

Heart failure is one of the major causes of death worldwide. Despite the development of several treatments for heart failure, such as β-blocker, angiotensin-converting enzyme inhibitor, and mineralocorticoid receptor antagonist, most severe heart failures irreversibly progress and cannot be cured without heart transplantation. In the excitation-contraction coupling in cardiomyocytes, Ca2+ reuptake into the sarcoendoplasmic reticulum (SR) through SERCA2a (SR Ca2+ ATPase 2a) is a key process in the relaxation of cardiomyocytes and in the proper storage of SR Ca2+ content for the next contraction.1 SERCA2a is downregulated in heart failure, and Ca2+ reuptake to SR is reduced in diseased cardiomyocytes. As a result, excitation-contraction coupling is impaired in systolic and diastolic phases, causing a vicious spiral of SERCA2a decrement and heart failure. Hence, correcting impaired intracellular Ca2+ homeostasis could be a therapeutic target. In heart failure animal models, overexpression of SERCA2a improved cardiac function,2 and gene therapy delivering the SERCA2a gene for heart failure treatment is expected to be highly successful in humans. Clinical studies, CUPID 13 and CUPID 2 (Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease),4 have already been conducted using adeno-associated virus carrying a SERCA2a gene, but the improvement in prognosis has not been clearly shown. This inconsistency in results may be attributable to the difference in study designs and factors such as patient characteristics, virus dosage, and the gene delivery system. Thus, these factors should be adjusted in future clinical trials. Moreover, a drug that could directly affect the SERCA2a function is also expected to be developed. SERCA2a activity is finely regulated by several mechanisms, and the modulation of SERCA2a activity is critical for efficient Ca2+ reuptake into SR. PLN (phospholamban) is highly expressed in cardiomyocytes and reversibly inhibits SERCA2a activity. Adeno-associated virus–mediated Pln knockdown rescued heart failure in an animal model.5 The activity and stability of SERCA2a is also regulated by post-translational modification, including the SUMO1 (small ubiquitin-related modifier 1). The level of SUMO1 is suppressed in heart failure, and Sumo1 overexpression rescued heart failure in an animal model.6 Those experiments lead to a better understanding of SERCA2a function; however, those findings have not yet been evaluated in humans. Further elucidation of the detailed regulatory mechanism of SERCA2a would lead to novel therapeutic concepts and contribute to drug development.
Article, see p 712
Cardiomyocytes respond to external stimuli by activating signal transduction cascades by enzymatic modifications, such as phosphorylation, acetylation, ubiquitination, and glycosylation. Among them, phosphorylation has a central role in cellular signaling, and kinases can be therapeutic targets because they can be controlled by chemical compounds. MLCKs (myosin light-chain kinases) are expressed in several types of myocytes, such as smooth muscle cells, skeletal muscle cells, and cardiac myocytes, and have an important role in cardiomyocyte function, including sarcomere organization.7 SPEG (striated muscle preferentially expressed protein kinase)—a serine/threonine kinase and member of MLCKs. SPEG is highly expressed in the developing heart. SPEG knockout mice showed dilated cardiomyopathy–like phenotype and perinatal death.8 The JMC (junctional membrane complex) between the plasma membrane and SR is an important structure for excitation-contraction coupling in cardiomyocytes. SPEG is associated with JMC proteins, and the expression of SPEG decreases in patients with heart failure. SPEG phosphorylates the JMC protein, JPH2 (junctophilin-2), and is essential for JMC integrity.9 Adult-onset cardiac-specific SPEG knockout mice showed a dilated cardiomyopathy–like phenotype and died. Moreover, SPEG mutation in humans also leads to centronuclear myopathy with dilated cardiomyopathy.10 The evidence presented clearly indicates that SPEG plays a crucial role in heart homeostasis, but the precise mechanism remains elusive.
With regard to this issue, Quan et al11 evaluated the function of SPEG by the identification of SPEG-binding partners and focused on the physical and functional interactions between SPEG and SERCA2a. Mass spectrometry analysis revealed that SPEG is physically associated with SERCA2a. Coexpression experiments showed that SPEG augments SERCA2a function, accelerating Ca2+ reuptake into SR. These data suggested that SPEG would directly affect the SERCA2a function, possibly through phosphorylation. SPEG has 2 serine/threonine kinase (SK) domains in the C-terminal region. SPEG increased SERCA2a oligomerization that enhances the transportation of Ca2+ to the SR that is independent of its ATPase activity.12 The authors focused on the potential phosphorylation targets of SERCA2a by SPEG. Mass spectrometry analysis identified a potential phosphorylation site of SERCA2a at Thr484. Moreover, knockdown of SPEG in rat cardiomyocytes decreased SERCA2a Thr484 phosphorylation and SERCA2a activity. Taken together, these data suggest that SPEG directly regulates SERCA2a Thr484 phosphorylation, induces SERCA2a oligomerization, and enhances SERCA2a activity. Next, the authors evaluated the function of SPEG in vivo and in adult cardiomyocytes by generating cardiac-specific SPEG knockout mice. They crossed SPEGfl/fl mice with cardiomyocyte-specific Cre expression mice, aMHC-Cre,13 producing SPEGfl/fl/αMHC-Cre. In these mice, phosphorylation of SERCA2a Thr484 was decreased, and a dilated cardiomyopathy–like phenotype was observed. As the SPEGfl/fl/aMHC-Cre mice showed cardiac dysfunction at around 2 weeks of age, the authors examined another mouse model—the adult-onset cardiac-specific SPEG knockout mice—in chronological order. They crossed SPEGfl/fl mice with cardiomyocyte-specific inducible Cre expression mice, Myh6-MerCreMer,14 producing SPEGfl/fl/Myh6-MCM, in which SPEG can be knocked out by tamoxifen injection. Thr484 phosphorylation and oligomerization of SERCA2a were significantly decreased in the heart of SPEGfl/fl/Myh6-MCM mice after tamoxifen injection, and left ventricular systolic function was apparently but gradually decreased, which suggests that SPEG may have a crucial role in regulating SERCA2a activity in vivo. Finally, the authors directly examined Ca2+ homeostasis in the heart and isolated cardiomyocytes of SPEGfl/fl/Myh6-MCM mice. The transcription factor NFAT (nuclear factor of activated T cells) is functionally regulated by Ca2+ and is involved in cardiac hypertrophy and heart failure.15 Rcan1.4 is one of the target genes of the NFAT pathway whose expression is correlated with cytosolic Ca2+. The heart of SPEGfl/fl/Myh6-MCM mice displayed normal gross morphology and function at around 4 weeks after tamoxifen treatment, but the Rcan1.4 level was increased in the heart of SPEGfl/fl/Myh6-MCM mice, suggesting that cytosolic Ca2+ was elevated. In the SPEG knockout primary cardiomyocytes, calcium reuptake into the SR was impaired at 4 weeks after tamoxifen treatment. Collectively, SPEG directly phosphorylates Thr484 SERCA2a, which induces SERCA2a oligomerization and SERCA2a activity enhancement. SPEG is indispensable for heart homeostasis in vivo, but SPEG is downregulated in heart failure. Moreover, SPEG may have a key role in the vicious spiral of SPEG decrement, SERCA2a functional impairment, and heart failure. Correcting impaired SPEG expression could, thus, be a therapeutic target.
Abnormality of Ca2+ homeostasis induces heart failure and fatal arrhythmia. This article showed that SPEG directly regulates the phosphorylation of Thr484 SERCA2a. Moreover, SPEG enhances the calcium reuptake activity of SERCA2a, and SPEG deficiency leads to heart failure in mice. SPEG is indispensable for heart homeostasis, but whether heart failure can be treated with SPEG gene transfer, SPEG activity augmentation, increased phosphorylation of Thr484 SERCA2a, or pThr484 SERCA2a mimic gene transfer remains unclear. Further investigation will facilitate the development of drugs to induce SPEG activity for heart failure treatment. The CUPID 2 trial failed to demonstrate the efficiency of gene therapy using adeno-associated virus/SERCA2a,4 but accumulating basic studies still encourage us to elucidate the undiscovered mechanisms of Ca2+ homeostasis and heart failure to perfect innovative therapies for heart failure.
Figure.
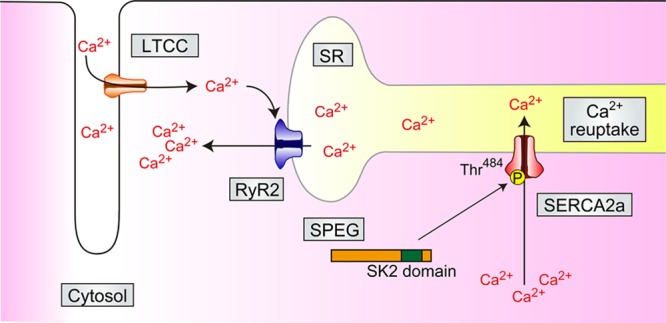
SPEG (striated muscle preferentially expressed protein kinase), which enhances calcium reuptake by SERCA2a (sarcoendoplasmic reticulum [SR] Ca2+ ATPase 2a) in cardiomyocytes. SPEG physically associates with SERCA2a, and SPEG-SK2 phosphorylates the Thr484 residue of SERCA2a. Thr484 SERCA2a facilitates SERCA2a oligomerization and enhances Ca2+ reuptake to SR. LTCC indicates L-type calcium channel; RyR2, ryanodine receptor 2; and SK2 domain, serine/threonine (Ser/Thr) kinase 2 domain.
Acknowledgments
We thank all of our laboratory members for their assistance.
Sources of Funding
This work was supported, in part, by grants from the Japan Society for the Promotion of Science (KAKENHI 16H05304).
Disclosures
K. Fukuda is a cofounder of, and has equity in, Heartseed, Inc. The other authors report no conflicts.
Footnotes
The opinions expressed in this article are not necessarily those of the editors or of the American Heart Association.
References
- 1.Kho C, Lee A, Hajjar RJ. Altered sarcoplasmic reticulum calcium cycling–targets for heart failure therapy. Nat Rev Cardiol. 2012;9:717–733. doi: 10.1038/nrcardio.2012.145. doi: 10.1038/nrcardio.2012.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Müller OJ, Lange M, Rattunde H, Lorenzen HP, Müller M, Frey N, Bittner C, Simonides W, Katus HA, Franz WM. Transgenic rat hearts overexpressing SERCA2a show improved contractility under baseline conditions and pressure overload. Cardiovasc Res. 2003;59:380–389. doi: 10.1016/s0008-6363(03)00429-2. [DOI] [PubMed] [Google Scholar]
- 3.Hajjar RJ, Zsebo K, Deckelbaum L, et al. Design of a phase ½ trial of intracoronary administration of AAV1/SERCA2a in patients with heart failure. J Card Fail. 2008;14:355–367. doi: 10.1016/j.cardfail.2008.02.005. doi: 10.1016/j.cardfail.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Desai AS, Barnard D, Bouchard A, Jaski B, Lyon AR, Pogoda JM, Rudy JJ, Zsebo KM. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet. 2016;387:1178–1186. doi: 10.1016/S0140-6736(16)00082-9. doi: 10.1016/S0140-6736(16)00082-9. [DOI] [PubMed] [Google Scholar]
- 5.Suckau L, Fechner H, Chemaly E, et al. Long-term cardiac-targeted RNA interference for the treatment of heart failure restores cardiac function and reduces pathological hypertrophy. Circulation. 2009;119:1241–1252. doi: 10.1161/CIRCULATIONAHA.108.783852. doi: 10.1161/CIRCULATIONAHA.108.783852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E, Park WJ, Hajjar RJ. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. 2011;477:601–605. doi: 10.1038/nature10407. doi: 10.1038/nature10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aoki H, Sadoshima J, Izumo S. Myosin light chain kinase mediates sarcomere organization during cardiac hypertrophy in vitro. Nat Med. 2000;6:183–188. doi: 10.1038/72287. doi: 10.1038/72287. [DOI] [PubMed] [Google Scholar]
- 8.Liu X, Ramjiganesh T, Chen YH, Chung SW, Hall SR, Schissel SL, Padera RF, Jr, Liao R, Ackerman KG, Kajstura J, Leri A, Anversa P, Yet SF, Layne MD, Perrella MA. Disruption of striated preferentially expressed gene locus leads to dilated cardiomyopathy in mice. Circulation. 2009;119:261–268. doi: 10.1161/CIRCULATIONAHA.108.799536. doi: 10.1161/CIRCULATIONAHA.108.799536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quick AP, Wang Q, Philippen LE, Barreto-Torres G, Chiang DY, Beavers D, Wang G, Khalid M, Reynolds JO, Campbell HM, Showell J, McCauley MD, Scholten A, Wehrens XH. SPEG (Striated Muscle Preferentially Expressed Protein Kinase) is essential for cardiac function by regulating junctional membrane complex activity. Circ Res. 2017;120:110–119. doi: 10.1161/CIRCRESAHA.116.309977. doi: 10.1161/CIRCRESAHA.116.309977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Agrawal PB, Pierson CR, Joshi M, et al. SPEG interacts with myotubularin, and its deficiency causes centronuclear myopathy with dilated cardiomyopathy. Am J Hum Genet. 2014;95:218–226. doi: 10.1016/j.ajhg.2014.07.004. doi: 10.1016/j.ajhg.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quan C, Li M, Du Q, Chen Q, Wang H, Campbell D, Fang L, Xue B, MacKintosh C, Gao X, Ouyang K, Wang HY, Chen S. SPEG controls calcium reuptake into the sarcoplasmic reticulum through regulating SERCA2a by its second kinase-domain. Circ Res. 2019;124:712–726. doi: 10.1161/CIRCRESAHA.118.313916. doi: 10.1161/CIRCRESAHA.118.313916. [DOI] [PubMed] [Google Scholar]
- 12.Chen LT, Yao Q, Soares TA, Squier TC, Bigelow DJ. Phospholamban modulates the functional coupling between nucleotide domains in Ca-ATPase oligomeric complexes in cardiac sarcoplasmic reticulum. Biochemistry. 2009;48:2411–2421. doi: 10.1021/bi8021526. doi: 10.1021/bi8021526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boisgérault F, Tieng V, Stolzenberg MC, Dulphy N, Khalil I, Tamouza R, Charron D, Toubert A. Differences in endogenous peptides presented by HLA-B*2705 and B*2703 allelic variants. Implications for susceptibility to spondylarthropathies. J Clin Invest. 1996;98:2764–2770. doi: 10.1172/JCI119102. doi: 10.1172/JCI119102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res. 2001;89:20–25. doi: 10.1161/hh1301.092687. [DOI] [PubMed] [Google Scholar]
- 15.Molkentin JD. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc Res. 2004;63:467–475. doi: 10.1016/j.cardiores.2004.01.021. doi: 10.1016/j.cardiores.2004.01.021. [DOI] [PubMed] [Google Scholar]