

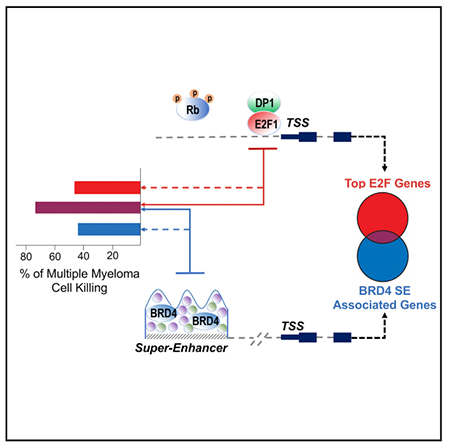
SUMMARY
The relationship between promoter proximal transcription factor-associated gene expression and super-enhancer-driven transcriptional programs are not well defined. However, their distinct genomic occupancy suggests a mechanism for specific and separable gene control. We explored the transcriptional and functional interrelationship between E2F transcription factors and BET transcriptional co-activators in multiple myeloma. We found that the transcription factor E2F1 and its heterodimerization partner DP1 represent a dependency in multiple myeloma cells. Global chromatin analysis reveals distinct regulatory axes for E2F and BETs, with E2F predominantly localized to active gene promoters of growth and/or proliferation genes and BETs disproportionately at enhancer-regulated tissue-specific genes. These two separate gene regulatory axes can be simultaneously targeted to impair the myeloma proliferative program, providing an important molecular mechanism for combination therapy. This study therefore suggests a sequestered cellular functional control that may be perturbed in cancer with potential for development of a promising therapeutic strategy.
Graphical Abstract
In Brief
Uncontrolled proliferation is a hallmark of tumorigenesis and is associated with perturbed transcriptomic profile. Fulciniti et al. explored the interrelationship between E2F transcription factors and BET transcriptional co-activators in multiple myeloma, reporting the existence of two distinct regulatory axes that can be synergistically targeted to impact myeloma growth and survival.
INTRODUCTION
The transcription of cell-cycle regulators is tightly controlled to ensure cellular fidelity: uncontrolled cell division emanating from deregulated and sustained proliferative signaling is a central hallmark of tumorigenesis (Hanahan and Weinberg, 2011). Chemotherapies that specifically target cell-cycle processes are effective anti-proliferative agents but fail to discriminate between tumor and normal proliferating cells (Johnstone et al., 2002). The adverse toxicity of chemotherapies necessitates targeted approaches to halt tumor cell proliferation. Here, advances in the selective inhibition of oncogenic growth factor signal transduction have proved highly effective, especially in tumors driven by deregulated growth factor signaling including lung and breast cancers (Downward, 2003; Paez et al., 2004). Unfortunately, resistance to both cytotoxic and targeted anti-proliferative therapies occurs commonly in metastatic tumors through adaptations that engage multiply redundant pathways converging on activation of master transcriptional regulators of growth and proliferation in the nucleus (Mellinghoff and Sawyers, 2002).
Multiple myeloma (MM) is a complex plasma cell malignancy driven by numerous genetic and epigenetic alterations that are acquired over time. Despite the advent of new drugs, relapse and refractory disease occurs in the vast majority of cases (Landgren and Iskander, 2017; Palumbo and Anderson, 2011) highlighting the need for novel therapeutic approaches. As in most malignancies, pathogenesis of MM is associated with deregulated expression and function of multiple key cellular genes controlling apoptosis, cell growth, and proliferation; therefore, targeting the transcriptional regulators of growth and proliferation represents an appealing option in this disease.
In mammalian cells, the E2F family of transcription factors (TFs) are master regulators of proliferation and drive the programmatic expression of genes required for cell-cycle progression (Müller and Helin, 2000). The multiple E2F proteins constitute a complicated regulatory network with diversified functions (Trimarchi and Lees, 2002), and their transcriptional output is the cumulative effect of the different family members that mediate both activator and repressor functions. Increased E2F activity is a common theme in MM pathogenesis as evidenced by common reciprocal translocations of the IgH enhancer to the E2F upstream activator cyclin D (CCND1, 2, 3) (Egan et al., 2012).
More commonly, E2F deregulation in cancer occurs through loss-of-function mutations to the RB family of pocket proteins (RB, p107, and p130) (Nevins, 2001; Sherr and McCormick, 2002). During G1, the activating E2F members (E2F1, E2F2, and E2F3a) are bound at target gene promoters in an inactive complex with their dimerization partner (DP1 or DP2) and the inhibitory RB complex (Weinberg, 1995). RB proteins act as a molecular scaffold, binding directly to E2F proteins and suppressing target gene transcription through the recruitment of chromatin modifier proteins and remodeling factors.
Recently, Liu et al. (2015) described RB loss contributing to the re-localization of E2F and MYC TFs at genes that are deregulated in RB mutant cells, providing a molecular mechanism by which E2F TFs may be “repurposed” and become essential in tumor cells with RB- and other tumor-suppressor-inactivating events. The idea that E2F function is essential for the control of cell proliferation has dominated several decades of experimentation (Wu et al., 2001). Incompatible with this view is the fact that mice deficient for individual or a combination of E2F genes do not have widespread defects in cell proliferation; on the other hand, E2F transgenic mice develop various tumors, and overexpression and/or amplification of E2F1 and/or additional E2F-activating members has been observed in different human cancers (Chen et al., 2009; Kent et al., 2017; Lee et al., 2010; Sharma et al., 2010). Taken together, these results suggest a differential requirement for E2Fs in the control of cell proliferation in oncogenic compared with normal environments.
Moreover, these studies establish MYC as a functional collaborator of E2F in addition to acting as an upstream activator. Indeed, chromatin immunoprecipitation (ChIP) experiments that examined the genome-wide distribution of E2Fs and proteins involved in the MYC network described a very close association between E2F and MYC binding sites and their target genes (Chen et al., 2008). MYC regulates E2F expression, and together they couple growth and proliferative gene expression programs by binding to the promoters of and driving many so-called “housekeeping” genes responsible for growth, metabolic, and biogenic functions (Secombe et al., 2004).
Despite recent findings that E2F may be required for tumor cell but not normal cell proliferation, the therapeutic targeting of E2F transcriptional activity has been minimally explored. Inhibition of E2F activity is often an indirect consequence of anti-proliferative agents that reengage cell-cycle check-point apparatuses. Consequently, mutations that remove these tumor suppressors occur frequently in relapsed and refractory disease (e.g., p53) (Lowe et al., 1994). Direct targeting of E2F is further complicated by the multiple redundant E2F family members capable of rescuing loss of any individual factor. For instance, E2F1−/− mice are viable, though they exhibit hyper-proliferation in the thyroid and deficiencies in gut epithelium proliferation and neurogenesis (Cooper-Kuhn et al., 2002; Field et al., 1996). Curiously, mice lacking DP1, the dimerization partner of all E2F TFs, are viable provided that DP1 is expressed in the extra-embryonic compartment (Kohn et al., 2003; Kohn et al., 2004). These observations suggest that cell proliferation can occur during development in the context of E2F deficiency thus potentiating a therapeutic window for targeted inhibition in cancer.
Here, we demonstrate that E2F1 and its heterodimerization partner DP1 are required for MM tumor growth. Integration of E2F1 and DP1 genome-wide profiling into the MM epigenome landscape reveals two non-overlapping regulatory axes controlled by promoter- and enhancer-driven processes, governing distinct biological functions. E2F is predominantly localized at the promoter of growth/proliferation genes, while BETs are disproportionately localized at enhancer-regulated tissue-specific genes. Dual chemical inhibition of E2F and BETs displays a superior activity against MM cell growth and viability both in vitro and in vivo compared to single perturbation alone. These data implicate that targeting these two non-overlapping vulnerabilities may provide important molecular mechanism for combination therapies in myeloma as well other malignancies.
RESULTS
E2F1 and DP1 Are Required for MM Growth and Proliferation
To determine the role of E2F in MM growth and proliferation, we perturbed E2F1 and DP1 in a panel of MM cell lines that all detectably express both genes (Figure S1A). Steady-state depletion of E2F1 or DP1 effectively inhibited cell proliferation in MM1.S cell line (Figures 1A and S1B) and across 8 different cell lines independent of their genetic background (Figures 1B and S1C). We confirmed the growth inhibitory effects of E2F/DP1 knockdown in 4 different MM cell lines using a 2D colony formation assay (Figures 1C and S1D). Cell-cycle analysis of E2F1 or DP1-depleted cells revealed an increase in G1 phase population indicative of cell-cycle arrest (Figure 1D) and subsequent apoptosis (Figure S1E). Conversely, overexpression of E2F1 increased proliferation rates in MM (Figure S1F), establishing a dose-dependent relationship between E2F activity and MM proliferation. To investigate the impact of E2F1 and DP1 depletion in normal cells, we performed similar knockdowns in normal peripheral blood mononuclear cells (PBMCs). The level of E2F knockdown was limited in PBMCs, but a significant DP1 knockdown had minimal impact on viability of PBMCs while having significant impact on MM1.S cell line used as positive control (Figure S1G) suggesting a MM-specific vulnerability to DP1 perturbation.
Figure 1. E2F1 and Heterodimerization Partner DP1 Are Required for MM Cell Growth In Vitro and In Vivo.
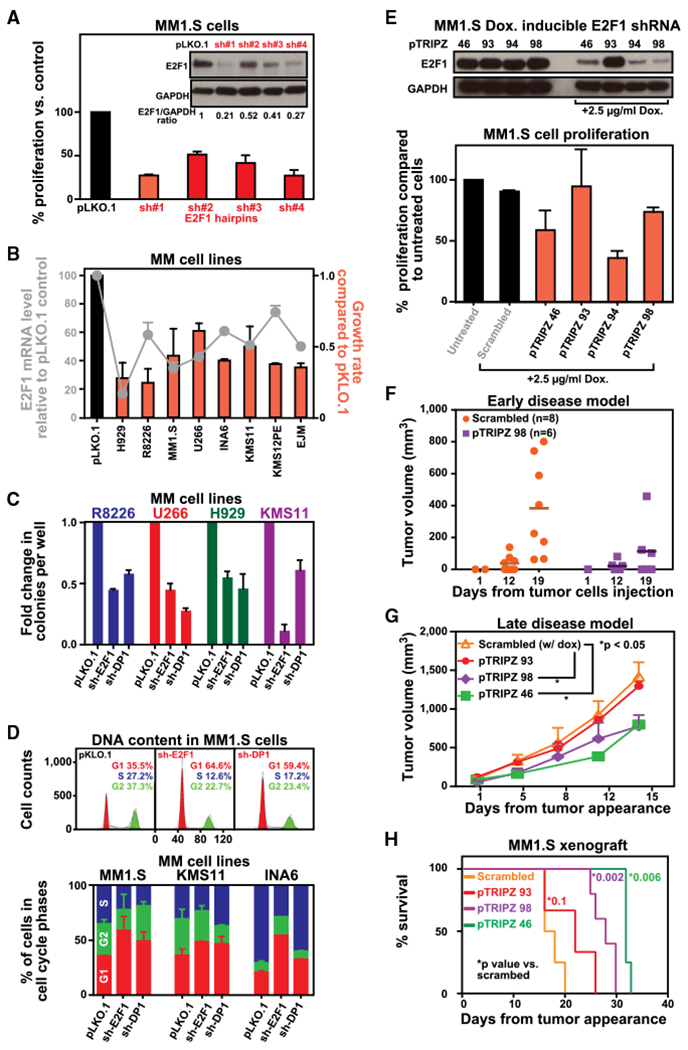
(A) MM1.S cells were infected with either scrambled (pLKO.1) or 4 E2F1-targeted shRNAs and selected with puromycin for 72 hr. Western blot analysis was performed to test knockdown efficiency, using GAPDH as a loading control. Transduced cells were analyzed for effect on cell growth by 3(H) thymidine uptake. Data are shown as the mean values ± SD of triplicates.
(B) sh-E2F1 #1 was used to knock down E2F1 expression in a panel of 7 MM cell lines. E2F1 mRNA levels and cell growth were evaluated after 3 days from puromycin selection by qPCR and thymidine uptake, respectively. The results are presented as mRNA (line) or cell growth (bars) changes from cells infected with pLKO.1. Data are shown as the mean values ± SD of triplicates.
(C) To test effects of E2F1 and DP1 silencing on the malignant phenotype of MM cells, we measured colony formation in semi-solid, methylcellulose media. Graphs depict average colony numbers (mean ± SD) for a panel of control and KD MM cell lines in methyl cellulose medium at day 21.
(D) Cell-cycle analysis was performed in MM cells infected with either scrambled (pLKO.1) or E2F1-targeted shRNA #1 for 72 hr after puromycin selection by propidium iodide (PI) staining followed by flow-cytometry acquisition. Analysis was performed using ModFit software. Representative images for MM1.S cells are shown in the upper panel. In the lower panel, graphs depict average of cell number in different phases of cell cycle for a panel of control and KD MM cell lines. Data are shown as the mean values ± SD of triplicates.
(E) Genetic depletion of E2F1 was achieved using 4 different tetracycline-inducible pTRIPz-Turbo-RFP vectors (Thermo Scientific, Pittsburgh, PA) containing the target sequence or scramble control. Transfected MM1.S cells were plated in growth medium in the absence or presence of 2.5 μg mL−1 doxycycline. Western blot (WB) analysis (top) was performed at day 3 confirming decreased E2F1 protein expression in cells expressing inducible E2F1 shRNAs. Cellular proliferation (bottom) was evaluated by 3(H)thymidine uptake (day 3) and presented as percentage of cell proliferation compared to untreated cells. In the medium containing doxycycline, reduced expression of E2F1 is accompanied by a reduction of cell growth rate compared to control cells. Data are shown as the mean values ± SD of triplicates.
(F) In vivo mouse xenograft studies were performed with MM cells harboring doxycycline-inducible shRNAs targeting E2F1. In the early intervention model, a cohort of mice was treated with irradiated 0.0625% doxycycline diet continuously starting 3 days after injection and monitored for tumor progression. Tumors were measured in two perpendicular dimensions using caliper measurements at the indicated time. Each dot in the graph represent a single mouse.
(G) In the late-intervention model, mice were treated with irradiated 0.0625% doxycycline diet continuously (1–6 mg of doxycycline per mouse per day) after tumor appearance and monitored for tumor progression using caliper measurements. Data are presented as mean values ± SD (n = 5/group); Student’s t test.
(H) Survival was evaluated from the first day of tumor appearance until death using the GraphPad analysis software. Survival curves (Kaplan-Meier) show prolongation of survival in mice with E2F1 depletion. Median survival for Scrambled, pTRIPZ93, pTRIPZ98, and pTRIPZ46 was 17, 22, 28, and 32 days, respectively.
See also Figure S1.
To investigate whether E2F1 or DP1 levels functionally impact in vivo tumor growth, we performed mouse xenograft studies with MM1.S cells stably expressing small hairpin RNAs(shRNAs) targeting either E2F1 or DP1. Depletion of either E2F1 or DP1 resulted in significant reduction in tumor growth over 9 weeks when compared to control cells (Figure S1H).
We have additionally transduced MM1.S cells with 4 different conditional E2F1 shRNAs to perform inducible E2F1 depletion in vitro and in vivo. The cells with most significant reduction in E2F1 protein following induction with doxycycline (pTRIPZ #46, #94, #98) showed the greatest inhibition of cell growth compared to scrambled cells (Figure 1E). Decreased E2F1 expression was also accompanied by a decrease in number of cells in the S phase and an increase in cells in G1 (data not shown).
To test whether inducible depletion of E2F1 in MM cells might affect their ability to form tumors in vivo, we utilized two models: the “early intervention model” where E2F depletion was induced at the time of tumor inoculation, and the “late-intervention model” where E2F depletion was induced after visible detection of tumor. In the early model, all mice injected with scrambled shRNA developed tumor approximately 3 weeks after cell injection. On the other hand, knockdown of E2F1 significantly delayed tumorigenesis in vivo, with only 50% of mice developing tumor 3 weeks from tumor cell injection (Figure 1F). The size of these tumors was also significantly smaller than the control mice. In the late-disease model, E2F1 depletion after tumor development significantly inhibited MM tumor growth (Figure 1G) with overall improvement in survival (Figure 1H). Importantly, the clone with the lowest reduction in E2F1 (pTRIPZ 93) had no significant effect on tumor development and growth as well as survival. We have also confirmed these observations using the U266 MM cell line (Figures S1I-S1K).
To evaluate the clinical significance of E2F, we analyzed its expression in purified myeloma cells from bone-marrow biopsy specimens from 172 newly diagnosed patients. We observed a lower progression-free and overall survival associated with high expression of activating E2F members (Figure 2A). Moreover, high E2Fs expression correlates with an expression-based proliferation index predictor of outcome (Hose et al., 2011) (Figure S2A), suggesting that high E2F demarcates a highly proliferative cohort of disease. Interestingly, the inverse correlation between expression and clinical outcome extended to the dimerization partner DP1 (Figure 2B), motivating an exploration of the functional importance and dependency of E2F TFs and their interaction with DP1 in MM cells.
Figure 2. Functional E2F Dependency in MM Cells.
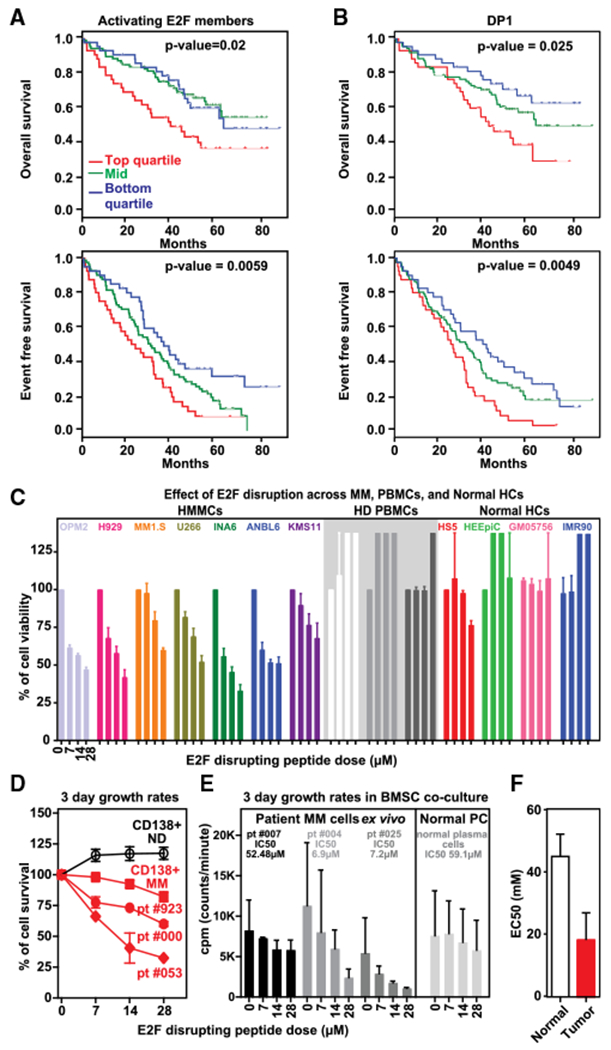
(A and B) Prognostic relevance of E2F activator members and DP1 expression on progression-free survival (PFS) and overall survival (OS) was investigated using Kaplan-Meier curves, log-rank tests, and Cox regression models in a dataset of 172 samples from uniformly treated MM patients. Red line indicates patient group with higher expression and shorter survival (4thQ); blue line represents group of patients with lower expression and longer survival (1stQ) whereas green line indicates group of patients with intermediate expression levels of E2F-activating members (A) and DP1 (B). See website https://www.ncbi.nlm.nih.gov/geo/ for gene expression data under accession number GSE39754.
(C) A panel of MM cell lines (n = 7), PBMCs from healthy donors (n = 3), and normal human cell lines (n = 4) were treated with different concentration of blocking peptide for 72 hr. Cell viability was assessed by CellTiter-Glo and presented as percentage of control cells (untreated cells). Data are shown as the mean values ± SD of three experiments performed in triplicates.
(D) Primary CD138+ MM cells from 3 patients as well as plasma cells from normal donor (ND) were treated with different concentrations of E2F-DP1 blocking peptide for 48 hr. Cell viability was assessed by CellTiter-Glo and presented as percentage of control cells (untreated cells).
(E) Primary CD138+ MM cells from 3 MM patients and NPC from healthy donor were cultured with bone marrow stromal cells (BMSC) with and without E2F-DP1 blocking peptide. Cell proliferation was assessed by (3H)thymidine uptake and expressed as cpm (counts per minute).
(F) Bone marrow from 2 myeloma patients was diluted with RPMI to seed 400–8,000 live cells per well into 96-well plates previously prepared with increasing concentration of peptide and were incubated for 48 hr. After red cell lysis, cells were stained with annexin V and CD138 mAb to identify viable myeloma and normal cells. EC50 analysis was performed using GraphPad analysis software.
See also Figure S2.
The heterodimerization of E2F-DP1 is essential for both high-affinity DNA binding and efficient transcriptional regulation (Bandara et al., 1993; Girling et al., 1993; Rubin et al., 2005). A polypeptide corresponding to residues 163–199 of DP1 has been identified to interrupt DP1-E2F interactions and therefore inhibit their transcriptional activity (Bandara et al., 1997). We used this polypeptide to abrogate E2F1-DP1 binding in MM cells (Figure S2E), which led to inhibition of cell viability in a dose-dependent manner in a panel of MM cell lines, with less significant effect on normal proliferating cell lines and healthy donor PBMC both with and without activation (Figures 2C and S2G). The antimyeloma activity of the blocking peptide was confirmed by analysis of DNA synthesis, where a significantly higher IC50 was indeed observed in normal cell lines as compared to myeloma cell lines (Figure S2F).
Importantly, genetic depletion of E2F1 and/or DP1 as well as abrogation of E2F/DP1 binding via blocking peptide was effective against MM cell growth even in the context of human myeloma bone-marrow milieu (Figures S2B and S2H), which up-regulates DP1 expression and E2F/DP1 DNA binding activity (Figures S2C and S2D). The impact of E2F/DP1 inhibition was confirmed in primary patient MM cells. We show significant inhibition of growth and survival of primary patient MM cells, both with and without the presence of stromal cells, while sparing normal CD138+ plasma cells (NPCs) isolated from bone-marrow aspirate of healthy individuals (Figures 2D and 2E). Additionally, we have evaluated the effect of the blocking peptide in primary myeloma cells from 2 patients cultured in the presence of their respective bone-marrow microenvironment using an automated flow cytometry platform for functional drug screening. We observed a significant impact on MM cell viability, while normal bone-marrow cells derived from the same patient resulted less sensitive to the treatment as suggested by EC50 analysis (Figure 2F). Finally, we confirmed caspases activation and induction of apoptosis after treatment with peptide (Figures S2I–S2J). Altogether, these data establish a functional E2F dependency in MM cells.
E2F1 and DP1 Co-occupy Active Promoters in MM
To better understand transcriptional influence of E2F on MM cells, we mapped the landscape of E2F genomic occupancy in the MM1.S and U266 MM cell lines. Previously, we and others have characterized the genomic occupancy of numerous transcription and chromatin regulators in the MM1.S system, making it one of the most well-characterized tumor epigenomes (Anders et al., 2014; Lovén et al., 2013). Integration of E2F1 and DP1 global occupancy into the MM1.S reference epigenome revealed specific co-occupancy of the factors at promoters of active genes marked by H3K4me3, with a strong positive correlation between E2F and RNA Polymerase II (RNA Pol II) levels at transcription start sites (Figures 3A, 3C, and S3A). In contrast, active enhancers, as defined by promoter distal Mediator (MED1) peaks and marked by H3K27ac (Hnisz et al., 2013; Lovén et al., 2013), show virtually no E2F binding genome-wide (Figure 3B). These trends are highlighted at the promoter-driven MDC1-TUBB and E2F1 loci (Figures 3C and S3A), and at the IRF4 proximal super-enhancer and BCL2L1 super-enhancer overlapping loci (Figures 3D and S3B). Notably, the transcriptional co-activator BET bromodomain (BRD4) is associated with both promoters and enhancers. Across the genome, E2F1-DP1 binds at more than half of all active promoters, but only at 10% of active enhancers (Figures 3E and 3F). From these data, we conclude that E2F and DP1 are depleted at enhancers and occupy the MM epigenome specifically at the promoters of active genes. Importantly, even enhancers with high CG content (>0.1 CG fraction) exhibit lower E2F occupancy than a matched set of active gene promoter regions (Figure S3C). Overall, E2F1 and DP1 exhibit similar ChIP sequencing (ChIP-seq) quality-control metrics (Figures S3D-S3F) and highly correlated occupancy patterns (Figures S3G-S3J).
Figure 3. E2F Binds to Promoters of Active Genes in MM Cells.
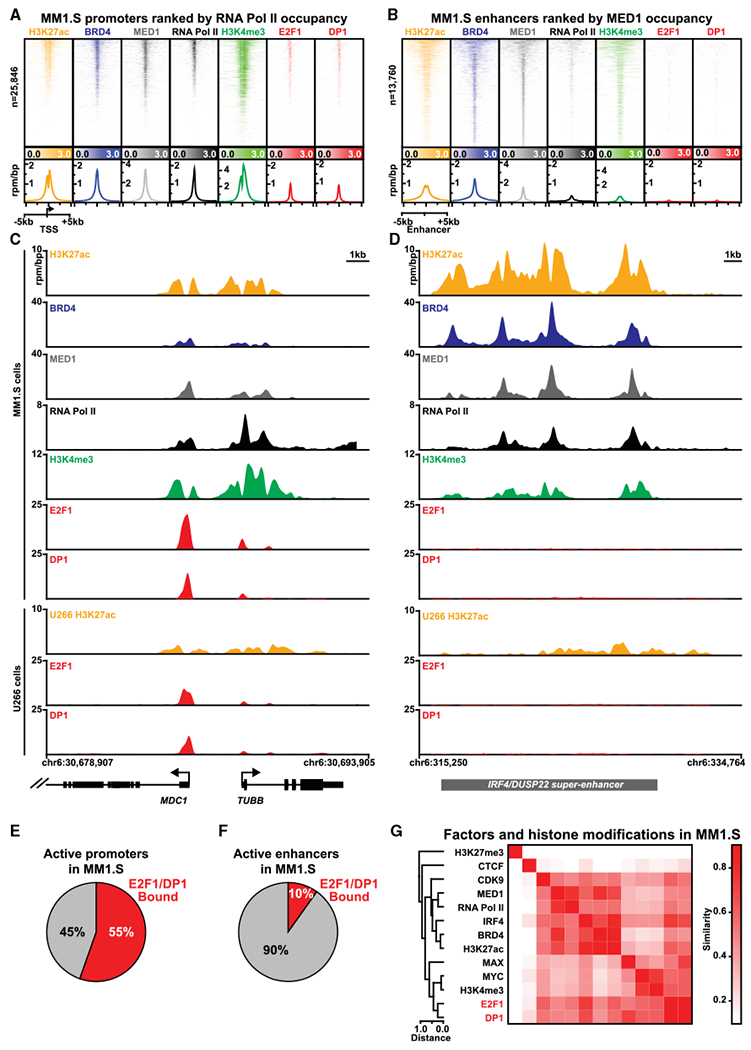
(A and B) Heatmaps of ChIP-seq occupancy of the ±5-kb region centered around transcription start sites (TSSs) (A) or enhancer centers (B). Each row of the heatmap is a specific region. Promoter(TSS) regions (A) are ordered by RNA Pol II occupancy. Enhancer regions (B) are ordered by MED1 occupancy. For each factor, ChIP-seq occupancy is shaded from light to dark in units of reads per million per base pair (rpm/bp). Lower panels show meta plots of average ChIP-seq occupancy summarized over active (RNA Pol II bound) promoters (A) or all enhancers (B) are shown underneath each heatmap with units of ChIP-seq occupancy in rpm/bp.
(C and D) Gene tracks showing ChIP-seq signal at individual loci for (C) MDC1 and TUBB and (D) IRF4 super-enhancer region. The x axis displays genomic coordinates with gene models depicted below. The y axis shows signal in units of rpm/bp.
(E and F) Pie charts showing the fraction of active promoters (E) or active enhancers (F) in MM1.S that are bound by E2F1 and DP1.
(G) Clustergram heatmap showing pairwise similarities between patterns of ChIP-seq occupancy genome-wide for assorted chromatin and TFs in MM1S. Pairwise similarities (Pearson correlation) are shaded from white to red.
Evaluation of high-resolution peak finding data reveal a spatial proximity between E2F1 and DP1 (14 bp median) that is consistent with other dimer-forming TFs (MYC-MAX), and tighter than between E2F1 and other TFs (IRF4) or RNA Pol II and proximal H3K4me3 nucleosomes (Figure S3K). These observations are consistent with X-ray crystallography structures of E2F1 and DP1 in complex and suggest that E2F1 and DP1 bind in tandem at promoters across the MM1.S genome (Rubin et al., 2005).
The co-localization of BET proteins with E2Fs at active promoters suggested a potential overlap in their gene regulatory programs and similar transcriptional consequences of their inhibition. We observe that, unlike other TFs and chromatin regulators that bind both promoters and enhancers, E2F1-DP1 signal is almost exclusively promoter proximal (Figure S3L). Indeed, BET bromodomains, although present at promoters, are disproportionately localized at enhancers and SEs (Lovén et al., 2013). Likewise, the effects of BET inhibition are most pronounced at these genes. From these observations, we hypothesize, that E2Fs act distinctly at the promoter gene regulatory axis.
To test this hypothesis, we first assembled genome-wide occupancy data for histone modifications, chromatin, and transcription regulators in MM1.S (Lovén et al., 2013). Using unbiased hierarchical clustering, we organized different factors and/or epigenetic modifications in MM1.S by spatial similarity of binding patterns. From this analysis, we identified two distinct active regulatory axes in MM1.S (Figure 3G). The first comprised Mediator, P-TEFb (CDK9), RNA Pol II, BRD4, IRF4, and H3K27ac– factors and/or epigenetic modifications that are found at both promoters and enhancers. The second comprised MYC-MAX, E2F1-DP1, and the transcription start site-specific histone modification H3K4me3. Although MYC is also found at enhancers in MM1.S (Lin et al., 2012), these data are consistent with MYC and E2F collaborative regulation of E2F target genes at promoters (Chen et al., 2008) and suggest a role for E2F in regulation of MM transcription separable from BETs.
E2F and BET Bromodomains Establish Distinct Oncogenic Regulatory Axes in MM
We next explored the programmatic gene control of the BET and E2F regulatory axes by quantifying the enhancer BRD4 signal and E2F (E2F1 and DP1 combined) promoter signal for all active genes in MM1.S (Figure 4A). We found that genes with high enhancer BRD4 signal (BRD4 super enhancer [SE] genes) had limited E2F promoter binding and vice versa confirming that these factors establish distinct target gene programs. At the extremes, we found less than 10% of genes were among the top 500 in BRD4 enhancer signal (i.e., SE regulated) and top 500 E2F promoter signal (Figure 4B). Interestingly, these included the histone HIST2H and HIST1H gene clusters that are highly occupied by both BRD4 and E2F. Genes governed by BRD4 SEs or high E2F exhibited divergent functionality with BRD4 SE-associated genes involved in cell signaling, apoptosis, and hypoxia and E2F-associated genes involved in cell-cycle regulation and canonical E2F-MYC regulation (Figure 4C) (Tables S1 and S2). Importantly, among BRD4 SE genes were MYC (by virtue of the IgH translocation) and cyclin D2 (CCND2), both activators of E2F (Figure 4A). These results were replicated using enhancer histone acetylation (H3K27ac) as a surrogate for enhancer activity in MM1.S and U266 cells (Figures S4A and S4B), and also observed in an additional B cell malignancy (diffuse large B cell lymphoma cells) (Figure S4C).
Figure 4. E2F and Super-enhancers Establish Distinct Regulatory Axes in Multiple Myeloma.
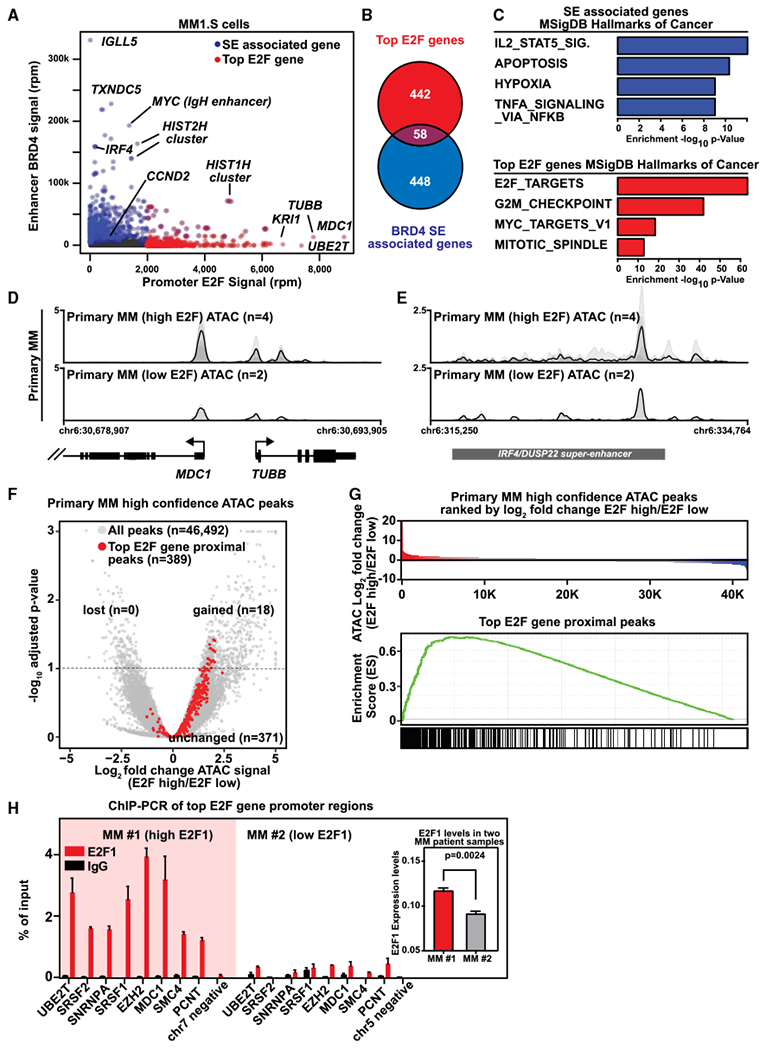
(A) Scatterplot showing the E2F promoter signal (x axis) and BRD4 enhancer signal (y axis) for all actively transcribed genes. E2F signal represents the average ChIP-seq occupancy (units of reads per million, rpm) of E2F1 and DP1 in the ±5-kb TSS region. BRD4 enhancer signal represents the total BRD4 ChIP-seqoccupancy (rpm) at all enhancers within 50 kb of the TSS. Super-enhancer (SE)-associated genes are colored in blue and the top 500 genes ranked by E2F promoter occupancy are shaded in red. Select genes are labeled.
(B) Venn diagram showing the overlap of super-enhancer (SE)-associated genes and top 500 E2F promoter occupancy genes.
(C) Bar plots showing the −log10 p value enrichments for the top four MSigDB Hallmarks of Cancer gene sets found for SE-associated genes (upper panel) or top E2F occupied genes (lower panel).
(D and E) Gene tracks showing ATAC-seq signal at individual loci for (D) MDC1 and TUBB and (E) DUSP2-IRF4 enhancer region in MM patient cells with high (n = 4) and low (n = 2) E2F expression levels, respectively. Individual replicates are plotted as translucent shapes, and the plotted line represents the mean signal.
(F) Volcano plot showing the log2 fold change of ATAC-seq signal compared using a generalized linear model (x axis) and false discovery rate (FDR)-adjusted p values (y axis) for a comparison of high-confidence ATAC-seq peaks between E2F high and low patients (n = 4 for E2F high, n = 2for E2F low). ATAC-seq peaks associated with the promoters of top E2F genes (n = 389) are highlighted in red. The dotted line represents a 0.1 FDR cutoff. 18 of 389 top E2F gene-associated ATAC-seq peaks are found to be statistically significantly gained in high E2F patients and enrichment of p = 3.3 × 10−06 (Fisher’s exact test).
(G) Upper panel: water fall of all high-confidence ATAC-seq peaks in primary MM ranked by average log2 fold change in ATAC-seq signal between E2F high and low patients (n = 4 for E2F high, n = 2 for E2F low). Lower panel: GSEA leading edge enrichment score for the top E2F gene-associated ATAC-seq peaks.
(H) E2F1 expression in CD138+ myeloma cells from two MM patients (MM#1 and MM#2) was evaluated by qPCR analysis. ChIP for E2F1 or control rabbit IgG followed by qPCR on a representative set of randomly chosen genes defined by ChIP-seq in MM1.S in MM#1 and MM#2 patient cells. Data are represented as percentage of input. Expression for E2F1 in each patient was determined using qPCR and shown in the right inset. Data are shown as absolute expression level and represent the mean values ± SD of triplicates. The statistical difference in expression is denoted as Welch’s two-tailed t test.
Together, these observations suggest BETs and E2F define separate gene regulatory axes that govern distinct biological functions in MM.
We then examined whether the E2F promoter axis defined in MM cell lines exhibits higher activity in patient MM cells. Using RNA sequencing (RNA-seq) data analysis performed on purified CD138+ myeloma cells from 409 newly diagnosed patients and unsupervised hierarchical clustering, we found a positive correlation between expression of E2F1 and top E2F target genes (Figure S4D). Moreover, in MM patient cells with elevated E2F expression, we observed increased promoter chromatin accessibility of the top E2F promoter gene regulatory axis as measured by ATAC sequencing (ATAC-seq) (Figures 4D, S3A, and S4E).
Although we observed robust differences in promoter ATAC-seq occupancy at top E2F genes, we also detected changes at super-enhancer regions (Figures 4E and S4F). We observed similar standard quality-control metrics (Figures S4G and S4H) and equivalent total aligned read GC% between low and high E2F ATAC-seq datasets with the distributions of reads being statistically similar (Figures S4I-S4L). We also observed similar QC metrics between ATAC-seq samples and E2F1-DP1 ChIP-seq datasets (Table S3). We next applied two approaches specifically controlling for batch variability. Using a generalized linear model, we find that of high-confidence ATAC-seq peaks, the top E2F-associated peaks are highly enriched among those statistically gained in E2F high patients (volcano plot; Fisher’s exact test p = 3.3 × 10−06) (Figure 4F). Additionally, a non-cutoff based leading edge analysis (gene set enrichment analysis [GSEA]) shows a strong enrichment for top E2F-associated peaks as gained in E2F high patients (Figure 4G). This evidence for high E2F binding in patient samples is corroborated by quantitative E2F1 ChIP-qPCR assay in low- and high-E2F1-expressing patient MM cells, where we confirmed a differential degree of E2F1 binding at the tested promoters based on the E2F1 expression levels in the tumor cells (Figure 4H).
These data demonstrate that top E2F-associated c/’s-regulatory elements exhibit increased chromatin accessibility in patients with high E2F levels, consistent with our overall model that E2F regulates expression of these genes. As expected, E2F1 and DP1 depletion by shRNA result in selective downregulation of only E2F target genes and not BRD4 SE genes (Figure S4I), suggesting selective targeting of this promoter-driven gene-regulatory program.
Significant Myeloma Growth Inhibitory Effects of Dual E2F and BET Inhibition
Prompted by these observations, we explored the ability of promoter modulation (through E2F inhibition) combined with enhancer modulation (through BET inhibition) to produce a more profound effect against MM cell growth and survival. We have used the inducible shE2F1 system characterized in Figure 1 to combine E2F1 depletion with single low-dose of the BET inhibitor JQ1 and assayed cell proliferation 72 hr after JQ1 administration. We observed a significant growth inhibitory effect of dual E2F and BET inhibition compared to individual inhibition in MM1.S and U266 cells (Figures 5A and S5A). These data were also confirmed using MM cells with a stable E2F knockdown (Figure S5B). RNA-seq analysis shows significant impact of combined E2F and BRD4 perturbation on expression of E2F top genes (Figure 5B), suggesting that at low JQ1 doses the addition of E2F1 depletion significantly affects cell viability and downregulates the promoter-controlled gene expression axis.
Figure 5. E2F1 Depletion Enhances Anti-myeloma Activity of the Bromodomain Inhibitor JQ1 In Vitro and In Vivo.
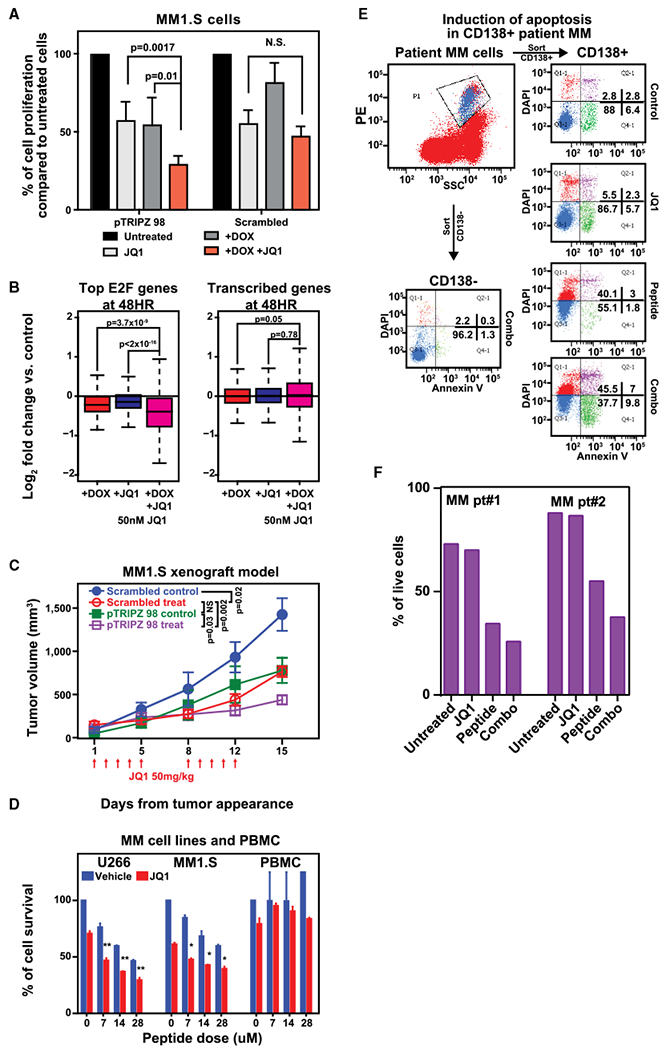
(A) Transfected MM1.S cells expressing tetracycline-inducible containing the E2F1 sequence or scramble control were plated in growth medium in the absence or presence of 2.5 μg mL−1 doxycycline in combination with IC50 dose of JQ1 (50 nM) for 3 days. Cell growth was evaluated by 3(H)-thymidine uptake and presented as the percentage of cell proliferation compared to control cells. Data are mean values ± SD of triplicates; Student’s t test.
(B) RNA-seq expression analysis log2 fold changes between treatment and control in MM1.S cells expressing inducible shRNA against E2F1. The red bar represents changes in cells treated with doxycycline (2.5 μg/mL); the blue bar represents changes in cells treated with JQ1 (50 nM), and the pink bar represents combination. Left panel shows top 500 E2F-associated genes; right panels are all expressed genes. Significance between distributions is denoted by a Welch’s two-tailed t test.
(C) MM1.S cells harboring doxycycline-inducible shRNAs targeting E2F1 or scramble control were inoculated subcutaneously in SCID mice. Following detection of tumor, mice were treated with irradiated 0.0625% doxycycline diet in the presence of either JQ1 (50 mg/kg) (treat) or placebo (control) intraperitoneal (i.p.) for 5 consecutive days/week for 2 weeks. Tumors were measured in two perpendicular dimensions using caliper. Baseline values before treatment were not significantly different among groups. Significance between groups was evaluated using one-way ANOVA test.
(D) MM1.S and U266 MM cell lines as well as PBMC from healthy donors were cultured in the presence of different doses of blocking peptide and JQ1 alone or in combination for 72 hr. Cell viability was assessed by CellTiter-Glo assay. Data are mean values ± SD of triplicates, Student’s t test.
(E and F) BM cells from MM patients were cultured with or without E2F-DP1 blocking peptide (14 μM) and/or JQ1 (10 nM) for 48 hr and analyzed using multichannel flow cytometry. Viability of CD138+ MM cells and CD138− normal BM stromal cells was determined by Annexin V and DAPI staining. A representative experiment (E) as well as effects of these agents alone or in combination on MM cells from 2 different patients (F) are shown.
See also Figure S5.
We next investigated how E2F1 inhibition modulates sensitivity to BET inhibition in vivo in mice subcutaneously inoculated with MM1.S cells harboring scrambled or doxycycline-inducible E2F1 shRNA. E2F1 depletion significantly inhibited MM cell growth compared with scrambled cells, and the antitumor effect was more pronounced upon treatment with JQ1 (Figure 5C).
This superior anti-MM effect of the dual E2F/BET inhibition was also observed when E2F functional impairment was achieved by using the dimerization inhibiting peptide. While significant effects were not observed in PBMCs, combination with JQ1 displayed synergistic effects against MM growth and survival relative to single-agent treatment (Figures 5D, S5C, and S5D) including the non-MYC-translocated MM (U266). In these cells, MYC or other upstream regulators of E2F activity are not present in the BET-regulated axis (Figure S4B), suggesting a potential increased therapeutic benefit in these patients achieved through direct targeting of E2F. Finally, we have confirmed ex vivo the impact of dual E2F1 and BET inhibition in primary MM cells in the presence of their bone-marrow microenvironment. As seen in Figures 5E and 5F, the effect of E2F/DP1 inhibition was amplified in the presence of JQ1 in CD138+ MM cells, with minimal effects on normal components of the bone marrow. Together these data implicate targeting both E2F and BET bromodomain regulatory axes as a potential therapeutic strategy in MM.
DISCUSSION
Through integrated chromatin and transcriptional studies, we have identified two distinct transcriptional programs in MM cells: a regulatory module, only at active promoters, with E2F-MYC-RNA Pol II-H3K4me3, confirming a very close association between E2F and MYC binding sites and their target genes with MYC as functional collaborator of E2F and Mediator-CDK9-BRD4-IRF4-H3K27ac regulatory axis at both promoters and enhancers. The highly E2F1 bound regions mark genes that are not SE associated, and consequently transcription of genes associated with SE is not perturbed by altered E2F1 and/or DP1-mediated transactivation. E2F1 binding to high GC regions that occur preferentially at promoters is predicted; however, it was previously unknown whether E2F1 binds enhancer regions. Although this is expected given the low GC content (C+G %) of enhancers, it is not obvious. In fact, it is well appreciated that a small subset of super-enhancers occurs at clustered CpG islands (Meng et al., 2014). Analysis of these regions that are distal to promoters and have high CG content reveal the absence of E2F1-DP1 binding.
These divergent BRD4 enhancer and E2F promoter axes are also observed in another malignancy, diffuse large B cell lymphoma, a tumor model that is sensitive to BRD4 inhibition (Chapuy et al., 2013) and where E2F deregulation is implicated (Monti et al., 2012). This demonstration of E2F and BET bromodomains each regulating distinct sets of genes with different oncogenic functions may therefore have significant implications for understanding innate oncogenic transforming activities of these factors as well as the therapeutic application of their inhibition.
Among oncogenic TFs commonly dysregulated in cancer, E2Fs are unique as they predominantly drive a limited number of genes through specific proximal transcription start site binding. As these genes tightly couple growth factor stimulation to cell-cycle entry, targeting E2F regulatory axis may present an attractive mechanism to selectively arrest tumor cell proliferation. Indeed, our finding that MM growth is dependent on E2F TFs, with limited effects on growth and viability of normal cells, suggests a higher threshold requirement of E2F activity and/or tumor-specific functions of E2F in MM. Our data show that depletion of either E2F1 or its cofactor, DP1, in MM cells results in cell-cycle arrest and apoptosis. In vivo, stable or inducible E2F1 knockdown had significant effect on tumor development and growth. These murine studies were not intended to address the question about toxicity but the relevance of E2F in supporting the myeloma growth. However, number of previous publications using mice deficient for individual or a combination of E2F genes do not have widespread defects in cell proliferation; on the other hand, E2F transgenic mice develop various tumors (Chen et al., 2009; Kent et al., 2017; Lee et al., 2010; Sharma et al., 2010), suggesting a differential requirement for E2Fs in the control of cell proliferation in oncogenic compared with normal environments. The observation that depletion of a single E2F family member (E2F1) results in potent anti-proliferative effects suggests the multiple redundancies of the E2F family are not sufficient to restore the necessary E2F activity in these tumor cells.
As E2F family TFs are thought to act as master regulators of cell-cycle progression and proliferation, at first glance, the anti-proliferative effects of E2F1 or DP1 depletion in MM are not entirely unexpected. However, given the ability of normal cells to proliferate in the presence of either E2F1 or DP1 knockdown, our data suggest that E2F may represent a tumor-specific vulnerability in MM, which is validated by our in vivo observations. These results are further corroborated by data from patient samples demonstrating elevated expression of the E2F regulatory axis in patients with high E2F levels and a statistically significant inverse correlation between expression of activating E2F family members and/or DP1 and overall and event-free survival in MM patients. Although it is possible that the poor prognosis observed with high E2F expression may be related with its effect on proliferation-related genes, it is also likely that this observation may be related with effects of E2F on number of other processes. In our previous study focusing on Sp1, another TF driving proliferation-related genes, its expression did not predict poor survival.
As a cofactor for E2Fs, DP1 has been reported to enhance the oncogenic function of E2F1 and cause transformation of cells indicating a proto-oncogenic potential (Wang et al., 2001). However, DP1 has been predominantly described as an E2F partner with limited, if any, direct role in cell biology, and its biological and functional role in a cancer-related disease was previously unknown. Altogether, our data provide interesting information on E2F activity to be further investigated as a possible target in MM. As for many TFs, direct pharmacologic inhibition of E2F remains an elusive challenge in drug discovery (Verdine and Walensky, 2007). However, E2F is not entirely “undruggable” as reported in recent papers focused on CDK4-6 inhibitor (Teh et al., 2018). Moreover, we have previously described the perturbation of the transcriptional program under control of the BET bromodomain BRD4 through the inhibitor JQ1 (Delmore et al., 2011).
The observation that the dependencies between super-enhancer and promoter-driven processes are non-overlapping has significant biological and clinical relevance. We have in fact explored the ability of promoter modulation (through E2F and BET inhibition) combined with enhancer modulation (through BET inhibition) to produce a synergistic effect against MM cell growth and survival. Here, we found that dual targeting of BETs and E2F enhances the anti-myeloma activity observed with either single perturbation in MM cell lines and primary patient cells. Although the dual inhibition leads to greater level of transcriptional inhibition, it is still expected to have a cell-type- and cell-state-specific effects compared to a general transcription inhibitor.
The aim of drug-combination studies is to achieve additive or synergistic therapeutic effects, by using subcytotoxic dosages. Our results show that, at low JQ1 doses, the addition of E2F1 depletion significantly affects cell viability and downregulates the promoter-controlled proliferation gene expression axis. Given emerging data regarding on target toxicity of BET inhibition in the clinic, synergistic strategies targeting non-overlapping dependencies such as E2F provide the opportunity to increase the therapeutic index at lower doses.
In conclusion, we describe a unique non-overlapping control of the transcriptome by promoter- and SE-associated dependencies in MM and suggest their role in maintenance of tumor cell state. These two vulnerabilities can be synergistically targeted, providing rationale for therapeutic translation in MM and other malignancies.
STAR*METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nikhil Munshi (nikhil_munshi@dfci.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and primary cells
The human cell lines used in the study were purchased from American Type Culture Collection (ATCC), German Collection of Tissue Culture or Japanese Collection of Research Bioresources Cell Bank. IL–6–dependent INA–6 cell line was provided by Dr. Renate Burger (University of Kiel, Germany). All cell lines were cultured in RPMI–1640 media containing 10% fetal bovine serum (FBS, GIBCO, Life technologies, Carlsbad, CA, United States), 2 mmol/L L-glutamine, 100 U/mL penicillin, and 100 mg/mL streptomycin (GIBCO, Life technologies, Carlsbad, CA, United States), with 2.5 ng/mL of IL–6 only in INA–6 cells. Bone marrow mononuclear cells (BMMNCs) and primary MM cells were isolated using Ficoll-Hypaque density gradient sedimentation from BM aspirates MM patients following informed consent and IRB (Dana-Farber Cancer Institute) approval. Peripheral blood mononuclear cells (PBMC) were isolated using Ficoll-Hypaque density gradient sedimentation from fresh buffy-coats from healthy donors and activated with 50 units/ml of IL-2.
Mouse Models
All animal experiments were approved by and conform to the relevant regulatory standards of the Institutional Animal Care and Use Committee at the Dana-Farber Cancer Institute. CB-17SCID-mice were subcutaneously injected with 1 × 106 MM.1S cells harboring non-targeting, E2F1 or DP1 shRNAs subcutaneously in the right flank in serum–free medium, and tumor growth was monitored with caliper measurements. For inducible knock-down experiment in vivo, scrambled, pTRIPZ 63, pTRIPZ93 and pTRIPZ98 MM1.S cells were inoculated in the right flank in serum-free medium. Induction of viral expression was obtained by treatment with irradiated 0.0625% Doxycycline Diet (Lab Diet) continuously, which provided 1–6 mg of Doxycycline per mouse/per day. Tumors were monitored and measured approximately twice weekly. Statistical significance was determined by Student’s t test. Survival of mice was measured by using the Prism GraphPad software (Systat Software, San Jose, CA).
METHOD DETAILS
Cell proliferation, viability and cell cycle assay
MM cell proliferation was measured by 3(H)thymidine (PerkinElmer, Boston, MA) incorporation assay. Cell viability was analyzed by CellTiter-Glo (CTG) (Promega). Cell cycle was evaluated by flow cytometric analysis following propidium iodide (PI) staining. Study of caspases activity was performed using Caspases 3–7, Caspase 8 and Caspase 9 Glo assay (Promega). Apoptosis was evaluated by flow cytometric analysis following Annexin-V staining. For primary MM cell studies, mononuclear cells that had been fraction separated by Ficoll gradients from the BM aspirates of consenting MM patients (in accordance with the Declaration of Helsinki) were plated at a cell density of 5 × 105 cells/mL in RPMI medium with 20% FBS plus the indicated concentrations of E2F/DP1 peptide and JQ1. After 48 hours in culture, cells were stained with anti-CD138-PE (PharMingen), FITC-conjugated annexin V (BioVision) and DAPI, and cell viability was assessed by flow cytometry. Exvitech® automated flow cytometry platform (Vivia biotech, Madrid, Spain) was used to evaluate activity of blocking peptide against primary myeloma cells in their microenvironment, as previously described. Statistical significance was determined by Student’s t test. Isobologram analysis was performed using the CalcuSyn software program (Biosoft, Ferguson, MO, and Cambridge, United Kingdom). A combination index (CI) less than 1.0 indicates synergistic activity.
E2F disrupting peptide
The 19-aa sequence is the H2 fragment derived from the DEF box region in DP1 (Bandara et al., 1997): R-R-R-V-Y-D-A-L-N-V-L-M-A-M-N-I-I-S-K
Peptide was purified by HPLC. Purity was greater than 90% (Celtek Bioscience, LLC).
Lentiviral-mediated stable gene knockdown
Hairpin-containing PLKO.1 plasmids were obtained from Sigma Mission. Packaged viral particles were used to infect MM cells using polybrene media (final concentration 8 mg/ml). Infected MM cells were selected by puromycin (0.5 μg/ml) for 48 hr (Sigma, St. Louis, MO), and then left to recover for 24 h. Knockdown efficacy was determined by qRT-PCR or western blotting and cells were used for functional studies as described above.
Inducible gene knockdown
Human TRIPZ E2F1 shRNA vectors were purchased from Thermo Scientific Bio (Tewksbury, MA, United States). shRNA expression was induced by adding 2.5 mg/ml doxycycline to the culturing media. The efficacy of the induction was confirmed by examining the cells microscopically for the presence of TurboRFP and by western blot analysis after 72 h of induction. Functional studies were performed as described above.
Stable overexpression
LentiORF clone of human E2F1 mGFP tagged (NM_005225) was purchased from Origene. MM cells were transduced in polybrene media (final concentration 8 μg/ml) for 8 hours and selected by sorting GFP positive cells.
Quantitative RT-PCR analysis
Expression of human E2F1 and DP1 transcripts were determined using real-time quantitative reverse transcriptase–polymerase chain reaction (qPCR) based on TaqMan fluorescence methodology, following manufacturer protocols (Applied Biosystems, Foster City, CA). Relative expression was calculated using the comparative delta delta (Ct) method.
Western blotting
MM cells were harvested and lysed using RIPA lysis buffer. Cell lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis SDS–PAGE, transferred to nitrocellulose membranes, and immunoblotted with different E2F1 (Cell Signaling Technology) and Dp1 (Santa Cruz) antibodies. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or B-actin were used as loading control (Santa Cruz Biotechnology).
Chromatin immunoprecipitation with massively parallel sequencing from cell lines (ChIP-Seq)
Briefly, 1×108 MM1.S and U266 cells, and 1×106 CD138+ MM cells were cross-linked with 1% formaldehyde for 10 minutes at 37°C. The cross-linked chromatin was then extracted, diluted with lysis buffer, and sheared by sonication. The chromatin was divided into equal samples for immunoprecipitation with specific antibodies. The immunoprecipitates were pelleted by centrifugation and incubated at 65°C to reverse the protein-DNA cross-linking. The DNA was extracted from the elute by the Qiaquick PCR purification kit (QIAGEN). Antibodies used were as follows: E2F1 (Cell Signaling #3742), DP1 (Santa Cruz), immunoglobulin G (negative control) polyclonal antibody (Abcam). In addition, a parallel sample of input DNA from the same cells was sequenced as a control. All samples were initially sequenced to generate a set of raw reads (each read has a length of 36 bp) from Illumina/Solexa GAII system ranging from ≈30 million to ≈100 million reads per sample. After mapping to UCSC Human HG18 assembly, a set of ≈50 million and ≈30 million mapped reads with unique genomic locations were obtained for DP1 and E2F1 respectively in both cell lines. ChIP-Seq quality control analysis is included in SF3D,F.
Chromatin immunoprecipitation followed by qPCR (ChIP-qPCR) in primary MM cells
1×106 CD138+ MM cells were cross-linked with 1% formaldehyde for 10 minutes at 37°C and processed as above. E2F1 ChIP and input DNA were analyzed using SYBR Green real-time PCR analysis (Applied Biosystems). Primers for ChIP-qPCR at given regions are listed in Table S5.
Patient multiple myeloma ATAC-Seq
10,000 CD138+ multiple myeloma cells were obtained (see above) from 4 patients with prior characterized gene expression levels. Three samples with high levels of E2F expression and two samples with low levels were chosen using the median average expression of E2F1, E2F2, E2F3, and TFDP1. For each sample, 10,000 cells were lysed for 10 minutes at 4°C in lysis buffer 10 mM Tris-HCl pH 7.4,10 mM NaCl, 3 mM MgCl2,0.1% IGEPAL CA-360). After lysis, the pellets were subject to a transposition reaction (37°C, 60 minutes) using the 2× TD buffer and transposase enzyme (Illumina Nextera DNA preparation kit, FC-121–1030). The transposition mixture was purified using a QIAGEN MinElute PCR purification kit. Library amplification was performed using custom Nextera primers and the number of total cycles determined by running a SYBR-dye based qPCR reaction and calculating the cycle number that corresponds to % the maximum. Amplified libraries were purified using a QIAGEN PCR purification kit and sequenced with paired end 75bp reads on an Illumina NextSeq. ATAC-Seq quality control analysis is included in SF4G-H
QUANTIFICATION AND STATISTICAL ANALYSIS
ChIP-Seq, ATAC-Seq, and RNA-Seq Data analysis
Genomic coordinates and gene annotation
All coordinates in this study were based on human reference genome assembly hg19, GRCh37 (https://www.ncbi.nlm.nih.gov/assembly/2758/). Gene annotations were based on Refseq annotation release 19 (https://www.ncbi.nlm.nih.gov/refseq/).
Calculating read density
We calculated the normalized read density of a ChIP-Seq dataset in any genomic region using the Bamliquidator (version 1.0) read density calculator (https://github.com/BradnerLab/pipeline/wiki/bamliquidator). Briefly, ChIP-Seq reads aligning to the region were extended by 200bp and the density of reads per base pair (bp) was calculated. The density of reads in each region was normalized to the total number of million mapped reads producing read density in units of reads per million mapped reads per bp (rpm/bp).
Identifying ChIP-Seq enriched regions
We used the MACS version 1.4.2 (Model based analysis of ChIP-Seq) (Zhang et al., 2008) peak finding algorithm to identify regions of ChIP-Seq enrichment over background. A p value threshold of enrichment of 1e-9 was used for all datasets. See Table S4 for the GEO accession number and background used for each dataset.
Identifying actively transcribed genes
In MM1.S, U266 and LY1, transcriptionally active genes were defined as those with a RNA Pol II enriched region within ± 1kb of the transcription start site (TSS).
Mapping typical enhancers and super-enhancers
Super-enhancers (SEs) and typical enhancers (TEs) in MM1.S, U266, and LY1 samples were mapped using the ROSE2 software package available at https://github.com/BradnerLab/pipeline and originally described in Brown et al. (2014). For MM1.S and U266, enhancers were defined in MM1.S using MED1 for Figure 3 to exactly match the original enhancer landscape published in Lovén et al. (2013). We and others have demonstrated that enhancers and super-enhancers can be mapped using MED1, BRD4, and H3K27ac (Brown et al., 2014; Hnisz et al., 2013; Lovén et al., 2013). To compare regulatory axes between BRD4 and E2F, we mapped super-enhancers in MM1.S using BRD4 (Figure 4). To enable comparison of enhancers between MM1.S and U266, enhancers and SEs were also mapped using H3K27ac (Figures S4A and S4B). Enhancers were also mapped in LY1 DLBCL cells using BRD4 (Figure S4C).
To map enhancers using a given factor, we first input a set of enriched regions peaks generated using MACS 1.4.2 (see above). The ROSE2 algorithm first determines an optimal distance to stitch together proximal peaks in order to maximize the number of consolidated regions while minimizing the inclusion of non-enriched genomic regions. After peak stitching, regions are filtered to keep enhancer regions. As enhancer regions can sometimes span the entire gene and/or promoter region, peaks are excluded only if they are contained entirely within the ± 2.5kb region flanking gene TSSs (using Refseq GRCh37 annotation). Regions are un-stitched if they overlap more than 3 distinct genes to prevent clusters of house-keeping genes from being included in the analysis. For each factor, background subtracted area under curve (in units of total rpm) are calculated for each region, and all regions are ranked by their overall AUC. To determine the cutoff between typical and super-enhancers, a graphical approach is used to determine the point at which the rank curve is tangential to a line with a slope of 1.
Creating heatmap and meta representations of ChIP-Seq occupancy
Heatmaps and meta plots of ChIP-Seq occupancy for various factors were created as in Lin et al. (2012). Heatmaps were created for all active promoters or all active enhancers. Each row plots the ± 5kb region flanking the TSS (for promoters) or the enhancer center. Rows are ranked by peak occupancy of RNA Pol II for promoters or by MED1 occupancy for enhancers.
ChIP-seq and ATAC profiles summary metric
The BAM files were read in R environment by readGAlignments function of GenomicAlignments Bioconductor package. The uniquely mapping reads in the BAM files were filtered by the XS tags. The counts were determined by the summarizeOverlaps function of GenomicAlignments Bioconductor package with mode as IntersectionNotEmpty. BSgenome.Hsapiens.UCSC.hg19 package was used for determining the GC content of the peak region.
Peaks proximity analysis
The top twenty percent high confidence peaks by auc for every factor were used. The distance was calculated from the summit of the peak of one factor to the summit of the peak of another factor. In case of distance from TSS, the distance was calculated from the summit of the peak to the nearest TSS. The peaks within 1kb were retained. All the RefSeq TSS were used in this analysis.
Clustering of chromatin modifications, regulators, transcription factors, and RNA Pol II in MM1S
To determine similarities of global occupancy patterns for various chromatin associated proteins, we quantified ChIP-Seq signal at the union of all enriched regions for H3K27me3, H3K4me3, H3K27ac, RNA Pol II, CTCF, CDK9, MED1, IRF4, BRD4, MAX, MYC, E2F1, and DP1. ChIP-Seq signal across all regions was median normalized for each factor and the similarity in occupancy was assessed using a Pearson correlation statistic. Factors were clustered based on patterns of similarity using unbiased hierarchical clustering. Figure 3F displays the relationships between factor occupancy patterns.
Determining distributions of distances between peaks
We sought to quantify the distances between E2F1 and DP1 peaks in comparison to RNA Pol II and H3K4me3. We iterated through all E2F1 peaks in MM1.S and measured the distance to the nearest DP1 peak (within 50kb). We collected similar measurements for E2F1 and DP1 in U266 and for RNA Pol II and H3K4me3 in MM1S. The distributions of peak distances are shown in Figure S3J.
Determining distribution of distances to nearest active promoter
We sought to investigate the proximity of various transcription factors to active gene promoters. For each transcription factor, we measured the distance (within 1mb) to the nearest active TSS (as previously defined). The distribution of distances to the nearest active TSS for various TFs is shown in Figure S3K.
Calculating enhancer signal and promoter E2F signal for all active genes
For all active genes in MM1.S, we quantified the BRD4 enhancer signal as the cumulative area under curve of all enhancers present within 50kb of the TSS. The promoter E2F signal was quantified as the average signal of E2F1 and DP1 (Figure 4A). To compare enhancer/promoter signal between MM1.S and U266, this analysis was repeated on the same set of active genes (genes active in MM1.S) using enhancers defined by H3K27ac in either MM1.S or U266 (Figures S4A and S4B).
Identifying enriched cancer hallmark gene sets of BRD4 SE associated genes or Top E2F genes
We defined BRD4SE associated genes as those with an SE within 50kb of the TSS. Top E2F genes were defined as the top 500 genes ranked by E2F signal in MM1S. These gene sets (BRD4 SE associated genes, n = 506 &Top E2F genes, n = 500) were queried using the MSigDB (http://software.broadinstitute.org/gsea/msigdb) to identify statistically enriched cancer hallmark gene sets. The top 4 enriched gene sets are shown in Figures 4C and 4D.
Quantifying changes in expression for BRD4 SE associated genes or Top E2F genes
To quantify changes upon JQ1 or E2F perturbation, we accessed JQ1 perturbation expression data from GSE44931 at 6 hours and 24 hours post 500nM treatment collected on Affymetrix Primeview 3′ UTR arrays (GPL16043). For E2F perturbation, we utilized data collected on Affymetrix exon arrays. Probe values were collapsed to common gene name and log2 expression values versus DMSO (for JQ1) or empty vector (for E2F) were utilized. Boxplots of gene expression values are compared in Figures 4E and S4F. The statistical significance of changes in distributions was assessed using a Welch’s two tailed t test.
Quantifying changes in ATAC-Seq promoter accessibility in patient MM with high and low E2F levels
Using ATAC-Seq datasets for patient MM samples with either high or low E2F expression (n of 2 each), we quantified promoter accessibility at Top E2F genes in the ± 1kb TSS region (Figure S4E). The statistical significance of changes in distributions was assessed using a Welch’s two tailed t test.
Quantifying changes in ATAC-Seq occupancy at individual loci
We first determined high-confidence ATAC-Seq peaks as those present in at least 2 of 4 samples. This resulted in 41,735 peaks in the analysis. Of these, 389 were proximal (+/− 1kb of the TSS) to a MM top E2F as determined by the intersection of the top 500 E2F genes in MM1.S and U266. A generalized linear model was used to normalize dataset variability and a fisher’s exact test was used to determine the statistical enrichment of top E2F genes in the set considered gained in E2F high patients (> log2 fold change and adjusted p value < 0.1) (Figure 4F).
Determining leading edge enrichment of top E2F genes
High confidence ATAC-Seq peaks associated with top E2F genes were determine as above. All high confidence ATAC-Seq peaks were ranked by their average log2 fold change between E2F high and E2F low patient samples (after normalization of signal using a generalized linear model). Using GSEA (Subramanian et al., 2005), we determined the leading-edge enrichment score for top E2F genes (Figure 4G).
Determining changes in gene expression in MM1.S with shE2F1 ± JQ1
Log2 fold changes in gene expression were calculated for MM1.S cells treated with either Dox, JQ1, or Dox & JQ1 at 48 hours compared to an average of 3 replicate untreated controls (24,48, 72 hours). The statistical significance of the difference in gene expression values was determined using a two-tailed t test (Figure 5B).
DATA AND SOFTWARE AVAILABILITY
Patient multiple myeloma data
Exon-array data from 172 multiple myeloma patient samples used in Figures 2A and 2B can be found in the NCBI GEO repository https://www.ncbi.nlm.nih.gov/geo/ under accession number GSE39754. An extended cohort of RNA-Seq data used in Figure S4D is currently under preparation (Cleynen et al., 2017).
E2F perturbation gene expression array data
Gene expression data after E2F depletion can be found in the Harvard Dataverse database using following link https://dataverse.harvard.edu/dataset.xhtml?persistentId=doi:10.7910/DVN/E7MAKN.
Multiple myeloma cell line ChIP-Seq
All ChIP-Seq data generated in this publication can be found in the NCBI GEO repository https://www.ncbi.nlm.nih.gov/geo/ under accession number GSE80661.
Patient multiple myeloma ATAC-Seq
ATAC-Seq data from patients multiple myeloma samples is submitted to the Harvard Dataverse https://dataverse.harvard.edu/dataset.xhtml?persistentId=doi:10.7910/DVN/E7MAKN.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-CD138-PE | PharMingen | CAT#: 552026; RRID:AB_394323 |
| Anti-E2F1 | Cell Signaling | CAT#: 3742; RRID:AB_2096936 |
| Anti-DP1 | Santa Cruz | sc-53642; RRID:AB_783104 |
| GAPDH | Santa Cruz | sc-47724; RRID:AB_627678 |
| β-actin | Santa Cruz | sc-47778; RRID:AB_626632 |
| Rabbit IgG polyclonal antibody | Abcam | ab37415; RRID:AB_2631996 |
| Anti-Histone H3 (acetyl K27) | Abcam | Ab4729; RRID:AB_2118291 |
| Biological Samples | ||
| Bone Marrow Mononuclear Cells | Isolated from Patients | NA |
| Primary CD138+ myeloma cells | Isolated from Patients | NA |
| Peripheral Blood Mononuclear Cells | Isolated from peripheral blood of healthy donors | NA |
| Chemicals, Peptides, and Recombinant Proteins | ||
| RPMI-1640 | GIBCO, Life technologies | CAT#: 11875-093 |
| Fetal Bovine Serum | GIBCO, Life technologies | CAT#: 10437-028 |
| L-glutamine | GIBCO, Life technologies | CAT#: 25030-081 |
| Pen Strep | GIBCO, Life technologies | CAT# 15140-122 |
| Interleukin-6 | BioLegend | Cat#: 570804 |
| Interleukin-2 | GIBCO, Life technologies | PHC0027 |
| Ficoll-Hypaque | GE Healthcare | CAT#: 17-1440-03 |
| 3(H)thymidine | PerkinElmer | NET027W001MC |
| DAPI | 3D Parmingen | CAT#: 564907 |
| JQ1 | Jun Qi (Harvard Medical School, USA) | NA |
| Puromycin | GIBCO, Life technologies | CAT#: A11138-03 |
| Polybrene | Santa Cruz | sc-134220 |
| Doxycycline Hyclate | Sigma | CAT#: D9891-1G |
| RIPA Lysis Buffer | Boston BioProducts | CAT#: BP-115DG |
| FITC-Conjugated Annexin V | BD Biosciences | CAT#: 556419 |
| RK-19 E2F disrupting peptide (Bandara et al., 1997) | Celtek Bioscience, LLC | NA |
| Formaldehyde 16% Solution | Electron Microscopy Sciences | CAT#: 15710 |
| SYBR Green PCR Master Mix | Applied Biosystems | CAT#: 4309155 |
| Critical Commercial Assays | ||
| CellTiter-Glo | Promega | CAT#: G7572 |
| Caspase-Glo 3/7 | Promega | CAT#: G8090 |
| Caspase-Glo 8 | Promega | CAT#: G8200 |
| Caspase-Glo 9 | Promega | CAT#: G8210 |
| Illumina Nextera DNA preparation kit | Illumina | FC-121-1030 |
| MinElute PCR purification kit | QIAGEN | 28004 |
| Qiaquick PCR purification kit | QIAGEN | 28104 |
| Experimental Models: Cell Lines | ||
| MM1S | ATCC | CRL-2974 |
| U266 | ATCC | TIB-196 |
| INA6 | Dr. R Burger (University of Kiel, Germany) | NA |
| KMS12PE | Japanese Collection of Research Bioresources Cell Bank | JCRB0430 |
| KMS11 | Japanese Collection of Research Bioresources Cell Bank | JCRB1179 |
| RPMI-8226 | ATCC | CCL-155 |
| EJM | DSMZ | ACC 560 |
| H929 | ATCC | CRL-9068 |
| Experimental Models: Organisms/Strains | ||
| Fox Chase SCID Congenic Mouse | Charles River Laboratories | C.B-17 SCID |
| Recombinant DNA | ||
| E2F1 MISSION shRNATRCN0000039659 | Sigma | SHCLNG-NM_005225 |
| E2F1 MISSION shRNATRCN0000000249 | Sigma | SHCLNG-NM_005225 |
| E2F1 MISSION shRNATRCN0000039658 | Sigma | SHCLNG-NM_005225 |
| E2F1 MISSION shRNATRCN0000039661 | Sigma | SHCLNG-NM_005225 |
| E2F1 MISSION shRNATRCN0000274180 | Sigma | SHCLNG-NM_005225 |
| E2F1 MISSION shRNATRCN0000019879 | Sigma | SHCLNG-NM_005225 |
| DP1 MISSION shRNATRCN0000274180 | Sigma | SHCLNG-NM_005225 |
| DP1 MISSION shRNATRCN0000019879 | Sigma | SHCLNG-NM_005225 |
| TRIPZ Human E2F1 shRNA V3THS_413546 | Thermo Scientific Bio | RHS4696-201895801 |
| TRIPZ Human E2F1 shRNA V3THS_393593 | Thermo Scientific Bio | RHS4696-201897811 |
| TRIPZ Human E2F1 shRNA V3THS_393594 | Thermo Scientific Bio | RHS4696-201904734 |
| TRIPZ Human E2F1 shRNA V2THS_43498 | Thermo Scientific Bio | RHS4696-200701670 |
| E2F1 (NM_005225) Human Tagged ORF Clone | Origene | RC208247L2 |
| Software and Algorithms | ||
| CalcuSyn | Biosoft | NA |
| Prism | GraphPad | NA |
| RefSeq Annotation | https://www.ncbi.nlm.nih.gov/refseq/ | NA |
| Bamliquidator (version 1.0) | https://github.com/BradnerLab/pipeline/wiki/bamliquidator | NA |
| MACS version 1.4.2 (Model based analysis of ChIP-Seq) | https://github.com/taoliu/MACS/ | NA |
| R environment | The R Foundation | NA |
| BSgenome.Hsapiens.UCSC.hg19 | https://github.com/BradnerLab/pipeline | NA |
| GenomicAlignments | https://doi.org/10.18129/B9.bioc.GenomicAlignments | NA |
| ROSE2 | https://github.com/linlabbcm/rose2 | NA |
| GSEA | http://software.broadinstitute.org/gsea/msigdb | NA |
| Deposited Data | ||
| Multiple myeloma cell line ChIP-Seq | This paper | GEO: GSE80661 |
| Multiple myeloma primary cells Atac-Seq | This paper | https://dataverse.harvard.edu/dataset.xhtml?persistentId=doi:10.7910/DVN/E7MAKN |
Highlights.
E2F1 and its heterodimerization partner DP1 are required for myeloma cell proliferation
E2F1-DP1 heterodimers regulate promoter proximal transcription
E2F1 and BRD4 establish distinct regulatory axes in multiple myeloma
Combined inhibition of BRD4 and E2F co-operatively reduces myeloma tumor growth
ACKNOWLEDGMENTS
We thank Jun Qi for providing JQ1 and Jennifer Perry for critical review of the manuscript. This work was supported by Department of Veterans Affairs Merit Review Award1 I01BX001584 (N.C.M.), NIH grants P01155258-07 (N.C.M., H.A.-L., R.A.Y., C.Y.L., M.F., and K.C.A.), P01-CA066996 (J.E.B.), and 1R01CA215452-01 (C.Y.L.), National Cancer Institute Spore 5P50CA100707-13 (M.F., C.Y.L., J.E.B., N.C.M., H.A.-L., and K.C.A.), Department of Defense CDMRP CA120184 (C.Y.L.), and Cancer Prevention Research Institute of Texas RR150093 (C.Y.L.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and five tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.12.016.
DECLARATION OF INTERESTS
K.C.A. serveson advisory boards for Celgene, Millennium, and Gilead Sciences and is a Scientific founder of OncoPep and C4 Therapeutics. R.A.Y. is a founder and share holder of Syros Pharmaceuticals, C4 Therapeutics, and OmegaTherapeutics. J.E.B. is now an executive and shareholder of Novartis AG and has been a founder and shareholder of SHAPE (acquired by Medivir), Acetylon (acquired by Celgene), Tensha (acquired by Roche), Syros, Regency, and C4 Therapeutics. N.C.M. serves on advisory boards to Millennium, Celgene, Abbvie, Oncopep, and Novartis; he is a Scientific founder of OncoPepand received research funding Celgene. All other authors declare no conflict of interest.
REFERENCES
- Anders L, Guenther MG, Qi J, Fan ZP, Marineau JJ, Rahl PB, Lovén J, Sigova AA, Smith WB, Lee TI, et al. (2014). Genome-wide localization of small molecules. Nat. Biotechnol 32, 92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandara LR, Buck VM, Zamanian M, Johnston LH, and La Thangue NB (1993). Functional synergy between DP-1 and E2F-1 in the cell cycle-regulating transcription factor DRTF1/E2F. EMBO J. 12, 4317–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandara LR, Girling R, and La Thangue NB (1997). Apoptosis induced in mammalian cells by small peptides that functionally antagonize the Rb-regulated E2F transcription factor. Nat. Biotechnol 15, 896–901. [DOI] [PubMed] [Google Scholar]
- Brown JD, Lin CY, Duan Q, Griffin G, Federation A, Paranal RM, Bair S, Newton G, Lichtman A, Kung A, et al. (2014). NF-κB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol. Cell 56, 219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, et al. (2013). Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 24, 777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. (2008). Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133, 1106–1117. [DOI] [PubMed] [Google Scholar]
- Chen HZ, Tsai SY, and Leone G (2009). Emerging roles of E2Fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 9, 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleynen A, Szalat R, Kemal Samur M, Robiou du Pont S, Buisson L, Boyle E, Chretien ML, Anderson K, Minvielle S, Moreau P, et al. (2017). Expressed fusion gene landscape and its impact in multiple myeloma. Nat. Commun 8, 1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper-Kuhn CM, Vroemen M, Brown J, Ye H, Thompson MA, Winkler J, and Kuhn HG (2002). Impaired adult neurogenesis in mice lacking the transcription factor E2F1. Mol. Cell. Neurosci 21, 312–323. [DOI] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. (2011). BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J (2003). Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 3, 11–22. [DOI] [PubMed] [Google Scholar]
- Egan JB, Shi CX, Tembe W, Christoforides A, Kurdoglu A, Sinari S, Middha S, Asmann Y, Schmidt J, Braggio E, et al. (2012). Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood 120, 1060–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin WG Jr., Livingston DM, Orkin SH, and Greenberg ME (1996). E2F-1 functions in miceto promote apoptosis and suppress proliferation. Cell 85, 549–561. [DOI] [PubMed] [Google Scholar]
- Girling R, Partridge JF, Bandara LR, Burden N, Totty NF, Hsuan JJ, and La Thangue NB (1993). A new component of the transcription factor DRTF1/E2F. Nature 365, 468. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: The next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andrd V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hose D, Reme T, Hielscher T, Moreaux J, Messner T, Seckinger A, Benner A, Shaughnessy JD Jr., Barlogie B, Zhou Y, et al. (2011). Proliferation is a central independent prognostic factor and target for personalized and risk-adapted treatment in multiple myeloma. Haematologica 96, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone RW, Ruefli AA, and Lowe SW (2002). Apoptosis: A link between cancer genetics and chemotherapy. Cell 108, 153–164. [DOI] [PubMed] [Google Scholar]
- Kent LN, Bae S, Tsai SY, Tang X, Srivastava A, Koivisto C, Martin c. K., Ridolfi E, Miller GC, Zorko SM, et al. (2017). Dosage-dependent copy number gains in E2f1 and E2f3 drive hepatocellular carcinoma. J. Clin. Invest 127, 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn MJ, Bronson RT, Harlow E, Dyson NJ, and Yamasaki L (2003). Dp1 is required for extra-embryonic development. Development 130, 1295–1305. [DOI] [PubMed] [Google Scholar]
- Kohn MJ, Leung SW, Criniti V, Agromayor M, and Yamasaki L (2004). Dp1 is largely dispensable for embryonic development. Mol. Cell. Biol 24, 7197–7205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgren O, and Iskander K (2017). Modern multiple myeloma therapy: Deep, sustained treatment response and good clinical outcomes. J. Intern. Med 281, 365–382. [DOI] [PubMed] [Google Scholar]
- Lee JS, Leem SH, Lee SY, Kim SC, Park ES, Kim SB, Kim SK, Kim YJ, Kim WJ, and Chu IS (2010). Expression signature of E2F1 and its associated genes predict superficial to invasive progression of bladder tumors. J. Clin. Oncol 28, 2660–2667. [DOI] [PubMed] [Google Scholar]
- Lin CY, Lovdn J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, and Young RA (2012). Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151, 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Tang X, Srivastava A, Pecot T, Daniel P, Hemmelgarn B, Reyes S, Fackler N, Bajwa A, Kladney R, et al. (2015). Redeployment of Myc and E2f1-3 drives Rb-deficient cell cycles. Nat. Cell Biol 17, 1036–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovdn J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE, Fisher DE, Housman DE, and Jacks T (1994). p53 status and the efficacy of cancer therapy in vivo. Science 266, 807–810. [DOI] [PubMed] [Google Scholar]
- Mellinghoff IK, and Sawyers CL (2002). The emergence of resistance to targeted cancer therapeutics. Pharmacogenomics 3, 603–623. [DOI] [PubMed] [Google Scholar]
- Meng FL, Du Z, Federation A, Hu J, Wang Q, Kieffer-Kwon KR, Meyers RM, Amor C, Wasserman CR, Neuberg D, et al. (2014). Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell 159, 1538–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti S, Chapuy B, Takeyama K, Rodig SJ, Hao Y, Yeda KT, Inguilizian H, Mermel C, Currie T, Dogan A, et al. (2012). Integrative analysis reveals an outcome-associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B cell lymphoma. Cancer Cell 22, 359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller H, and Helin K (2000).The E2F transcription factors: Key regulators of cell proliferation. Biochim. Biophys. Acta 1470, M1–M12. [DOI] [PubMed] [Google Scholar]
- Nevins JR (2001). The Rb/E2F pathway and cancer. Hum. Mol. Genet 10, 699–703. [DOI] [PubMed] [Google Scholar]
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et al. (2004). EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 304, 1497–1500. [DOI] [PubMed] [Google Scholar]
- Palumbo A, and Anderson K (2011). Multiple myeloma. N. Engl. J. Med 364, 1046–1060. [DOI] [PubMed] [Google Scholar]
- Rubin SM, Gall AL, Zheng N, and Pavletich NP (2005). Structure of the Rb C-terminal domain bound to E2F1-DP1: A mechanism for phosphorylation-induced E2F release. Cell 123, 1093–1106. [DOI] [PubMed] [Google Scholar]
- Secombe J, Pierce SB, and Eisenman RN (2004). Myc: A weapon of mass destruction. Cell 117, 153–156. [DOI] [PubMed] [Google Scholar]
- Sharma A, Yeow WS, Ertel A, Coleman I, Clegg N, Thangavel C, Morrissey C, Zhang X, Comstock CE, Witkiewicz AK, et al. (2010). The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J. Clin. Invest 120, 4478–4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ, and McCormick F (2002). The RB and p53 pathways in cancer. Cancer Cell 2, 103–112. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teh JLF, Cheng PF, Purwin TJ, Nikbakht N, Patel P, Chervoneva I, Ertel A, Fortina PM, Kleiber I, HooKim K, et al. (2018). In vivo E2F reporting reveals efficacious schedules of MEK1/2-CDK4/6 targeting and mTOR-S6 resistance mechanisms. Cancer Discov. 8, 568–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimarchi JM, and Lees JA (2002). Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell Biol 3, 11–20. [DOI] [PubMed] [Google Scholar]
- Verdine GL, and Walensky LD (2007). The challenge of drugging undruggable targets in cancer: Lessons learned from targeting BCL-2 family members. Clin. Cancer Res 13, 7264–7270. [DOI] [PubMed] [Google Scholar]
- Wang D, Russell J, Xu H, and Johnson DG (2001). Deregulated expression of DP1 induces epidermal proliferation and enhances skin carcinogenesis. Mol. Carcinog 31, 90–100. [DOI] [PubMed] [Google Scholar]
- Weinberg RA (1995). The retinoblastoma protein and cell cycle control. Cell 81, 323–330. [DOI] [PubMed] [Google Scholar]
- Wu L, Timmers C, Maiti B, Saavedra HI, Sang L, Chong GT, Nuckolls F, Giangrande P, Wright FA, Field SJ, et al. (2001). The E2F1-3 transcription factors are essential for cellular proliferation. Nature 414, 457–462. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Patient multiple myeloma data
Exon-array data from 172 multiple myeloma patient samples used in Figures 2A and 2B can be found in the NCBI GEO repository https://www.ncbi.nlm.nih.gov/geo/ under accession number GSE39754. An extended cohort of RNA-Seq data used in Figure S4D is currently under preparation (Cleynen et al., 2017).
E2F perturbation gene expression array data
Gene expression data after E2F depletion can be found in the Harvard Dataverse database using following link https://dataverse.harvard.edu/dataset.xhtml?persistentId=doi:10.7910/DVN/E7MAKN.
Multiple myeloma cell line ChIP-Seq
All ChIP-Seq data generated in this publication can be found in the NCBI GEO repository https://www.ncbi.nlm.nih.gov/geo/ under accession number GSE80661.
Patient multiple myeloma ATAC-Seq
ATAC-Seq data from patients multiple myeloma samples is submitted to the Harvard Dataverse https://dataverse.harvard.edu/dataset.xhtml?persistentId=doi:10.7910/DVN/E7MAKN.