co-workers also demonstrated a synergistic effect between β2 and β5 inhibition of the P. falciparum proteasome. Their β5-selective inhibitor did not yield a significant reduction in parasitemia in vivo on its own, but when combined with the β2-selective inhibitor WLW-vs, reduced parasitemia by 95%.52 We also found that while the P1 and P3 positions are the major determinants of selectivity, the P2 and capping group also influence the selectivity subtly, especially when combined with specific P3 and P1 variants. In fact, the substitution of the morpholinoacetyl cap with a 2-Me-5-thiazole ring led to an increase in β5 inhibition. To provide a structural understanding of the specificity determinants, a study of high-resolution electron microscopy structure of the P. falciparum proteasome with our new potent and selective inhibitor bound in multiple active sites is in progress.
We have found that the vinyl sulfone is an ideal reactive electrophile for development of parasite-selective inhibitors. Swapping of the vinyl sulfone on our highly selective lead molecule for an epoxyketone or boronic acid shows that the high degree of reactivity of these electrophiles effectively negates all the specificity gained by design of an optimal peptide sequence. The epoxyketone and boronic acid groups also have additional liabilities that may prevent their effective use as antimalarial agents. Human proteasome inhibitors such as carfilzomib and oprozomib are rapidly metabolized and cleared via epoxide hydrolysis within minutes31–34 and the boronic acid is a highly efficient inhibitor for diverse enzymes that use nucleophilic hydroxyl residues. Our in vitro metabolism study with compound 7 demonstrated that the vinyl sulfone is stable in microsomes. Given that the vinyl sulfone functions by irreversible inhibition, it is possible that our lead compounds could induce prolonged pharmacological effects in spite of rapid clearance. Furthermore, the vinyl sulfone has been shown to have a limited spectrum of possible target, all of which are cysteine protease such as the cathepsins. This small family of potential off targets are not particularly problematic as they can be inhibited without causing short-term toxic effects.53,54
The drug exposure of our optimized Plasmodium-specific proteasome inhibitor 28 following oral administration in a rat PK study is promising although the overall oral bioavailability needs to be further improved. Previous studies have demonstrated that toxicity of irreversible proteasome inhibitors correlates with the Cmax values rather than the AUC.31,55 The fact that compound 28 showed a low peak plasma concentration therefore might not be problematic, especially if the compound is used in combination with ART where complete inhibition of the proteasome is not required to enhance ART-mediated parasite killing.
Inhibitor 28 was able to reduce parasite growth in vivo. Given the large therapeutic window and no noticeable toxic effect or other signs of sickness in the mice, we conclude that the Pf-specific proteasome inhibitor 28 could be used to attenuate parasite growth without appreciable toxicity to the host. More importantly, the strong synergy displayed between the proteasome inhibitors and ART derivatives in both ART-sensitive and -resistant lines suggests that highly selective Pf proteasome inhibitors may be most beneficial when used in combination with ART for malaria treatment. Furthermore, because the proteasome activity is critical for the onset of a delayed response to ART, even low-level inhibition of the parasite proteasome (i.e., at levels not sufficient to kill parasites through proteasome inhibition alone) may be sufficient to enhance ART killing and prevent the onset of resistance. Therefore, the compounds described here already have the potential to be used as safe adjunct therapies for the effective treatment of complicated cases of malaria.
EXPERIMENTAL SECTION
Fluorogenic Substrate Assay.
Substrate assays were performed in black 96 or 384-well Costar plates. For these assays, purified human (purchased from Boston Biochem) or P. falciparum 20S proteasomes38 were activated using human PA28α. Each enzyme was added to a final concentration of 2 nM and the reaction proceeded in the presence of substrates (100 μM) for 1 h at 37 °C. The fluorescent signal (RFU, relative florescence unit) of ACC was measured at 380 nm (excitation) and 460 nm (emission). The slope over the linear range of the reaction (RFU/s) was determined for each of the enzymes. All experiments were performed at least in triplicate.
P. falciparum Replication Assays.
P. falciparum W2 cultures were maintained, synchronized, and lysed as previously described.38 P. falciparum W2 was obtained originally from the Malaria Research and Reference Reagent Resource Center. Parasite cultures were grown in human erythrocytes (no blood type discrimination) purchased from the Stanford Blood Center (approved by Stanford University). Ring stage P. falciparum culture at 2% parasitemia and 1% hematocrit was added to 96-well plates spotted with compounds. The P. falciparum culture was incubated with each compound for either 1 or 72 h. After compound treatment for 1 h, the inhibitor was removed, and the culture was washed three times in media, before fresh media was added for a further 71 h of growth. After incubation, the culture was fixed in a final concentration of 1% paraformaldehyde (in PBS) for 30 min at room temperature. The nuclei stain YOYO-1 was then added to a final concentration of 50 nM and incubated overnight at 4 °C. Parasite replication was monitored by observation of a YOYO1-positive population (infected) and YOYO-1-negative population (uninfected) using a BD Accuri C6 automated flow cytometer. Values in Tables 1–3 and S3 are the average; n = 6 parasite cultures from two independent experiments with triplicates.
Cell Viability Assay in Mammalian Cells.
HFFs used in all experiments were nontransformed primary cells (obtained from J. Boothroyd) derived from human foreskin tissues and tested for Mycoplasma contamination. HFFs were plated at 2500 cells (nonconfluent) per well <24 h before addition of the compound. Compounds were diluted for dose–response concentrations in media and added to the cells for either 1 or 72 h. Cell viability was measured using the CellTiter-Blue Assay (Promega) as per the manufacturer’s instructions. Values in Tables 1–3 and S3 are the average n = 9 cell cultures from three independent experiments of triplicates for HFF treatment.
Activity-Based Probe Labeling of Mammalian and P. falciparum Proteasome.
BMV03736 was used at a final concentration of 10 μM to label P. falciparum proteasome activity, and MV15141 was used at a final concentration of 2 μM to label mammalian proteasome activity. Purified proteasomes (human or P. falciparum, 10 nM) were incubated with each inhibitor for 1 h at 37 °C before adding BMV037 or MV151 for a further 2 h at 37 °C. Samples were denatured by addition of SDS sample buffer, boiled briefly and run on a 12% SDS PAGE. Gels were scanned on the Cy5 channel (for BMV037) or the Cy3 channel (for MV151) on a Typhoon Scanner (GE Healthcare).
Assessment of DHA Synergism against ART-Sensitive and -Resistant P. falciparum.
Tightly synchronized 0–3 h postinvasion rings were exposed to combinations of the drugs (with fixed ratios of 1:0, 4:1, 2:1, 1:1, 1:2, 1:4, and 0:1 of 16× IC50 drug stocks) for 3 h. Drugs were then removed with three washes, and the wells were incubated with fresh drug-free media. Parasitemias were determined following an additional 69 h of culture. Data were collected from two separate biological repeats (indicated as different colors), each with technical replicates.
In Vivo Assessment.
CD1 mice were infected by intraperitoneal (IP) injection with 107 P. berghei ANK strain MRA-868 parasites isolated from an infected mouse on Day 0. WLL-vs, 3, 7, and 28 (50 mg/kg), or vehicle (10% DMSO, 40% PEG-400, 10% (2-hydroxypropyl)-β-cyclodextrin in PBS) was administered by intravenous injection for 4 consecutive days. For each drug dose, three to five mice were used. Due to the technical challenge of 4 consecutive days of tail vein injections, some doses were administered as retro-orbital injection when required. Treatment with 50 mg/kg compounds for all mice was administered as tail vein injection (day 0 and 3) and retro-orbital injections (day 1 and 2). During drug treatment, the health of the mice was assessed daily by observing their physical appearances and activities of mice. Parasitemia was monitored daily by Geimsa-stained thin blood smears obtained from the tail vein and quantified by light microscope counting, containing at least 1000 red blood cells.
Synthesis of Fluorogenic Substrate Library.
The preparation of ACC and Fmoc-ACC-resin was carried out as described previously.30 The substitution level of the resin (0.5 mmol/g) was determined by a spectrophotometric Fmoc quantitative assay. The synthesis of the library was performed using a Syro II 96-tip synthesis module (Biotage). To prepare the P1 library, each of 45 of single Fmoc-amino acids 10 equiv (50 μmol) with 10 equiv of HATU (50 μmol), 10 equiv of collidine (50 μmol) in DMF (180 μL/tip) was added to the same tips as ACC-resin (10 mg, 5 μmol). The reaction was carried out for 24 h. Then, resin was washed five times with DMF and the reaction was repeated using above reagents. After washing with DMF, Fmoc protecting group was removed using 20% piperidine (v/v) in DMF. For fixed positions, 5 eq of single amino acid (Fmoc-Ser(Me)-OH at P2 and Fmoc-Hfe–OH at P3) was used. Reactions were carried out with 5 equiv of HATU and 5 equiv of collidine in DMF for 3 h. N-terminus capping was carried out using 5 equiv of morpholin-4-yl-acetic acid with 5 eq of HATU and 5 equiv of collidine in DMF for 3 h. The resin was washed with DMF and then with CH2Cl2. The substrates were cleaved from the resin by treating with a solution of trifluoroacetic acid: triisopropylsilane: H2O (95:2.5:2.5, 200 μL/tip) for 1 h, and the collected material was precipitated with cold diethyl ether (Et2O, 2 mL). After precipitation, the mixture was centrifuged and washed with Et2O (1 mL). After centrifugation, a white precipitate was dissolved in acetonitrile/H2O and lyophilized. The final products were dissolved in DMSO to a concentration of 20 mM and used without further purification. For the P2, and P3 libraries, Fmoc-Leu-OH bound to ACC-resin was prepared. The resin was dried and then split into the tips (5 μmol/tip, 50 tips per each library). In the same manner as described above, P2 and P3 libraries were synthesized by coupling 50 of single Fmoc-amino acid residues to P2 (using Fmoc-Hfe-OH at P3) and respectively P3 position (using Fmoc-Ser(Me)-OH at P2).
Analog Synthesis.
The synthesis of compounds was divided into two parts, the amino acid-vs part (P1-vs) and the peptide part that contains the cap-P3-P2 moieties. Each of these two parts was synthesized separately according to a previously established procedure.13 The synthesis was completed by attaching the P1-vs moiety to the desired dipeptide moiety (Cap-P3-P2-OH) using simple HBTU/ HOBt coupling to afford the desired analogs. The crude compounds were purified on RP-HPLC to afford pure final products.
Kinetic Solubility.
The compounds were added to 96-well plates in duplicate, from their respective 10 mM stocks. A MiVac sample evaporator was then used to dry down the DMSO under vacuum. Buffer at pH 2 and pH 7.4 was then added to the dried-down wells. The plates were then incubated at 25 °C for 24 h with shaking. The plates were then centrifuged and 30 μL of samples was carefully transferred to an analysis plate. Finally, 270 μL of acetonitrile containing 100 nM carbamazepine as internal standard was added to the analysis plate. Six calibration standards and five quality control samples were prepared by adding DMSO after the dry-down step to give final concentrations between 5–250 μM and were processed similarly. The positive controls hydrocortisone and reserpine were prepared similarly, but with a 3-point calibration curve between 10–220 μM.
Microsomal Stability.
Metabolic stability was performed in duplicate in a 96-well microtiter plate. The test compounds (1 μM) were incubated individually in mouse, rat, and pooled human liver microsomes (0.4 mg/mL) at 37 °C for predetermined time points, in the presence or absence of the cofactor NADPH (1 mM). Reactions were quenched by adding 300 μL of ice cold acetonitrile containing internal standard (carbamazepine, 0.0236 μg/mL). Test compounds in the supernatant were analyzed by means of LC-MS/MS (Agilent Rapid Resolution HPLC, AB SCIEX 4000 QTRAP MS). Metabolite searches were not conducted during the metabolic stability assay.
In Vitro Incubations with Mouse and Human Plasma.
The test compound (10 μM) was incubated at 37 °C in human or mouse plasma for 6 h while shaking. The samples were then prepared by ice-cold acetonitrile precipitation, centrifuged, and filtered for LC-MS/ MS analysis. Vinpocetine and Procaine (10 μM) were incubated concomitantly as positive controls.
In Vitro Incubations with Mouse Liver Microsomes (MLM) and Human Liver Microsomes (HLM).
The test compound (10 μM) was incubated at 37 °C in a solution containing 1 mg/mL microsomes (MLM, male mouse CD1, Lot No. 1510043, Xenotech; HLM; mixed gender, Lot No. 1410013 Xenotech), magnesium chloride (5 mM), and NADPH (1 mM) in potassium phosphate buffer (100 mM, pH 7.4) for 60 min while shaking. The samples were then prepared by ice-cold acetonitrile precipitation, centrifuged, and filtered for LC-MS/MS analysis. Controls containing all the sample constituents (not incubated), and in which NADPH, microsomes, or the test compound were individually excluded were also prepared and handled similarly to the test sample. Propranolol (10 μM) was incubated concomitantly as a positive control.
Metabolite Identification.
Metabolites were identified by comparison of the chromatograms of the incubated samples to those at T0 using Lightsight 2.3. The tentative identity of the metabolites was deduced by comparison of the product ion spectra of the [M + H]+ ions of the metabolites with that of the parent compound using Analyst 1.6.
Rat Pharmacokinetics.
Sprague–Dawley rats, weighing approximately 235 to 265 g were obtained and used for all of the pharmacokinetic (PK) studies. Animals were allowed to acclimate for a minimum of 3 days before experimentation and allowed food and water ad libitum. Compound 28 was formulated in 10% DMSO, 40% PEG-400, 10% (2-hydroxypropyl)-β-cyclodextrin in PBS (pH 7.4). For intravenous bolus administration, five animals received a single dose of 28 via retro-orbital injection at 50 mg/kg. For oral administration, six rats received 28 via oral gavage at 100 mg/kg. Blood samples were collected at 0.25, 0.5, 1, 2, 4, and 8 h postdose and processed to plasma. Compound concentrations were determined by LC-MS/MS.
Supplementary Material
ACKNOWLEDGMENTS
We thank Warren Olifant and Nesia Barnes for guidance on solubility and metabolic stability assays. We thank Christopher Kirk (Kezar Life Sciences) for helpful discussion. We thank Jeremy Burrows and Stephen Brand (Medicines for Malaria Venture) for advice and help with in vitro DMPK studies. We also thank Ellen Yeh for access to the BD Accuri flow cytometer. Funding for this work was provided in part by R21 AI127581 (to M.B. and D.A.F.).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b06656.
Substrate assay data of the P. falciparum and human proteasome 20S using a focused library of fluorogenic substrates, additional data of inhibition for activity-based probe labeling of P. falciparum and human constitutive and immune-proteasome 20S, metabolite identification of compound 7, kinetic solubility, Log D, rat hepatocyte, human liver microsomal stability, and calculated physicochemical properties of analogues, in vivo efficacy of selected lead compounds, cell viability data of P. falciparum Dd2 strain and HepG2 cells for selected inhibitors, inhibition for cleavage processing of the fluorogenic substrates of human proteasome 20S, in vivo PK parameters of compound 28, and detailed experimental procedures including synthesis, characterization (PDF)
REFERENCES
- (1).World Malaria Report; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- (2).Blasco B; Leroy D; Fidock DA Nat. Med 2017, 23, 917–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Straimer J; Gnadig NF; Witkowski B; Amaratunga C; Duru V; Ramadani AP; Dacheux M; Khim N; Zhang L; Lam S; Gregory PD; Urnov FD; Mercereau-Puijalon O; BenoitVical F; Fairhurst RM; Menard D; Fidock DA Science 2015, 347, 428–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ariey F; Witkowski B; Amaratunga C; Beghain J; Langlois AC; Khim N; Kim S; Duru V; Bouchier C; Ma L; Lim P; Leang R; Duong S; Sreng S; Suon S; Chuor CM; Bout DM; Menard S; Rogers WO; Genton B; Fandeur T; Miotto O; Ringwald P; Le Bras J; Berry A; Barale JC; Fairhurst RM; Benoit-Vical F; Mercereau-Puijalon O; Menard D Nature 2014, 505, 50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ashley EA; Dhorda M; Fairhurst RM; Amaratunga C; Lim P; Suon S; Sreng S; Anderson JM; Mao S; Sam B; Sopha C; Chuor CM; Nguon C; Sovannaroth S; Pukrittayakamee S; Jittamala P; Chotivanich K; Chutasmit K; Suchatsoonthorn C; Runcharoen R; Hien TT; Thuy-Nhien NT; Thanh NV; Phu NH; Htut Y; Han K-T; Aye KH; Mokuolu OA; Olaosebikan RR; Folaranmi OO; Mayxay M; Khanthavong M; Hongvanthong B; Newton PN; Onyamboko MA; Fanello CI; Tshefu AK; Mishra N; Valecha N; Phyo AP; Nosten F; Yi P; Tripura R; Borrmann S; Bashraheil M; Peshu J; Faiz MA; Ghose A; Hossain MA; Samad R; Rahman MR; Hasan MM; Islam A; Miotto O; Amato R; MacInnis B; Stalker J; Kwiatkowski DP; Bozdech Z; Jeeyapant A; Cheah PY; Sakulthaew T; Chalk J; Intharabut B; Silamut K; Lee SJ; Vihokhern B; Kunasol C; Imwong M; Tarning J; Taylor WJ; Yeung S; Woodrow CJ; Flegg JA; Das D; Smith J; Venkatesan M; Plowe CV; Stepniewska K; Guerin PJ; Dondorp AM; Day NP; White NJ N. Engl. J. Med 2014, 371, 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Saunders DL; Vanachayangkul P; Lon CN Engl. J. Med 2014, 371, 484–485. [DOI] [PubMed] [Google Scholar]
- (7).Kreidenweiss A; Kremsner PG; Mordmuller B Malar. J 2008, 7, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Prudhomme J; McDaniel E; Ponts N; Bertani S; Fenical W; Jensen P; Le Roch K PLoS One 2008, 3, e2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Czesny B; Goshu S; Cook JL; Williamson KC Antimicrob. Agents Chemother 2009, 53, 4080–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lindenthal C; Weich N; Chia YS; Heussler V; Klinkert MQ Parasitology 2005, 131, 37–44. [DOI] [PubMed] [Google Scholar]
- (11).Gantt SM; Myung JM; Briones MR; Li WD; Corey EJ; Omura S; Nussenzweig V; Sinnis P Antimicrob. Agents Chemother 1998, 42, 2731–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Aminake MN; Schoof S; Sologub L; Leubner M; Kirschner M; Arndt HD; Pradel G Antimicrob. Agents Chemother 2011, 55, 1338–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Li H; O’Donoghue AJ; van der Linden WA; Xie SC; Yoo E; Foe IT; Tilley L; Craik CS; da Fonseca PC; Bogyo M Nature 2016, 530, 233–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Dogovski C; Xie SC; Burgio G; Bridgford J; Mok S; McCaw JM; Chotivanich K; Kenny S; Gnadig N; Straimer J; Bozdech Z; Fidock DA; Simpson JA; Dondorp AM; Foote S; Klonis N; Tilley L PLoS Biol. 2015, 13, e1002132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).O’Donoghue AJ; Eroy-Reveles AA; Knudsen GM; Ingram J; Zhou M; Statnekov JB; Greninger AL; Hostetter DR; Qu G; Maltby DA; Anderson MO; Derisi JL; McKerrow JH; Burlingame AL; Craik CS Nat. Methods 2012, 9, 1095–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Li H; Bogyo M; da Fonseca PC FEBS J. 2016, 283, 4238–4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).LaMonte GM; Almaliti J; Bibo-Verdugo B; Keller L; Zou BY; Yang J; Antonova-Koch Y; Orjuela-Sanchez P; Boyle CA; Vigil E; Wang L; Goldgof GM; Gerwick L; O’Donoghue AJ; Winzeler EA; Gerwick WH; Ottilie SJ Med. Chem 2017, 60, 6721–6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhou HJ; Aujay MA; Bennett MK; Dajee M; Demo SD; Fang Y; Ho MN; Jiang J; Kirk CJ; Laidig GJ; Lewis ER; Lu Y; Muchamuel T; Parlati F; Ring E; Shenk KD; Shields J; Shwonek PJ; Stanton T; Sun CM; Sylvain C; Woo TM; Yang JJ Med. Chem 2009, 52, 3028–3038. [DOI] [PubMed] [Google Scholar]
- (19).Thornberry NA; Rano TA; Peterson EP; Rasper DM; Timkey T; Garcia-Calvo M; Houtzager VM; Nordstrom PA; Roy S; Vaillancourt JP; Chapman KT; Nicholson DW J. Biol. Chem 1997, 272, 17907–17911. [DOI] [PubMed] [Google Scholar]
- (20).Harris JL; Backes BJ; Leonetti F; Mahrus S; Ellman JA; Craik CS Proc. Natl. Acad. Sci. U. S. A 2000, 97, 7754–7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Choe Y; Leonetti F; Greenbaum DC; Lecaille F; Bogyo M; Bromme D; Ellman JA; Craik CS J. Biol. Chem 2006, 281, 12824–12832. [DOI] [PubMed] [Google Scholar]
- (22).Backes BJ; Harris JL; Leonetti F; Craik CS; Ellman JA Nat. Biotechnol 2000, 18, 187–193. [DOI] [PubMed] [Google Scholar]
- (23).Schneider EL; Craik CS Methods Mol. Biol 2009, 539, 59–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Patterson AW; Wood WJL; Ellman JA Nat. Protoc 2007, 2, 424. [DOI] [PubMed] [Google Scholar]
- (25).Chapelat J; Berst F; Marzinzik AL; Moebitz H; Drueckes P; Trappe J; Fabbro D; Seebach D Eur. J. Med. Chem 2012, 57, 1–9. [DOI] [PubMed] [Google Scholar]
- (26).Gladysz R; Lambeir AM; Joossens J; Augustyns K; Van der Veken P ChemMedChem 2016, 11, 467–476. [DOI] [PubMed] [Google Scholar]
- (27).Kasperkiewicz P; Poreba M; Snipas SJ; Parker H; Winterbourn CC; Salvesen GS; Drag M Proc. Natl. Acad. Sci. U. S. A 2014, 111, 2518–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Poreba M; Mihelic M; Krai P; Rajkovic J; Krezel A; Pawelczak M; Klemba M; Turk D; Turk B; Latajka R; Drag M Amino Acids 2014, 46, 931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Poreba M; Salvesen GS; Drag M Nat. Protoc 2017, 12, 2189–2214. [DOI] [PubMed] [Google Scholar]
- (30).Maly DJ; Leonetti F; Backes BJ; Dauber DS; Harris JL; Craik CS; Ellman JA J. Org. Chem 2002, 67, 910–915. [DOI] [PubMed] [Google Scholar]
- (31).Yang J; Wang Z; Fang Y; Jiang J; Zhao F; Wong H; Bennett MK; Molineaux CJ; Kirk CJ Drug Metab. Dispos 2011, 39, 1873–1882. [DOI] [PubMed] [Google Scholar]
- (32).Wang Z; Yang J; Kirk C; Fang Y; Alsina M; Badros A; Papadopoulos K; Wong A; Woo T; Bomba D; Li J; Infante JR Drug Metab. Dispos 2013, 41, 230–237. [DOI] [PubMed] [Google Scholar]
- (33).Wang Z; Fang Y; Teague J; Wong H; Morisseau C; Hammock BD; Rock DA; Wang Z Drug Metab. Dispos 2017, 45, 712–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Fang Y; Wang Z; Zhang T; Teague J; Wang Z Contribution of epoxide hydrolase and cytochrome P450 (CYP) enzymes on oprozomib disposition. Annual Meeting of American Association of Pharmaceutical Scientists; Orlando, Florida, USA, October 25–29, 2015, AAPS 2015–001707. [Google Scholar]
- (35).Gramec D; Peterlin Masic L; Sollner Dolenc M Chem. Res. Toxicol 2014, 27, 1344–1358. [DOI] [PubMed] [Google Scholar]
- (36).Li H; van der Linden WA; Verdoes M; Florea BI; McAllister FE; Govindaswamy K; Elias JE; Bhanot P; Overkleeft HS; Bogyo M ACS Chem. Biol 2014, 9, 1869–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Britton M; Lucas MM; Downey SL; Screen M; Pletnev AA; Verdoes M; Tokhunts RA; Amir O; Goddard AL; Pelphrey PM; Wright DL; Overkleeft HS; Kisselev AF Chem. Biol 2009, 16, 1278–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Li H; Ponder EL; Verdoes M; Asbjornsdottir KH; Deu E; Edgington LE; Lee JT; Kirk CJ; Demo SD; Williamson KC; Bogyo M Chem. Biol 2012, 19, 1535–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Huber EM; Basler M; Schwab R; Heinemeyer W; Kirk CJ; Groettrup M; Groll M Cell 2012, 148, 727–738. [DOI] [PubMed] [Google Scholar]
- (40).Ferrington DA; Gregerson DS Prog. Mol. Biol. Transl. Sci 2012, 109, 75–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Verdoes M; Florea BI; Menendez-Benito V; Maynard CJ; Witte MD; van der Linden WA; van den Nieuwendijk AM; Hofmann T; Berkers CR; van Leeuwen FW; Groothuis TA; Leeuwenburgh MA; Ovaa H; Neefjes JJ; Filippov DV; van der Marel GA; Dantuma NP; Overkleeft HS Chem. Biol 2006, 13, 1217–1226. [DOI] [PubMed] [Google Scholar]
- (42).Blackburn C; Gigstad KM; Hales P; Garcia K; Jones M; Bruzzese FJ; Barrett C; Liu JX; Soucy TA; Sappal DS; Bump N; Olhava EJ; Fleming P; Dick LR; Tsu C; Sintchak MD; Blank JL Biochem. J 2010, 430, 461–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Mok S; Ashley EA; Ferreira PE; Zhu L; Lin Z; Yeo T; Chotivanich K; Imwong M; Pukrittayakamee S; Dhorda M; Nguon C; Lim P; Amaratunga C; Suon S; Hien TT; Htut Y; Faiz MA; Onyamboko MA; Mayxay M; Newton PN; Tripura R; Woodrow CJ; Miotto O; Kwiatkowski DP; Nosten F; Day NP; Preiser PR; White NJ; Dondorp AM; Fairhurst RM; Bozdech Z Science 2015, 347, 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Robert A; Cazelles J; Meunier B Angew. Chem., Int. Ed 2001, 40, 1954–1957. [DOI] [PubMed] [Google Scholar]
- (45).Dambuza NS; Smith P; Evans A; Norman J; Taylor D; Andayi A; Egan T; Chibale K; Wiesner L Malar. J 2015, 14, 505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Bross PF; Kane R; Farrell AT; Abraham S; Benson K; Brower ME; Bradley S; Gobburu JV; Goheer A; Lee SL; Leighton J; Liang CY; Lostritto RT; McGuinn WD; Morse DE; Rahman A; Rosario LA; Verbois SL; Williams G; Wang YC; Pazdur R Clin. Cancer Res 2004, 10, 3954–3964. [DOI] [PubMed] [Google Scholar]
- (47).Chauhan D; Hideshima T; Mitsiades C; Richardson P; Anderson KC Mol. Cancer Ther 2005, 4, 686–692. [DOI] [PubMed] [Google Scholar]
- (48).McBride A; Klaus JO; Stockerl-Goldstein K Am. J. Health-Syst. Pharm 2015, 72, 353–360. [DOI] [PubMed] [Google Scholar]
- (49).Kubiczkova L; Pour L; Sedlarikova L; Hajek R; Sevcikova SJ Cell. Mol. Med 2014, 18, 947–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Moreau P; Richardson PG; Cavo M; Orlowski RZ; San Miguel JF; Palumbo A; Harousseau JL Blood 2012, 120, 947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Tschan S; Mordmuller B; Kun JF Expert Opin. Ther. Targets 2011, 15, 365–378. [DOI] [PubMed] [Google Scholar]
- (52).Kirkman LA; Zhan W; Visone J; Dziedziech A; Singh PK; Fan H; Tong X; Bruzual I; Hara R; Kawasaki M; Imaeda T; Okamoto R; Sato K; Michino M; Alvaro EF; Guiang LF; Sanz L; Mota DJ; Govindasamy K; Wang R; Ling Y; Tumwebaze PK; Sukenick G; Shi L; Vendome J; Bhanot P; Rosenthal PJ; Aso K; Foley MA; Cooper RA; Kafsack B; Doggett JS; Nathan CF; Lin G Proc. Natl. Acad. Sci. U. S. A 2018, 115, E6863–e6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Joyce JA; Baruch A; Chehade K; Meyer-Morse N; Giraudo E; Tsai FY; Greenbaum DC; Hager JH; Bogyo M; Hanahan D Cancer Cell 2004, 5, 443–453. [DOI] [PubMed] [Google Scholar]
- (54).Joyce JA; Hanahan D Cell Cycle 2004, 3, 1516–1619. [DOI] [PubMed] [Google Scholar]
- (55).Papadopoulos KP; Siegel DS; Vesole DH; Lee P; Rosen ST; Zojwalla N; Holahan JR; Lee S; Wang Z; Badros AJ Clin. Oncol 2015, 33, 732–739. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.