

Abstract
Mutual conformational selection and population shift followed by minor induced fit optimization is the key mechanism in biomolecular recognition; and monomers and small oligomers binding to amyloid seeds in fibril growth is a molecular recognition event. Here, we describe amyloid aggregation, preferred species, cross-species barriers and transmission within the broad framework of molecular recognition. Cross seeding of amyloid species is governed by conformational selection of compatible (complementary) states. If the dominant conformations of two species are similar, they can cross-seed each other; on the other hand, if they are sufficiently different, they will grow into different fibrils, reflecting species barriers. Such a scenario has recently been observed for the tau protein which has four repeats. While a construct consisting of repeats 1, 3 and 4 can serve as a seed for the entire 4-repeat tau segment, the inverse does not hold. On the other hand, the tau protein repeats with the characteristic U-turn shape can cross-seed Alzheimer’s Aβ, and similarly the Islet Amyloid Polypeptide (IAPP). Within this framework, we suggest that the so-called ‘central dogma’ of amyloid formation, where aggregation takes place through non-specific backbone hydrogen bonding interactions which are common to all peptides and proteins, is a simple reflection of the heterogeneous, polymorphic free energy landscape of amyloid species. Here, we review available data and make some propositions addressing this key problem. In particular, we argue that recent theoretical and experimental observations support the key role of selective molecular recognition in amyloidosis and in determining cross-species barriers and transmission.
Keywords: beta-amyloid oligomers, amyloid fibril structures, Alzheimer’s disease, prion, conformational selection, nucleation polymerization, energy landscape, conformational diseases
Graphical Abstract
1. Introduction
Conformational diseases, such as Parkinson’s and Alzheimer’s, are related to proteins that unfold abnormally and form nonnative oligomeric or polymeric aggregates, which largely consist of β-sheet structures. Common characteristics of conformational diseases display late or episodic onset due to gradual accumulation of these aggregates and the acceleration of their formation by stress.1 However, increasing evidence indicates that soluble oligomers of amyloidogenic proteins are both responsible for amyloidosis2; 3 and are the toxic agents.4–6 Some data suggest that their final large aggregates can also lead to cytotoxicity.7; 8 Thus, a complex picture emerges, where oligomers, fibrils, and fibril fragments/fibrilar oligomers can all be involved in cytotoxicity, albeit through different mechanisms.9; 10
The two most studied amyloidogenic proteins are the prion protein in prion (mad cow) disease and amyloid β peptide in Alzheimer’s disease. Prion diseases can cause fatal neurodegenerative infections that affect human and animals. The conformational changes of the prion protein underlie the biology of prion infection and strains. The normal prion protein (PrPc) itself is not toxic; however, a conformational isoform of the prion protein (PrPsc) is able to polymerize and is neurotoxic. PrPc has a partially structured N-terminal segment (1–100) and a well folded C-terminus (121–230) with short antiparallel β-sheets and three longer α-helices. There is no structural information for the isolated PrPsc protein, but its polymerized form is an amyloid fibril dominated by β-sheet conformation. The infectious prion protein is related to the PrPsc isoform, and can catalyze the conversion of the normal PrPc form to the PrPsc isoform, and the autocatalysis of PrPc to PrPsc is the prion replication process. The toxic form of the prion protein derives from either small oligomers, which are formed during the polymerization of the PrPsc isoform, or from intermediate species produced during the conformation conversion from PrPc to PrPsc. Since the kinetics of the autocatalysis has a long lag time, large scale spread of the PrPsc isoform in the infected host only occurs years after infection.
Amyloid β peptide (Aβ peptide) has been a paradigm for studies of amyloid formation and conformations. The full-length Aβ peptide has 40–42 residues when cut from its precursor protein. Aβ aggregates are observed in brain tissues of Alzheimer’s patients,11 and it is generally believed that Aβ peptide oligomerization is the major mechanism leading to neuron-cell death.7 In addition to the full-length Aβ peptides, a number of in vitro-generated β-amyloid fragments were also observed to form amyloid fibrils. The sequences cover all Aβ peptide regions: the N-terminus, middle, and C-terminus. Together, the full-length β-amyloid and its fragments constitute a warehouse of sequences prone to amyloid formation with a gigantic polymorphic range.12 Experimentally, different full-length Aβ fibril conformations are preferred under different incubation conditions. A broad range of soluble Aβ forms have also been observed in AD and Lewy body (DLB) brains, and mouse models. For example, the amyloid β-Derived Diffusible Ligands (ADDLs) were proposed to have different morphologies and to exert their toxicity through different mechanisms.13 The molecular weight of globular amyloid β-peptide ‘globulomer’14 is about 60 KDa. Another smaller soluble form, Aβ*56 causes memory deficiency in middle-aged mice.15 Finally, ‘amylospheroids’ (ASPDs), which are larger soluble oligomers16; 17 with higher mass (more than 100 kDa) were reported to have sizes ~10–15 nm.
Amyloids fold through diverse mechanisms which involve competing folding and binding events.18 Formation of oligomers and aggregates of amyloidogenic peptides make it difficult to predict and control their polymorphic range, preferred aggregate states, pathways leading to these, timescales, and the subsequent mechanisms of toxicity. Here, we view amyloid formation as a molecular recognition event. Within this broad framework, we discuss the conformational changes involved in amyloid growth within the same or across species and their recognition scenarios and consequences. The underlying mechanism of molecular recognition is (mutual) conformation selection with subsequent optimization, whether side chains and minor backbone movements in proteins; or bases and backbones in RNA/DNA; or in membrane lipid heads and tails. Below, we first describe the mechanism in the healthy functional state in general terms with some examples. We follow with amyloid polymerization. Overall, because of the clear relevance to disease, we expect research in the coming years to increasingly focus on transmission (self-replication) and cross-species barriers.
2. Conformational selection in functional proteins
Stable, folded proteins in crystal structures have well defined three-dimensional structures, which are not as dynamic as those in solution. In solution, proteins exist as conformational ensembles and their populations follow thermodynamic distributions.19–21 Local energy fluctuations in water range between 10 to 20 kcal/mol (16.8 to 33.6 kT at 300K ),22 which is enough to perturb a well folded protein to a vast number of states. The conformational differences can be small or large; and some conformations are more populated than others (Figure 1A). The more flexible are the proteins the larger the number of their populated states.
Figure 1.
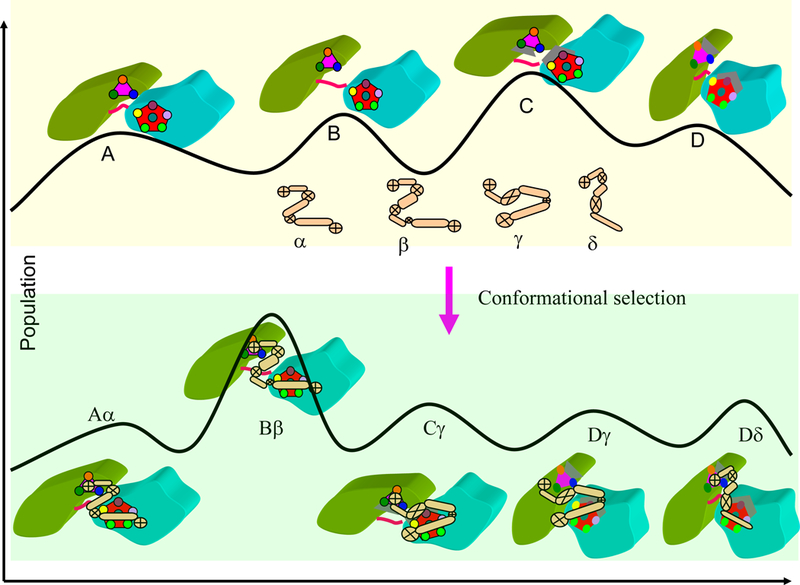
An illustration of conformational selection and population shift in protein recognition. Top: four protein conformations with different populations are shown as A, B, C, and D. The ligand may have conformation α, β, γ and δ. The mutual conformational selection between protein and ligands may generate five (roughly typical of real scenario’s) leading conformers: Aα, Bβ, Cγ, Dγ, and Dδ. The Bβ conformer has the highest population in final complex.
The distribution of protein conformations derives from their polymeric nature; however, it is optimized by evolution to fit their functions. Proteins function through their interaction with other molecules. The traditional ‘lock and key’ mechanism states that a protein has to have an exact match with its ligand to form a functional complex. The ‘induced fit’ hypothesis23 argues that the fact that protein complexes often have different conformations from those of their unbound protein constituents, suggests that the bound conformation is ‘induced’ by the binding partner. The ‘conformational selection and population shift’ model24–27 provides a more detailed description of the real molecular mechanism which prevails in cellular life. The conformational selection concept recognizes that both binding partners are flexible and have conformational distributions. During binding, higher energy (lower population) conformers which are most complementary to some pre-existing ligand conformations can be selected and the equilibrium shifts toward these conformers (Figure 1B).24–27 Increasing experimental evidence obtained from single molecule, NMR, and other techniques has validated the conformational selection mechanism.28–31
Nature has used and optimized conformational selection to facilitate normal functions, for example, in enzyme catalysis with step-wise conformational selection32; 33 and in transcription factor binding34–36 to help recognition of specific DNA regulatory elements; it has used it for functions of folded proteins as well as functions of the disordered states.37; 38 For the natively disordered states, the highly flexible protein conformations similarly have preferred conformational ensembles.39 As shown in Figure 1B, the conformation that fits some pre-existing receptor conformation will be selected, get stabilized, with a subsequent equilibrium shift toward this conformation. As described in the next section, such mechanism also applies to mis-folded, denatured (or disordered) states and can lead to amyloid formation.
A recent example illustrates conformational selection in a multi-domain protein, with a hinge in its linker40; 41 which can present large scale conformational changes over high barriers. The tandem RNA recognition motif domains of U2AF65 are efficiently recruited to the intron during spliceosome assembly, and have two different domain arrangements in the absence and presence of a high affinity ligand. Conformational selection and population shift between the closed and open conformations of U2AF65 account for recognition of sequence variation,42 and the equilibrium between the two conformations quantitatively correlates with the natural variability in the polypyrimidine tract. The Suc1 protein is an example of conformational selection and population shift for two native states, a monomer and a domain-swapped dimer. Single point mutations shift the equilibrium between the states which lead to altered ligand binding preferences.43 Another example, which like amyloids also presents a broad heterogeneous landscape, relates to DNA sequence recognition by a transcription factor (TF). The DNA contains a very large number of response elements (REs) which can be recognized by a TF, with each relating to regulation of a specific gene. How does the TF know which to bind?44 The affinity (conformational compatibility) appears to play a key role: if the concentration of the TF is low, it will bind only to high affinity REs. On the other hand, if the TF concentration is high, it will bind all its respective REs so long as they are exposed (not covered by nucleosomes in the compact, folded chromatin).34 Current data suggest that TF concentration in the nucleus is high, and all exposed REs are bound to their corresponding TFs. In amyloids, the concentration is expected to always be very high, which suggests that all polymorphic seeds will be populated and grow, although as we discuss below, at different time scales which depend on the distribution of the populations. In regulation, subsequent events will differ: Because the REs have slightly different sequences, the bound TF will (allosterically) display a slightly different surface, which will be recognized by another protein cofactor. Selective molecular recognition has a physical basis similar to that taking place in crystallization processes.
3. Nucleated polymerization and conformational selection in homo-molecular amyloid formation
Conformational diseases start with perturbation of normal folded proteins, which lead to some conformational changes. Following cleavage from the APP precursor protein and getting away from the membrane environment, the Aβ peptide is mostly disordered. Polyglutamine in Huntington disease is also a natively disordered protein. However, not all amyloidogenic proteins are completely unfolded. β2-microglobulin is a good example of a protein which forms amyloids either from the completely denatured state under highly acidic conditions or through less disrupted states.45 The long and straight amyloid (LS) fibril forms through a nucleation-dependent mechanism, leading to parallel, in-register β-sheet.46 In less acidic conditions and high ionic strength, β2-microglobulin spontaneously forms worm-like (WL) fibril via a non-nucleated pathway.47
In general, proteins in disordered states are more like synthetic organic polymers, which typically follow nucleation-dependent pathways, with long time scales for accumulation of aggregates and absence of lag times in the presence of ‘seeds’. Protein aggregation from highly disordered states can be viewed as analogous to aggregation of colloidal particles, with certain unique polymer dynamics.48 The aggregation of the Aβ peptide is surprisingly similar to the self-assembly of block copolymers into nanofiber micelles.49 Depending on the solvent properties, the phase diagrams of block copolymers can present nano-structures with chain-like, spheres, and vesicles morphologies, which are comparable to the linear fibrils, nanospheres, and ring-like structures observed for Aβ peptides. In a certain sense, the sequence of full length Aβ peptide resembles a block copolymer with a highly polar and charged N-terminal and a non-polar C-terminal segment. Some studies indicate that the neurotoxicity of Aβ42 is mediated by a nucleated polymerization process rather than by discrete Aβ42 species.50
Unlike the synthetic block polymer, the protein backbone atoms are able to form hydrogen bonds; thus, the nucleation of amyloidogenic proteins is not a simply random micelle aggregation. Secondary structure features can be expected to accompany initial nucleation processes; and to be stabilized, pre-nucleation intermediates need to sample intramolecular β-sheets.51 At the pentamer level, TEM (transmission electron microscopy) and AFM (atomic force microscopy) images indicate that Aβ42 pentamers are round disk-like particles.52 However, the loosely packed pentamer is unlikely to be stable.53 H/D exchange protection experiments indicated that there are turns in the His13-Gln15, Gly25-Gly29, and Gly37-Gly38 regions,52 which suggest an intra-chain β-hairpin structure for the pentamer. Mixtures of intra-chain β-hairpin turns and α-helical regions can account for most of the experimental H/D exchange protection data.53 Consistently, several kinetic models considered the nucleation of nanosized amyloid fibrils to be composed of successively layered β-sheets at the molecular level, which make it possible for (β-strand) segments to directly interact with, and extend the β-sheets.54; 55 Nucleation and oligomer formation at this stage may resemble a ‘molten globule’56 which has native-like secondary structures; but without a tightly packed protein interior, whose formation may follow via conformational selection.31 Nucleation polymerization can be viewed as multiple such molecular recognition processes which allow fast seed growth (Figure 2, top panel).57 In the prion protein, localized small sequence segments were observed to be able to initiate distinct intermolecular recognition events that drive the formation of different amyloid assemblies. These segments control the nucleation and templating of soluble protein structures, catalyzing their transformation into prion amyloid strains.58 Further, templating can take place even in the earliest events of aggregate nucleation.56
Figure 2.
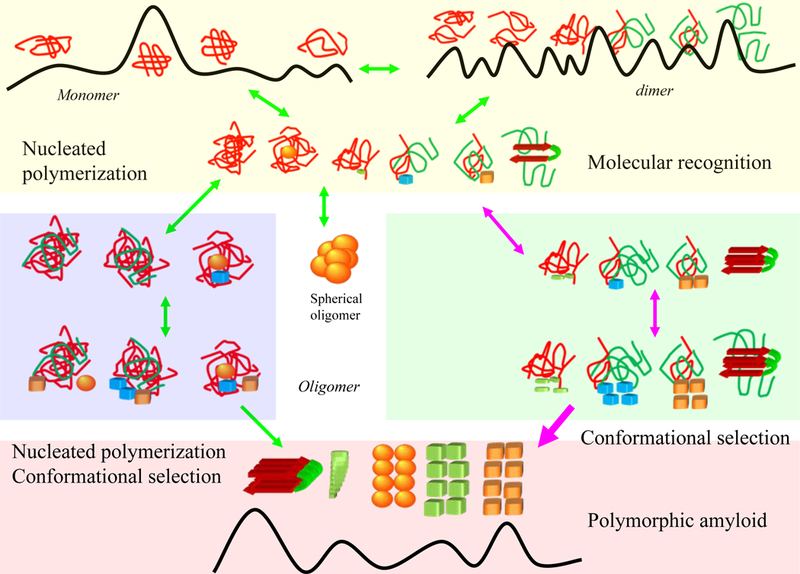
Conformational selection mechanism may operate in the early and late stages of amyloid polymerization. The top yellow box highlights that the initial perturbation or unfolding of the native protein structure can lead to the formation of small oligomers (top yellow box); the formation of nucleation site for further polymerization may take long time for total disordered monomers, with many rounds of association-disassociation events. Partially unfolded proteins, as in the case of domain swapping, may accelerate the formation through direct molecular recognition. The middle panels relate to the formation of larger oligomers. The blue box is the nucleation polymerization pathway and the green box depicts molecular recognition. The middle panel also highlights the formation of spherical particles. In the bottom panel, molecular recognition dominates fibril growth, with possibly leading to polymorphic amyloid states. Molecular recognition takes place via mutual conformation selection and population shift.
Some amyloidogenic proteins form amyloids near their native states.59; 60 S. solfataricus acylphosphatase remains in a native-like conformation which persists even when the protein forms the initial aggregates.60 The spontaneous formation of worm-like (WL) β2-microglobulin fibrils via non-nucleated pathways47 can provide an example of conformational selection in the early amyloid formation stage. A (relatively minor) perturbation of a folded β2-microglobulin can generate certain conformations that can act as templates. Deletion of the first six residues of β2-microglobulin can shift the equilibrium toward amyloidogenic conformations, which can serve as templates to select (catalyze) amyloid formation by wild-type β2-microglobulin.61 Proline cis-trans isomerization may also accelerate conformational changes of β2-microglobulin.62 It is possible that highly amyloidogenic conformations relate to domain swapping. Single domain antibody can trap a domain swapped dimer of ΔN6 β2-microglobulin.63 Domain opening of β2-microglobulin was observed in early molecular dynamics simulations, even when the disulfide bond was left intact.64 When the disulfide bond is rearranged, swapping leads to β2-microglobulin fibrillation.65 Domain swapping has been proposed to be one of the general mechanisms for amyloid formation.66; 67 Other amyloidogenic proteins, like p13suc1,68 human stefins,69 and human cystatin C70 were proposed to form amyloids via domain swapping. A recent MD simulation suggested that the aggregation of γ-crystallins associated with human cataracts may also follow a domain swapping mechanism.71
Figure 2 summarizes the initial nucleation processes. The top yellow box highlights the initial perturbation or unfolding of the native protein structure which leads to the formation of small oligomers; the blue box in the middle panels illustrates the nucleation polymerization pathway and the green box symbolizes molecular recognition. Even though two pathways (nucleation polymerization and molecular recognition) are depicted, they are not necessarily entirely distinct. In the middle panel, we also highlight the formation of spherical oligomers. Stable spherical oligomers may not convert to amyloids directly. Some oligomers can have homogenous distributions. For example, it has been estimated that the Aβ42 globulomer has exactly 12 monomers.14 Even though their formation may be under kinetic control and depend on solution conditions;72; 73 spherical oligomers may need to be thermodynamically stable and are unable to grow through conformational selection. Molecular dynamics simulations suggested that the Aβ42 dodecamer globulomer may have orthogonal β-sheets,74 although under different environment, other globulomer forms are possible.75
Figure 3 illustrates two Aβ42 globulomer models (GO1 and GO3). The correlation of the NH solvation factors of the GO3 model with experimental H/D exchange protection ratio for the Aβ42 fibril may suggest that the globulomer model has a biological role.53; 74 As shown in the figure, GO1 could be the most stable conformer for Aβ42 dodecamers, satisfying the thermodynamic stability condition. In both GO1 and GO3 the edge β-strands are blocked, indicating that further growth of the globulomer beyond dodecamer is unlikely. Molecular recognition may play a role in the initial globulomer formation, because the Aβ42 globulomers can be promoted by fatty acids.14 The orthogonal β−sheets conformation in globulomer models (GO1 and GO3) resemble the fatty acids proteins binding motif (Figure 4III), and it is possible that binding between fatty acids and the Aβ42 globulomer selectively favors orthogonal β−sheet conformations.
Figure 3.
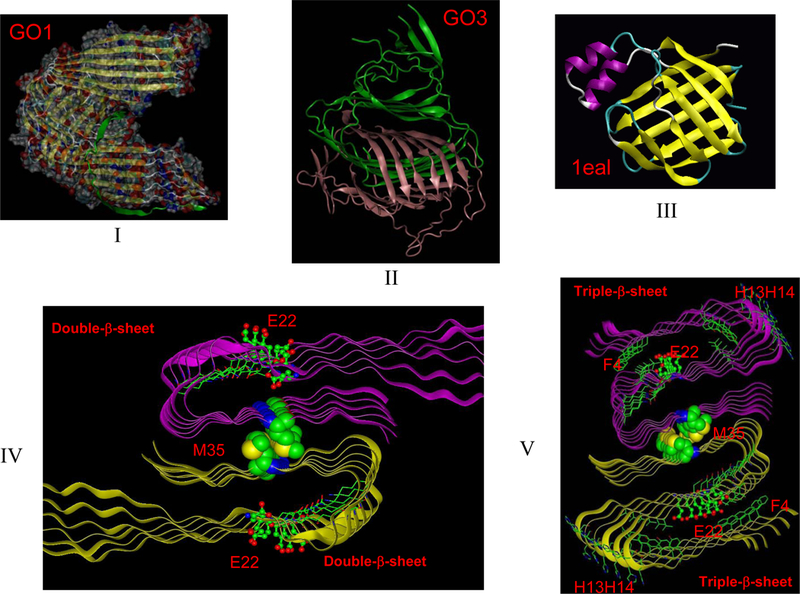
Unlike amyloid seed oligomer, globulomers cannot recruit additional monomers to extend their size through conformation selection. (I) Aβ42 globulomer model with parallel β-sheets (GO1 model). (II) Aβ42 globulomer model with antiparalle β-sheets (GO3 model). The interlocking of two hexamer β-sheets makes the globulomer unable to bind incoming β-strands. (III) The orthogonal β−sheets conformation of the globulomer models is similar to the motif of ‘classical’ fatty acids binding proteins (pdb code: 1eal). (IV). Structure of a traditional amyloid motif with double β−sheets conformation. (V) Structure of a traditional amyloid motif with a triple β−sheets conformation, which may explain E22Q Dutch mutation in Alzeheimer’s Disease53.
Figure 4.
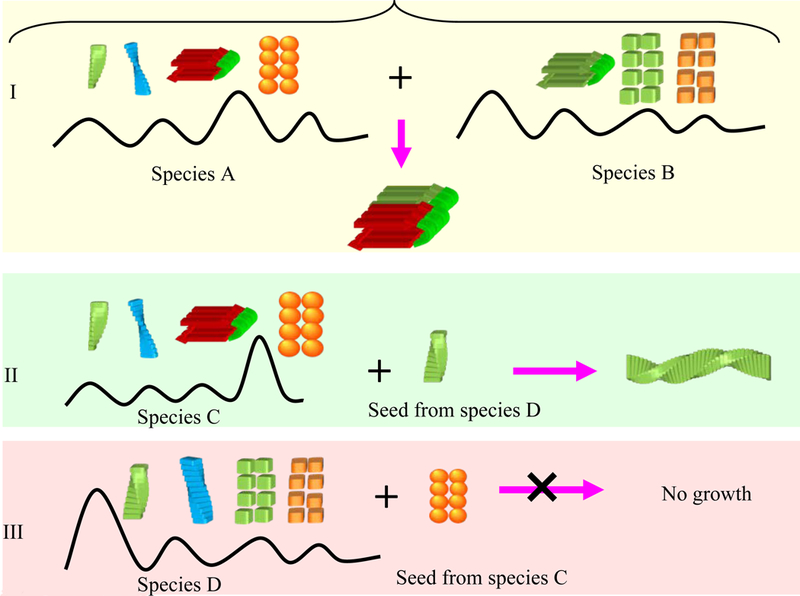
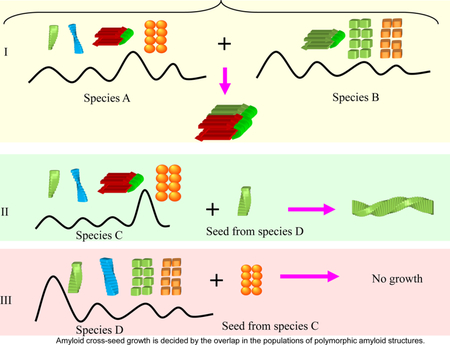
Amyloid cross-seed growth is decided by the overlap in the populations of polymorphic amyloid structures. (I) This is the case where both species A and B share a similar structure with high population. The amyloid formation should be transmittable freely between species A and B. (II) If species C has some polymorphic population of an amyloid form which is similar to the seeds from species D, then seeds from species D can recruit and catalyze the amyloid formation of species C. (III) However, because the major amyloid form of species C does not have corresponding polymorphic population in species D, the seeds from species C recruit species D to form amyloid.
Following the lag period, amyloid growth is likely to be dominated by molecular recognition (pink box, Fig 2). For Aβ42, because the C-terminal β-sheet in proto-oligomers is largely curved, which impedes fast growth along the fibril axis, the conformation of the seed may consist of two stacked U-shaped proto-oligomer β-sheets.74; 76 In Figure 3, we illustrate two fibril seed conformers of Aβ42, one with ‘traditional’ double-β-sheet conformation;7; 52; 77; 78 the other with the recently proposed triple-β-sheet conformation, which may be favored for the E22Q Dutch mutant in Alzeheimer’s Disease.53 The edge β-strands in the seed would select an approaching Aβ42 peptide with similar conformation, which would catalyze fibril formation. This conformation selection mechanism was termed a ‘dock and lock’ mechanism for the deposition of soluble monomers onto a growing amyloid.79 The ‘dock and lock’ mechanism is mediated by two distinct kinetic processes: in the first ‘dock’ (conformational selection) phase, the addition of Aβ to amyloid seeds can be reversible, while in the second ‘lock’ (minor induced fit optimization) phase, the deposited peptide becomes irreversibly associated with the template in a time-dependent manner.80 Thus, conformational selection is consistent with the energy landscape description that Massi and Straub used to explain Aβ amyloid growth by a ‘dock-lock’ mechanism79. The monomer does not necessarily have a stable β-sheet conformation; however, the selected conformation can react and reorganize at the growing fiber tip.81 Conformational selection of fibril morphology through multiple rounds of seeding is also commonly used to generate samples for structural studies and may also be important in biological processes.
4. Molecular recognition in hetero-molecular and cross-species amyloid formation
Prion strain-specific properties create prion transmission species barriers. Prions isolated from one species are often less infectious to others (i.e., incurring longer incubation time and reduced attack rates). The explanation relates to the differences in the amino acid sequence which delay the initial conversion process from the PrPc form (species A) to the PrPsc isoform (species B). Different strains may co-exist; however, with different tendencies to induce toxic effects in different organs. Prion strains have been one of the most puzzling phenomena.82 Even though it has been suggested that prion strains derive from different PrPsc isoform conformations, their structural basis has been elusive. To have specific replication properties, the conformation must be sufficiently stable and this is only possible with the oligomerized PrPsc isoforms. The compatibility between conformations of two co-existing sequences in the ensemble decides the cross-species transmission. Recent studies indicated that the stability of amyloid oligomer correlates with the conformational stability of the prion strain.83; 84 Sequence differences alone cannot selectively account for species barriers if the seeds are prepared following identical procedure,85 which can lead to amyloids with similar structures. For example, two HET-s prions with only 38% sequence identity may have highly similar structures, and consequently can efficiently cross-seed each other.86
The structural basis of prion strain variants was proposed to relate to different amyloid cores formed from different amino acid recognition segments.83 Atomic level evidence has also emerged. Three X-ray structures were solved for the same segment from human, mouse, and hamster PrP. This segment is critical for forming amyloid and confers species specificity in PrP seeding experiments. The structures reveal that as expected, sequence differences can accompany structural variability, which gives rise to transmission barriers, such as those that protect humans from acquiring bovine spongiform encephalopathy (BSE) and chronic wasting disease (CWD).87 This supports the mechanistic role of conformational selection in transmission barriers, where the barriers relate to overlap of the populations of the species (Figure 4I).88
Structural similarities can lead to amyloids composed of different sequences. The U-shaped β-sheet, which was initially found for Aβ peptides, has been observed to constitute a common amyloid structural motif, and consequently, Aβ peptides may interact with other molecules in amyloid formation. Experimentally, such cross-sequence interaction between Aβ40 and IAPP peptides has been observed, leading to amyloid hetero-assembly.89 This cross interaction may provide the molecular explanation for the apparent link between AD and type II diabetes.90 Aβ42 also interacts with prion proteins in the Alzheimer brain,91 and with tau protein, to form soluble complexes that may promote self-aggregation, in both cases into the insoluble forms observed in Alzheimer’s.92 A computational study indicated that Aβ42 may interact with three domains in the tau protein, the R2, R3, and R4 repeats; all with high β-structure propensity. Among these, Aβ42 oligomers are likely to interact with the R2 domain to form a stable complex with better alignment in the turn region and the β-structure domain93. Further, it is known that there is synergistic fibrillation between tau and the α-synuclein protein.94 However, it still remains to be seen if Aβ42 or Aβ40 can trigger amyloid formation of α-synuclein. The high potential of the Aβ42 peptide to induce other proteins to form amyloids makes us echo the question ‘is Alzeimer’s disease infectious?’95
It is interesting that cross amyloid seeding may be a ‘one way street’ in some cases. The tau protein exists in two major isoforms: one with four microtubule binding repeats; the other without the second repeat. A peptide construct with all four repeats is called K18 peptide, and the construct with three repeats (R1, R3 and R4) is called K19. K18 and K19 can mix and co-assemble into heterogeneous filaments.96 However, when mixing preformed seeds from individual solutions of K18 or K19, the K19 seeds can recruit both K19 and K18 to form amyloids, while K18 seeds can only recruit K18, but not K19.97 The asymmetric barrier between K18 and K19 indicates a population-dependent88 cross-species transmission of amyloid structure. As shown in Figure 4II, if the polymorphic amyloid population of species C has some forms which are similar to the seeds from species D, then seeds from species D can recruit and catalyze amyloid formation of species C. However, because the major amyloid form of species C does not have corresponding polymorphic population in species D, the seeds from species C cannot recruit species D to form amyloids. The K18 and K19 tau isoforms might be exactly such a case, which indicates that a sequence and structural compatibility with an amyloid structure with higher population will eventually control the transmission rate. This suggests that it could be more relevant to study an ensemble of amyloid motifs rather than focus on one specific structure.
Conclusions
The ‘central dogma’ of amyloid formation has long held that aggregation takes place through non-specific backbone hydrogen bonding interactions which are common to all peptides and proteins. However, when we consider that the β-pleated sheet structure, which is the building block of amyloid fibers, is the thermodynamically most stable supramolecular arrangement of all the possible peptide dimers and oligomers,98 and the broadly heterogeneous, polymorphic free energy landscape of amyloid species, these nonspecific hydrogen bonds may well be an integrated reflection across all such β-sheet containing species. These generic interactions do not explain the species barriers. It is challenging to figure out the key interactions and their organization which would allow seeding and cross-seeding (Figure 4), and currently it is still unclear whether a quantum mechanical treatment98 would be needed, or a classical molecular dynamics approach99; 100 would already be able to obtain coherent clues. Protein conformational dynamics and transformation into amyloid species are built into the amino acid sequences. The high energy fluctuations in water make the protein exist as an ensemble of states. A change in the solvation environment, mutational events, binding of small molecules,101 or a shift in the biological balance in the human body can lead to nonnative, populated protein conformations. These conformations may be selected to form polymeric aggregates which underlie the molecular mechanisms for many conformational diseases.
Selective macromolecular recognition governs biological processes; and conformational selection and population shift with subsequent, minor, optimization is the key recognition mechanism.102 The fundamental role played by molecular recognition of amyloid surfaces103 was recently highlighted in the case of a B10 antibody fragment, which displays conformation-specific binding to amyloid fibrils. Remarkably, among the highly variable amyloid surfaces present in the ensemble, B10 binds amyloid fibrils from D- or L-amino acid peptides and non-proteinaceous biopolymers which present highly regular and anionic surface properties, thus illustrating discrimination of distinct fibril populations. These data establish that B10 binding does not depend on an amyloid-specific or protein-specific backbone structure. Instead, it involves the recognition of a highly regular and anionic surface pattern.104 Even though it was suggested that conformational selection could be a mechanism for prion infection, the molecular and atomic underpinnings for this earlier proposal were missing. In this review, we address molecular recognition events in key stages in amyloid formation: nucleation polymerization and seeded growth. We further relate these events to prion strain variance, infectivity and cross-species transmission. We suggest that prion behavior is not unique in this sense, as recently shown by cross-seeding of tau protein repeats isoforms by Margittai and his colleagues.97 The asymmetric cross-seeding observed for the tau repeats constructs underscores the generality of this concept. Cross seeding of amyloids is controlled by selective recognition, where higher population will eventually govern the transmission rate. Even though two species may present similar amyloid organization, the differences in their dominant and most populated conformers may be responsible for the asymmetric transmission barrier. The challenge is to figure out the recognition sequences and motifs; the prevailing populations; and the range of their conformational states. The potential occurrence of cross seeding in disease, in addition to prion and tau proteins, as also for example for Aβ and IAPP,90 and albumin,105 if the species populate the same organs or environment, emphasizes these problems. The highly complex free energy landscapes of amyloids, the enormously heterogeneous population which can easily shift in experiment and under different simulation protocols, make these studies difficult. Further, subtle differences in the conformations and their compatibility can lead to different outcomes. The sequence and structural basis of molecular recognition mechanisms in amyloid formation and propagation follow the principles of functional processes in the cell. It has been suggested that within a set of select proteins, prion-like transmission of biological information can arise spontaneously; yet specifically. These metastable proteins can be intricately linked to stress-response pathways.106 Together, these critical aspects of amyloid behavior challenge our imagination.
Highlight:
Conformational selection is the key mechanism in biomolecular recognition.
Mayloid seed formation may be classified by the type of association events that take place: random nucleation coupled with molecular recognition followed by rearrangement; and directly via conformational selection without nucleation.
Amyloid growth is similarly a molecular recognition event. Cross seeding of amyloid species is also governed by conformational selection of compatible (complementary) states.
Acknowledgements
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carrell RW & Lomas DA (1997). Conformational disease. Lancet 350, 134–8. [DOI] [PubMed] [Google Scholar]
- 2.Kirkitadze MD, Bitan G & Teplow DB (2002). Paradigm shifts in Alzheimer’s disease and other neuro degenerative disorders: The emerging role of oligomeric assemblies. Journal of Neuroscience Research 69, 567–577. [DOI] [PubMed] [Google Scholar]
- 3.Walsh DM & Selkoe DJ (2004). Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept Lett 11, 213–28. [DOI] [PubMed] [Google Scholar]
- 4.Hardy J & Selkoe DJ (2002). Medicine - The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- 5.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW & Glabe CG (2003). Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489. [DOI] [PubMed] [Google Scholar]
- 6.Bitan G (2006). Structural study of metastable amyloidogenic protein oligomers by photo-induced cross-linking of unmodified proteins. Methods Enzymol 413, 217–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D & Riek R (2005). 3D structure of Alzheimer’s amyloid-beta(1–42) fibrils. Proc Natl Acad Sci U S A 102, 17342–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshiike Y, Akagi T & Takashima A (2007). Surface structure of amyloid-beta fibrils contributes to cytotoxicity. Biochemistry 46, 9805–12. [DOI] [PubMed] [Google Scholar]
- 9.Xue WF, Hellewell AL, Gosal WS, Homans SW, Hewitt EW & Radford SE (2009). Fibril fragmentation enhances amyloid cytotoxicity. J Biol Chem 284, 34272–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, Margol L, Wu J, Breydo L, Thompson JL, Rasool S, Gurlo T, Butler P & Glabe CG (2007). Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pitschke M, Prior R, Haupt M & Riesner D (1998). Detection of single amyloid beta-protein aggregates in the cerebrospinal fluid of Alzheimer’s patients by fluorescence correlation spectroscopy. Nat Med 4, 832–4. [DOI] [PubMed] [Google Scholar]
- 12.Miller Y, Ma B and Nussinov R (2010). Polymorphism in Alzheimer Aβ amyloid organization reflects conformational selection in a rugged energy landscape. Chem. Review 110, 4820–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA & Klein WL (1998). Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A 95, 6448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barghorn S, Nimmrich V, Striebinger A, Krantz C, Keller P, Janson B, Bahr M, Schmidt M, Bitner RS, Harlan J, Barlow E, Ebert U & Hillen H (2005). Globular amyloid beta-peptide oligomer - a homogenous and stable neuropathological protein in Alzheimer’s disease. J Neurochem 95, 834–47. [DOI] [PubMed] [Google Scholar]
- 15.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M & Ashe KH (2006). A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440, 352–7. [DOI] [PubMed] [Google Scholar]
- 16.Hoshi M, Sato M, Matsumoto S, Noguchi A, Yasutake K, Yoshida N & Sato K (2003). Spherical aggregates of beta-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3beta. Proc Natl Acad Sci U S A 100, 6370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Noguchi A, Matsumura S, Dezawa M, Tada M, Yanazawa M, Ito A, Akioka M, Kikuchi S, Sato M, Ideno S, Noda M, Fukunari A, Muramatsu SI, Itokazu Y, Sato K, Takahashi H, Teplow DB, Nabeshima YI, Kakita A, Imahori K & Hoshi M (2009). Isolation and characterization of patient-derived, toxic, high-mass amyloid {beta}-protein (A{beta}) assembly from Alzheimer’s disease brains. J Biol Chem [DOI] [PMC free article] [PubMed]
- 18.Eichner T & Radford SE (2011). A diversity of assembly mechanisms of a generic amyloid fold. Mol Cell 43, 8–18. [DOI] [PubMed] [Google Scholar]
- 19.Ansari A, Berendzen J, Bowne SF, Frauenfelder H, Iben IET, Sauke TB, Shyamsunder E & Young RD (1985). Protein States and Protein Quakes. Proceedings of the National Academy of Sciences of the United States of America 82, 5000–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller DW & Dill KA (1997). Ligand binding to proteins: the binding landscape model. Protein Sci 6, 2166–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dill KA & Chan HS (1997). From Levinthal to pathways to funnels. Nature Structural Biology 4, 10–19. [DOI] [PubMed] [Google Scholar]
- 22.Ohmine I, Tanaka H & Wolynes PG (1988). Large local energy fluctuations in water .2. cooperative motions and fluctuations. Journal of Chemical Physics 89, 5852–5860. [Google Scholar]
- 23.Koshland DE (1958). Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc Natl Acad Sci U S A 44, 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma B, Kumar S, Tsai CJ & Nussinov R (1999). Folding funnels and binding mechanisms. Protein Eng 12, 713–20. [DOI] [PubMed] [Google Scholar]
- 25.Tsai CJ, Kumar S, Ma B & Nussinov R (1999). Folding funnels, binding funnels, and protein function. Protein Sci 8, 1181–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsai CJ, Ma B & Nussinov R (1999). Folding and binding cascades: shifts in energy landscapes. Proc Natl Acad Sci U S A 96, 9970–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar S, Ma B, Tsai CJ, Sinha N & Nussinov R (2000). Folding and binding cascades: dynamic landscapes and population shifts. Protein Sci 9, 10–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boher DD, Nussinov R & Wright PE (2009). The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol 5, 789–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lampe JN, Brandman R, Sivaramakrishnan S & de Montellano PR (2010). Two-dimensional NMR and all-atom molecular dynamics of cytochrome P450 CYP119 reveal hidden conformational substates. J Biol Chem 285, 9594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henzler-Wildman KA, Lei M, Thai V, Kerns SJ, Karplus M & Kern D (2007). A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 450, 913–6. [DOI] [PubMed] [Google Scholar]
- 31.Kjaergaard M, Teilum K & Poulsen FM (2010). Conformational selection in the molten globule state of the nuclear coactivator binding domain of CBP. Proc Natl Acad Sci U S A 107, 12535–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma B, Kumar S, Tsai CJ, Hu Z & Nussinov R (2000). Transition-state ensemble in enzyme catalysis: possibility, reality, or necessity? J Theor Biol 203, 383–97. [DOI] [PubMed] [Google Scholar]
- 33.Ma B & Nussinov R (2010). Enzyme dynamics point to stepwise conformational selection in catalysis. Curr Opin Chem Biol 14, 652–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan Y, Tsai CJ, Ma B & Nussinov R (2010). Mechanisms of transcription factor selectivity. Trends Genet 26, 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma B, Tsai CJ, Pan Y & Nussinov R (2010). Why does binding of proteins to DNA or proteins to proteins not necessarily spell function? ACS Chem Biol 5, 265–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuan P, Liang K, Ma B, Zheng N, Nussinov R & Huang J (2011). Multiple-targeting and conformational selection in the estrogen receptor: computation and experiment. Chem Biol Drug Des 78, 137–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsai CJ, Ma BY, Sham YY, Kumar S & Nussinov R (2001). Structured disorder and conformational selection. Proteins-Structure Function and Genetics 44, 418–427. [DOI] [PubMed] [Google Scholar]
- 38.Gunasekaran K, Tsai CJ, Kumar S, Zanuy D & Nussinov R (2003). Extended disordered proteins: targeting function with less scaffold. Trends in Biochemical Sciences 28, 81–85. [DOI] [PubMed] [Google Scholar]
- 39.Eliezer D (2009). Biophysical characterization of intrinsically disordered proteins. Curr Opin Struct Biol 19, 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma B, Shatsky M, Wolfson HJ & Nussinov R (2002). Multiple diverse ligands binding at a single protein site: a matter of pre-existing populations. Protein Sci 11, 184–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma B, Tsai CJ, Haliloglu T & Nussinov R (2011). Dynamic allostery: linkers are not merely flexible. Structure 19, 907–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mackereth CD, Madl T, Bonnal S, Simon B, Zanier K, Gasch A, Rybin V, Valcarcel J & Sattler M (2011). Multi-domain conformational selection underlies pre-mRNA splicing regulation by U2AF. Nature 475, 408–11. [DOI] [PubMed] [Google Scholar]
- 43.Schymkowitz JW, Rousseau F, Wilkinson HR, Friedler A & Itzhaki LS (2001). Observation of signal transduction in three-dimensional domain swapping. Nat Struct Biol 8, 888–92. [DOI] [PubMed] [Google Scholar]
- 44.Pan Y, Tsai CJ, Ma B & Nussinov R (2009). How do transcription factors select specific binding sites in the genome? Nat Struct Mol Biol 16, 1118–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Konuma T, Chatani E, Yagi M, Sakurai K, Ikegami T, Naiki H & Goto Y (2011). Kinetic intermediates of beta(2)-microglobulin fibril elongation probed by pulse-labeling H/D exchange combined with NMR analysis. J Mol Biol 405, 851–62. [DOI] [PubMed] [Google Scholar]
- 46.Debelouchina GT, Platt GW, Bayro MJ, Radford SE & Griffin RG (2010). Intermolecular Alignment in beta(2)-Microglobulin Amyloid Fibrils. J Am Chem Soc [DOI] [PMC free article] [PubMed]
- 47.Smith DP, Woods LA, Radford SE & Ashcroft AE (2011). Structure and Dynamics of Oligomeric Intermediates in beta(2)-Microglobulin Self-Assembly. Biophys J 101, 1238–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pappu RV, Wang X, Vitalis A & Crick SL (2008). A polymer physics perspective on driving forces and mechanisms for protein aggregation. Arch Biochem Biophys 469, 132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qian J, Zhang M, Manners I & Winnik MA (2010). Nanofiber micelles from the self-assembly of block copolymers. Trends Biotechnol 28, 84–92. [DOI] [PubMed] [Google Scholar]
- 50.Jan A, Adolfsson O, Allaman I, Buccarello AL, Magistretti PJ, Pfeifer A, Muhs A & Lashuel HA (2011). Abeta42 neurotoxicity is mediated by ongoing nucleated polymerization process rather than by discrete Abeta42 species. J Biol Chem 286, 8585–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ruschak AM & Miranker AD (2009). The role of prefibrillar structures in the assembly of a peptide amyloid. J Mol Biol 393, 214–26. [DOI] [PubMed] [Google Scholar]
- 52.Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, Elliott JI, Van Nostrand WE & Smith SO (2010). Structural conversion of neurotoxic amyloid-beta(1–42) oligomers to fibrils. Nat Struct Mol Biol 17, 561–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ma B & Nussinov R (2011). Polymorphic triple beta-sheet structures contribute to amide H/D exchange protection in Alzheimer’s amyloid beta42. J Biol Chem [DOI] [PMC free article] [PubMed]
- 54.Cabriolu R & Auer S (2011). Amyloid fibrillation kinetics: insight from atomistic nucleation theory. J Mol Biol 411, 275–85. [DOI] [PubMed] [Google Scholar]
- 55.Kashchiev D & Auer S (2010). Nucleation of amyloid fibrils. J Chem Phys 132, 215101. [DOI] [PubMed] [Google Scholar]
- 56.Liang Y, Lynn DG & Berland KM (2010). Direct observation of nucleation and growth in amyloid self-assembly. J Am Chem Soc 132, 6306–8. [DOI] [PubMed] [Google Scholar]
- 57.Cohen SI, Vendruscolo M, Dobson CM & Knowles TP (2011). Nucleated polymerization with secondary pathways. II. Determination of self-consistent solutions to growth processes described by non-linear master equations. J Chem Phys 135, 065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tessier PM & Lindquist S (2007). Prion recognition elements govern nucleation, strain specificity and species barriers. Nature 447, 556–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Corazza A, Rennella E, Schanda P, Mimmi MC, Cutuil T, Raimondi S, Giorgetti S, Fogolari F, Viglino P, Frydman L, Gal M, Bellotti V, Brutscher B & Esposito G (2010). Native-unlike long-lived intermediates along the folding pathway of the amyloidogenic protein beta2-microglobulin revealed by real-time two-dimensional NMR. J Biol Chem 285, 5827–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pagano K, Bemporad F, Fogolari F, Esposito G, Viglino P, Chiti F & Corazza A (2010). Structural and dynamics characteristics of acylphosphatase from Sulfolobus solfataricus in the monomeric state and in the initial native-like aggregates. J Biol Chem 285, 14689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eichner T, Kalverda AP, Thompson GS, Homans SW & Radford SE (2011). Conformational conversion during amyloid formation at atomic resolution. Mol Cell 41, 161–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fogolari F, Corazza A, Varini N, Rotter M, Gumral D, Codutti L, Rennella E, Viglino P, Bellotti V & Esposito G (2011). Molecular dynamics simulation of beta-microglobulin in denaturing and stabilizing conditions. Proteins 79, 986–1001. [DOI] [PubMed] [Google Scholar]
- 63.Domanska K, Vanderhaegen S, Srinivasan V, Pardon E, Dupeux F, Marquez JA, Giorgetti S, Stoppini M, Wyns L, Bellotti V & Steyaert J (2011). Atomic structure of a nanobody-trapped domain-swapped dimer of an amyloidogenic beta2-microglobulin variant. Proc Natl Acad Sci U S A 108, 1314–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ma B & Nussinov R (2003). Molecular dynamics simulations of the unfolding of β2-microglobulin and its variants. Protein Eng 16, 561–75. [DOI] [PubMed] [Google Scholar]
- 65.Liu C, Sawaya MR & Eisenberg D (2011). beta-microglobulin forms three-dimensional domain-swapped amyloid fibrils with disulfide linkages. Nat Struct Mol Biol 18, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nelson R & Eisenberg D (2006). Recent atomic models of amyloid fibril structure. Curr Opin Struct Biol 16, 260–5. [DOI] [PubMed] [Google Scholar]
- 67.Zerovnik E, Stoka V, Mirtic A, Guncar G, Grdadolnik J, Staniforth RA, Turk D & Turk V (2011). Mechanisms of amyloid fibril formation--focus on domain-swapping. FEBS J 278, 2263–82. [DOI] [PubMed] [Google Scholar]
- 68.Rousseau F, Wilkinson H, Villanueva J, Serrano L, Schymkowitz JW & Itzhaki LS (2006). Domain swapping in p13suc1 results in formation of native-like, cytotoxic aggregates. J Mol Biol 363, 496–505. [DOI] [PubMed] [Google Scholar]
- 69.Zerovnik E, Staniforth RA & Turk D (2010). Amyloid fibril formation by human stefins: Structure, mechanism & putative functions. Biochimie 92, 1597–607. [DOI] [PubMed] [Google Scholar]
- 70.Wahlbom M, Wang X, Lindstrom V, Carlemalm E, Jaskolski M & Grubb, (2007). Fibrillogenic oligomers of human cystatin C are formed by propagated domain swapping. J Biol Chem 282, 18318–26. [DOI] [PubMed] [Google Scholar]
- 71.Das P, King JA & Zhou R (2011). Aggregation of gamma-crystallins associated with human cataracts via domain swapping at the C-terminal beta-strands. Proc Natl Acad Sci U S A 108, 10514–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sahoo B, Nag S, Sengupta P & Maiti S (2009). On the stability of the soluble amyloid aggregates. Biophys J 97, 1454–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Krebs MR, Domike KR & Donald AM (2009). Protein aggregation: more than just fibrils. Biochem Soc Trans 37, 682–6. [DOI] [PubMed] [Google Scholar]
- 74.Ma B & Nussinov R (2010). Polymorphic C-terminal {beta}-sheet interactions determine the formation of fibril or ADDL-like globulomer for the Alzheimer A{beta}42 dodecamer. J Biol Chem 286, 37102–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yu X & Zheng J (2011). Polymorphic structures of Alzheimer’s beta-amyloid globulomers. PLoS One 6, e20575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu JW, Breydo L, Isas JM, Lee J, Kuznetsov YG, Langen R & Glabe C (2010). Fibrillar oligomers nucleate the oligomerization of monomeric amyloid beta but do not seed fibril formation. J Biol Chem 285, 6071–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ma B & Nussinov R (2006). Simulations as analytical tools to understand protein aggregation and predict amyloid conformation. Curr Opin Chem Biol 10, 445–52. [DOI] [PubMed] [Google Scholar]
- 78.Petkova AT, Yau WM & Tycko R (2006). Experimental constraints on quaternary structure in Alzheimer’s beta-amyloid fibrils. Biochemistry 45, 498–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Massi F & Straub JE (2001). Energy landscape theory for Alzheimer’s amyloid beta-peptide fibril elongation. Proteins 42, 217–29. [DOI] [PubMed] [Google Scholar]
- 80.Esler WP, Stimson ER, Jennings JM, Vinters HV, Ghilardi JR, Lee JP, Mantyh PW & Maggio JE (2000). Alzheimer’s disease amyloid propagation by a template-dependent dock-lock mechanism. Biochemistry 39, 6288–95. [DOI] [PubMed] [Google Scholar]
- 81.Pease LF 3rd, Sorci M, Guha S, Tsai DH, Zachariah MR, Tarlov MJ & Belfort G (2010). Probing the nucleus model for oligomer formation during insulin amyloid fibrillogenesis. Biophys J 99, 3979–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aguzzi A, Heikenwalder M & Polymenidou M (2007). Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol 8, 552–61. [DOI] [PubMed] [Google Scholar]
- 83.Toyama BH, Kelly MJ, Gross JD & Weissman JS (2007). The structural basis of yeast prion strain variants. Nature 449, 233–7. [DOI] [PubMed] [Google Scholar]
- 84.Sun Y, Makarava N, Lee CI, Laksanalamai P, Robb FT & Baskakov IV (2008). Conformational stability of PrP amyloid fibrils controls their smallest possible fragment size. J Mol Biol 376, 1155–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Makarava N, Lee CI, Ostapchenko VG & Baskakov IV (2007). Highly promiscuous nature of prion polymerization. J Biol Chem 282, 36704–13. [DOI] [PubMed] [Google Scholar]
- 86.Wasmer C, Zimmer A, Sabate R, Soragni A, Saupe SJ, Ritter C & Meier BH (2010). Structural similarity between the prion domain of HET-s and a homologue can explain amyloid cross-seeding in spite of limited sequence identity. J Mol Biol 402, 311–25. [DOI] [PubMed] [Google Scholar]
- 87.Apostol MI, Wiltzius JJ, Sawaya MR, Cascio D & Eisenberg D (2011). Atomic structures suggest determinants of transmission barriers in mammalian prion disease. Biochemistry 50, 2456–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Collinge J & Clarke AR (2007). A general model of prion strains and their pathogenicity. Science 318, 930–6. [DOI] [PubMed] [Google Scholar]
- 89.Andreetto E, Yan LM, Tatarek-Nossol M, Velkova A, Frank R & Kapurniotu A (2010). Identification of hot regions of the Abeta-IAPP interaction interface as high-affinity binding sites in both cross- and self-association. Angew Chem Int Ed Engl 49, 3081–5. [DOI] [PubMed] [Google Scholar]
- 90.Nicolls MR (2004). The clinical and biological relationship between Type II diabetes mellitus and Alzheimer’s disease. Curr Alzheimer Res 1, 47–54. [DOI] [PubMed] [Google Scholar]
- 91.Zou WQ, Xiao X, Yuan J, Puoti G, Fujioka H, Wang X, Richardson S, Zhou X, Zou R, Li S, Zhu X, McGeer PL, McGeehan J, Kneale G, Rincon-Limas DE, Fernandez-Funez P, Lee HG, Smith MA, Petersen RB & Guo JP (2011). Amyloid-beta42 interacts mainly with insoluble prion protein in the Alzheimer brain. J Biol Chem 286, 15095–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guo JP, Arai T, Miklossy J & McGeer PL (2006). Abeta and tau form soluble complexes that may promote self aggregation of both into the insoluble forms observed in Alzheimer’s disease. Proc Natl Acad Sci U S A 103, 1953–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Miller Y, Ma B & Nussinov R (2011). Synergistic interactions between repeats in tau protein and Abeta amyloids may be responsible for accelerated aggregation via polymorphic states. Biochemistry 50, 5172–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ & Lee VM (2003). Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300, 636–40. [DOI] [PubMed] [Google Scholar]
- 95.Reis HJ, Mukhamedyarov MA, Rizvanov AA & Palotas A (2010). A new story about an old guy: is Alzheimer’s disease infectious? Neurodegener Dis 7, 272–8. [DOI] [PubMed] [Google Scholar]
- 96.Siddiqua A & Margittai M (2010). Three- and four-repeat Tau coassemble into heterogeneous filaments: an implication for Alzheimer disease. J Biol Chem 285, 37920–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dinkel PD, Siddiqua A, Huynh H, Shah M & Margittai M (2011). Variations in filament conformation dictate seeding barrier between three- and four-repeat tau. Biochemistry 50, 4330–6. [DOI] [PubMed] [Google Scholar]
- 98.Perczel A, Hudaky P & Palfi VK (2007). Dead-end street of protein folding: thermodynamic rationale of amyloid fibril formation. J Am Chem Soc 129, 14959–65. [DOI] [PubMed] [Google Scholar]
- 99.Berryman JT, Radford SE & Harris SA (2011). Systematic examination of polymorphism in amyloid fibrils by molecular-dynamics simulation. Biophys J 100, 2234–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zanuy D & Nussinov R (2003). The sequence dependence of fiber organization. A comparative molecular dynamics study of the islet amyloid polypeptide segments 22–27 and 22–29. J Mol Biol 329, 565–84. [DOI] [PubMed] [Google Scholar]
- 101.Ladiwala AR, Lin JC, Bale SS, Marcelino-Cruz AM, Bhattacharya M, Dordick JS & Tessier PM (2010). Resveratrol selectively remodels soluble oligomers and fibrils of amyloid Abeta into off-pathway conformers. J Biol Chem 285, 24228–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Csermely P, Palotai R & Nussinov R (2010). Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem Sci 35, 539–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Glabe CG (2004). Conformation-dependent antibodies target diseases of protein misfolding. Trends Biochem Sci 29, 542–7. [DOI] [PubMed] [Google Scholar]
- 104.Haupt C, Bereza M, Kumar ST, Kieninger B, Morgado I, Hortschansky P, Fritz G, Rocken C, Horn U & Fandrich M (2011). Pattern recognition with a fibril-specific antibody fragment reveals the surface variability of natural amyloid fibrils. J Mol Biol 408, 529–40. [DOI] [PubMed] [Google Scholar]
- 105.Milojevic J & Melacini G (2011). Stoichiometry and affinity of the human serum albumin-Alzheimer’s Abeta peptide interactions. Biophys J 100, 183–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Alberti S, Halfmann R, King O, Kapila A & Lindquist S (2009). A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 137, 146–58. [DOI] [PMC free article] [PubMed] [Google Scholar]