

Abstract
Object recognition memory (ORM) confers the ability to discriminate the familiarity of previously encountered items. Reconsolidation is the process by which reactivated memories become labile and susceptible to modifications. The hippocampus is specifically engaged in reconsolidation to integrate new information into the original ORM through a mechanism involving activation of brain-derived neurotrophic factor (BDNF) signaling and induction of LTP. It is known that BDNF can control LTP maintenance through protein kinase Mζ (PKMζ), an atypical protein kinase C isoform that is thought to sustain memory storage by modulating glutamatergic neurotransmission. However, the potential involvement of PKMζ in ORM reconsolidation has never been studied. Using a novel ORM task combined with pharmacological, biochemical, and electrophysiological tools, we found that hippocampal PKMζ is essential to update ORM through reconsolidation, but not to maintain the inactive recognition memory trace stored over time, in adult male Wistar rats. Our results also indicate that hippocampal PKMζ acts downstream of BDNF and controls AMPAR synaptic insertion to elicit reconsolidation and suggest that blocking PKMζ activity during this process deletes active ORM.
SIGNIFICANCE STATEMENT Object recognition memory (ORM) is essential to remember facts and events. Reconsolidation integrates new information into ORM through changes in hippocampal plasticity and brain-derived neurotrophic factor (BDNF) signaling. In turn, BDNF enhances synaptic efficacy through protein kinase Mζ (PKMζ), which might preserve memory. Here, we present evidence that hippocampal PKMζ acts downstream of BDNF to regulate AMPAR recycling during ORM reconsolidation and show that this kinase is essential to update the reactivated recognition memory trace, but not to consolidate or maintain an inactive ORM. We also demonstrate that the amnesia provoked by disrupting ORM reconsolidation through PKMζ inhibition is due to memory erasure and not to retrieval failure.
Keywords: memory, PKMζ, reconsolidation
Introduction
To persist, long-term memories (LTMs) must overcome the turnover of their molecular substrates. There are several hypotheses about the mechanisms responsible for LTM maintenance, including, to name but a few examples, suggestions that LTM is stored as modifications in DNA methylation/sequence (Vanyushin et al., 1974; Crick, 1984), prion-like changes in synaptic growth (Bailey et al., 2004), or proteins able to auto-perpetuate their active state (Fukunaga and Miyamoto, 2000; Lisman and McIntyre, 2001). More recently, Sacktor (2008) proposed that PKMζ, an atypical PKC isoform expressed in neural tissue and enriched in the hippocampus, is sufficient to maintain LTM. In vertebrates, PKMζ is transcribed from an internal promoter within the PKCζ gene to produce mRNAs lacking PKCζ auto-inhibitory domain. Therefore, although PKMζ is persistently active, under basal conditions, translation of its mRNA is repressed (Bal et al., 2016). This block on PKMζ synthesis can be released through neurotrophin signaling (Adasme et al., 2011), allowing PKMζ to perpetuate LTM by promoting AMPAR expression at postsynaptic sites (Migues et al., 2010). As with other hypotheses about memory persistence, the notion that PKMζ sustains LTM is controversial (Lisman, 2012), has been validated (Cai et al., 2011; Wang et al., 2016; Hu et al., 2017) and refuted (Wu-Zhang et al., 2012; Lee et al., 2013; Volk et al., 2013) repeatedly, and fails to accommodate comfortably the fact that consolidated LTMs are not immutable but can incorporate information at the time of reactivation (RA) through a protein-synthesis-dependent reconsolidation process (Nader and Hardt, 2009). It has been reported that PKMζ inhibition can cause amnesia when memory is reactivated within a certain period of time after PKMζ antagonist administration (Parsons and Davis, 2011), suggesting that, in addition to inactive memory maintenance, this kinase may also mediate active memory reconsolidation (Kwapis and Helmstetter, 2014). However, the few reports published so far about the putative involvement of PKMζ in reconsolidation have been inconclusive (Crespo et al., 2012; Levitan et al., 2016), mainly because they used behavioral tasks that cannot distinguish clearly between reconsolidation and other phenomena induced by memory RA, such as extinction.
Object recognition memory (ORM) is a core component of declarative memory. Its failure is one of the earliest signs of Alzheimer's disease, which is also associated with impaired hippocampal BDNF signaling (O'Bryant et al., 2009). ORM consolidation and retrieval require the functional participation of the hippocampus (DeVito and Eichenbaum, 2010; Cohen and Stackman, 2015). However, ORM reconsolidation only recruits this brain region when memory RA occurs concomitantly with novelty detection (Rossato et al., 2007; Winters et al., 2011). When induced by presentation of a novel object, reconsolidation mediates integration of new information into the reactivated ORM trace and is controlled by BDNF (Radiske et al., 2017b) and accompanied by a brief early post-RA depotentiation period followed by a late stage of synaptic efficacy enhancement (Clarke et al., 2010). Because PKMζ controls the facilitatory effect of BDNF on hippocampal LTP (Mei et al., 2011) and both PKMζ and BDNF regulate AMPAR surface expression during synaptic potentiation (Caldeira et al., 2007; Yao et al., 2008; Jourdi and Kabbaj, 2013), which seems necessary for reconsolidation (Rao-Ruiz et al., 2011; Bhattacharya et al., 2017), we hypothesized that PKMζ modulates ORM reconsolidation by mediating the interplay between BDNF and AMPAR recycling, so its inhibition after retrieval in the presence of a novel object impairs ORM reconsolidation and causes amnesia by deleting the reactivated recognition memory trace.
Here, we confirm that inactive ORM does not require hippocampal PKMζ activity to persist (Hardt et al., 2010) and demonstrate that it becomes vulnerable to PKMζ inhibition following RA in the presence of a novel object, suggesting that PKMζ is necessary to update ORM through reconsolidation, but not to consolidate or maintain the dormant recognition trace. We also present evidence that hippocampal PKMζ acts downstream of BDNF to control AMPAR recycling during reconsolidation and that blocking this process with PKMζ inhibitors erases ORM.
Materials and Methods
Subjects
All experiments were performed in accordance with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals and the local institutional ethics committee recommendations (Comissão de Ética no Uso de Animais, CEUA, UFRN). A total of 1174 adult male Wistar rats (3 months old; 300–350 g) were used; 150 were used to revalidate the ORM task and 1024 to test our hypotheses. Rats were housed in groups of 5 per cage with ad libitum access to food and water and kept in the institutional vivarium on a 12 h lights on/off schedule (lights on at 6:00 A.M.) at 23°C. All behavioral experiments were performed during the light phase of the cycle. Researchers were blinded as to the rat's treatment condition.
Stereotaxic surgery for cannula and multielectrode array implants
Rats were anesthetized with ketamine (80 mg/kg)/xylazine (10 mg/kg) and bilaterally implanted with 22-gauge stainless steel guides aimed to the CA1 region of the dorsal hippocampus (AP −4.2; LL, ±3.0; DV, −3.0) and/or the entorhinal cortex (EC) (AP −6.8; LL ±5.0; DV −8.1). Some rats were also implanted with electrode arrays aimed to the dorsal hippocampus (AP −3.6; LL +2.4; DV −3.6 mm). Coordinates were taken from Paxinos and Watson (2007). Arrays were made of 16 tungsten electrodes (50 μm, PFA coated; A-M Microsystems) organized in two rows spaced by 250 μm. Ground screw electrodes were localized in the parietal bone. Meloxicam (0.2 mg/kg) was administered by subcutaneous injection at the end of all surgical procedures as an analgesic. Rats implanted with electrode arrays were housed individually. Rats were allowed a recovery period of at least 7 d to regain presurgery weight before behavioral procedures. During this period, the rats were handled daily for 1–2 min.
Experimental design and statistical analysis
Novel object recognition (NOR) task.
ORM was assessed using a NOR task based on the spontaneous exploratory behavior of rats (Ennaceur and Delacour, 1988). If animals are put in the presence of a familiar and a novel object in an open-field arena, they will preferentially explore the novel one. The NOR task was conducted in a gray plywood open-field arena (60 × 60 × 60 cm) placed in a dim-light illuminated room acclimatized at 23–24°C. Rats were handled and allowed to freely explore the training arena in the absence of stimuli objects for 20 min/d during 4 d (habituation sessions). One day after the last habituation session, rats were exposed to two novel stimuli objects (objects A and B) for 5 min in the training arena (training session; TR). Memory RA was conducted by reexposing the rats to one of the objects presented during TR (object A) together with a novel object (object C) for 5 min in the arena (RA session). RA sessions were performed 1 or 7 d after TR. ORM retention was evaluated 3 h, 1 d, or 7 d after RA by exposing the rats to a novel object D alongside familiar objects A, B, or C for 5 min (test session). One hour before the experimental sessions, rats were transported from the vivarium to the experimental anteroom. From there, each rat was individually brought to the experiment room in a transport cage. At the end of each session, rats were return to the experimental anteroom and stayed there for an additional hour before being transported back to the vivarium. Stimuli objects were made of metal, glass, or glazed ceramic and had no significant innate preference for rats (Table 1). The open-field arena and the stimuli objects were cleaned with ethanol (50%) before each trial to ensure the absence of olfactory cues. Object exploration was defined as sniffing and touching the stimuli objects with the muzzle and/or forepaws. Sitting on or turning around the objects was not considered exploratory behavior. Digital video cameras fixed above the open-field arenas were used for tracking the rat's position and behavior. Video data were acquired at 30 frames/s and analyzed using the ObjectScan system (CleverSys). We validated the performance of the ObjectScan system by comparing it with human scores before performing the experiments reported here. The discrimination index (DI) was calculated as follows: (time exploring novel object − time exploring familiar object)/total object exploration time, considering data from the 5 min session (Akirav and Maroun, 2006; Maroun and Akirav, 2008). Naive rats discriminate between novel and familiar objects throughout the entire retention test session (Table 2). DI can vary between −1 and +1; positive DI score indicates preference for the novel object, whereas a DI score close to zero indicates that rats explored both objects to the same extent, suggesting absence of discrimination.
Table 1.
Objects utilized in the NOR task have no natural significance and are behaviorally equivalent for naive adult male Wistar rats
| Object pair | Object exploration time (s) |
DI | pa | n | ||
|---|---|---|---|---|---|---|
| Object (1) | Object (2) | Total | ||||
| AB | 28.69 ± 3.72 | 29.04 ± 4.54 | 57.73 ± 7.27 | −0.0057 ± 0.0784 | 0.9431 | 9 |
| AC | 25.18 ± 4.11 | 22.87 ± 3.52 | 48.05 ± 7.31 | −0.0467 ± 0.0685 | 0.5124 | 10 |
| AD | 24.20 ± 2.73 | 25.36 ± 2.58 | 49.57 ± 4.58 | 0.0286 ± 0.0552 | 0.6166 | 10 |
| AE | 27.50 ± 3.68 | 26.19 ± 3.06 | 53.66 ± 5.60 | −0.0300 ± 0.0745 | 0.6959 | 10 |
| BC | 26.14 ± 2.31 | 28.74 ± 3.65 | 54.88 ± 5.18 | 0.0271 ± 0.0564 | 0.6431 | 9 |
| BD | 28.59 ± 2.26 | 26.47 ± 1.51 | 55.06 ± 2.88 | −0.0286 ± 0.0481 | 0.5669 | 10 |
| BE | 26.79 ± 2.81 | 26.83 ± 4.35 | 53.63 ± 5.92 | −0.0255 ± 0.0819 | 0.7632 | 9 |
| CD | 31.19 ± 3.13 | 29.83 ± 2.79 | 61.02 ± 4.72 | −0.0192 ± 0.0626 | 0.7667 | 9 |
| CE | 29.86 ± 2.56 | 29.45 ± 3.72 | 59.30 ± 5.72 | −0.0292 ± 0.0506 | 0.5777 | 10 |
| DE | 27.83 ± 3.62 | 25.44 ± 2.26 | 53.27 ± 5.56 | −0.0255 ± 0.0391 | 0.5306 | 10 |
Shown are mean exploration time and DI (±SEM) for naive animals during spontaneous object exploration in the training session of the NOR task. Total exploration time did not differ between object pairs (F(9,86) = 0.5161, p = 0.8592). DIs for each object pair exploration are shown.
ap in one-sample Student's t test with theoretical mean = 0.
Table 2.
Adult male Wistar rats trained in the NOR task discriminate between novel and familiar objects during the whole retention test session
| 1st min | 2nd min | 3rd min | 4th min | 5th min | |
|---|---|---|---|---|---|
| DI | 0.276 ± 0.043 | 0.312 ± 0.060 | 0.221 ± 0.059 | 0.209 ± 0.075 | 0.320 ± 0.067 |
| pa | <0.001 | <0.001 | <0.001 | <0.01 | <0.001 |
| Object exploration time (s) | 15.76 ± 0.65 | 13.55 ± 0.79 | 11.37 ± 0.75 | 10.92 ± 0.79 | 11.24 ± 0.89 |
Shown are mean ± SEM DI and total exploration time for each consecutive minute of a 5-min-long ORM retention test session in the presence of familiar object A and novel object C performed 24 h after training in the presence of two different novel objects (A and B).
ap in one-sample Student's t test with theoretical mean = 0 (n = 50).
Morris water maze (MWM) task.
The MWM was a black circular pool (200 cm in diameter) conceptually divided in four equal imaginary quadrants from which rats learned to escape onto a hidden platform that was 1.5 cm beneath the water surface. Water temperature was regulated at 24–25°C. The maze was located in a well lit white room with several posters and other distal visual stimuli hanging on the walls to provide spatial cues. A curtain separated the maze from the area where the computer was set up and the rats temporarily housed during the behavioral sessions. Training was performed over 5 consecutive days as described previously (Rossato et al., 2007). On each day, rats received eight consecutive training trials during which the hidden platform was kept in a constant location. A different starting location was used on each trial, which consisted of a swim followed by a 30 s platform sit. The experimenter guided any rat that did not find the platform within 60 s to it. The intertrial interval was 30 s. Memory retention was evaluated in a 60 s probe trial performed in the absence of the escape platform 24 h after the last training session. Video data were acquired at 30 frames/s and analyzed online using the TopScan system (CleverSys).
Step-down inhibitory avoidance (IA) task.
The training apparatus was a 50 × 25 × 25 cm Plexiglas box with a 5-cm-high, 8-cm-wide, 25-cm-long platform on the left end of a series of bronze bars that made up the floor of the box. For training, rats were placed on the platform facing the left rear corner of the training box and, when they stepped down and placed their four paws on the grid, received a 2 s, 0.4 mA scrambled foot shock. Memory retention was evaluated in a non-reinforced test session performed 24 h after TR. The test session finished when the rat stepped down to the grid or after 300 s.
Drugs and infusion procedures
All drug doses used in this work were based on previous studies and pilot experiments. Anisomycin (ANI; 160 μg/μl), autocamptide-2 related inhibitor peptide (AIP; 5 nmol/μl), dynasore hydrate (DYN; 120 pmol/μl), lidocaine (LID; 4%), muscimol (MUS; 0.1 μg/μl), and recombinant human BDNF (0.25 μg/μl) were from Sigma-Aldrich. Pep2m (PEP; 5 pmol//μl), myristoylated ζ-inhibitory peptide (ZIP; 1 nmol/μl), and myristoylated scrambled ZIP (Scr-ZIP; 1 nmol/μl) were from Tocris Bioscience. PKMζ antisense (ASO; 5′-CTCTTG GGA AGGCATGA-3′; 2 nmol/μl) and PKMζ scrambled antisense oligonucleotides (sASO; 5′-AACAATGGGTCGTCTCG-3′; 2 nmol/μl) were from GBT-Oligos. ASO and sASO were phosphorothioated on the three terminal bases to avoid nuclease degradation. sASO had ASO base composition in a scrambled order and did not match any mammalian sequence in the GenBank database. ICAP (0.01–1 nmol/μl) was a generous gift from Dr. Robert V. Farese (University of South Florida). Drugs and oligonucleotides were dissolved upon arrival and stored at −20°C until use. On the day of the experiment, stock aliquots were thawed and diluted to working concentration in sterile saline (pH 7.2; vehicle, VEH). At the time of drug delivery, infusion cannulas were fitted into the guides and injections (1 μl/side at a rate of 0.5 μl/min) performed using a Hamilton syringe coupled to an infusion pump (Harvard Apparatus). Infusion cannulas were left in place for 1 additional minute to minimize backflow. Placement of the cannulas was verified postmortem. All infusions took place in a room separated from the experimental room and anteroom.
Immunoblotting
Rats were killed by decapitation without anesthesia and immediately thereafter the CA1 region of the dorsal hippocampus was dissected out and homogenized in ice-chilled homogenization buffer (10 mm Tris-HCl, pH 7.4, containing 0.32 m sucrose, 1 mm EDTA, 1 mm EGTA, 1 mm PMSF, 10 μg/ml aprotinin, 15 μg/ml leupeptin, 10 μg/ml bacitracin, 10 μg/ml pepstatin, 50 mm NaF, and 1 mm sodium orthovanadate). Dorsal CA1 microdissection was performed on an ice-chilled surface with the help of a stereo zoom microscope. Subcellular fractions were prepared at 4°C. To do that, hippocampal homogenates were centrifuged at 1000 × g for 10 min and the supernatant transferred to a new tube for a second centrifugation round at 10,000 × g during 15 min to obtain a crude synaptosomal fraction (CSF) enriched in synaptic plasma membranes. The CSF pellet was washed twice and finally resuspended in 10 mm Tris-HCl, pH 7.4, 1 mm EDTA, 1 mm EGTA, 1 mm PMSF, 10 μg/ml aprotinin, 15 μg/ml leupeptin, 10 μg/ml bacitracin, 10 μg/ml pepstatin, 50 mm NaF, and 1 mm sodium orthovanadate. Protein concentration was determined using the BCA protein assay kit (Pierce). SDS/PAGE was performed under reducing conditions and proteins were electrotransferred onto PVDF membranes (Immobilon-P; Merck Millipore). Blots were blocked for 2 h in TTBS (100 mm Tris-HCl, 0.9% NaCl, 0.1% Tween 20, pH 7.4) and incubated overnight at 4°C with anti-pThr286CaMKII (1:5000 dilution; rabbit polyclonal; Cell Signaling Technology), anti-pThr305CaMKII (1:5000 dilution; rabbit polyclonal; Sigma-Aldrich), anti-αCaMKII (1:5000 dilution; goat polyclonal; Santa Cruz Biotechnology), anti-PKMζ (1:80,000 dilution; rabbit polyclonal; Sigma-Aldrich), anti-GluA1 (1:10,000 dilution; rabbit polyclonal; Santa Cruz Biotechnology), anti-GluA2/3 (1:10,000 dilution; rabbit polyclonal; Santa Cruz Biotechnology) or anti-β-tubulin (1:40,000; rabbit polyclonal; Abcam) antibodies. Then, blots were washed with TTBS and incubated with HRP-coupled anti-IgG antibody. The West-Pico enhanced chemiluminescence kit (GE Healthcare) was used to detect immunoreactivity. Blots were quantified using the GE Healthcare Imager 600 RGB system.
Electrophysiological recordings
LFPs were recorded continuously using a Cerebus Neural Signal Processor system (Blackrock Microsystems). Signals were amplified, digitized, filtered at cutoff frequencies of 0.3 and 250 Hz, and sampled at 1 kHz. Data were analyzed offline using built-in (Signal Processing Toolbox) and custom-written routines in MATLAB (RRID:SCR_001622). The CA1 pyramidal cell layer was identified by stereotaxic coordinates and standard electrophysiological parameters (Brankack et al., 1993; Bragin et al., 1995), as described in Radiske et al. (2017a). To evaluate hippocampal oscillatory activity during object exploration periods, LFPs were continuously recorded throughout training and test sessions in the NOR task. Digital video cameras fixed above the arenas were used for tracking the rat's position. Video data were acquired at 30 frames/s. Active exploration was defined as sniffing and touching the stimuli objects with the muzzle and/or forepaws,and was detected using ObjectScan system software (CleverSys). LFP epochs of object A active exploration were extracted, merged, and analyzed (Fig. 6A is a schematic diagram of the procedure). Data from adjacent exploration epochs separated by <0.5 s were merged and considered as a single event. Data from exploration periods <0.5 s were excluded from analysis. We used the Welch periodogram method (3 s Hamming windows, 75% overlap) for power spectra computing. Band power of theta, beta, and slow-gamma were defined as the average power in the frequency range of 5–10 Hz, 13–20 Hz, and 30–55 Hz, respectively. To determine power changes during object A exploration in TR and test sessions, we normalized data from each subject using a power ratio that indicates power per unit frequency normalized by power during 60-s-long baseline epoch. Baseline LFPs were acquired in the recording cage 30 min before TR and test sessions. For cross-frequency coupling analysis, theta phases and slow-gamma amplitude were computed from the Hilbert transform of the filtered versions of each frequency band. Theta phases were binned into 18 intervals of 20°. Mean gamma amplitude was computed for each theta phase bin and normalized by the sum of amplitude values over all bins. The modulation index (MI) was used to express coupling strength. MI indicates the Kullback–Leiber distance between the uniform distribution and the probability function derived from mean amplitude per phase distribution (Tort et al., 2010). Co-modulation maps were obtained by expressing the MI of several frequency band pairs (4 Hz bandwidths, 1 Hz steps for phase frequencies; 10 Hz bandwidths, 5 Hz steps for amplitude frequencies) in a bidimensional pseudocolor plot (Tort et al., 2010). Mean MI was defined as the average MI values in the (5–10 Hz) × (30–55 Hz) regions of the co-modulation maps. MIs were calculated from 10-s-long epochs of object A exploration with equalized theta and gamma power between training and test sessions to ensure that changes in MI values were not a result of differences in theta or gamma power. A 10-s-long period was chosen given that it is long enough to average out fluctuations in gamma amplitude that are not coupled to the theta cycle (Zhang et al., 2016). Slow-gamma events, defined as time intervals when gamma power surpassed by 2 SDs the time-averaged power, were identified and the theta phase associated with them was determined. Time intervals with power >6 SDs were excluded to avoid analysis of artifactual events. Episodes separated by <100 ms were merged and considered as a single event. Theta phase at the time points corresponding to the maximum of each gamma event was extracted and the circular mean was computed, obtaining a single-phase value associated to the occurrence of high-gamma amplitude. To evaluate the effect of intra-CA1 ZIP infusion (1 nmol/μl) on spontaneous hippocampal oscillatory activity, we recorded LFPs for 30 min before drug infusion (baseline) and for ∼60 min after intra-CA1 administration of VEH, ZIP, or LID. In this analysis, we evaluated changes in theta, beta, and the entire gamma range, defining gamma-band power as the average power in the frequency range of 30–90 Hz. Band power and LFP root mean square (RMS) were computed using epochs of 5 min corresponding to baseline, 15 min, and 60 min after drug infusion. To construct spectrograms, power spectrum was computed using sliding windows of 4 and 2 s steps. Multiunit activity was estimated by band-pass filtering (500–3000 Hz) recordings sampled at 30 kHz and computing RMS as described in Sharma et al. (2012). To evaluate the effect of intra-CA1 drug infusions, we computed the ratio of after infusion to preinfusion hippocampal neural activity.
Figure 6.

ORM reacquisition after ZIP-induced amnesia recapitulates the electrophysiological correlates of original learning. A, Rats were trained in NOR using two different stimuli objects (A and B, day 1, TR) and, 24 h after TR (day 2), were submitted to a 5-min-long ORM RA in the presence of familiar object A and novel object C. Immediately after RA, rats received bilateral intra-CA1 infusions (1 μl/side) of VEH (saline) or ZIP (1 nmol/μl). One day later (day 3, TEST), rats were exposed to familiar object A and novel object D for 5 extra minutes to evaluate ORM retention. LFP signals from dorsal CA1 were recorded during TR and TEST. LFP activity from epochs of object A exploration was analyzed. We show a representative diagram of instantaneous speed and periods of active exploration during TR from one rat. B, Left, DI during TR and TEST (mean ± SEM). #p < 0.05 in one-sample Student's t test with theoretical mean = 0. Dashed line represents chance level. Right, Mean speed during exploration of object A in TR and TEST. During active exploration periods, rats remained motionless or moved slowly (mean speed < 6 cm/s). Mean speed did not change across TR and TEST. C, Mean power ratio (1–100 Hz) showing alterations in hippocampal oscillatory activity during TR and TEST; bold lines represent group mean and shaded areas represent SEM. D, Mean power ratio for theta (5–10 Hz), beta (13–20 Hz), and slow-gamma (30–55 Hz) frequency bands during TR and TEST. *p < 0.05 in one-sample Student's t test with theoretical mean = 1. E, Example of filtered dorsal-CA1 LFP recordings during object A exploration in TEST for VEH- and ZIP-treated rats. F, Representative phase-amplitude comodulograms during object A exploration in TR and TEST. G, Boxplot distribution of mean theta-slow-gamma modulation indexes during object A exploration in TR and TEST. Modulation indexes were calculated using 10-s-long epochs in which theta and slow-gamma power did not differ between TR and TEST. *p < 0.05, unpaired Student's t test. H, Representative circular histograms showing the distribution of gamma events over theta phases during object A exploration in TR and TEST. γS, slow-gamma. 0° was defined as the peak of the theta cycle. n = 5 per group.
Data analysis
Statistical analyses were performed using GraphPad Prism 6 software (RRID:SCR_002798). Significance was set at p < 0.05. NOR data were analyzed using one-sample t test with theoretical mean = 0, unpaired Student's t test, or one-way/two-way ANOVA followed by Bonferroni's multiple comparisons. MWM data were analyzed using one-sample t test with theoretical mean = 25 and unpaired Student's t test. IA data were analyzed using two-tailed Mann–Whitney U test. Data from immunoblots were analyzed using repeated-measures ANOVA followed by Bonferroni's multiple-comparisons test or Student's t test. Electrophysiological data were analyzed using repeated-measures two-way ANOVA followed by Bonferroni's multiple-comparisons test, Student's t test, one-sample t test with theoretical mean = 1, or Rayleigh test.
Cannula and electrode placement
To verify cannula placement, 2–4 h after the end of the behavioral tests, we infused 1 μl of 4% methylene-blue as described above and rats were killed 30 min thereafter. The dye spread was taken as indication of the diffusion of the drug previously injected. Only data from rats with correct cannula implants (96%) were included in statistical analysis. To verify electrode placement, rats were anesthetized and perfused intracardially with 4% paraformaldehyde, pH 7.2. Immediately after that, brains were removed and left in 30% sucrose for 48 h before cutting into coronal sections (50 μm). Relevant slices were stained with cresyl violet to confirm electrode tracks.
Results
ORM reconsolidation requires PKMζ activity in the dorsal hippocampus
To analyze the effect of hippocampal PKMζ inhibition on ORM reconsolidation, we trained naive adult male Wistar rats in a NOR task involving exploration of two different but behaviorally equivalent novel objects (objects A and B) in an open-field arena. In this task, the hippocampus is engaged in reconsolidation only when the consolidated recognition trace is reactivated in the presence of a novel object (Rossato et al., 2007). Therefore, to induce hippocampus-dependent ORM reconsolidation, 1 d after TR, we reexposed the rats for 5 min to one of the objects presented during the training session (object A) together with a novel object C to reactivate the ORM trace. Immediately after that, the rats received bilateral intra-CA1 infusions of VEH (saline), the PKMζ inhibitor ZIP (1 nmol/μl; Parsons and Davis, 2011), or its inactive analog Scr-ZIP (1 nmol/μl). ORM retention was tested 24 h later. To do that, we randomly assigned the rats to different experimental groups and exposed them to novel object D in the presence of familiar object A, B, or C. Rats that received VEH or Scr-ZIP immediately after ORM RA discriminated novel object D from objects A, B, and C during the retention session, reflecting that they remembered objects A and B, which had been presented during the training session, and also that they acquired memory for object C during the RA session. Conversely, rats that received ZIP right after ORM RA did not discriminate objects A and C from object D, indicating that ZIP hampered retention of the memory for object A and blocked formation of the memory for object C. Memory for familiar object B, which was absent during the RA session, was unaffected by ZIP (Fig. 1A; test AD: F(2,25) = 9.334, p = 0.0009 for treatment effect; t(25) = 3.667, p < 0.01 for VEH vs ZIP; t(25) = 3.842, p < 0.01 for Scr-ZIP vs ZIP; test CD: F(2,25) = 7.078, p = 0.0037 for treatment effect; t(25) = 3.667, p < 0.01 for VEH vs ZIP; t(25) = 3.842, p < 0.01 for Scr-ZIP vs ZIP). ZIP also hindered memory for objects A and C when given 6 h, but not 12 h, after RA (Fig. 1B, left; test AD: t(16) = 3.390, p < 0.01; test CD: t(15) = 3.864, p < 0.01). The amnesia caused by ZIP was independent of the time elapsed between training and RA sessions and between RA and retention test sessions (Fig. 1C, left, test AD: t(18) = 2.199, p < 0.05; test CD: t(18) = 2.382, p < 0.05; right, test AD: t(18) = 5.243, p < 0.001; test CD, t(18) = 4.316, p < 0.001). ZIP did not affect memory for object A when given immediately after ORM RA with two familiar objects (Fig. 1D) or when injected either after exploration of the training arena in the absence of stimuli objects (Fig. 1E) or in the presence of two novel ones (Fig. 1F). Intra-CA1 administration of ZIP immediately after memory RA in the presence of a novel object did not impair ORM retention when tested 3 h later (Fig. 1G). Moreover, pre-RA intra-CA1 infusion of MUS (0.1 μg/μl) impeded both ORM retrieval and the amnesia caused by ZIP (Fig. 1H; RA session: t(46) = 6.734, p < 0.0001; Test session: F(1,44) = 9.335, p = 0.0038 for pre-RA treatment effect; F(1,44) = 6.975, p = 0.0114 for after RA treatment effect; F(1,44) = 13.90, p = 0.0005 for interaction; t(44) = 4.505, p < 0.001 for VEH-VEH vs VEH-ZIP; t(44) = 4.028, p < 0.01 for MUS-VEH vs VEH-ZIP; t(44) = 4.797, p < 0.001 for VEH-ZIP vs MUS-ZIP), whereas ORM RA in the presence of a novel object, but not in the presence of two familiar ones, increased PKMζ levels (Fig. 1I; F(5,15) = 4.300, p = 0.0126 for treatment effect; t(15) = 4.130, p < 0.01 TR vs 360 min after RA). Importantly, ZIP had no effect on ORM when given in dorsal CA1 immediately, 24 h, or 168 h after TR in the absence of specific contextual stimuli (Fig. 1J). These results demonstrate that ORM is immune to intrahippocampal ZIP administration up to 7 d after TR unless it is reactivated in the presence of a novel object. The amnesia caused by ZIP took >3 h to develop, was contingent on ORM RA in the presence of a novel object, and was specific for that object and the familiar one present during the RA session, suggesting that PKMζ is necessary to update ORM through reconsolidation, but not to consolidate or maintain the dormant recognition trace.
Figure 1.

ZIP impairs ORM reconsolidation but does not affect ORM consolidation. A, Rats were trained in NOR using two different objects (A and B, day 1, TR) and 24 h later (day 2) submitted to an ORM RA session in the presence of familiar object A and novel object C. Immediately after RA, rats received intra-CA1 infusions of VEH (saline), ZIP, or Src-ZIP. One day later (day 3, TEST), rats were exposed to a familiar object (A, B, or C) and novel object (D) to evaluate retention. Data (mean ± SEM) are presented as DI during TR, RA, and TEST, n = 9–10 per group. Rats explored objects equally during TR (A = 28.81 ± 1.14 s, B = 30.6 ± 1.13 s, t(83) = 1.548, p = 0.125) and discriminated them during RA (A = 21.29 ± 0.961 s, C = 35.74 ± 1.285 s, t(83) = 19.10, p < 0.001). Total object exploration time did not differ among TR, RA, and TEST (F(2,249) = 1.257, p = 0.286). B, Rats were treated as in A, but received intra-CA1 VEH or ZIP 6 h (left) or 12 h (right) after RA. Data (mean ± SEM) are presented as DI during TR, RA, and TEST, n = 8–10 per group. Rats explored objects equally during TR (left: A = 27.12 ± 1.68 s, B = 28.85 ± 1.51 s, t(51) = 1.111, p = 0.2717; right: A = 31.87 ± 1.242 s, B = 33.31 ± 1.31 s, t(57) = 0.967, p = 0.338) and discriminated them during RA (left: A = 21.84 ± 1.26 s, C = 38.02 ± 1.76 s, t(51) = 12.61, p < 0.001; right: A = 25.60 ± 0.95 s, C = 40.93 ± 1.38 s, t(57) = 16.33, p < 0.001). Total object exploration time did not differ among TR, RA, and TEST (left: F(2,153) = 2.357, p = 0.0981; right: F(2,171) = 2.885, p = 0.0586). C, Rats were treated as in A, but RA was performed 7 d after TR (left) or TEST was performed 7 d after RA (right). Data (mean ± SEM) are presented as DI during TR, RA, and TEST, n = 9–10 per group. Rats explored objects equally during TR (left: A = 24.94 ± 1.24 s, B = 26.86 ± 1.08 s, t(57) = 1.60, p = 0.115; right: A = 27.48 ± 1.07 s, B = 28.09 ± 1.12 s, t(57) = 0.484, p = 0.63) and discriminated them during RA (left: A = 19.31 ± 0.71 s, C = 31.14 ± 1.06 s, t(57) = 16.85, p < 0.001; right: A = 21.0 ± 0.77 s, C = 35.23 ± 1.16 s, t(57) = 16.96, p < 0.001). Total object exploration time did not differ among TR, RA, and TEST (left: F(2,171) = 2.33, p = 0.101; right: F(2,171) = 2.71, p = 0.067). D, Rats were treated as in A, but RA was performed in the presence of familiar objects A and B. Data (mean ± SEM) are presented as DI during TR, RA, and TEST, n = 10 per group. Rats explored objects equally during TR (A = 26.89 ± 1.88 s, B = 29.39 ± 1.71 s, t(19) = 1.35, p = 0.193) and did not discriminate them during RA (A = 23.11 ± 1.7 s, C = 24.52 ± 1.70 s, t(19) = 0.95, p = 0.354). Total object exploration time did not differ among TR, RA, and TEST (F(2,57) = 1.95, p = 0.152). E, Rats were treated as in A, but 24 h after TR, they explored the training arena in the absence of objects. Data (mean ± SEM) are presented as DI during TR and TEST, n = 8–9 per group. Rats explored objects equally during TR (A = 27.87 ± 1.30 s, B = 29.37 ± 1.17 s, t(50) = 1.149, p = 0.256). Total object exploration time did not differ between TR and TEST (t(100) = 1.252, p = 0.214). F, Rats were treated as in A, but 24 h after TR, they explored two novel objects (C and D; TR2). Data (mean ± SEM) are presented as DI during TR, TR2, and TEST, n = 8–10 per group. Rats explored objects equally during TR (A = 31.25 ± 1.36 s, B = 31.34 ± 1.43 s, t(70) = 0.0596, p = 0.953) and TR2 (C = 32.14 ± 1.56 s, D = 30.54 ± 1.52 s, t(70) = 0.935, p = 0.353). Total object exploration time did not differ among TR, TR2, and TEST (F(2,210) = 1.55, p = 0.215). G, Rats were treated as in A, but ORM was evaluated 3 h after RA. Data (mean ± SEM) are presented as DI during TR, RA, and TEST, n = 9–10 per group. Rats explored objects equally during TR (A = 36.65 ± 3.06 s, B = 34.98 ± 2.05 s, t(18) = 0.530, p = 0.602) and discriminated them during RA (A = 25.7 ± 2.00 s, C = 42.88 ± 2.47 s, t(18) = 10.07, p < 0.001). Total object exploration time did not differ among TR, RA, and TEST (F(2,54) = 0.115, p = 0.892). H, Rats were treated as in A, but 15 min before RA, received intra-CA1 infusions of VEH or MUS and, immediately after RA, were given VEH or ZIP in the dorsal CA1. Data (mean ± SEM) are presented as DI during TR, RA, and TEST, n = 12 per group. Rats explored objects equally during TR (A = 22.55 ± 0.81 s, B = 22.21 ± 0.73 s, t(47) = 0.402, p = 0.67). Rats that received VEH before RA (A = 15.47 ± 0.87 s, C = 28.57 ± 1.39 s, t(23) = 7.65, p < 0.001), but not those given MUS (A = 21.88 ± 1.71 s, C = 18.55 ± 1.46, t(47) = 1.62, p = 0.119), discriminated objects during RA. Total object exploration time did not differ among TR, RA, and TEST (F(2,141) = 1.69, p = 0.188). I, Rats were trained as in A and, 24 h later, submitted to RA in the presence of familiar object A and novel object C. At different times after RA, rats were killed and the CA1 region of the dorsal hippocampus was dissected out and homogenized to determine PKMζ levels by immunoblot. PKMζ levels were also evaluated 360 min after ORM RA in the presence of familiar objects A and B (AB360). Data are expressed as mean ± SEM, n = 4. TUB, β-tubulin. J, Rats were trained as in A and immediately (0 h; left), 24 h (center), or 168 h (right) after TR received intra-CA1 infusions of VEH or ZIP. ORM retention was evaluated 24 h later. Data (mean ± SEM) are presented as DI during TR and TEST, n = 10–11 per group. Rats explored objects equally during TR (left: A = 22.86 ± 1.61 s, B = 25.78 ± 2.84 s, t(19) = 1.257, p = 0.224; center: A = 30.7 ± 1.97 s, B = 29.08 ± 1.8 s, t(19) = 0.84, p = 0.412; right: A = 25.78 ± 1.29 s, B = 27.39 ± 1.15 s, t(21) = 1.265, p = 0.22). Total object exploration time did not differ between TR and TEST (left: t(38) = 0.646, p = 0.523; center: t(38) = 1.47, p = 0.149; right: t(42) = 1.82, p = 0.076). In all graphs, dashed lines represent chance level; #p < 0.05 in one-sample Student's t test with theoretical mean = 0; *p < 0.05, **p < 0.01, and ***p < 0.001 in unpaired t test or Bonferroni's multiple-comparisons test after one-way or two-way ANOVA.
ZIP does not affect spontaneous oscillatory activity or basal CaMKII autophosphorylation
There is an ongoing debate over whether the effect of ZIP on memory is due to PKMζ inhibition or if it results from nonspecific mechanisms of action (Lisman, 2012, 2017). Actually, PKMζ knock-out mice can form ZIP-sensitive memories (Lee et al., 2013; Volk et al., 2013) and ZIP induces seizures and reduces spontaneous hippocampal LFPs in anesthetized rats when used at concentrations 10 or more times higher than in our experiments (LeBlancq et al., 2016). At high concentrations, ZIP can also inhibit CaMKII and PKCι/λ (Pastalkova et al., 2006; Jalil et al., 2015), which can control AMPAR function and transport and compensate for PKMζ in PKMζ knock-out mice, respectively (Ren et al., 2013; Tsokas et al., 2016). At the dose used in our experiments (1 nmol/μl), we did not observe seizure-like episodes following ZIP administration. Moreover, amnesic rats that received ZIP after ORM RA were later able to acquire and express a spatial preference in the water maze (Fig. 2A, left, training: F(1,16) = 1.427, p = 0.2496 for treatment effect, F(4,64) = 31.45, p < 0.0001 for session effect) and to learn an inhibitory avoidance response (Fig. 2A, right), two memory types that require the integrity of the hippocampal formation (Paratcha et al., 2000; Martin et al., 2005), suggesting that ZIP did not trigger any lingering deleterious effect on hippocampal function.
Figure 2.

ZIP does not affect spontaneous oscillatory activity or CaMKII autophosphorylation. A, Rats that received intra-CA1 infusions of VEH (saline) or ZIP immediately or 6 h after ORM RA were trained in the spatial version of the MWM or in the IA task 48 h later. Bottom left, Escape latency during MWM TR sessions shown in blocks of eight trials/session. Bottom center, Percentage of time spent in target quadrant (TQ) during a 60-s-long probe test performed 24 h after the last training session. Data are presented as mean ± SEM, n = 9 per group. Bottom right, Step-down latency during IA TR and a retention TEST session performed 24 h later; data are expressed as median ± interquartile range, n = 8 per group. B, LFP signals were simultaneously recorded from the hippocampus before (baseline) and after intra-CA1 infusions of VEH, ZIP (1 nmol/μl), or LID (4%). Five-minute-long recording periods were analyzed. C, Representative power spectrum density plots from rats that received VEH, ZIP, or LID. D, Representative hippocampal raw LFP traces and spectrograms from rats that received VEH, ZIP, or LID. Baseline and 15 min and 60 min posttreatment time points are shown. E, Normalized hippocampal theta, beta, and gamma power and RMS values from rats that received intra-CA1 infusions of VEH, ZIP, or LID. Baseline and 15 min and 60 min posttreatment time points are shown. F, Mean speed and traveled distance from rats that received intra-CA1 infusions of VEH, ZIP, or LID. Baseline and 15 min and 60 min posttreatment time points are shown. G, Peak frequency of theta, beta, and gamma oscillations from rats that received intra-CA1 infusions of ZIP. Baseline and 15 min and 60 min posttreatment time points are shown. H, Hippocampal multiunit activity (MUA) from rats that received intra-CA1 infusions of ZIP. Baseline and 15 min and 60 min posttreatment time points are shown. Data from electrophysiological recordings are presented as mean ± SEM, n = 4 per group. I, Rats received intra-CA1 infusions of VEH or ZIP and 30 min afterward were killed by decapitation and the CA1 region of the dorsal hippocampus dissected out and homogenized to determine total, pT286, and pT305 αCaMKII levels by immunoblot. Data are expressed as mean ± SEM, n = 4 per group. **p < 0.01 and ***p < 0.001 in paired t test or Bonferroni's multiple-comparisons test after one-way or two-way ANOVA, as appropriate.
Analysis of spontaneous hippocampal oscillatory activity in freely moving rats after ZIP (1 nmol/μl) or LID (4%) administration showed that LID reduced LFP power and RMS values 15 min after infusion, but ZIP did not modify ongoing LFP (Fig. 2B–E; theta: F(2,18) = 20.49, p < 0.0001 time effect, F(2,9) = 4.450, p = 0.0453 treatment effect; t(18) = 4.857, p < 0.001 for LID 15′ vs baseline; beta: F(2,18) = 9.069, p = 0.0019 time effect, F(2,9) = 1.940, p = 0.1993 treatment effect; t(18) = 3.465, p < 0.01 LID 15′ vs baseline; RMS: F(2,18) = 21.36, p < 0.0001 for time effect, F(2,9) = 3.107, p = 0.094 for treatment effect, F(4,18) = 7.893, p = 0.0007 for interaction; t(18) = 5.364, p < 0.001 for LID 15′ vs baseline). ZIP also did not affect theta, beta, or gamma peak frequencies (Fig. 2G) or multiunit activity (Fig. 2H). In addition, immunoblots of dorsal CA1 homogenates showed that intra-CA1 infusion of ZIP (1 nmol/μl) did not alter basal αCaMKII autophosphorylation levels (Fig. 2I).
PKMζ knock-down, but not PKCι/λ inhibition, mimics the amnesic effect of ZIP
We also found that intra-CA1 administration of the PKCι/λ inhibitor ICAP (0.01–1 nmol/μl; Tsokas et al., 2016) immediately after ORM RA had no effect on retention (Fig. 3A), suggesting that the amnesia caused by ZIP was indeed triggered by PKMζ inhibition. Supporting this claim, knocking down hippocampal PKMζ expression with ASO (2 nmol/μl) mimicked the amnesic effect of ZIP. PKMζ sASO (2 nmol/μl) did not affect ORM (Fig. 3B; t(14) = 3.227, p = 0.0061 for sASO vs ASO).
Figure 3.
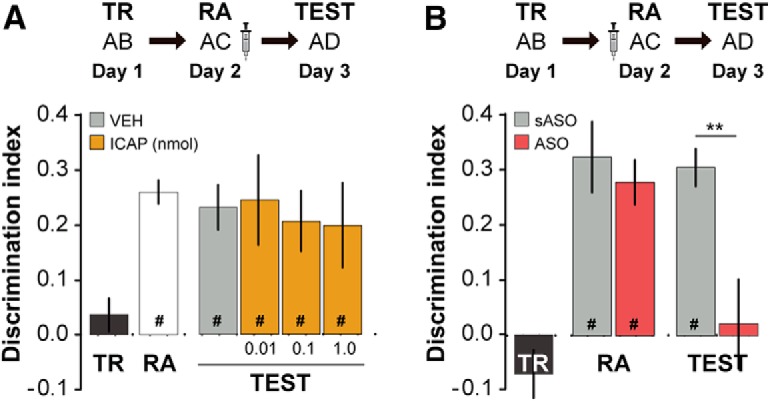
PKMζ knock-down, but not PKCι/λ inhibition, mimics the amnesic effect of ZIP. A, Rats were trained in NOR using two different objects (A and B, day 1, TR) and 24 h later (day 2) submitted to an ORM RA session in the presence of familiar object A and novel object C. Immediately after RA, rats received bilateral intra-CA1 infusions (1 μl/side) of VEH or the specific PKCι/λ inhibitor ICAP. One day later (day 3, TEST), rats were exposed to familiar object A and novel object D to evaluate ORM retention. Data (mean ± SEM) are presented as DI during TR, RA, and TEST, n = 8–9 per group. Rats explored objects equally during TR (A = 25.96 ± 1.23 s, B = 28.55 ± 1.68 s, t(33) = 1.15, p = 0.13) and discriminated them during RA (A = 19.61 ± 1.30 s, C = 32.73 ± 1.82 s, t(33) = 10.61, p < 0.001). Total object exploration time did not differ among TR, RA, and TEST (F(2,99) = 0.201, p = 0.82). B, Rats were treated as in A, but 20 min before RA, received intra-CA1 infusions of PKMζ ASO or sASO. Data (mean ± SEM) are presented as DI during TR, RA, and TEST, n = 8 per group. Rats explored objects equally during TR (A = 27.1 ± 2.12 s, B = 24.82 ± 3.04 s, t(15) = 1.102, p = 0.288) and discriminated them during RA (sASO, A = 19.00 ± 3.32 s, C = 39.06 ± 6.31 s, t(7) = 3.63, p < 0.01; ASO, A = 17.74 ± 1.94 s, C = 31.94 ± 3.16, t(7) = 5.84, p < 0.001). Total object exploration time did not differ among TR, RA, and TEST (F(2,45) = 0.239, p = 0.788). In both graphs, dashed lines represent chance level; #p < 0.05 in one-sample Student's t test with theoretical mean = 0; **p < 0.01 in unpaired t test.
PKMζ acts downstream of BDNF to control AMPAR recycling during reconsolidation
ORM reconsolidation induces LTP in the hippocampus (Clarke et al., 2010) and PKMζ sustains both LTP and nondeclarative LTM by regulating N-ethylmaleimide-sensitive factor (NSF)/GluA2-dependent AMPAR trafficking (Ling et al., 2006; Migues et al., 2010). Because BDNF modulates PKMζ turnover (Kelly et al., 2007) to maintain hippocampal LTP even when protein synthesis was blocked (Mei et al., 2011) and rescues reactivated ORM from the amnesia induced by protein synthesis inhibitors (Radiske et al., 2017b), we posited that BDNF controls PKMζ function to regulate AMPAR synaptic insertion during ORM reconsolidation. In agreement with this hypothesis, we found that BDNF coinfusion (0.25 μg/μl) reversed the amnesia caused by post-RA intra-CA1 administration of the protein synthesis inhibitor ANI (160 μg/μl), as described previously (Radiske et al., 2017b), but not that induced by ZIP (Fig. 4A; F(5,50) = 11.11, p < 0.001 for treatment effect; t(50) = 4.468, p < 0.001 for VEH vs ANI; t(50) = 4.051, p < 0.01 for VEH vs ZIP; t(50) = 3.289, p < 0.05 for VEH vs BDNF-ZIP; t(50) = 4.820, p < 0.001 for ANI vs BDNF-VEH; t(50) = 4.993, p < 0.001 for ANI vs BDNF-ANI; t(50) = 4.407, p < 0.001 for ZIP vs BDNF-VEH; t(50) = 4.575, p < 0.001 for ZIP vs BDNF-ANI; t(50) = 3.665, p < 0.01 for BDNF-VEH vs BDNF-ZIP; t(50) = 3.833, p < 0.01 for BDNF-ANI vs BDNF-ZIP).
Figure 4.

PKMζ acts downstream of BDNF to regulate CSF-associated AMPAR levels during ORM reconsolidation. A, Rats were trained in NOR using two different stimuli objects (A and B, day 1, TR) and, 24 h after TR (day 2), submitted to a 5-min-long ORM RA session in the presence of familiar object A and novel object C. Immediately after RA, rats received intra-CA1 infusions of VEH (saline), ZIP, ANI (−BDNF) or coinfusions of BDNF with VEH, ZIP, or ANI (+BDNF). One day later (day 3, TEST), rats were exposed to familiar object A and novel object D for 5 extra minutes to evaluate ORM retention. Data (mean ± SEM) are presented as DI during TR, RA, and TEST, n = 9–10 per group. Rats explored objects equally during TR (A = 28.04 ± 1.32 s, B = 26.05 ± 1.04 s, t(55) = 1.64, p = 0.107) and discriminated them during RA (A = 19.79 ± 0.764 s, C = 33.93 ± 1.17 s, t(55) = 17.16, p < 0.001). Total object exploration time did not differ among TR, RA, and TEST (F(2,165) = 2.81, p = 0.063). B, Rats were treated as in A, except that they received bilateral intra-CA1 infusions of VEH or PEP immediately after RA. One day later, rats were exposed to a familiar object (A, B, or C) and novel object D for 5 min to evaluate ORM retention. Data (mean ± SEM) are presented as DI during TR, RA, and TEST, n = 8–9 per group. Rats explored objects equally during TR (A = 28.75 ± 0.90 s, B = 28.63 ± 0.96 s, t(52) = 0.129, p = 0.898) and discriminated them during RA (A = 19.84 ± 0.73 s, C = 33.72 ± 1.04 s, t(52) = 18.43, p < 0.001). Total object exploration time did not differ among TR, RA, and TEST (F(2,156) = 1.625, p = 0.20). C, left. Rats received intra-CA1 infusions of VEH or DYN and, 30 min later, were killed by decapitation and the CA1 region of the dorsal hippocampus was dissected out, homogenized, and processed to purify a CSF to determine GluA1 and GluA2 levels by immunoblot. Data are expressed as mean ± SEM, n = 4 per group. Right, Densitometry quantification and representative immunoblot showing the relative enrichment of PSD-95 in a CSF purified from dorsal CA1. HT, Dorsal CA1 total homogenate. Data are expressed as mean ± SEM, n = 3 per group. D, Rats were treated as in A except that, immediately after RA, they received intra-CA1 infusions of VEH, ZIP, DYN, or a coinfusion of DYN and ZIP. Data (mean ± SEM) are presented as DI during TR, RA, and TEST, n = 9–10 per group. Rats explored objects equally during TR (A = 31.04 ± 1.78 s, B = 30.10 ± 1.50 s, t(37) = 0.584, p = 0.563) and discriminated them during RA (A = 21.65 ± 1.56 s, C = 38.62 ± 2.34 s, t(37) = 10.58, p < 0.001). Total object exploration time did not differ among TR, RA, and TEST (F(2,111) = 2.455, p = 0.090). E, Rats were trained as in A and, 24 h later, submitted to RA in the presence of familiar object A and novel object C. At different times after RA (30, 90, 180, 360, or 720 min), rats were killed by decapitation and the CA1 region of the dorsal hippocampus was dissected out, homogenized, and processed to purify a CSF to determine GluA1 and GluA2 levels by immunoblot (left). GluA1 and GluA2 levels were also evaluated 180 min and 360 min after ORM RA in the presence of two familiar objects (A and B; right). Data are expressed as mean ± SEM, n = 4 per group. F, Rats were treated as in E except that, immediately after RA, they received intra-CA1 infusions of VEH, ZIP, or PEP and, 180 min or 360 min later were killed by decapitation and the CA1 region of the dorsal hippocampus was dissected out, homogenized, and processed to purify a CSF to determine GluA1 and GluA2 levels by immunoblot. Data are expressed as mean ± SEM, n = 4 per group. Dashed lines represent chance level. #p < 0.05 in one-sample Student's t test with theoretical mean = 0. *p < 0.05, **p < 0.01, and ***p < 0.001 in paired t test, unpaired t test, or Bonferroni's multiple-comparisons test after one-way ANOVA, as appropriate. TUB, β-tubulin.
We also found that Pep2m (PEP; 5 pmol/μl), a peptide that disrupts NSF/GluA2 interaction and reduces AMPAR surface expression (Ralph et al., 2001), was amnesic when given in dorsal CA1 immediately after ORM RA in the presence of a novel object (Fig. 4B; test AD: t(16) = 3.430, p = 0.0034; test CD: t(16) = 3.084, p = 0.0071). Conversely, post-RA intra-CA1 infusion of DYN (120 pmol/μl), a cell-permeable dynamin inhibitor that impedes AMPAR internalization (Ferreira et al., 2015) and increases GluA1 and GluA2 expression in a CSF enriched in synaptic-associated proteins (Fig. 4C, left, GluA1: t(3) = 3.879, p = 0.0303 for VEH vs DYN; GluA2: t(3) = 6.635, p = 0.007 for VEH vs DYN; right, t(2) = 8.217, p = 0.014 for HT vs CSF), reversed the amnesia caused by ZIP (Fig. 4D; F(3,34) = 8.496, p = 0.0002 for treatment effect; t(34) = 3.962, p < 0.01 for VEH vs ZIP; t(34) = 4.152, p < 0.01 for ZIP vs DYN; t(34) = 4.130, p < 0.01 for ZIP vs ZIP-DYN). Moreover, ORM RA in the presence of a novel object, but not in the presence of two familiar ones, increased CSF-associated GluA1 and GluA2 levels in dorsal CA1 (Fig. 4E; GluA1: F(5,15) = 4.113, p = 0.0149 for treatment effect; t(15) = 3.276, p < 0.05 for TR vs RA180; GluA2: F(5,15) = 7.096, p = 0.0014 for treatment effect; t(15) = 3.632, p < 0.05 for TR vs RA360), an effect prevented by ZIP and PEP (Fig. 4F; GluA1: F(3,9) = 9.567, p = 0.0037 for treatment effect; t(9) = 3.529, p < 0.05 for VEH vs TR; t(9) = 4.108, p < 0.01 for VEH vs ZIP; t(9) = 4.985, p < 0.01 for VEH vs PEP; GluA2: F(3,9) = 8.365, p = 0.0057 for treatment effect; t(9) = 4.868, p < 0.01 for VEH vs TR; t(9) = 3.019, p < 0.05 for VEH vs ZIP; t(9) = 3.421, p < 0.05 for VEH vs PEP).
Relearning after ZIP-induced amnesia recapitulates ORM consolidation
The question of whether the amnesia caused by reconsolidation impairment results from permanent storage deficit or reversible retrieval failure remains unsolved (Lee et al., 2017). Interestingly, research on the nature of the amnesia triggered by PKMζ inhibition has caused similar dispute (Glanzman, 2013). We reasoned that, if impairment of ORM reconsolidation with ZIP erased the recognition trace, then subsequent representation of the forgotten object should induce repetition of the consolidation process because the rat would have to relearn that object anew. Therefore, treatments able to specifically hinder consolidation should block ORM reacquisition upon retraining. Conversely, if the amnesic effect of ZIP were due to retrieval impairment, then retraining should not induce repetition of consolidation, so consolidation blockers would not affect ORM reinstatement. To address this problem, we took advantage of the fact that ORM consolidation, but not reconsolidation, requires protein synthesis in the entorhinal cortex (Lima et al., 2009; Fig. 5A,B). We found that rats rendered amnesic by ZIP infused into dorsal CA1 after ORM RA reacquired memory for forgotten object A upon retraining. However, consistent with the notion that PKMζ inhibition erases the reactivated recognition trace, intra-entorhinal cortex injection of ANI after retraining abolished relearning, as if memory for object A had to be consolidated again (Fig. 5C; test 1: t(34) = 4.286, p = 0.0002 for VEH vs ZIP; test 2: F(1,32) = 15.29, p = 0.0005 for pre-RA treatment effect; F(1,32) = 14.32, p = 0.0006 for post-RA treatment effect; F(1,32) = 6.661, p = 0.00146 for interaction; t(32) = 5.441, p < 0.001 for VEH-VEH vs ZIP-ANI; t(32) = 4.501, p < 0.001 for ZIP-VEH vs ZIP-ANI; t(32) = 4.590, p < 0.001 for VEH-ANI vs ZIP-ANI).
Figure 5.

PKMζ inhibition erases the reactivated recognition trace. A, Rats were trained in NOR using two different stimuli objects (A and B, day 1, TR) and immediately thereafter received bilateral infusions of VEH (saline) or ANI into the EC. One day later (day 2, TEST), rats were exposed to familiar object A and novel object D for 5 min to evaluate ORM retention. Data (mean ± SEM) are presented as DI during TR and TEST, n = 12 per group. Rats explored objects equally during TR (A = 28.7 ± 2.83 s, B = 29.79 ± 3.05 s, t(23) = 0.480, p = 0.635). Total object exploration time did not differ between TR and TEST (t(46) = 0.222, p = 0.825). B, Rats were trained as in A and, 24 h after TR (day 2), were submitted to a 5-min-long ORM RA in the presence of two familiar objects (A and B) or in the presence of familiar object A and novel object C. Immediately after RA, rats received bilateral intra-EC infusions of VEH or ANI. One day later (day 3, TEST), rats were exposed to familiar object A and novel object D for 5 min to evaluate ORM retention. Data (mean ± SEM) are presented as DI during TR, RA, and TEST, n = 11 per group. Rats explored objects equally during TR (left: A = 24.57 ± 2.56 s, B = 23.54 ± 1.81 s, t(21) = 0.371, p = 0.714; right: A = 24.07 ± 2.59 s, B = 23.37 ± 2.71 s, t(21) = 0.432, p = 0.670). Rats submitted to RA in the presence of familiar objects (AB) did not discriminate between them during that session (A = 21.90 ± 0.66 s, B = 20.05 ± 1.27 s, t(21) = 1.50, p = 0.148). Rats submitted to RA in the presence of familiar object A and novel object C discriminated the objects during that session (A = 21.77 ± 2.34 s, B = 32.87 ± 2.75 s, t(21) = 5.47, p < 0.001). Total object exploration time did not differ among TR, RA, and TEST (left: F(2,63) = 1.81, p = 0.172; right: F(2,63) = 0.721, p = 0.490). C, Rats were trained as in A and, 24 h after TR (day 2), were submitted to a 5-min-long ORM RA in the presence of familiar object A and novel object C. Immediately after RA, rats received bilateral intra-CA1 infusions of VEH or ZIP. One day later (day 3, TEST 1), rats were exposed to familiar object A and novel object D for 5 extra minutes to evaluate ORM retention and, immediately thereafter, received bilateral infusions of VEH or ANI in the EC. One day later (day 4, TEST 2), rats were exposed to familiar object A and novel object E for 5 min to evaluate ORM retention. Data (mean ± SEM) are presented as DI during TR, RA, TEST1, and TEST2, n = 9 per group. Rats explored objects equally during TR (A = 25.77 ± 1.16 s, B = 29.59 ± 1.88 s, t(35) = 1.98, p = 0.055) and discriminated them during RA (A = 20.66 ± 1.30 s, B = 34.59 ± 1.79 s, t(35) = 10.42, p < 0.001). Total object exploration time did not differ between TR, RA, TEST1, and TEST2 (F(3,140) = 0.105, p = 0.957). In all graphs, dashed lines represent chance level; #p < 0.05 in one-sample Student's t test with theoretical mean = 0; ***p < 0.001 in unpaired t test or Bonferroni's multiple-comparisons test after two-way ANOVA.
To further investigate whether the amnesia caused by ZIP returned the amnesic brain to its prelearning state, we analyzed whether relearning following ZIP-induced reconsolidation impairment recapitulated the electrophysiological correlates of ORM acquisition. It is known that declarative memory acquisition increases theta and slow-gamma power in the hippocampus, whereas retrieval is usually linked to theta/slow-gamma cross-frequency coupling (Chang and Huerta, 2012; Colgin, 2015; Trimper et al., 2017).
ORM acquisition augmented theta (5–10 Hz) and slow-gamma (30–55 Hz) band power without modifying beta oscillations (13–20 Hz) and theta/slow-gamma coupling or slow-gamma amplitude distribution over theta phases in dorsal CA1 (Fig. 6A–D; t(9) = 3.477, p = 0.0070 for theta power; t(9) = 3.316, p = 0.0090 for slow-gamma power). Conversely, ORM retrieval did not modify theta amplitude but increased beta power (13–20 Hz) and slow-gamma power (Fig. 6A–D; t(4) = 4.453, p = 0.0112 for beta power; t(4) = 8.340, p = 0.0011 for slow-gamma power). We also found that ORM retrieval induced strong theta/slow-gamma coupling (Figs. 6E–G; t(8) = 3.033, p = 0.0162) with slow-gamma events concentrated on the late ascending portion of the theta cycle (Fig. 6H; 328.5° ± 38.84; nonuniform phase distribution, z = 3.98, p = 0.0186). Consistent with the hypothesis that impairing reconsolidation through PKMζ signaling disruption deletes the reactivated ORM trace, rats rendered amnesic with ZIP showed prominent theta and slow-gamma oscillatory activity and low modulation indexes during a retraining session (Fig. 6D,G; t(4) = 2.999, p = 0.0400 for theta power; t(4) = 10.86, p = 0.0004 for slow-gamma power), resembling the oscillatory pattern typical of the ORM acquisition process. During active exploration periods, rats remained motionless or moved slowly (mean speed <6 cm/s).
Discussion
Our findings confirm that PKMζ plays a prominent role in mnemonic processing and endorse the hypothesis that its inhibition deletes memory (Pastalkova et al., 2006). However, our data also indicate that hippocampal PKMζ activation mediates ORM reconsolidation rather than ORM maintenance. This is in agreement with a previous report showing that intrahippocampal ZIP does not affect ORM consolidation (Hardt et al., 2010), but is at odds with extensive literature showing that PKMζ is essential for storage of different memory types from mollusks to mammals (Sacktor, 2012).
ZIP has been crucial to study the participation of PKMζ on memory (Sacktor and Hell, 2017). Nonetheless, ZIP specificity has been questioned by studies showing that, at high concentrations, it impairs plasticity in PKMζ knock-out mice, suppresses brain oscillations, and hinders CaMKII/PKCι/λ activity (Kwapis and Helmstetter, 2014). We overcame these confounders by using a dose of ZIP much lower than that typically used and by performing a series of control experiments showing that, in our preparation, ZIP did not alter spontaneous oscillatory activity or basal CaMKII autophosphorylation and that its amnesic effect was mimicked by PKMζ ASOs, but not by PKCι/λ inhibition.
Regardless of the suggestion that low doses of ZIP may selectively disrupt reactivated associations (Sacktor, 2012), it is unlikely that the differential effect of ZIP on ORM reconsolidation and consolidation that we observed was due to the low concentration of ZIP that we used because reconsolidation is usually less sensitive than consolidation to metabolic inhibitors (Debiec et al., 2002). Moreover, it was shown previously that ZIP does not impair ORM consolidation when injected in the hippocampus at 10× the concentration used in our experiments (Hardt et al., 2010).
The lack of effect of ZIP in ORM consolidation does not mean that the hippocampus does not participate in this process. Although it is true that, based mainly on lesion studies, it has previously been suggested that the involvement of the hippocampus in ORM is restricted to the processing of spatial and contextual information instead of recognition memory per se (Ainge et al., 2006), it has lately become clear that this region is critical for ORM consolidation even when all spatial and contextual components of the task are omitted (Liu et al., 2016; Stackman et al., 2016; Canto de Souza et al., 2017; Cercato et al., 2016; Lymer et al., 2017). This is particularly evident when rodents are exposed to two identical or two different but behaviorally equivalent objects in a familiar open-field arena. Under these training conditions, temporary inactivation of the hippocampus hinders ORM consolidation (de Lima et al., 2006), which requires protein synthesis, NMDAR, and proteasome activation (Rampon et al., 2000; Baker and Kim, 2002; Figueiredo et al., 2015) and increases hippocampal c-fos (Tanimizu et al., 2017) and extracellular glutamate levels (Cohen et al., 2013). In any case, we found that 1 nmol/μl ZIP was sufficient to prevent acquisition of the memory for a new object provided that this object was presented alongside a familiar one. This result not only indicates that the dose of ZIP that we used is enough to inhibit PKMζ activity, but also that reconsolidation, and not consolidation, mechanisms bring about active ORM updating in the hippocampus, which is of the outmost importance because it has been reported that the opposite is true for nondeclarative memories (Tronel et al., 2005). Nevertheless, the majority of studies show that ZIP causes amnesia in the absence of memory RA, suggesting that the synaptic plasticity involved in ORM is different from that in most other forms of memory. Indeed, unlike the rapid effect that it causes on the maintenance of other memory types (Pastalkova et al., 2006; Hardt et al., 2010), ZIP administration following ORM RA results in delayed amnesia. In fact, ORM processing, and in particular ORM reconsolidation, depends on the integrity of a rather specific set of pathways and networks that span various brain regions and their interactions (Winters et al., 2011; Rossato et al., 2013, 2015). For example, contrary to other memory types (Lee et al., 2004), ORM reconsolidation requires hippocampal BDNF signaling (Radiske et al., 2017b), which, as demonstrated here, controls the integration of new information into active ORM through a protein-synthesis-independent mechanism acting upstream PKMζ to modulate hippocampal AMPAR synaptic insertion, in agreement with findings showing that ORM reconsolidation induces synaptic potentiation in the hippocampus (Clarke et al., 2010) and that BDNF facilitates LTP in the absence of protein synthesis through PKMζ (Mei et al., 2011; Schuette et al., 2016). Therefore, our results should not be generalized to indicate that all memories are sensitive to PKMζ inhibition only when reactivated. However, future studies might benefit from taking into account the possibility that, in addition to impairing memory maintenance, PKMζ inhibition can also affect memory reconsolidation. This suggestion may be particularly useful for studies dealing with recent memories because memories become increasingly resistant to reconsolidation with age (Frankland et al., 2006), as well as for those analyzing memories that are sensitive to PKMζ blockers and reconsolidation inhibitors at the same posttraining times. It is important to stress that reconsolidation can result not only from exposure to obvious reminders, but also to inconspicuous signals unable to produce discernible behavioral outputs (Gisquet-Verrier and Riccio, 2012; Soeter and Kindt, 2015). The only other study that analyzed the involvement of PKMζ in reconsolidation thoroughly showed that retrieval protects conditioned taste aversion memory (CTA) from the amnesic effect of intrainsular cortex (IC) ZIP administration during the same amount of time it takes ZIP to be able to impair consolidation, supposedly as a result of reconsolidation induction (Levitan et al., 2016). It is necessary to point out, though, that the differences between CTA and ORM are so fundamental that such contradictory findings should come as no surprise. CTA is a nondeclarative memory prone to be extinguished upon retrieval (Maroun et al., 2012), whereas ORM is an inextinguishable declarative memory. Therefore, CTA non-reinforced RA may induce two competing, opposite, and overlapping processes requiring IC protein synthesis and, therefore, different from ORM, the effect of post-RA PKMζ inhibition on CTA persistence cannot be unequivocally attributed to either reconsolidation or extinction modulation. Moreover, CTA maintenance requires IC PKMζ activity late after TR, but ORM consolidation is unaffected by hippocampal PKMζ inhibition, which precludes ORM reconsolidation and ZIP-sensitive mechanisms related to ORM persistence from being mutually exclusive, as proposed for CTA.
A great deal of effort has been dedicated to determining whether forgetting caused by inhibition of memory maintenance mechanisms is due to storage or retrieval failure. Most studies on the subject involve treating rats with agents that affect the mechanisms under scrutiny at different postlearning or postretrieval times and later, once the agent has washed out, verify the occurrence of amnesia as evidenced by impaired performance. If amnesia is observed, then some intervention is given, usually in the form of a reminder cue, and performance is reassessed in a subsequent retention test. If memory recovers, then retrieval failure is assumed to be responsible for amnesia. Conversely, absence of recovery is commonly accepted as evidence of storage impairment. However, absence of recovery does not unequivocally demonstrate memory erasure because it could also reflect failure to retrieve an otherwise intact memory (Hardt et al., 2009). In addition, a memory that cannot be retrieved at a given time can theoretically be retrieved later. Successful memory recovery does not prove retrieval failure either because reminder cues may cause apparent recovery by promoting new learning indistinguishable from the original one. This matter is especially important in the case of PKMζ because it has been suggested that its effect on memory maintenance results from memory erasure rather than retrieval impairment because the amnesia caused by PKMζ inhibition persists even after removal of its inhibitor (Pastalkova et al., 2006). To investigate this issue for the amnesia caused by the intrahippocampal administration of ZIP following ORM RA in the presence of a novel object, we used a twofold strategy. First, we exploited the fact that the rat's brain engages different mechanisms to consolidate and to reconsolidate ORM. ORM consolidation requires protein synthesis in EC, whereas ORM reconsolidation does not. Therefore, we analyzed whether rats that received intrahippocampal ZIP after ORM RA were able to reacquire ORM upon retraining and, if so, whether EC protein synthesis inhibition blocked reacquisition. The result of this experiment was straightforward. Relearning was blocked by ANI given in EC after retraining as if the amnesic rats were consolidating the forgotten ORM again. Second, we analyzed whether ORM reacquisition recapitulates the electrophysiological signature of original learning. To do that, we took advantage of the fact that ORM acquisition and retrieval are associated with different hippocampal oscillatory patterns. Again, the result was clear-cut. During retraining, the oscillatory pattern of rats that received ZIP after ORM RA was indistinguishable from that recorded during original training, as if these rats were learning the forgotten object anew.
In conclusion, our results show that hippocampal PKMζ acts downstream of BDNF to regulate AMPAR recycling and ORM updating during reconsolidation and indicate that its inhibition deletes the reactivated memory trace.
Footnotes
This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brazil). A.R. and M.C.G. are Postdoctoral Research Fellows supported by CAPES and CNPq, respectively. G.A. holds a CNPq M.Sc. Research Fellowship through Programa de Pós-Graduação em Neurociencias at Universidade Federal do Rio Grande do Norte (UFRN, Brazil).
The authors declare no competing financial interests.
References
- Adasme T, Haeger P, Paula-Lima AC, Espinoza I, Casas-Alarcón MM, Carrasco MA, Hidalgo C (2011) Involvement of ryanodine receptors in neurotrophin-induced hippocampal synaptic plasticity and spatial memory formation. Proc Natl Acad Sci U S A 108:3029–3034. 10.1073/pnas.1013580108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ainge JA, Heron-Maxwell C, Theofilas P, Wright P, de Hoz L, Wood ER (2006) The role of the hippocampus in object recognition in rats: examination of the influence of task parameters and lesion size. Behav Brain Res 167:183–195. 10.1016/j.bbr.2005.09.005 [DOI] [PubMed] [Google Scholar]
- Akirav I, Maroun M (2006) Ventromedial prefrontal cortex is obligatory for consolidation and reconsolidation of object recognition memory. Cereb Cortex 16:1759–1765. 10.1093/cercor/bhj114 [DOI] [PubMed] [Google Scholar]
- Bailey CH, Kandel ER, Si K (2004) The persistence of long-term memory: a molecular approach to self-sustaining: changes in learning-induced synaptic growth. Neuron 44:49–57. 10.1016/j.neuron.2004.09.017 [DOI] [PubMed] [Google Scholar]
- Baker KB, Kim JJ (2002) Effects of stress and hippocampal NMDA receptor antagonism on recognition memory in rats. Learn Mem 9:58–65. 10.1101/lm.46102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal NV, Susorov D, Chesnokova E, Kasianov A, Mikhailova T, Alkalaeva E, Balaban PM, Kolosov P (2016) Upstream open reading frames located in the leader of protein kinase Mζ mRNA regulate its translation. Front Mol Neurosci 9:103. 10.3389/fnmol.2016.00103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Kimble W, Buabeid M, Bhattacharya D, Bloemer J, Alhowail A, Reed M, Dhanasekaran M, Escobar M, Suppiramaniam V (2017) Altered AMPA receptor expression plays an important role in inducing bidirectional synaptic plasticity during contextual fear memory reconsolidation. Neurobiol Learn Mem 139:98–108. 10.1016/j.nlm.2016.12.013 [DOI] [PubMed] [Google Scholar]
- Bragin A, Jandó G, Nádasdy Z, van Landeghem M, Buzsáki G (1995) Dentate EEG spikes and associated interneuronal population bursts in the hippocampal hilar region of the rat. J Neurophysiol 73:1691–1705. 10.1152/jn.1995.73.4.1691 [DOI] [PubMed] [Google Scholar]
- Brankack J, Stewart M, Fox SE (1993) Current source density analysis of the hippocampal theta rhythm: associated sustained potentials and candidate synaptic generators. Brain Res 615:310–327. 10.1016/0006-8993(93)90043-M [DOI] [PubMed] [Google Scholar]
- Cai D, Pearce K, Chen S, Glanzman DL (2011) Protein kinase M maintains long-term sensitization and long-term facilitation in Aplysia. J Neurosci 31:6421–6431. 10.1523/JNEUROSCI.4744-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldeira MV, Melo CV, Pereira DB, Carvalho R, Correia SS, Backos DS, Carvalho AL, Esteban JA, Duarte CB (2007) Brain-derived neurotrophic factor regulates the expression and synaptic delivery of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunits in hippocampal neurons. J Biol Chem 282:12619–12628. 10.1074/jbc.M700607200 [DOI] [PubMed] [Google Scholar]
- Canto de Souza L, Provensi G, Vullo D, Carta F, Scozzafava A, Costa A, Schmidt SD, Passani MB, Supuran CT, Blandina P (2017) Carbonic anhydrase activation enhances object recognition memory in mice through phosphorylation of the extracellular signal-regulated kinase in the cortex and the hippocampus. Neuropharmacology 118:148–156. 10.1016/j.neuropharm.2017.03.009 [DOI] [PubMed] [Google Scholar]
- Cercato MC, Vázquez CA, Kornisiuk E, Aguirre AI, Colettis N, Snitcofsky M, Jerusalinsky DA, Baez MV (2016) GluN1 and GluN2A NMDA receptor subunits increase in the hippocampus during memory consolidation in the rat. Front Behav Neurosci 10:242. 10.3389/fnbeh.2016.00242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EH, Huerta PT (2012) Neurophysiological correlates of object recognition in the dorsal subiculum. Front Behav Neurosci 6:46. 10.3389/fnbeh.2012.00046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke JR, Cammarota M, Gruart A, Izquierdo I, Delgado-García JM (2010) Plastic modifications induced by object recognition memory processing. Proc Natl Acad Sci U S A 107:2652–2657. 10.1073/pnas.0915059107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SJ, Stackman RW Jr (2015) Assessing rodent hippocampal involvement in the novel object recognition task: a review. Behav Brain Res 285:105–117. 10.1016/j.bbr.2014.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SJ, Munchow AH, Rios LM, Zhang G, Asgeirsdóttir HN, Stackman RW Jr (2013) The rodent hippocampus is essential for nonspatial object memory. Curr Biol 23:1685–1690. 10.1016/j.cub.2013.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgin LL. (2015) Theta-gamma coupling in the entorhinal-hippocampal system. Curr Opin Neurobiol 31:45–50. 10.1016/j.conb.2014.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo JA, Stöckl P, Ueberall F, Jenny M, Saria A, Zernig G (2012) Activation of PKCzeta and PKMzeta in the nucleus accumbens core is necessary for the retrieval, consolidation and reconsolidation of drug memory. PLoS One 7:e30502. 10.1371/journal.pone.0030502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. (1984) Memory and molecular turnover. Nature 312:101. 10.1038/312101a0 [DOI] [PubMed] [Google Scholar]
- Debiec J, LeDoux JE, Nader K (2002) Cellular and systems reconsolidation in the hippocampus. Neuron 36:527–538. 10.1016/S0896-6273(02)01001-2 [DOI] [PubMed] [Google Scholar]
- de Lima MN, Luft T, Roesler R, Schröder N (2006) Temporary inactivation reveals an essential role of the dorsal hippocampus in consolidation of object recognition memory. Neurosci Lett 405:142–146. 10.1016/j.neulet.2006.06.044 [DOI] [PubMed] [Google Scholar]
- DeVito LM, Eichenbaum H (2010) Distinct contributions of the hippocampus and medial prefrontal cortex to the “what-where-when” components of episodic-like memory in mice. Behav Brain Res 215:318–325. 10.1016/j.bbr.2009.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennaceur A, Delacour J (1988) A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav Brain Res. 31:47–59. [DOI] [PubMed] [Google Scholar]
- Ferreira JS, Schmidt J, Rio P, Águas R, Rooyakkers A, Li KW, Smit AB, Craig AM, Carvalho AL (2015) GluN2B-containing NMDA receptors regulate AMPA receptor traffic through anchoring of the synaptic proteasome. J Neurosci 35:8462–8479. 10.1523/JNEUROSCI.3567-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo LS, Dornelles AS, Petry FS, Falavigna L, Dargél VA, Köbe LM, Aguzzoli C, Roesler R, Schröder N (2015) Two waves of proteasome-dependent protein degradation in the hippocampus are required for recognition memory consolidation. Neurobiol Learn Mem 120:1–6. 10.1016/j.nlm.2015.02.005 [DOI] [PubMed] [Google Scholar]
- Frankland PW, Ding HK, Takahashi E, Suzuki A, Kida S, Silva AJ (2006) Stability of recent and remote contextual fear memory. Learn Mem 13:451–457. 10.1101/lm.183406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga K, Miyamoto E (2000) A working model of CaM kinase II activity in hippocampal long-term potentiation and memory. Neurosci Res 38:3–17. 10.1016/S0168-0102(00)00139-5 [DOI] [PubMed] [Google Scholar]
- Gisquet-Verrier P, Riccio DC (2012) Memory reactivation effects independent of reconsolidation Learn Mem 19:401–409. 10.1101/lm.026054.112 [DOI] [PubMed] [Google Scholar]
- Glanzman DL. (2013) PKM and the maintenance of memory. F1000 Biol Rep 5:4. 10.3410/B5-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardt O, Wang SH, Nader K (2009) Storage or retrieval deficit: the yin and yang of amnesia. Learn Mem 16:224–230. 10.1101/lm.1267409 [DOI] [PubMed] [Google Scholar]
- Hardt O, Migues PV, Hastings M, Wong J, Nader K (2010) PKMzeta maintains 1-day- and 6-day-old long-term object location but not object identity memory in dorsal hippocampus. Hippocampus 20:691–695. 10.1002/hipo.20708 [DOI] [PubMed] [Google Scholar]
- Hu J, Adler K, Farah CA, Hastings MH, Sossin WS, Schacher S (2017) Cell-specific PKM isoforms contribute to the maintenance of different forms of persistent long-term synaptic plasticity. J Neurosci 37:2746–2763. 10.1523/JNEUROSCI.2805-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalil SJ, Sacktor TC, Shouval HZ (2015) Atypical PKCs in memory maintenance: the roles of feedback and redundancy. Learn Mem 22:344–353. 10.1101/lm.038844.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdi H, Kabbaj M (2013) Acute BDNF treatment upregulates GluR1-SAP97 and GluR2-GRIP1 interactions: implications for sustained AMPA receptor expression. PLoS One 8:e57124. 10.1371/journal.pone.0057124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MT, Crary JF, Sacktor TC (2007) Regulation of protein kinase mzeta synthesis by multiple kinases in long-term potentiation. J Neurosci 27:3439–3444. 10.1523/JNEUROSCI.5612-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwapis JL, Helmstetter FJ (2014) Does PKM(zeta) maintain memory? Brain Res Bull 105:36–45. 10.1016/j.brainresbull.2013.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlancq MJ, McKinney TL, Dickson CT (2016) ZIP it: neural silencing is an additional effect of the PKM-zeta inhibitor zeta-inhibitory peptide. J Neurosci 36:6193–6198. 10.1523/JNEUROSCI.4563-14.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AM, Kanter BR, Wang D, Lim JP, Zou ME, Qiu C, McMahon T, Dadgar J, Fischbach-Weiss SC, Messing RO (2013) Prkcz null mice show normal learning and memory. Nature 493:416–419. 10.1038/nature11803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JLC, Nader K, Schiller D (2017) An update on memory reconsolidation updating. Trends Cogn Sci 21:531–545. 10.1016/j.tics.2017.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JL, Everitt BJ, Thomas KL (2004) Independent cellular processes for hippocampal memory consolidation and reconsolidation. Science 304:839–843. 10.1126/science.1095760 [DOI] [PubMed] [Google Scholar]
- Levitan D, Fortis-Santiago Y, Figueroa JA, Reid EE, Yoshida T, Barry NC, Russo A, Katz DB (2016) Memory retrieval has a dynamic influence on the maintenance mechanisms that are sensitive to ζ-inhibitory peptide (ZIP). J Neurosci 36:10654–10662. 10.1523/JNEUROSCI.1568-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima RH, Rossato JI, Furini CR, Bevilaqua LR, Izquierdo I, Cammarota M (2009) Infusion of protein synthesis inhibitors in the entorhinal cortex blocks consolidation but not reconsolidation of object recognition memory. Neurobiol Learn Mem 91:466–472. 10.1016/j.nlm.2008.12.009 [DOI] [PubMed] [Google Scholar]
- Ling DS, Benardo LS, Sacktor TC (2006) Protein kinase mzeta enhances excitatory synaptic transmission by increasing the number of active postsynaptic AMPA receptors. Hippocampus 16:443–452. 10.1002/hipo.20171 [DOI] [PubMed] [Google Scholar]
- Lisman J. (2012) Memory erasure by very high concentrations of ZIP may not be due to PKM-zeta. Hippocampus 22:648–649. 10.1002/hipo.20980 [DOI] [PubMed] [Google Scholar]
- Lisman J. (2017) Criteria for identifying the molecular basis of the engram (CaMKII, PKMzeta). Mol Brain 10:55. 10.1186/s13041-017-0337-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, McIntyre CC (2001) Synaptic plasticity: a molecular memory switch. Curr Biol 11:R788–R791. [DOI] [PubMed] [Google Scholar]
- Liu Y, Du S, Lv L, Lei B, Shi W, Tang Y, Wang L, Zhong Y (2016) Hippocampal activation of Rac1 regulates the forgetting of object recognition memory. Curr Biol 26:2351–2357. 10.1016/j.cub.2016.06.056 [DOI] [PubMed] [Google Scholar]
- Lymer J, Robinson A, Winters BD, Choleris E (2017) Rapid effects of dorsal hippocampal G-protein coupled estrogen receptor on learning in female mice. Psychoneuroendocrinology 77:131–140. 10.1016/j.psyneuen.2016.11.019 [DOI] [PubMed] [Google Scholar]
- Maroun M, Akirav I (2008) Arousal and stress effects on consolidation and reconsolidation of recognition memory. Neuropsychopharmacology 33:394–405. 10.1038/sj.npp.1301401 [DOI] [PubMed] [Google Scholar]
- Maroun M, Kavushansky A, Holmes A, Wellman C, Motanis H (2012) Enhanced extinction of aversive memories by high-frequency stimulation of the rat infralimbic cortex. PLoS One 7:e35853. 10.1371/journal.pone.0035853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SJ, de Hoz L, Morris RG (2005) Retrograde amnesia: neither partial nor complete hippocampal lesions in rats result in preferential sparing of remote spatial memory, even after reminding. Neuropsychologia 43:609–624. 10.1016/j.neuropsychologia.2004.07.007 [DOI] [PubMed] [Google Scholar]
- Mei F, Nagappan G, Ke Y, Sacktor TC, Lu B (2011) BDNF facilitates L-LTP maintenance in the absence of protein synthesis through PKMζ. PLoS One 6:e21568. 10.1371/journal.pone.0021568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migues PV, Hardt O, Wu DC, Gamache K, Sacktor TC, Wang YT, Nader K (2010) PKMzeta maintains memories by regulating GluR2-dependent AMPA receptor trafficking. Nat Neurosci 13:630–634. 10.1038/nn.2531 [DOI] [PubMed] [Google Scholar]
- Nader K, Hardt O (2009) A single standard for memory: the case for reconsolidation. Nat Rev Neurosci 10:224–234. 10.1038/nrn2590 [DOI] [PubMed] [Google Scholar]
- O'Bryant SE, Hobson V, Hall JR, Waring SC, Chan W, Massman P, Lacritz L, Cullum CM, Diaz-Arrastia R; Texas Alzheimer's Research Consortium (2009) Brain-derived neurotrophic factor levels in Alzheimer's disease. J Alzheimers Dis 17:337–341. 10.3233/JAD-2009-1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paratcha G, Furman M, Bevilaqua L, Cammarota M, Vianna M, de Stein ML, Izquierdo I, Medina JH (2000) Involvement of hippocampal PKCbetaI isoform in the early phase of memory formation of an inhibitory avoidance learning. Brain Res 855:199–205. 10.1016/S0006-8993(99)02323-9 [DOI] [PubMed] [Google Scholar]
- Parsons RG, Davis M (2011) Gone but not forgotten. Front Behav Neurosci 5:51. 10.3389/fnbeh.2011.00051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton AA, Sacktor TC (2006) Storage of spatial information by the maintenance mechanism of LTP. Science 313:1141–1144. 10.1126/science.1128657 [DOI] [PubMed] [Google Scholar]
- Paxinos GW, Watson C (2007) The rat brain in stereotaxic coordinates, Ed 6 San Diego: Academic. [Google Scholar]
- Radiske A, Gonzalez MC, Conde-Ocazionez SA, Feitosa A, Köhler CA, Bevilaqua LR, Cammarota M (2017a) Prior learning of relevant nonaversive information is a boundary condition for avoidance memory reconsolidation in the rat hippocampus. J Neurosci 37:9675–9685. 10.1523/JNEUROSCI.1372-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radiske A, Rossato JI, Gonzalez MC, Köhler CA, Bevilaqua LR, Cammarota M (2017b) BDNF controls object recognition memory reconsolidation. Neurobiol Learn Mem 142:79–84. 10.1016/j.nlm.2017.02.018 [DOI] [PubMed] [Google Scholar]
- Ralph GS, Bienemann A, Ma J, Tan HK, Noel J, Henley JM, Uney JB (2001) Disruption of the GluR2-NSF interaction protects primary hippocampal neurons from ischemic stress. Mol Cell Neurosci 17:662–670. 10.1006/mcne.2000.0959 [DOI] [PubMed] [Google Scholar]
- Rampon C, Tang YP, Goodhouse J, Shimizu E, Kyin M, Tsien JZ (2000) Enrichment induces structural changes and recovery from nonspatial memory deficits in CA1 NMDAR1-knockout mice. Nat Neurosci 3:238–244. 10.1038/72945 [DOI] [PubMed] [Google Scholar]
- Rao-Ruiz P, Rotaru DC, van der Loo RJ, Mansvelder HD, Stiedl O, Smit AB, Spijker S (2011) Retrieval-specific endocytosis of GluA2-AMPARs underlies adaptive reconsolidation of contextual fear. Nat Neurosci 14:1302–1308. 10.1038/nn.2907 [DOI] [PubMed] [Google Scholar]
- Ren SQ, Yan JZ, Zhang XY, Bu YF, Pan WW, Yao W, Tian T, Lu W (2013) PKCλ is critical in AMPA receptor phosphorylation and synaptic incorporation during LTP. EMBO J 32:1365–1380. 10.1038/emboj.2013.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossato JI, Bevilaqua LR, Myskiw JC, Medina JH, Izquierdo I, Cammarota M (2007) On the role of hippocampal protein synthesis in the consolidation and reconsolidation of object recognition memory. Learn Mem 14:36–46. 10.1101/lm.422607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossato JI, Radiske A, Kohler CA, Gonzalez C, Bevilaqua LR, Medina JH, Cammarota M (2013) Consolidation of object recognition memory requires simultaneous activation of dopamine D1/D5 receptors in the amygdala and medial prefrontal cortex but not in the hippocampus. Neurobiol Learn Mem 106:66–70. 10.1016/j.nlm.2013.07.012 [DOI] [PubMed] [Google Scholar]
- Rossato JI, Köhler CA, Radiske A, Bevilaqua LR, Cammarota M (2015) Inactivation of the dorsal hippocampus or the medial prefrontal cortex impairs retrieval but has differential effect on spatial memory reconsolidation. Neurobiol Learn Mem 125:146–151. 10.1016/j.nlm.2015.09.001 [DOI] [PubMed] [Google Scholar]
- Sacktor TC. (2008) PKMzeta, LTP maintenance, and the dynamic molecular biology of memory storage. Prog Brain Res 169:27–40. 10.1016/S0079-6123(07)00002-7 [DOI] [PubMed] [Google Scholar]
- Sacktor TC. (2012) Memory maintenance by PKMζ: an evolutionary perspective. Mol Brain 5:31. 10.1186/1756-6606-5-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacktor TC, Hell JW (2017) The genetics of PKMζ and memory maintenance. Sci Signal 10:eaao2327. 10.1126/scisignal.aao2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuette SR, Fernández-Fernández D, Lamla T, Rosenbrock H, Hobson S (2016) Overexpression of protein kinase Mζ in the hippocampus enhances long-term potentiation and long-term contextual but not cued fear memory in rats. J Neurosci 36:4313–4324. 10.1523/JNEUROSCI.3600-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma AV, Nargang FE, Dickson CT (2012) Neurosilence: profound suppression of neural activity following intracerebral administration of the protein synthesis inhibitor anisomycin. J Neurosci 32:2377–2387. 10.1523/JNEUROSCI.3543-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeter M, Kindt M (2015) Retrieval cues that trigger reconsolidation of associative fear memory are not necessarily an exact replica of the original learning experience. Front Behav Neurosci 9:122. 10.3389/fnbeh.2015.00122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stackman RW Jr, Cohen SJ, Lora JC, Rios LM (2016) Temporary inactivation reveals that the CA1 region of the mouse dorsal hippocampus plays an equivalent role in the retrieval of long-term object memory and spatial memory. Neurobiol Learn Mem 133:118–128. 10.1016/j.nlm.2016.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimizu T, Kenney JW, Okano E, Kadoma K, Frankland PW, Kida S (2017) Functional connectivity of multiple brain regions required for the consolidation of social recognition memory. J Neurosci 37:4103–4116. 10.1523/JNEUROSCI.3451-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tort AB, Komorowski R, Eichenbaum H, Kopell N (2010) Measuring phase-amplitude coupling between neuronal oscillations of different frequencies. J Neurophysiol 104:1195–1210. 10.1152/jn.00106.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimper JB, Galloway CR, Jones AC, Mandi K, Manns JR (2017) Gamma oscillations in rat hippocampal subregions dentate gyrus, CA3, CA1, and subiculum underlie associative memory encoding. Cell Rep 21:2419–2432. 10.1016/j.celrep.2017.10.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronel S, Milekic MH, Alberini CM (2005) Linking new information to a reactivated memory requires consolidation and not reconsolidation mechanisms. PLoS Biol 3:e293. 10.1371/journal.pbio.0030293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokas P, Hsieh C, Yao Y, Lesburguères E, Wallace EJC, Tcherepanov A, Jothianandan D, Hartley BR, Pan L, Rivard B, Farese RV, Sajan MP, Bergold PJ, Hernández AI, Cottrell JE, Shouval HZ, Fenton AA, Sacktor TC (2016) Compensation for PKMζ in long-term potentiation and spatial long-term memory in mutant mice. Elife 5:e14846. 10.7554/eLife.14846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanyushin BF, Tushmalova NA, Guskova LV (1974) Brain DNA methylation as an index of genome participation in mechanism of memory formation. Doklady Akademii Nauk SSSR 219:742–744. [Google Scholar]
- Volk LJ, Bachman JL, Johnson R, Yu Y, Huganir RL (2013) PKM-ζ is not required for hippocampal synaptic plasticity, learning and memory. Nature 493:420–423. 10.1038/nature11802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sheng T, Ren S, Tian T, Lu W (2016) Distinct roles of PKCι/λ and PKMζ in the initiation and maintenance of hippocampal long-term potentiation and memory. Cell Rep 16:1954–1961. 10.1016/j.celrep.2016.07.030 [DOI] [PubMed] [Google Scholar]
- Winters BD, Tucci MC, Jacklin DL, Reid JM, Newsome J (2011) On the dynamic nature of the engram: evidence for circuit-level reorganization of object memory traces following reactivation. J Neurosci 31:17719–17728. 10.1523/JNEUROSCI.2968-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu-Zhang AX, Schramm CL, Nabavi S, Malinow R, Newton AC (2012) Cellular pharmacology of protein kinase Mζ (PKMζ) contrasts with its in vitro profile: implications for PKMζ as a mediator of memory. J Biol Chem 287:12879–12885. 10.1074/jbc.M112.357244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Kelly MT, Sajikumar S, Serrano P, Tian D, Bergold PJ, Frey JU, Sacktor TC (2008) PKM zeta maintains late long-term potentiation by N-ethylmaleimide-sensitive factor/GluR2-dependent trafficking of postsynaptic AMPA receptors. J Neurosci 28:7820–7827. 10.1523/JNEUROSCI.0223-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Ma X, Chen G, Barkai E, Lin L (2016) Theta rhythmic clock-like activity of single units in the mouse hippocampus. J Neurosci 36:4415–4420. 10.1523/JNEUROSCI.3851-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]