

Summary
DNA polymerase stalling activates the ATR checkpoint kinase, which in turn suppresses fork collapse and breakage. Herein, we describe use of ATR inhibition (ATRi) as a means to identify genomic sites of problematic DNA replication in murine and human cells. Over 500 high-resolution ATR-dependent sites were ascertained using two distinct methods (RPA-ChIP and BrITL). The genomic feature most strongly associated with ATR dependence was repetitive DNA that exhibited high structure-forming potential. Repeats most reliant on ATR for stability included structure-forming microsatellites, inverted retroelement repeats, and quasi-palindromic AT-rich repeats. Notably, these categories of repeats differed both in structure formation and in their ability to stimulate RPA accumulation and breakage, implying that the causes and character of replication fork collapse under ATR inhibition can vary in a DNA structure-specific manner. Collectively, these studies identify key sources of endogenous replication stress that rely on ATR for stability.
eTOC blurb:
Shastri et al. have identified new classes of difficult-to-replicate sequences in the mouse and human genomes that are highly dependent on ATR function for stability during DNA replication. Structure-forming short tandem repeats, inverted retroelements, and quasi-palindromic AT-rich repeats characterize the sites for fork collapse caused by ATR inhibition.
Graphical Abstract
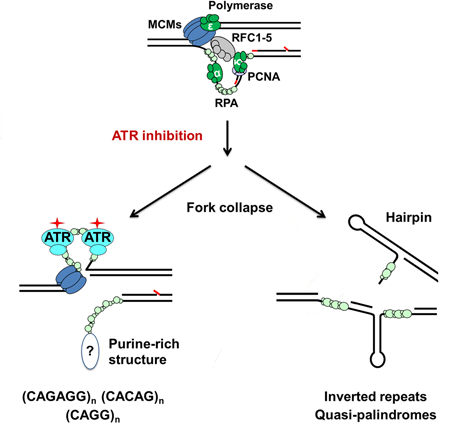
Introduction
Numerous sensory and repair networks are required to safeguard genome integrity during duplication. These networks operate when progressing replication forks encounter impediments to polymerase progression, such as damaged bases and difficult-to-replicate sequences. Replication protein A (RPA), is an immediate responder to such events by protecting the unwound template strands. RPA-coated ssDNA recruits and activates numerous DNA repair and cell cycle checkpoint regulators, including the ATR checkpoint kinase. ATR activation at stalled replication forks stabilizes these intermediates and prevents progression into M phase (Saldivar et al., 2017). However, while steady progress has been made in defining the signal transduction cascades that regulate ATR and operate downstream of it, substantially less is known about the genomic sequences that promote fork stalling and make ATR an essential gene.
While it has long been known that ATR loss of function is sufficient to cause chromosome breaks (Brown and Baltimore, 2000), pinpointing the sites of genomic breakage has primarily relied on candidate approaches. For example, ATR suppression increases chromosome breakage at common fragile sites (CFS), which were originally defined as breaks observed in mitotic cells following partial DNA synthesis inhibition (Casper et al., 2002; Glover et al., 2017). Other candidate genomic features examined for ATR dependence include expanded microsatellite repeats. CAG/CTG trinucleotide repeats, the causative feature in Huntington’s disease and Myotonic Dystrophy, form non-B DNA structures (Mirkin, 2007; Neil et al., 2017) and are stabilized by the ATR ortholog, Mec1, in S. cerevisae (Lahiri et al., 2004). A similar dependence has been reported for CGG/CCG repeats, expansion of which causes Fragile X syndrome (Entezam and Usdin, 2008). Finally, telomere repeats are reliant on ATR for stability, putatively through replication perturbation at G-quadruplexes (Johnson et al., 2008; Sfeir et al., 2009). Nevertheless, ATR-dependent stability of these repeat sequences was examined due to proven or speculated inhibitory effects on DNA replication, not through an unbiased screen for sequences that are the most reliant on ATR for stability.
Herein, we use ATR inhibition and two distinct methods (RPA-ChIP and BrITL) to detect replication fork collapse. We have defined over 500 sites of problematic replication across the mouse and human genomes. While numerous genomic and transcriptional features have been proposed to cause a dependence on ATR, we find that the element most commonly associated with fork collapse is the presence of microsatellite repeats and inverted repeats that form stable secondary structures. The repeats most reliant on ATR for stability are distinct from those previously examined and include microsatellite repeats that form purine-rich non-B DNA structures and inverted transposable elements (SINE, LINE, LTR, Alu), and quasi-palindromic AT-rich minisatellite repeats that have the potential to form stable hairpin structures. Notably, the type of structure formed by these repeats impacts the propensity to cause breakage and accumulate RPA, indicating that the character of fork collapse differs in a repeat-specific manner. These observations have defined not only new sites of problematic DNA replication that rely on ATR for stability, but also distinct categories of replication fork collapse that differ in a manner associated with the DNA structure formed. These findings indicate that such repeats may comprise part of the mechanism of action of ATR inhibitors, which have entered clinical trials as cancer therapeutics.
Results
Genome-wide identification of RPA-enriched sites following ATR inhibition
RPA accumulates at sites of replication fork collapse (Fig. 1A). To identify such sites following ATR inhibition, RPA-ChIP Seq (Yamane et al., 2013) was performed on passage-immortalized ATRflox/-Cre-ERT2+ mouse embryonic fibroblasts (Ruzankina et al., 2007; Smith et al., 2009) that were either treated or left untreated with ATR inhibitor and a partially inhibitory concentration of the DNA polymerase antagonist, aphidicolin (0.2 μM), as a fork stalling enhancer (ATRi+aph18hrs). As shown (Fig. 1B), chromatin was sonicated to relatively large fragment sizes (500–2,000 bps) prior to RPA-ChIP retrievals to increase the opportunity to map regions of repetitive DNA using adjacent unique sequences. Isolated DNA was subsequently re-sonicated (200–300 bps), subjected to NGS, and mapped to the reference genome (Fig. 1B). Several measures were taken to assure validity of ATR-dependent sites, including: 1) normalization of retrievals by genomic representation (pre-ChIP inputs); 2) selection of sites with a minimum read enrichment of >4-fold over input and a p-value of <10−20; and 3) identification of these sites in two independent biological replicates. The peaks called in both biological replicates and not observed in DMSO controls were defined as ATR-dependent.
Figure 1.
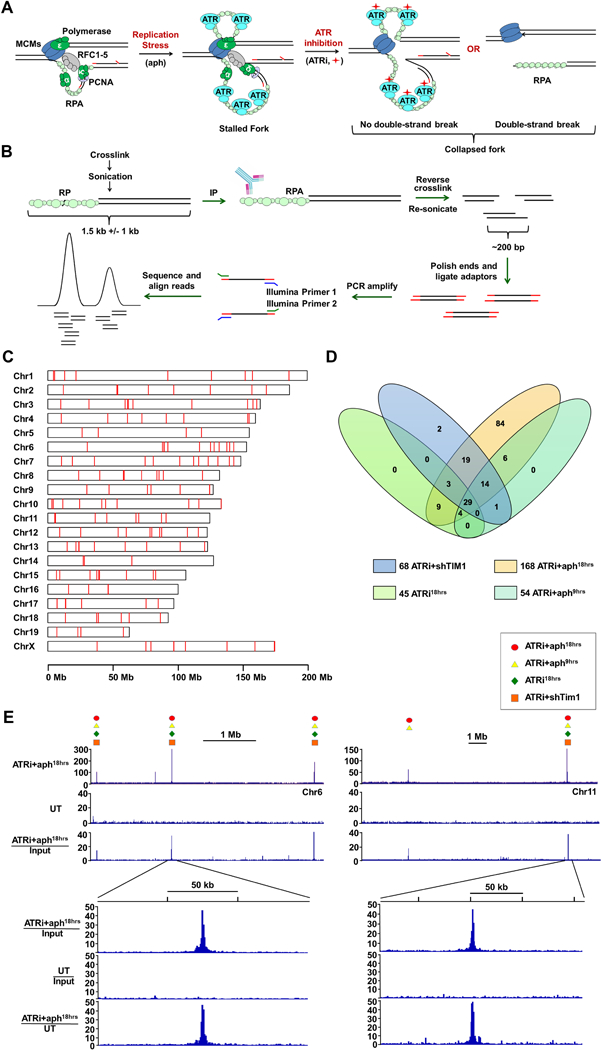
Genome-wide Identification of Fork Collapse Sites by RPA-ChIP Seq (A) RPA-ChIP Seq detection of replication fork collapse from ATR inhibition (ATRi, red diamond). (B) Schematic of RPA-ChIP Seq experimental approach. Cross-linked chromatin was sonicated into large fragments (1.5 kb average) prior to immunoprecipitation with RPA2 antibody. Retrieved DNA was sonicated into smaller fragments (200–300 bp) for NGS. (C) RPLs identified in the mouse genome (red marks). (D) Venn diagram depicting overlap of peaks identified from different conditions. (E) Representative peaks in RPA-ChIP Seq coverage and ratio tracks (ATRi+aph18hrs and DMSO-treated control, UT). Symbols above select peaks indicate identification under additional experimental conditions. See also Supplemental Table S1 and S2.
A total of 168 sites of significant and specific RPA enrichment were identified in the ATRi+aph18hrs condition with an average peak signal intensity of 13.6-fold over background (Fig. 1C, D). Notably, the average peak footprint (~1.5 kb) corresponded well to the size of the sonicated chromatin fragments used for RPA-ChIP, and the cumulative peaks comprised a small fraction of the total murine genome (10−4). These metrics indicated a high degree of resolution and specificity in ATR-dependent site identification.
To expand and prioritize identified ATR-dependent sites, three additional ATRi conditions were examined by RPA-ChIP: 1) 9-hour ATRi + aph treatment (ATRi+aph9hrs), 2) ATRi treatment alone (ATRi18hrs), and 3) ATRi combined with suppression of the replication fork protection complex factor, TIMELESS (ATRi+shTIM1, Smith et al., 2009). Notably, of the 87 sites observed using these additional conditions, 84 (97%) overlapped with those identified with ATRi+aph18h treatment (Fig. 1D, E, Supplemental Table S1) and co-occurrence correlated well with RPA signal intensity. Peaks observed in all 4 conditions (29 sites, ATRi29) exhibited an average signal intensity that was 22.5-fold over background in the ATRi+aph18h condition. Those peaks found in 2–3 conditions (56 sites, ATRi56) or exclusively in the ATRi+aph18h exhibited a downward trend in signal intensity (13.3-fold and 10.8-fold over background, respectively; Fig. 1D, Supplemental Table S1 and S2). RPA-ChIP Seq on ATR-deleted cells also led to the preferential identification of these higher-priority ATRi29 and ATRi56 sites (Supplemental Table S2). Identification of these ATR-dependent RPA-accumulation sites, 171 in total (ATRi171), and their prioritization by signal intensity and common occurrence provided a means to investigate the dominant causes of replication fork collapse following ATR inhibition. Because these sites met two key criteria of defective replication – RPA accumulation and dependence on ATR for stability – they were collectively termed Replication Perturbed Locations (RPLs).
RPLs and large-scale genomic features
To investigate the mechanisms underlying dependence on ATR, RPLs were examined for their overlap with large-scale genomic characteristics: chromatin state (euchromatic, heterochromatic, and boundary elements), gene location (transcription start sites and gene bodies), and sequence elements (transcription factor binding sites and repetitive sequences). RPL sites were found outside of gene bodies, promoters, and terminators at a frequency that roughly mirrors the aggregate percentage of non-coding DNA in the mammalian genome (Fig. 2A). Similarly, there was no significant correlation with the transcription state of coding genes, as determined by co-incidence with H3K4me3 and H3K27ac (Fig. 2A). Collectively, these data indicate that the transcriptional state of coding genes does not strongly influence the localization of RPLs.
Figure 2.
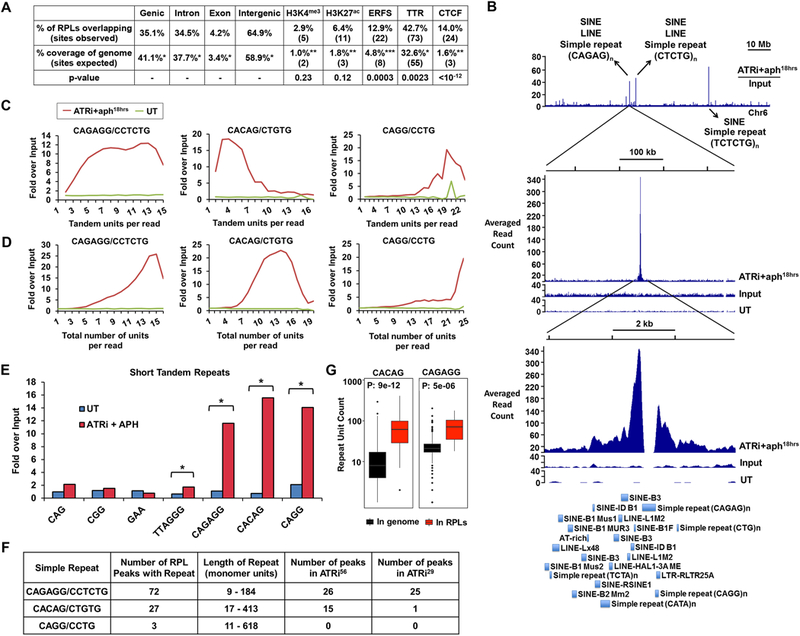
Short Tandem Repeats are Enriched in RPLs (A) Genomic features associated with RPLs. Percent and number of RPLs overlapping with noted features compared to expected overlap based on fraction of the genome comprised by these features is shown. Statistical significance (p value) was calculated by permutation test. (B) Example of repetitive DNA in RPL peaks. Top track: Representative ratio track of RPA-ChIP Seq reads over input reads from ATRi+aph18hrs-treated cells. Arrows detail examples of repetitive elements present. Middle and bottom track: Zoomed-in RPL peak. First track: RPA-ChIP of ATRi+aph18hrs; second track: input of ATRi+aph18hrs; third track: RPA-ChIP of DMSO-treated control (UT). Bottom: RepeatMasker annotations of repetitive elements within the peak region. (C, D) Quantification of tandem and total repeat units in RPA-ChIP Seq reads by REQer. X-axis depicts the (C) tandem repeat units and (D) total repeat units counted within the total RPA-ChIP Seq reads (ATRi+aph18hrs and DMSO control, UT) normalized by repeat occurrence in respective inputs. (E) Fold enrichment of tandem repeat occurrences in RPA-ChIP Seq NGS reads over input (average of 5 data points) in ATRi+aph18hrs and DMSO control (UT) is shown. *, p < 0.001, Student’s T-test. (F) Repeats most frequently observed as enriched in ATRi+aph18hrs RPA-ChIP Seq reads and their association with RPLs. (G) Lengths of CACAG and CAGAGG repeats in the mouse genome and in RPL peaks according to the reference genome. See also Figure S2 and S3.
In addition, while statistically significant, overlap of RPLs with Early Replicating Fragile Sites (ERFS, Barlow et al., 2013) and replication Timing Transition Regions (TTRs, (Yue et al., 2014) was minimal considering their broad genomic coverage (Fig. 2A). Indeed, only 22 out of the 171 RPLs (12.9%) were found within ERFS, with 8 expected from random co-occurrence (Fig. 2A), and no sub-peaks in the broad ERFS footprint (208 kb) were observed in alignment with RPLs (Supplemental Fig. S1). Similarly, the greater-than-expected overlap of RPLs with TTRs was relatively small given the genomic coverage of TTRs (Fig. 2A). Nevertheless, upon querying the association of RPLs with known characteristics of TTRs (Yue et al., 2014), one TTR marker in particular, CTCF, showed significant overlap with RPLs, suggesting it as the underlying cause of TTR association (Fig. 2A).
CTCF, which functions in transcription, chromatin anchoring, and boundaries between topologically-associated domains (TADs), is enriched in the early replicating and midpoint regions of TTRs (Pope et al., 2014; Yue et al., 2014). Notably, 24 of the 171 RPL sites were superimposable with previously identified CTCF-binding sites in MEFs (Fig. 2A, Supplemental Fig. S2A), and this association was highly significant (p-value <10−12). Of the 24 CTCF-associated RPL sites, 10 (42%) were within TTRs, which would be sufficient to account for the non-random fraction of RPLs associated with TTRs. While a small fraction of RPLs were strongly associated with CTCF binding sites, it is important to note that >99.9% of CTCF binding sites throughout the genome do not overlap with RPLs, indicating that CTCF binding is not sufficient to cause dependence on ATR for stability.
In search a search for alternative associations within these CTCF sites, we noted that 21 of the 24 RPLs that overlapped with CTCF binding were characterized by centrally located microsatellite repeats that did not fit the typical consensus for CTCF binding: (CACAG)n, (CAGAGG)n, or similar microsatellite repeats (Supplemental Fig. S2A). Notably, most CTCF-associated RPLs that contained repeats were observed under multiple ATRi conditions (5 in the ATRi29 subgroup and 12 in the ATRi56 subgroup), indicating that such repeats correlate well with RPL detection. Collectively, the association of RPLs with these repeat-containing CTCF sites was stronger than association with any other large-scale chromatin feature.
An ATR-TIMELESS-dependent RPL in rDNA is linked to microsatellites
Previous studies have demonstrated an important role for ATR in fork stability following TIMELESS suppression (Smith et al., 2009), a finding in accord with TIMELESS functions in limiting ssDNA formation at replication forks (Katou et al., 2003; Smith et al., 2009). Consistent with these studies, fork collapse sites identified by ATRi+shTIM1 overlap significantly with ATRi+aph18hrs sites (Fig. 1, Supplemental Table S1). However, deletion of the TIMELESS orthologs TOF1 and SWI1 in S. cerevisiae and S. pombe, respectively, has also been shown to abrogate replication fork barrier function at the rRNA transcriptional terminator, FOB1, which has been proposed to increase genomic instability through replication-transcription conflicts (Krings and Bastia, 2004; Mohanty et al., 2006). Notably, while no enrichment of RPA was observed in the rRNA transcription unit, one RPL was found in the intergenic spacer region of the 45 kb rDNA repeat, and its signal intensity was 10-fold greater in the ATRi+shTIM1 condition than in any other ATRi condition (Supplemental Fig. S2B). Once again, this rDNA-associated RPL was characterized by a centrally located microsatellite, (GAAA/TTTC)25, which may stall replication through the formation of H-DNA (Follonier et al., 2013; Mirkin, 2007) or stem-loop structures with the extensive TTT-rich repeats downstream. These data are consistent with previous findings indicating that TIMELESS orthologs in yeast counteract replication fork stalling and fragility at structure-forming repeats (Voineagu et al., 2008; Zhang et al., 2012) and suggest a specific role for TIMELESS in fostering replication through this type of microsatellite. Collectively, the localization of RPLs correlated better with short tandem repeats than any other genomic feature analyzed.
Strong association of RPLs with microsatellite repeats
The association of CTCF- and rDNA-RPL sites with microsatellite repeats suggested that repetitive DNA may be the main mechanism underlying ATR dependence at these sites. As shown in Fig. 2B, a high frequency of various repetitive sequences were observed within RPA-ChIP Seq enrichment sites, with microsatellite repeats frequently observed near the peak center (Fig. 2B). STR-occupied regions were often characterized by a gap in accumulation due to standard filtering of reads that align to multiple genomic locations (Fig. 2B, “CAGAG” repeat). However, the position of microsatellite repeats at the center of RPL peaks suggested that such features might strongly contribute to replication fork collapse upon ATR inhibition.
To determine which repeats found within RPL peaks most consistently associated with RPA accumulation, the number of reads containing these sequences were quantified in RPA-ChIP retrievals and in sequenced input DNA. Consistent with prior studies in yeast (Admire et al., 2006; Szilard et al., 2010), rDNA-encoding sequences were slightly enriched in RPA-ChIP retrievals from ATR-inhibited cells. However, other well-known satellite repeats and retrotransposable elements on the whole were not enriched in RPA-ChIP retrievals following ATR inhibition (Supplemental Fig. S3A). While other factors might influence the stability of a subset of these elements, such as an inverted orientation, these elements did not appear to be sufficient to drive RPA accumulation in ATR-inhibited cells.
Microsatellite repeats found within RPA-accumulation regions were also queried for enrichment in RPA-ChIP retrievals. To do so, we developed a repeat-counting program called REQer (Repeat Enrichment Quantifier) that evaluates the number of repeat units in tandem or in aggregate within NGS reads. This approach prevents artifacts associated with quantifying repeats from the reference genome, which is subject to polymorphisms in repeat length and incomplete genomic assemblies. Using REQer, the number of reads containing high numbers of repeat units were counted in RPA-ChIP retrievals and normalized by similar quantifications of sequenced input DNA (Fig. 2C, D).
As expected, unexpanded trinucleotide microsatellites were not significantly enriched in ATRi+aph18hrs RPA-ChIP reads (Fig. 2E, Supplemental Fig. S3B). Telomere repeats (TTAGGG)n, were enriched approximately 2.5-fold in RPA-ChIP DNA following ATR inhibition (Fig. 2E, Supplemental Fig. S3B). Strikingly, however, the microsatellites that exhibited greatest enrichment following ATR inhibition had not previously been reported as difficult to replicate (Fig. 2C-E, Supplemental Fig. S3C). These repeats included the hexameric and pentameric microsatellites (CAGAGG/CCTCTG)n and (CACAG/CTGTG)n. Other variants of CAGAGG, such as (CAGAGAGG/CCTCTCTG)n, (CAGAGGG/CCCTCTG)n and (CAGG/CCTG)n, were also enriched in RPLs, the latter of which is associated with myotonic dystrophy type 2 (Liquori et al., 2001). Most repeats enriched in RPLs occurred in tandem iterations without interruptions, with the exception of (CACAG/CTCTG)n, which showed a precipitous decline in enrichment at 5–9 tandem units compared to total overall units per read (Fig. 2C, D, Supplemental Fig. S3C-D).
Importantly, (CACAG/CTGTG)n and (CAGAGG/CCTCTG)n were also present in the greatest number and most commonly occurring RPLs (Fig. 2F). All ATRi56 sites exhibited abundant repetitive sequences, with (CAGAGG/CCTCTG)n and (CACAG/CTGTG)n comprising 26 and 15 RPLs in this category, respectively. Moreover, 25 of the ATRi29 subgroup were dominated by (CAGAGG/CCTCTG)n repeats, indicating this repeat as one of the strongest sensitizers to ATR inhibition. Notably, the lengths of (CAGAGG/CCTCTG)n and (CACAG/CTGTG)n repeats within RPLs, as defined by the reference genome, correlated with their identification by RPA-ChIP in ATR-inhibited cells (Fig. 2G), suggesting that longer lengths of these repeats increases dependence on ATR.
The short tandem repeats observed in RPLs form stable intrastrand structures
We hypothesized that the microsatellite repeats identified in RPLs form stable intramolecular secondary structures, which in turn could limit replicative polymerase progression. Synthetic single-stranded oligos of RPL repeats (Fig. 2, Supplemental Table S3) were first examined by native polyacrylamide gel electrophoresis (PAGE) after a single heating and cooling cycle (Fig. 3A). With the exception of (CAGAGT)15, a repeat variant found imbedded within (CAGAGG)n repeats, repeat-containing oligos migrated as single-dominant bands with mobilities greater than expected based on their lengths (Fig. 3A), consistent with the formation of compact, uniquely-folded structures. Notably, (CAGG)22, (CAGAGG)15, and (CAGAGAGG)11 exhibited greater electrophoretic mobility than oligos encoding their complementary strands (Fig. 3A), suggesting that the purine-rich strands of these repeats have more structure-forming potential than their pyrimidine-rich complements.
Figure 3.
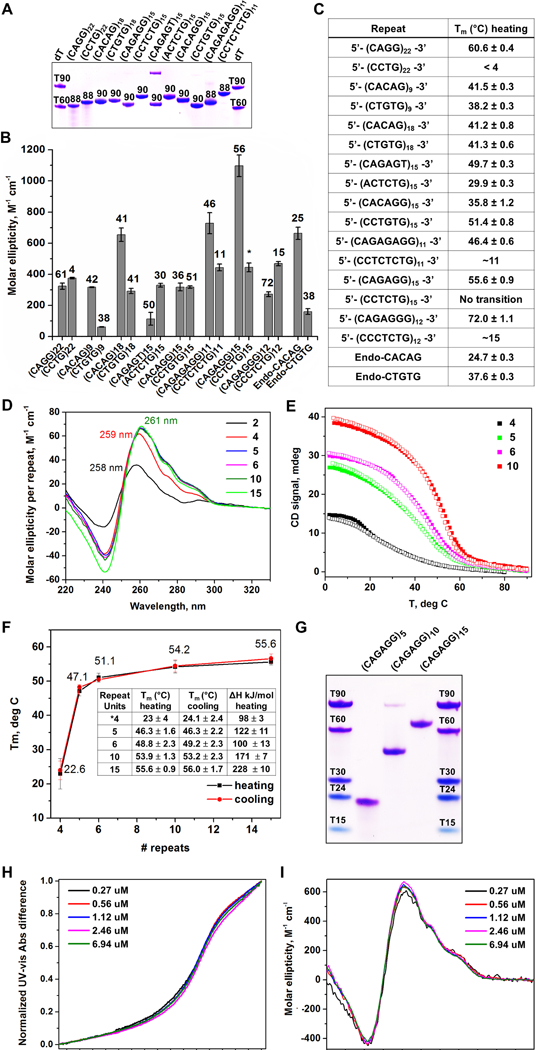
RPL-Associated Repeats Form Unique Intrastrand Secondary Structures (A) Non-denaturing PAGE gel of oligonucleotide repeats. (B) CD molar ellipticity peak values as a proxy for DNA folding. Melting temperatures, shown above bars, were obtained using a non-linear fit assuming two-state system. (*) indicates melting transition is characterized by a non-sigmoidal melting curve. (C) List of simple repeat sequences analyzed and respective melting temperatures. (D) Repeat-normalized CD wavelength scans of (CAGAGG)n, with n = 2, 4, 5, 6, 10, and 15. (E) Representative CD melting and cooling curves for (CAGAGG)n, n = 4, 5, 6, and 10. (F) Graph of melting temperatures obtained by UV-vis with different CAGAGG monomer lengths. Tm values and change in enthalpy are summarized in the embedded table. (G) Non-denaturing PAGE gel of (CAGAGG)n, n = 5, 10, and 15. Overlay of (H) normalized UV-vis melting and (I) CD scans (4°C) of (CAGAGG)10 at varying oligonucleotide concentrations. For (B), the data are represented as mean +/− SEM. For (A-I), all samples were prepared in 10 mM lithium cacodylate pH 7.2, 100 mM KCl and 2 mM MgCl2 buffer. For (A) and (G), DNA bands were visualized with Stains All. See also Supplemental Fig. S4and Table S3.
To determine the strength and types of secondary structures formed, repeat-containing oligos were then examined by circular dichroism (CD), UV-vis spectrometry, and thermal difference spectra (TDS). Oligos that exhibited the highest molar ellipticity, a proxy for the extent of DNA folding, were (CAGAGG)15 (Δε = 1100 ± 70 M−1cm−1), (CAGAGAGG)11 (Δε = 730 ± 70 M−1cm−1) and (CACAG)18 (Δε = 650 ± 40 M−1cm−1, Fig. 3B, Supplemental Fig. S4, Supplemental Table S3). The reversibility of melting transitions (Supplemental Fig. S4) also supported the formation of unimolecular (intrastrand) structures. Purine-rich strands of these repeats also exhibited high thermal stabilities, as did other RPL-associated repeats (Fig. 3B, C, Supplemental Fig. S4, Supplemental Table S3). Consistent with PAGE analysis, oligos encoding the complementary strand of these repeats were neither well-folded nor stable (Fig. 3A-C, Supplemental Table S3), indicating that the structure-forming potential of these RPL repeats resides primarily in their purine-rich strands.
The (CAGAGG)n repeat demonstrated stable structure formation (Fig. 3B, C, Supplemental Table S3,) and was observed most frequently in RPL sites (Fig. 2C-F). Thus, this repeat was further examined for length dependence and biophysical characteristics. These studies demonstrated that five tandem units of CAGAGG were sufficient to generate stable-secondary structures, and furthermore, structure formation, CD signature, and stability were not significantly affected by additional units of up to 15 (Fig. 3D-G). Finally, CD spectral and thermal stability across a >20-fold concentration range (Fig. 3H, I) and analytical ultracentrifugation (not shown) further supported the unimolecular nature of the (CAGAGG)n structure. Notably, the CD signature of (CAGAGG)n, which includes a major peak at ~260 nm and two prominent shoulders at 276 and 291 nm, is not expected from either B-form or G-quadruplex DNA, indicating a potentially novel secondary structure. Collectively, these results demonstrate that purine-rich strands of RPL microsatellites form intrastrand secondary structure that could impact DNA replication.
RPL-associated microsatellite impedes DNA replication
We next investigated if the structure-forming repeat most commonly observed in RPLs, (CAGAGG)n, is sufficient to slow DNA replication in vitro and in cultured cells. For in vitro studies, purine-rich (CAGAGG)15, complementary pyrimidine-rich (CCTCTG)15, and scrambled control sequences (two purine-rich, and two pyrimidine-rich) were examined for their ability to act as processive templates for the DNA polymerase δ holoenzyme (Polδ4-PCNA-RFC; Pol δHE). Pol δHE synthesis pausing was quantified as an increased accumulation of reaction products within specific regions of the vector with respect to the location of the inserted sequences: far upstream (68–11 nt), immediately upstream (10–1 nt), within the insert, and downstream (3’) of the insert (Fig. 4A, (Hile and Eckert, 2004, 2008).
Figure 4.
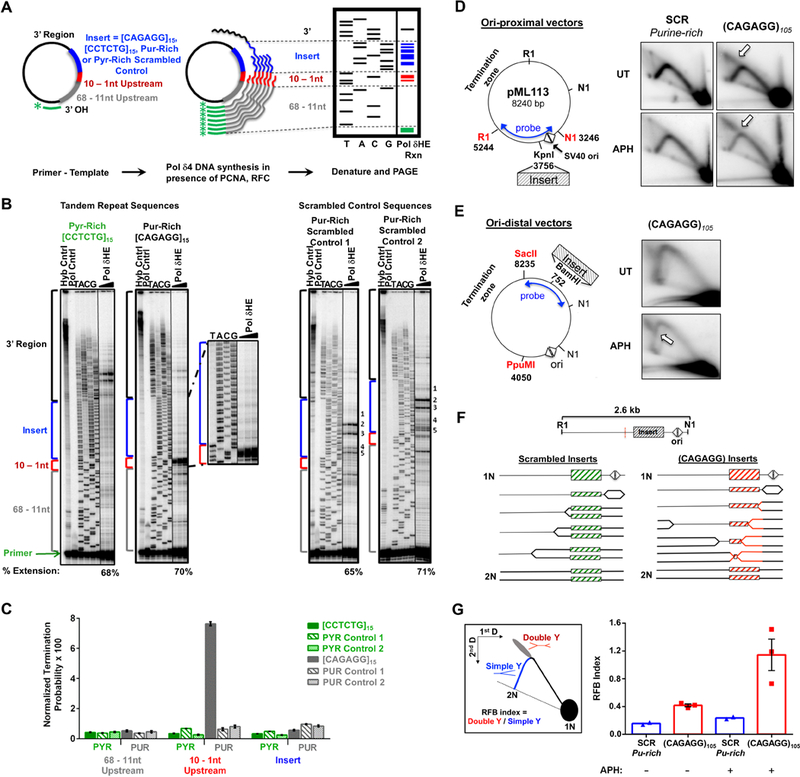
CAGAGG Repeats Impede DNA synthesis (A) Schematic of in vitro Pol δHE primer-extension assay. (B) Representative images of Pol δHE reaction products. Pol δHE DNA synthesis products from ssDNA templates containing (CAGAGG)15, (CCTCTG)15, or scrambled control inserts (purine-rich or pyrimidine-rich) with increasing reaction times (3 – 15 minutes, triangle) were separated by denaturing PAGE alongside a dideoxynucleotide sequencing of the same template (TACG). Left: (CCTCTG)15 and (CAGAGG)15 insert-containing templates; Right: two distinct purine-rich scrambled control insert-containing templates. Control lanes are indicated (-Pol, No Polymerase; Hyb, Primer-template hybridization). Percent Extension, extended DNA over extended plus unextended primer-bound DNA. Also see Supplemental Figure S5A for pyrimidine-rich scrambled control. (C) Pol δHE termination probability. Termination probability, normalized by the number of nucleotides in each region, was quantified as the ratio of DNA molecules within a specific region over these plus all longer DNA molecules. (D) Effect of (CAGAGG)n repeats on plasmid DNA synthesis in cells. Left: (CAGAGG)105 or a scrambled sequence of the same nucleotide composition and length (SCR) was inserted proximal to the bidirectional SV40 origin (triangles) SV40 large T-antigen (TAg) (Follonier et al., 2013). Right: Representative 2D gels. Plasmid transfected cells were either untreated (UT) or treated with 0.6 μM aphidicolin (APH) for 24 hours. Isolated episomal DNA was digested with DpnI, EcoRI (RI) and Eco NI (NI) and replication intermediates were resolved by 2D neutral-neutral gel electrophoresis with Southern hybridization to the indicated probe. Arrows denote the point of divergence of the double-Y structure from the simple-Y arc. (E) Replication intermediates of plasmids containing origin-distal (CAGAGG)105. Left: Schematic of the ori-distal vectors(2.7 kB from the origin). Right: Representative 2D gels. Experiment was carried out as described in (A), except that the purified DNAs were digested with DpnI, PpuMI, and SacII and detected with the indicated probe. (F) Schematic of replication through ori-proximal vectors and the formation of double-Y structures. Dashed red line indicates the center of the RI-NI fragment, the expected apex of the simple-Y arc. (G) Left: Schematic of replication fork barrier (RFB) index quantitation. The RFB index is the number of double Y structures (red) divided by the number present in >1.5N simple-Y structures (blue). Right: Quantitation of the RFB index in CAGAGG)105 and SCR ori-proximal vectors from 2D gels (E). For (C), the data are represented as mean +/− SEM of three independent polymerase reactions for each template. ****, p < 0.0001. For (G), the data are represented as mean +/− SEM, with individual data points representing independent biological replicates. See also Figure S5.
While DNA synthesis from the (CCTCTG)15 pyrimidine-rich template occurred without pausing either before or within the repeat, a substantial accumulation of reaction products was observed immediately upstream of the purine-rich (CAGAGG)15 template (Fig. 4B). No pause sites were observed within the repeat region itself, indicating that polymerase blockage at the repeat interface was substantial (Fig. 4B). Termination of the Pol δHE complex immediately upstream of the (CAGAGG)15 insert was 22-fold greater than that observed upstream of the complementary (CCTCTG)15 repeat, and 9- to 12-fold greater than that of the purine-rich scrambled controls (Fig. 4C; p<0.0001, 2 way ANOVA). Notably, no increase in Pol δHE termination was observed within the repetitive (CAGAGG)15 inserts themselves (Fig. 4C), indicating that DNA replication was not slowed randomly throughout the repeat region but rather was barred specifically at the start of the folded structure. In contrast, low-level sequence-specific Pol δHE pausing was observed within the purine-rich scrambled insert (Fig. 4B, Supplemental Fig. S5A), as expected based on nucleotide content (Walsh et al., 2013). Importantly, these data indicate that (CAGAGG)15 repeats are fundamentally different from other microsatellite sequences, which cause replicative polymerase pausing within the repeat sequence, not at the start of it (Hile and Eckert, 2004, 2008; Walsh et al., 2013).
To determine whether the duplex (CAGAGG/CCTCTG)n repeat impedes DNA replication in cells, a 105 unit repeat was amplified from an endogenous RPL (Chr7:35159697–35161220, mm10) and subcloned into the pML113 SV40 plasmid replication system (Follonier et al., 2013) at two distinct sites (Fig. 4D, E, Supplemental Fig. S5B-D). Plasmids containing scrambled (CAGAGG/CCTCTG)n synthetic sequences of similar length were also generated as controls. Plasmids were replicated in large T antigen-expressing human osteosarcoma cells (U2OS), and Dpn I-resistant replication intermediates were resolved by neutral-neutral 2D gel electrophoresis.
Simple Y structures of normal DNA replication through the scramble control insert were observed both with and without low-dose aph treatment (Fig. 4D). However, replication of the (CAGAGG/CCTCTG)105-containing plasmids generated distinct replication intermediates at the top of the simple Y arc, regardless of the repeat orientation (Fig. 4D, Supplemental Fig. S5B). These intermediates are consistent with the formation of double-Y structures (Huberman, 1997), which demonstrates that fork stalling at the (CAGAGG/CCTCTG)105 repeat near the SV40 origin was sufficiently persistent to allow the opposite-moving fork to replicate around the plasmid and ultimately stall on the other side of the (CAGAGG/CCTCTG)105 repeat (Fig. 4F). Consistent with this interpretation, the point of divergence from simple Y arc to the descending arm was altered when the (CAGAGG/CCTCTG)105 repeat was inserted in an origin-distal location (Fig. 4E, Supplemental Fig. S5D).
Quantification of these replication fork barrier effects showed that the (CAGAGG/CCTCTG)105 repeat was sufficient to impede fork progression by 2- to 3-fold over scrambled control inserts, and this inhibition was enhanced 5-fold by aph treatment (Fig. 4G). Importantly, treatment of transfected cells with low-dose aph increased the abundance of double-Y migration products only in (CAGAGG/CCTCTG)105 repeat-containing vectors, not scrambled controls (Fig. 4D, E, Supplemental Fig. S5B-D). This selectivity suggests that polymerase slowing may increase (CAGAGG)n structure formation, which further limits polymerase progression. In aggregate, these findings demonstrate that the (CAGAGG)n repeat is sufficient to cause replicative polymerase stalling and impeded fork progression.
Development of BrITL: a genome-wide method to identify sites of DSB formation
To validate RPL sites and to identify additional sites that accumulate little RPA, we developed a highly selective method that labels accessible DNA ends in the context of intact chromatin (Fig. 5A). This method, abbreviated BrITL for Breaks Identified by TdT Labeling, attaches biotin-nucleotide adducts to 3’ ends of breaks in permeabilized cells, thus eliminating background-inducing chemical fixation and long incubation steps that generate breaks through depurination and β-elimination. Similar to RPA-ChIP, BrITL retrievals are performed on DNA that is sonicated to relatively large fragment sizes (200–2,000 bp), thus increasing opportunities to map repeat-containing sites of breakage through co-retrieval of adjacent unique sequences (Fig. 5A).
Figure 5.
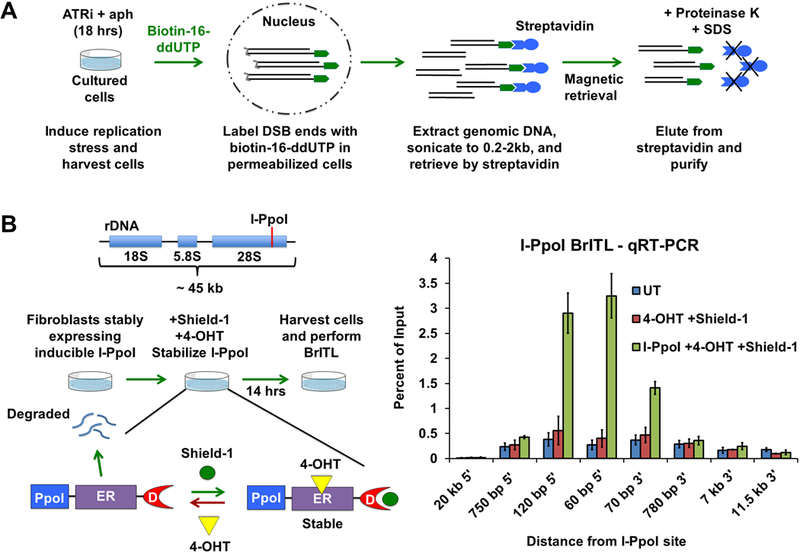
Development of BrITL (A) Schematic of the BrITL procedure. Treated cells are permeabilized and incubated with terminal deoxynucleotidyl transferase (TdT) and biotin-16-ddUTP. Extracted genomic DNA is then sonicated to 0.2–2 kb and subjected to streptavidin retrieval for analysis by qRT-PCR or NGS. (B) Validation of DSB detection by BrITL. Left: Genomic DSBs at I-PpoI recognition sites (red line) were conditionally generated by expression of I-PpoI (PpoI) fused to a destabilized FKBP12 (D) and a tamoxifen-specific form of the estrogen receptor (ER), followed by fusion protein stabilization and nuclear localization by Shield-1 and 4-hydroxytamoxifen (4-OHT) treatment. Right: qRT-PCR analysis of BrITL retrievals. Quantification of retrieved biotin-labeled fragments, normalized as a percent of input, is shown at specified distances from the rDNA I-PpoI site relative to the start of transcription. Conditions include UT (DMSO treatment), 4-OHT + Shield-1, and I-PpoI fusion expression + 4-OHT + Shield-1. For (B), the data are represented as mean +/− SEM.
To test this method, a I-PpoI endonuclease fusion protein was introduced into passage-immortalized MEFs, treated to stabilize and induce I-PpoI, and subjected to BrITL. The genomic regions proximal to the I-PpoI cleavage site were quantified by qRT-PCR with input DNA serving as a control, thus quantifying the amount of retrieved DNA relative to the total amount of genomic material present. I-PpoI induction led to a substantial increase in the BrITL detection of genomic DNA nearest the I-PpoI site (Fig. 5B). Regions 20 kb away from the I-PpoI endonuclease site were not readily detected by BrITL, even in I-PpoI-induced cells, and the breadth of I-PpoI-enhanced qRT-PCR signal approximated the expected retrieved fragment size of 200 bp to 2,000 bp (Fig. 5B). These data indicate that BrITL is capable of retrieving DSB ends both efficiently and specifically.
BrITL confirms ATRi-driven instability of key RPLs
Genome-wide BrITL-Seq was performed on ATRi+aph18h-treated cells to expand identification of ATR-dependent sites and validate RPA accumulation sites. Using similar bioinformatic criteria (>4-fold over background, IDR p-value <10−8, subtraction of DMSO controls), a total of 223 BrITL sites were identified as ATR-dependent (Supplemental Table S4). Importantly, RPL sites of highest signal intensity were validated as break sites using BrITL (Fig. 6A, C). BrITL detection of RPL sites was directly proportional to RPL signal intensity and their detection using various treatment conditions (Fig. 6C). Furthermore, sites harboring the most common RPL-associated repeat, (CAGAGG/CCTCTG)n, were prominently detected by BrITL-Seq and qRT-PCR, and enrichment of this repeat in BrITL-Seq reads from ATR inhibited cells was directly detected by REQer (Fig. 6B, C).
Figure 6.
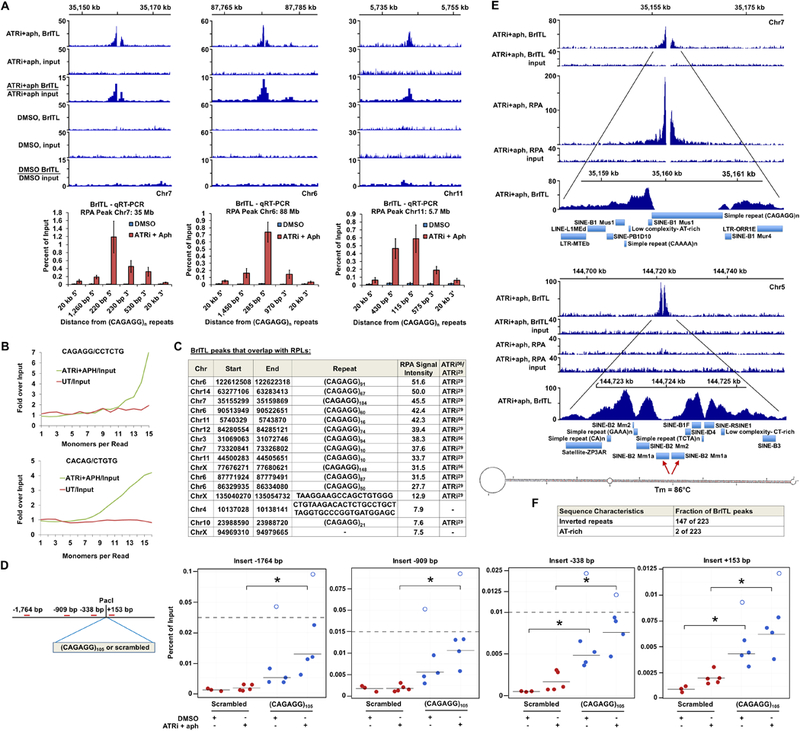
BrITL Sites Overlap with RPLs and Inverted Repeats (A) Top: Coverage and ratio tracks of BrITL retrievals and inputs of ATRi+aph18hrs and DMSO- treated cells at RPLs. Bottom: BrITL-qRT-PCR detection of RPL sites adjacent to peak-centric (CAGAGG/CCTCTG)n repeats. (B) Quantification of total repeat units in BrITL retrieval reads by REQer. X-axis depicts the total repeat units counted within the total BrITL reads (ATRi+aph18hrs and DMSO control, UT) normalized by repeat occurrence in respective inputs. (C) Table listing RPLs that overlap with BrITL peaks and the repeats associated with these sites. (D) Left: Schematic of (CAGAGG)105-containing vector for stable genomic integration and primer sets used in BrITL qRT-PCR analysis. Right: qRT-PCR analysis of genomic BrITL retrievals (ATRi+aph18hrs and DMSO control, UT) at indicated distances from the (CAGAGG)105 and 630 bp scrambled control insertion sites. Data points represent independent biological replicates; red: scrambled 630 bp insert; blue: (CAGAGG)105 insert. Hollow dots represent outliers. *, p < 0.05, Student’s T-test. (E) Representative coverage tracks of RPA-ChIP and BrITL retrievals and inputs at RPA-positive and RPA-negative BrITL sites following ATRi+aph18hrs. RepeatMasker annotations of repetitive elements as well as a representative inverted retroelement repeat and its M-fold-predicted stem-loop structure are shown below. (F) Inverted repeat and AT-rich sequence frequency in ATRi+aph18hrs BrITL peaks. For (A), the data are represented as mean +/− SEM. See also Figures S6 and S7 and Supplemental Table S4 and S5.
To determine if (CAGAGG/CCTCTG)n is sufficient to cause collapse into DSBs, the (CAGAGG/CCTCTG)105 and scrambled control inserts used for 2D gel analysis were subcloned into the GFP-expressing HFUW vector to generate stably-transfected cells (Fig. 6D). Stable lines were then treated with ATRi+aph for 18 hours and assayed for breaks by BrITL-qRT-PCR (Fig. 6D). While scrambled control inserts were not significantly affected by ATRi+aph18hrs treatment, this treatment caused a significant increase in vector sequence detection by BrITL in (CAGAGG/CCTCTG)105-transfected cells. Interestingly, instability of the (CAGAGG/CCTCTG)105 repeat was observed even in the absence of ATRi+aph18hrs treatment, which was reflected both by increased BrITL detection of vector sequences and reproducible selection against high-copy (CAGAGG/CCTCTG)n integrants (Fig. 6D, Supplemental Fig. S6). These data indicate that (CAGAGG/CCTCTG)n repeats are sufficient to cause DSB formation when placed outside of their normal genomic context, and this breakage is amplified by ATR inhibition.
While association of BrITL sites with various large-scale genomic features mirrored that of RPL sites, one exception to this similarity was the identification of new sites of ATR-dependent containing inverted repeats, which were not detected by RPA-ChIP (Fig. 6E, F, Supplemental Fig. S7). Indeed, 147 of the 223 BrITL sites identified following ATRi treatment harbored long inverted repeats and quasi-palindromes with predicted melting temperatures of >70°C. Notably, these repeats were mainly comprised of complementary retrotransposable elements, including a variety of SINEs, LINEs, and LTRs (Fig. 6E, Supplemental Fig. S7, Supplemental Table S5). Inverted repeats have long been known to be unstable through their ability to form stable stem-loop structures, and that breakage can occur at such structures in a manner that produces hairpin ends, which can alter processing (Lobachev et al., 2007). Indeed, sensitivity of quasi-palindromic retroelement repeats to checkpoint loss is consistent with engineered reporter system studies in yeast (Zhang et al., 2013). While we cannot exclude the possibility that BrITL might also detect regressed replication forks, such events would still indicate replication abnormalities under ATR inhibition. These data suggest that not all ATR-dependent sites accumulate RPA and that inverted repeats and quasi-palindromes comprise an additional category of vulnerable sites. Collectively, the use of two independent genome-wide methodologies indicate that replication fork collapse caused by ATR inhibition is primarily directed by structure-forming repetitive sequences.
ATR-dependent BrITL sites in human cells are associated with structure-forming repeats
Our findings demonstrate that purine-rich structure forming repeats, such as (CAGAGG/CCTCTG)n, and inverted repeats are key drivers of replication fork collapse following ATR inhibition in murine cells. Although it is possible a structural analog may exist, extensive tandem iterations of (CAGAGG/CCTCTG)n are not common in the human genome (ENCODE). Therefore, we asked what repeat or feature might dominate sensitivity to ATR inhibition in human cells.
Human triple-negative breast cancer cells (MDA-MB-231) were treated with ATRi+aph for 9 hours (ATRi+aph9hrs) and subjected to genome-wide BrITL-Seq. A total of 167 highly significant ATR-dependent sites of expected peak breadth were identified (Supplemental Table S6). Similar to murine sites, human BrITL sites were not strongly associated with coding genes (Table 1). Nevertheless, a greater-than-expected fraction of human BrITL sites overlapped with CFS (25 expected, 35 observed) and with H3K4me3 enrichment regions (1 expected, 15 observed), a marker of gene promoters (Table 1, Supplemental Table S7). The 15 H3K4me3-overlapping BrITL sites represented only a minute fraction (<0.1%) of total H3K4me3 enrichment sites, indicating that this histone modification on its own does not strongly influence ATR sensitivity. However, analogous to the association of RPLs with CTCF sites in murine cells, the association of BrITL sites with both H3K4me3 and CFS correlated best with the presence of peak-centric repeats, including minisatellites, microsatellites, and inverted repeats (Fig. 7A-C, Supplemental Table S7). Such repeats were observed in 11 of 15 H3K4me3-overlapping BrITL sites and 24 of 35 of BrITL sites that overlapped with CFS. (Supplemental Table S7). Thus, similar to the murine genome, the dependence on ATR for stabilization of the human genome correlated best with the presence of repetitive DNA.
Table 1.
Genomic Features of BrITL sites in human breast cancer cells
| Genic | Inter- genic |
H3 K4me3 |
H3 K27ac |
H3 K36me3 |
TTRs | CTCF | CFS | ||
|---|---|---|---|---|---|---|---|---|---|
| Intron | Exon | ||||||||
| % of BrITL sites overlapping (sites observed) | 37.7% | 62.3% | 9.0% (15) | 3.0% (5) | 0% (0) | 15.6% (26) | 6.0% (10) | 21.0% (35) | |
| 26.3% | 11.3% | ||||||||
| % coverage of human genome (sites expected) | 53.3%* | 46.7%* | 0.34% (1) ** | 0.78% (1) ** | 1.4% (2) ** | 11.6% (19) * | 0.36% (1) ** | 14.9% (25) *** | |
| 49.4* | 3.9%* | ||||||||
| p value | - | - | <10−4 | 0.11 | 1.0 | 0.07 | 0.08 | 0.02 | |
Figure 7.
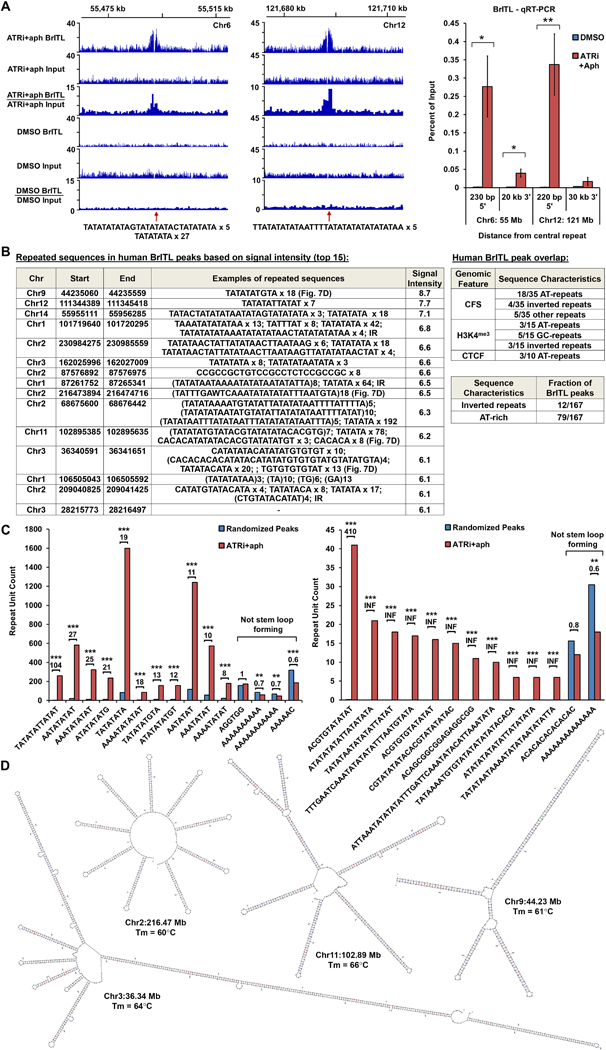
ATRi-Driven Breakage in Human Cells is Associated with Structure-Forming Repeats. (A) Left: Coverage tracks of BrITL retrievals and inputs from ATRi+aph9hrs and DMSO-treated cells. Right: qRT-PCR analysis of BrITL retrievals at specified distances from central AT-rich repeats. *, p < 0.05; **, p < 0.002. (B) Left: Top 15 ATRi+aph9hrs BrITL peaks and associated repeats. Right: Repeat sequences observed in ATRi+aph9hrs BrITL peaks that overlap with specific genomic features. Bottom right: Fraction of ATRi+aph9hrs BrITL peaks associated with inverted repeats or AT-rich repeats. (C) Bar graphs quantifying repeat motifs identified by MISA and HOMER2 within ATRi+aph9hrs BrITL peaks and randomly generated pseudo-peaks of similar size. *, p < 0.002; **, p < 0.0001; ***, p < 0.000001. (D) M-fold-predicted structures and Tm of notable AT-rich repeats in BrITL peaks. For (A), the data are represented as mean +/− SEM. See also Supplemental Table S6 and S7.
Although DNA repeats were a common feature of ATRi sensitivity, the repeat sequences found to be vulnerable in human BrITL peaks were different from those identified in the mouse (Fig. 7B, C). Indeed, nearly half of BrITL-associated minisatellite and microsatellite repeats were distinctly AT-rich, as defined by a greater-than-expected frequency of AT/TA content with respect to average genomic frequency (Fig. 7B, p < 0.05, Chi-Squared test). Such content was not a prevailing characteristic of murine BrITL sites (Fig. 6F). Non-AT-rich microsatellite and minisatellite repeats were also observed within human BrITL peaks; however, these repeats were less commonly associated compared to AT-rich repeats (Fig. 7B, C). Furthermore, in contrast to the dominance of tandem (CAGAGG/CCTCTG)n repeats in murine RPL and BrITL sites, AT-rich satellites in human BrITL sites were more pleomorphic, sometimes occurring tandemly in only one BrITL site. These observations begged the question whether specific motifs are enriched within these larger satellites and if peak-centric repeats observed had structure-forming potential.
The motif-finding program, HOMER2, and the microsatellite prediction tool, MISA (Beier et al., 2017), were used to identify repeated sequences within BrITL peaks in an unsupervised fashion. Identified motifs were then quantified within ATR-dependent BrITL peaks and within 10 independent sets of randomly selected “peak” regions of similar footprint size to assess their average representation within the genome (Fig. 7C). This unbiased approach once again identified AT-rich motifs as the most common sequence feature in human BrITL peaks (Fig. 7 C). For example, the TATATATTATAT/ATATAATATATA and ACGTGTATATAT/ATATATACACGT motifs were enriched 104-fold and 410-fold in BrITL peaks over randomly selected pseudo peaks (Fig. 7C). Remarkably, many of these BrITL-enriched motifs were found to be components within larger minisatellites. For example, the TATATATGTA/TACATATATA motif, which is highly repeated as a unit in BrITL peak Chr9:44.23 Mb, is also observed in composite with another MISA-identified motif, ACGTGTATATAT/ATATATACACGT, in the larger tandem minisatellite (TATATATGTATACGTATATATACACGTG)7 in BrITL peak Chr11:102.89 Mb (Fig. 7B, C). Many times, AT-rich repeats and other BrITL peak sequences were quasi- or perfect palindromes (e.g. TATATATAGTATATATACTATATATA x 5, BrITL peak Chr6:55.49 Mb), suggesting that the genomic sequences corresponding to these motifs form stable hairpin structures.
Mfold (Zuker, 2003) was used to examine the structure-forming potential of repeat-containing sequences identified within BrITL peaks. Overall, these repeat sequences were predicted to form large and stable hairpin structures with melting temperatures greater than 55°C as determined by Mfold (Fig. 7B-D and data not shown). Notably, the MISA- and HOMER2-identified sequence motifs found to be enriched in ATR-dependent BrITL peaks were also capable of forming stable secondary structures. In contrast, motifs not enriched in ATR-dependent BrITL peaks over randomly selected regions exhibited little hairpin-forming potential on their own (Fig. 7C). Indeed, some motifs, such as tandem polyA and polyT repeats, which do not form hairpins without a complementary sequence in cis, were significantly depleted from ATR-dependent BrITL peaks (Fig. 7C), indicating that such stretches on their own do not cause dependence on ATR for stability.
Consistent with stem-loop structures playing a role in ATRi-driven fork collapse in humans as well as mice, inverted repeats and quasi-palindromes, comprised of Alu elements and similar families of repeats, were observed in 12 human BrITL sites (Fig. 7B and Supplemental Table S7). The sensitivity of these sites to ATR inhibition is akin to that of inverted retrotransposable elements in murine ATR-dependent BrITL sites (Fig. 6E, F, Supplemental Table S5). Notably, the large hairpin-forming structures generated by AT-rich BrITL peaks show numerous similarities to the inverted and palindromic AT-rich repeats (PATRRs) that are associated with translocation and deletion hotspots in cancer and developmental disorders, such as DiGeorge and Emanuel Syndromes (Bacolla et al., 2016; Kato et al., 2012). Indeed, a sequence motif observed frequently in PATRRs, TATAATATA (Delihas, 2015), is enriched 46-fold in ATR-dependent BrITL peaks (461 occurrences) compared to randomly chosen pseudo-peaks (p < 10−20). Overall, these data indicate that human ATR-dependent BrITL sites, similar to murine sites, are strongly associated with repetitive sequences that form stable secondary structures.
Discussion
Using two distinct detection methods in mouse and human cells, we have shown that ATR inhibition causes localized replication fork collapse preferentially at repetitive DNA. While a variety of endogenous stresses can activate ATR, structure-forming repeats were the genomic feature most strongly associated with site-specific breakage under ATR inhibition. Collectively, the amplitude of peaks observed under various conditions, the types of repeats associated with collapse, and the optimal means of peak detection (RPA vs. BrITL) have created a diverse prioritized catalog of ATR-dependent sites in the mammalian genome. These sites can serve as specific genomic readouts of ATR dysfunction and biomarkers of response to ATR inhibition as a cancer therapy. In summary, these findings have provided a more precise understanding of the role of ATR in genome stabilization as well as new tools to study it.
Dependence of structure-forming repeats on ATR for stability
The repeat categories most dependent on ATR for stability were purine-rich structure-forming repeats and those that generate stem-loop structures. Purine-rich structure-forming repeats exhibited a similar sequence pattern: CN2–6G, with the intervening regions made up of (AG)n or ACA segments (e.g. CAGG, CAGAGG, CAGAGAGG, CACAG). Repeats of these sequences generate strand-selective intramolecular structures that impede DNA replication (Figs. 3 and 4). Notably, increased polymerase-helicase uncoupling and ssDNA generation through partial polymerase inhibition (aph) may facilitate the formation of such purine-rich structures, which further inhibits polymerase progression and increases fork collapse under ATR inhibition (Figs. 1, 2 and 4). Interestingly, secondary structure formation and polymerase blockage on the purine-rich strand would not necessarily impede synthesis from the complementary pyrimidine-rich strand, producing daughter strand gaps ahead of the purine-rich repeat that would lack available complementary sequence for reannealing. Accordingly, significant repeat-strand bias was observed in 16 of 18 RPLs where asymmetric RPA-ChIP signal intensity was observed around the central (CAGAGG/CCTCTG)n repeat (Supplemental Fig. S4F, p < 0.0009). It seems likely that strand-selective structure formation may be the underlying cause of robust RPA accumulation upon collapse at this type of repeat, but other models are certainly plausible.
The structures formed by purine-rich repeats are non B-form according to CD and TDS studies (Fig. 3, Supplemental Fig. S4) and are unlikely to involve G quartets given G-quadruplex-promoting ions (e.g. Li) and small molecules did not foster structure formation (data not shown). Principal component analysis of the CD signatures of over 60 known DNA structures were not consistent with the (CAGAGG)n signature (personal communication, Dr. J. Brad Chaires, University of Louisville). However, while this structure remains to be determined, a somewhat related purine-rich repeat in humans, (AGCGAGGG)n, forms a tetrahelical structure (Kocman and Plavec, 2014, 2017) that exhibits many biophysical similarities to murine (CAGAGG)n, suggesting that the structures may be conserved between species.
In contrast to the unusual strand-specific structures formed by purine-rich repeats, the other main category of ATR-dependent repeats, namely inverted repeats and AT-rich palindromes and quasi-palindromes, are predicted to form structures on both complementary strands (Fig. 6, 7 and Supplemental Fig. S7). These hairpin structures are well known to be sufficient to cause genomic instability (Kato et al., 2012; Lobachev et al., 2007). Notably, similar sequences not expected to form hairpins, such as poly(A)n, poly(T)n and head-to-tail LINE, SINE, LTR and Alu elements, were not enriched in BrITL retrievals after ATR inhibition (Fig. 7C, Supplemental Fig. S3A and data not shown). This correlation indicates that structural formation is a key aspect of dependence on ATR for stability. Additional research is required to determine the mechanism by which BrITL-associated repeats cause fork collapse and whether structure formation is sufficient for ATR dependence or requires other associated characteristics.
Notably, hairpin-forming sites in mouse and human cells did not accumulate substantial amounts of RPA in comparison to purine-rich sites, but were easily detected by BrITL (Fig. 6, 7 and Supplemental Fig. S7). These findings suggest that DNA resection is impaired at hairpin-forming sites, putatively by the close-ended structures themselves. Alternatively, it is possible that purine-rich repeats and strand-selective structure formation somehow fosters unusually high levels of RPA accumulation compared to other breakage sites. In either case, the distinct structural characteristics of these two main categories of ATR-dependent repeats (strand-selective structures vs. hairpin-forming) correlates with preferential detection by RPA or BrITL. This correlation indicates that both the structural causes and biochemical consequences of fork collapse at these two categories of repeats may be distinct.
RPLs, BrITL and Common Fragile Sites
CFS are associated with incomplete synthesis in M phase, which can lead to breaks either through endonuclease cleavage or physical strain at ultrafine bridges (Glover et al., 2017; Letessier et al., 2011; Wyatt et al., 2017; Ying et al., 2013). Accordingly, low origin density and difficult-to-replicate sequences have been proposed to cause CFS breakage (Glover et al., 2017, Letessier et al., 2011). In regards to the latter possibility, ATR-dependent BrITL sites were marginally enriched in human CFS (25 expected and 35 observed, Supplementary Table S7), with eight sites located in most expressive breast epithelial CFS (FRA1D, FRA2I, FRA5E, Hosseini et al., 2013). However, it is prudent to note that this enrichment is only 10 sites more than expected by chance (Table 1 and Supplementary Table S7). In addition, RPL and BrITL sites were not enriched in highly expressed murine CFS (14 expected and 9 observed, Supplementary Table S7). While it is possible that spreading of RPA and BrITL signals across these large regions were too weak to call, another possibility is that ATRi-driven fork collapse is not substantially more common in CFS than in other parts of the genome. Notably, non-peak-associated sensitivity to ATRi (ATRi vs. DMSO, 20–30kb from peak regions, Fig. 6A, 7A and data not shown) was not remarkably different from the effect of ATR suppression on CFS breakage (Casper et al., 2002). This viewpoint is in line with similar replication rates in CFS and the genome at large and that CFS breakage best correlates with large inter-origin distances (Letessier et al., 2011). Regardless of these potential mechanisms of CFS breakage, our unbiased identification of sites that are most sensitive to ATR inhibition pinpoints new, specific vulnerabilities both within CFS and throughout the genome.
Potential impact of RPL and BrITL sites on aging and cancer treatment
A variety of recent studies have implicated “replication stress” as a cause of age-related pathologies (Burhans and Weinberger, 2007; Flach et al., 2014; Ruzankina et al., 2007). Although the molecular causes of such stress has remained relatively obscure (Burhans and Weinberger, 2007), both triplet repeat expansions and ribosomal repeat instability have been associated with age-related pathologies (Flach et al., 2014; Orr and Zoghbi, 2007). It is interesting to speculate that the difficult-to-replicate sequences identified herein might contribute to such replicative stress, where replication fork collapse and the ensuing DNA damage response would degrade regenerative capacity. Identification of these ATR-dependent sites and causative repeat sequences provides tools to study their effects on tissue homeostasis.
Identification of ATR-dependent sites also has significant implications for cancer treatment. ATR inhibitors have entered clinical trials for the treatment of a variety of cancers. Although it is evident that replication fork collapse and cell cycle checkpoint abrogation are key components of the mechanism of ATRi action, difficult-to-replicate sequences that rely on ATR for stability are similarly part of that mechanism. Given that the lengths of microsatellite and minisatellite repeats are frequently polymorphic, it is intriguing to speculate that cancer cell-associated expansions of such repetitive sequences may help predict benefit from ATRi-based therapies. Accordingly, clinical response to ATRi would not be solely based on lethal interactions with defective gene products, but also with the state of RPL and BrITL-associated repeats.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eric Brown (brownej@upenn.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines –
Female ATRflox/-Cre-ERT2+ mouse embryonic fibroblasts (4–3 cells) (Smith et al., 2009); Tim knockdown (shTIM1) 4–3 cells (Smith et al., 2009); I-PpoI 4–3 cells were generated by transducing retrovirus expressing the I-PpoI restriction enzyme from the pBabe-ddIPpoI plasmid (Addgene plasmid #49052, (Goldstein et al., 2013)) into 4–3 cells. 4–3 cells used for BrITL were stably transduced with retrovirus expressing Bcl-xL (Addgene plasmid #8790, 3541 pMIG Bcl-XL) to decrease the amount of apoptotic cells collected. MDA-MB-231 cells (Laboratory of Andy J. Minn, University of Pennsylvania). U-2OS (HTB-96) osteosarcoma cells (female) were purchased from and validated by American Tissue Type Collection.
Treatment - 4–3
cells were treated with either DMSO or 1 μM ATR-45 (Charrier et al., 2011) and 0.2 μM aphidicolin (Calbiochem, CAS 38966–21-1) for 18 hrs; 1μM ATR-45 and 0.2 μM aphidicolin for 9 hrs; 1 μM ATR-45 for 18 hrs; and 0.2 μM aphidicolin for 18 hrs. I-PpoI 4–3 cells were treated with 1 μM Shield-1 (Laboratory of Tom Wandless, Stanford), and 0.5 μM 4-hydroxytamoxifen (4-OHT, EMD Chemicals/Calbiochem) for 14 hrs to induce nuclear expression of I-PpoI. Parental 4–3 cells were similarly treated with 1 μM Shield-1 and 0.5 μM 4-OHT for 14 hrs as a control. MDA-MB-231 cells were treated with either DMSO or 0.5 μM VE-822 (Selleck Chemicals, S7102) and 0.2 μM aphidicolin (Calbiochem, CAS 38966–21-1) for 9 hrs.
Culture –
All cells were grown in Dulbecco’s Modified Eagle’s medium (DMEM, Mediatech) supplemented with 10% fetal bovine serum (FBS, Benchmark, Gemini BioProducts), L-glutamine (2 mM, Mediatech), and streptomycin/penicillin (100 U/ml, Thermo Fisher Scientific) at 37°C/5% CO2. U-2OS ells were cultured in buffered Dulbecoo’s Modified Eagle Medium (Gibco), supplemented with 10% FBS (Hyclone), at 37˚C/5% CO2.
METHOD DETAILS
RPA ChIP-Seq –
This assay was performed on 4–3 cells under the following conditions: 1) 1 μM ATR-45 + 0.2 μM aphidicolin for 18 hrs; 2) 1 μM ATR-45 + 0.2 μM aphidicolin for 9 hrs; 3) 1 μM ATR-45 for 18 hrs; 4) 0.2 μM aphidicolin for 18 hrs; 5) DMSO; and 6) Tim knockdown 4–3 cells treated with 1 μM ATR-45 for 18 hrs. For each condition described, two biological replicates were performed, except for 1), in which three replicates were obtained.
For each immunoprecipitation reaction, 15 × 106 cells were trypsinized, collected, spun down and re-suspended in 25 mL PBS. Cells were fixed in 1% formaldehyde for 10 minutes at 37˚C and the reaction was stopped by adding glycine to 1% final concentration. The cell pellet was washed in 10 ml PBS and subsequently re-suspended in 1 ml cold PBS. The pellet was then lysed in lysis buffer (50 mM HEPES pH 7.9, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NPZ40, 0.25% Triton X-100) for 10 minutes on ice. The nuclei were recovered by spinning and washing twice (10 mM Tris-Cl pH 8.1, 200 mM NaCl, 1 mM EDTA pH 8.0, 0.5 mM EGTA pH 8.0). The nuclei were re-suspended in 1 ml shearing buffer (0.1% SDS, 1 mM EDTA, 10 mM Tris pH 7.6), and chromatin sheared using Covaris S220 to <4 kb using parameters according to the company hand book. Buffer (0.1% SDS, 1 mM EDTA, 10 mM Tris pH 7.6, 11% Triton X-100, 1.1% Na-DOC) was added to 1/10 volume to keep the sample in Radioimmunoprecipitation assay (RIPA) buffer. Dynabeads Protein A beads were pre-bound the night before by mixing 1 ml PBS, 10 μl 100 mg/ml BSA in PBS, 20 μg anti-RPA32 antibody (NA19L, Millipore), and 10 μg bridging antibody, rotating overnight at 4˚C. The next day, the beads were washed as follows: 2x with 1 ml of RIPA buffer, 2x with 1 ml of RIPA buffer + 0.3 M NaCl, 2x with 1 ml of LiCl buffer (0.25 M LiCl, 0.5% NP-40, 0.5% NaDOC, stored at 4˚C), 1x with 1 ml of TE (pH 8.0) + 0.2 % Triton X-100, 1x with 1 ml of TE (pH 8.0). The beads were then incubated with Proteinase K at 65˚C to reverse cross-link. DNA was extracted using phenol/chloroform and precipitated with ethanol/sodium acetate. Pellets were re-suspended in TE (pH 8.0) and prepared into libraries for NGS analysis, described below.
BrITL –
This assay was performed on 4–3 Bcl-xL cells treated with 1 μM ATR-45 (ATRi) + 0.2 μM aphidicolin, or DMSO (control), for 18 hrs and on MDA-MB-231 cells treated with 0.5 μM VE-822 (ATRi) + 0.2 μM aphidicolin, or DMSO (control), for 9 hrs. Two biological replicates of each condition were performed and prepared into libraries for NGS analysis, described below. This assay was also performed on I-PpoI-transduced 4–3 cells and parental 4–3 cells treated with 1 μM Shield-1 and 0.5 μM 4-hydroxytamoxifen, or DMSO (control), for 14 hrs. Three biological replicates of each condition were performed and processed for qRT-PCR analysis, described below.
For each BrITL reaction, ~2 × 106 cells were trypsinized and collected in an Eppendorf tube. Cells were washed with PBS, permeabilized in 0.1% Triton-X-100 in PBS for 5 minutes on ice and subsequently washed with 0.01% Triton-X-100 in PBS. Cells were incubated in a reaction containing 20 μM ddNTPs (Affymetrix, 77126) in 1x NEBuffer 2 for 5 minutes at 37°C. The reaction was stopped with 20 mM EDTA. Cells were washed four times with 0.01% Triton-X-100 in PBS before resuspending the cell pellet in a reaction mixture containing 2.5 mM CoCl2 (Roche, 11243306001) and 27 μM biotin-16-ddUTP (Enzo Life Sciences, ENZ-42813) in 1x TdT buffer (Roche, 11243276001). Upon addition of 150 units of TdT (Roche, 03333566001), the end-labeling reaction proceeded for 1 hour at 37°C. Cells were then washed twice with 50 mM EDTA in 0.01% Triton-X-100 in PBS. To lyse the cell pellet, TNE buffer (50 mM Tris-HCl pH 7.4, 100 mM NaCl, 0.1 μM EDTA) was added together with 10% SDS and 10 mg/ml Proteinase K for incubation overnight at 37°C. The next day, genomic DNA was extracted using phenol/chloroform followed by ethanol/sodium acetate precipitation. The pellet was re-suspended in TE (pH 8.0). Sonication occurred in the Biorupter (Diagenode) for 2 minutes on medium setting to obtain 0.2–2 kb fragments. After sonication, the samples were purified with Ampure XP beads (Beckman Coulter, A63880), utilizing 0.8x SPRI:DNA ratio. Washed and dried beads were incubated with EB buffer and left at room temperature for up to an hour before placing at 4°C overnight. The next day, the eluate was retrieved from the beads and brought up to 100 μl volume with TE. From this volume, 15 μl was aliquoted into a separate tube containing 85 μl TE and stored at 4°C to serve as the input. The rest of the sample was brought up to 200 μl with TE and proceeded to the next steps for retrieval.
Selection of biotin-labeled DNA fragments was performed with the Dynabeads KilobaseBinder kit (Life Technologies, 601–01). For this, 25 μl of streptavidin-coated magnetic beads were washed twice with 200 μl Binding buffer containing 5 μg/ml tRNA. The beads were then mixed in 200 μl sample plus 200 μl Binding buffer and left at room temperature on a rotating wheel for 2 hrs. The samples were then placed against a magnetic stand and the supernatant discarded. The beads were washed twice with wash buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA, 2 M NaCl) with 5-minute rotations at room temperature for each wash. The beads were then transferred to a new tube containing wash buffer with 4 μg/ml tRNA and subsequently washed in distilled autoclaved water twice. The washed and dried beads were then re-suspended in TE. To these samples, 20 μg boiled RNase A was added. Samples were incubated at 37°C for 30 minutes to remove RNA contaminants. Elution of biotinylated fragments bound to streptavidin-coated magnetic beads occurred by adding 1% SDS and 1 mg/ml Proteinase K to the samples and incubating at 55°C overnight. The next day, DNA was purified by sequential phenol, phenol/chloroform, and chloroform extraction before subsequent ethanol/sodium acetate precipitation. The DNA pellet was re-suspended in 50 μl of TE.
qRT-PCR analysis –
Real-time PCR was performed on the Applied Biosystems 7900HT Sequence Detection System. All PCR reactions were performed in duplicate for each sample with 0.9 μM of the forward and reverse primers in a final volume of 10 μl using SYBR Green PCR Master Mix (Applied Biosystems, 4309155). Primer sets are described in Key Resource Table.
NGS library preparation –
Libraries were prepared for both input and IP DNA according to the NEBNext kit. Briefly, DNA was sonicated to ~200 bp. DNA was end-repaired using a combination of T4 DNA polymerase, Klenow DNA polymerase and T4 polynucleotide kinase. The blunt, phosphorylated ends were treated with Klenow fragment (3’ to 5’ exo minus) and dATP to yield a protruding ‘A’ base for ligation of Illumina’s adapters which have a single ‘T’ base overhang at the 3’ end. After adapter ligation, DNA fragments of ~200 bp (insert plus adaptor) were band-isolated from a 2% agarose gel. The purified DNA was PCR amplified with Illumina primers for 18 cycles. The purified DNA was captured on an Illumina flow cell for cluster generation and sequenced on Illumina HiSeq 100 bp single-end following the manufacturer’s protocols.
Transfection of (CAGAGG)105 or scrambled sequence vectors –
HFUGW lentiviral expression vectors containing either the (CAGAGG)105 repeat or a scrambled sequence containing the same nucleotide content and length (Key Resource Table) were linearized by digestion with NdeI and transfected into 4–3 cells using lipofectamine transfection (Thermo Fisher Scientific, L3000001). GFP+ cells were sorted on a FACSAria machine to select for cells containing integrated HFUGW vector sequences, which express EGFP through the UBC promoter.
Southern Blot –
Southern blot performed on HFUGW-transfected cells utilized a biotinylated probe (Biotin DecaLabel DNA Labeling Kit; Thermo Fisher Scientific, K0651) between the PstI sites of the HFUGW vector. Briefly, 10 μg of genomic DNA from transfected cells were digested with 60 units of PstI for 2 hours at 37°C. After phenol/chloroform extraction and ethanol/sodium acetate precipitation of digested DNA, DNA was run on an 0.8% TBE gel overnight at 20 V. Gel was soaked in denaturization buffer (0.5 M NaOH, 1.5 M NaCl, pH 13.0) for 30 minutes twice. The gel was then rinsed with dH2O before being soaked in neutralization buffer (0.5 M Tris-HCl (pH 7.5), 1.5 M NaCl) for 15 minutes twice. Gel was rinsed with dH2O before being soaked in 20X SSC buffer for 30 minutes. Transfer to Hybond N+ membrane (Fisher Scientific, 45–000-927) occurred overnight in 20X SSC buffer. The membrane was UV-crosslinked (120,000 μJ). The blot was then pre-hybridized with boiled 100 μl of 10 mg/ml salmon sperm in 15 mL of pre-hybridization buffer (5X SSC, 5X Denhardt’s solution, 0.5% SDS) for 3 hours at 65°C. Probe mix was made by boiling 20 μg of mouse COT-1 DNA with 1 μg of biotinylated probe for 10 minutes, placed on ice for 2 minutes, and incubated with 1 mL pre-hybridization buffer for 1 hour at 37°C. The probe mix was then added to the blot and rotated overnight at 65°C. The next day, the membrane was washed with 0.4X SSC, 0.1% SDS for 15 minutes at 65°C and three more times for 30 minutes each at 65°C. Hybridized biotinylated probe on the membrane was detected using the Biotin Chromogenic Detection kit (Thermo Fisher Scientific, K0661).
Bioinformatics –
Peak-calling:
ChIP and BrITL libraries from at least 2 biological replicates of each experimental condition and their respective inputs were sequenced through Illumina HiSeq, generating 100 bp single-end sequencing reads. Adapter contamination in reads were trimmed using trimmomatic (Bolger et al., 2014) and reads were checked for quality control using fastqc (Leggett et al., 2013). Alignment was made to the mm10 reference genome for mouse samples or hg19 reference genome for human samples using STAR-2.5.2a Aligner with at most 3 mismatches (Dobin et al., 2013). Reads that contain non-unique sequences were initially allowed to be placed in up to 100 different genomic regions in order to later measure differences in regional read accumulation between multi-mapping of a single read with up to 100 placements and mapping of a single read to its most likely genomic placement. In the context of these experimental regions, which consist of non-unique repetitive genomic regions, measuring the difference between tracks with reads that are mapped up to 100 different placements and those with reads that are placed in their most likely home will reveal enrichment bias toward low complexity regions (i.e. if it is solely due to the low complexity nature of these regional sequences and not due to the experimental enrichment). Reads were then filtered by mapq score 10 to keep the high-confident read mappings. De-duplication of reads in the aligned tracks took place to increase the complexity of the read population. Additional alignment-specific quality-control metrics were conducted, including strand-cross-correlation (Landt et al., 2012), finger-plots (Ramirez et al., 2016) to gauge mutual back-level of enrichment across samples, Pearson and Spearman correlations of genomic and enriched regions across samples (0.6), principal component analysis (PCA) for clustering assessment, and a non-arbitrary estimate of ChIP signal over input using an NCIS-generated normalization ratio (Liang and Keles, 2012). Black-listed regions in the mm10 and hg19 genome were filtered out prior to peak-calling.
For enrichment analysis, the biological replicates and inputs of each experimental condition underwent an irreproducibility rate (IDR) analysis (Landt et al., 2012) from the ENCODE project with the MACS2 peak-calling program (Zhang et al., 2008) to give the final peak list per condition. IDR thresholds of >0.05 were used for self-consistency within each biological replicate and for comparison between biological replicates, and >0.005 for pooled-consistency analysis. Peaks that passed IDR thresholds and that were within a defined region (up to 5 kb) were merged. For RPA ChIP-Seq on 4–3 cells, merged peaks were further selected for p-value <10−20 and >4-fold signal enrichment over input. For BrITL on 4–3 cells, merged peaks were selected for p-value < 10−8 and >4-fold signal enrichment over input. For BrITL on MDA-MB-231 cells, merged peaks were selected for p-value <10−6 and >4-fold signal enrichment over input. Peaks were selected that were greater than or equal to 500 bp, which is the average fragment size for BrITL sonication and retrieval. For all experiments, the final peak list was generated by subtraction of peaks that were also identified in the DMSO-controls. Ratio tracks were generated using deepTools: bigwigCompare, 500 bp bin size.
Enrichment of complex repeats in RPA ChIP samples:
Trimmed fastq reads from each RPA ChIP-Seq sample that overlapped with different families of complex repeats (LSU_rRNA, SSU_rRNA, tRNA, etc.) were counted for each family of repeats. These numbers were then divided by the total number of reads with at least one reported alignment in each sample. Values from different biological replicates in each condition (ATRi+aph18hrs and DMSO (UT)) were averaged and normalized by the values calculated in the respective input samples. The resulting fold over input values for each family of complex repeats were graphed for each condition.
REQer:
To understand simple repeat sequences that may be enriched in the experimental conditions relative to input, an assessment of the sequence presence within individual reads was performed. Reads in fastq files were labeled according to how many times a repeat occurs as a single unit (monomer), or as different tandem units based upon the maximum length of the repeat in a 100 bp read, using a python script that incorporated regular expressions. This program was called REQer.
Tandem simple repeat analysis that measured repeats occurring purely in tandem was performed on trimmed and de-duplicated RPA-ChIP Seq reads from combined biological replicates of each condition (ATRi+aph18hrs and DMSO (UT)). The analysis was conducted by counting the number of different tandem lengths of a particular repeat sequence present in the reads and measuring the frequency of each length by dividing by the total count of the repeat sequence present in the reads. At each progressive tandem length of the repeat, the ratio of its frequency in ATRi+aph18hrs and in UT over the frequency in their respective inputs was calculated to obtain fold over input enrichment.
Total simple repeat analysis of trimmed and de-duplicated RPA-ChIP Seq reads from combined biological replicates of each condition (ATRi+aph18hrs and DMSO (UT)) was conducted by categorizing reads by the total unit count of a particular repeat sequence within a read. The frequency was calculated as the fraction of reads within the total number of reads from the combined replicates that contained the specified count of repeat units. At each progressive total repeat count per read, the ratio of its frequency in ATRi+aph18hrs and in UT over the frequency in their respective inputs was calculated to obtain fold over input enrichment.
Fork-pausing Experiments –
Plasmid constructions:
630 bp of CAGAGG tandem repeats were cloned into the BspEI and BssHII site of the pML113 plasmid in opposite orientations (Follonier et al., 2013) for the origin-proximal insertion, and into the BamHI site for the origin-distal insertion. Randomized controls of the same nucleotide composition and length were similarly constructed.
In vitro assay:
Templates for polymerase reactions were created by cloning [CAGAGG]15 repeats into the MCS/BamH1 site of the pGEM3Zf(−) vector (Promega, P2261). Inserts in two orientations were isolated in order to purify ssDNA templates of both strands. As controls, randomized sequences of the same nucleotide composition and length were similarly cloned. Subsequently, a C to A mutation at the 5’ BamHI site (GGATCC) and a G to T mutation at the 3’ BamHI site (GGATCC) flanking the repeat insert were introduced, in order to disrupt the potential for G-quadruplex formation between the vector and insert sequences. For each construct, single-stranded DNA was isolated after R408 helper phage (Promega, P2291) infection of plasmid-bearing SURE cells (e14-(McrA-), Δ(mcrCB-hsdSMR-mrr)171, endA1, gyrA96, thi-1, supE44, relA1, lac, recB, recJ, sbcC, umuC::Tn5 (Kanr) uvrC [F’ proAB lacIqZΔM15 Tn10 (Tetr) Amy Camr]; Agilent Technologies, 200152). Log phase plasmid-bearing SURE cells in 2XYT media were infected with 1/50th volume of R408 (titer of phage stock was >1 × 1011 plaque forming units (pfu)/mL) and incubated in a 37°C shaker for 3 hours to overnight. An overnight incubation was necessary for optimal yields of ssDNA from the [CAGAGG]15 strand. After pelleting the bacterial cells, virus particles in the supernatant were precipitated on ice for 30 min with a polyethylene glycol (Sigma, P5413)/ammonium acetate solution at final concentrations of 4% and 0.75M, respectively. Virus was pelleted and resuspended in an appropriate volume of Phenol Extraction Buffer (PEB; 100mM Tris, pH 8.0, 300mM NaCl, 1mM EDTA, pH 8.0). DNA was extracted one time with two volumes of phenol (Affymetrix/Thermo-Fisher, AAJ75829AN) saturated with PEB, one time with one volume of phenol, and one time with half volume 24:1 chloroform: isoamyl alcohol. After extraction, DNA was precipitated with ammonium acetate at 2.0M final concentration and 2 volumes of ethanol and resuspended in 10mM Tris and 1mM EDTA, pH 8.0 (Eckert and Gestl, 2010). Small ssDNA preparations from independent clones were sequenced to verify integrity of the insert prior to large scale purification of ssDNA templates. Repeat lengths longer than 15 units precluded our ability to rescue ssDNA of the correct sequence and/or length.
DNA synthesis templates were created by 32P end-labeling (γ32P ATP (6000Ci/mmol); Perkin-Elmer, BLU002Z001MC) a PAGE-purified 43mer oligonucleotide (G94–43mer, Integrated DNA Technologies) using T4 Polynucleotide Kinase (Thermo-Fisher, 18004010) according to the manufacturer’s instructions and hybridizing to ssDNA at a 1:1 molar ratio in 1X SSC buffer (150mM NaCl and 15mM sodium citrate). The G94–43mer oligonucleotide initiates synthesis 68 nucleotides downstream of the repeat inserts. To remove unincorporated radionucleotide, the hybridized primer-templates were purified over illustra Microspin G-50 columns (GE Healthcare, 27–5330-01). Primer extension reactions contained 100 fmol of primed ssDNA substrate, 400 fmol human recombinant PCNA (Xu et al., 2001), 1700 fmol yeast RFC (Thompson et al., 2012), 20 mM Tris HCl, pH 7.5, 8 mM MgCl2, 5 mM DTT, 40 μg/ml BSA, 150 mM KCl, 5% glycerol, 0.5 mM ATP, and 250 μM dNTPS, and were preincubated at 37°C for 3 min. Synthesis was initiated upon addition of 100 fmol purified 4-subunit recombinant human Pol δ4 (Zhou et al., 2012). Aliquots were removed at 3, 7, and 15 minutes, quenched in 1 volume STOP dye (Formamide, 5mM EDTA pH 8.0, 0.1% xylene cylanol, 0.1% bromophenol blue) and reaction products were separated on an 8% denaturing polyacrylamide gel and quantitated using a Molecular Dynamics STORM 860 Phosphoimager. A control for the percent of primers productively hybridized to each primer-template substrate (% Hyb) was performed using excess Exo- Klenow polymerase (Affymetrix/Thermo-Fisher, 70057Z), and a background control for primer impurities (no Pol) was performed by incubating unextended primer-template substrate in reaction buffer without addition of polymerase. Dideoxy sequencing reactions were carried out simultaneously with the Pol δHE reactions, using the same primer-template substrates and Sequenase 2.0 (Affymetrix/Thermo-Fisher, 70775Y). Total percent extension was calculated as the amount of total extended DNA molecules (corrected for percent hybridization and background) divided by this number plus the amount of corrected primer molecules. The number of DNA molecules within four regions were determined from the 15 minute reaction using ImageQuant 5.2 software quantitation: (R1) 68–11 bases 5’ to the insert; (R2) 10–1 bases 5’ to the insert; (R3) the insert; and (R4) all bases 3’ to the insert up to and including the well. After background correction, the termination probability within each region was calculated as the [number of molecules within the region ÷ by the number of molecules within the region plus all longer molecules]. To normalize for the different sizes of Regions 1–3, each region’s termination probability was divided by the number of nucleotides under consideration. For example, the termination probability/nt for Region 1 = [molecules in R1 ÷ molecules in R1+R2+R3+R4] ÷ 58 nucleotides.
Ex vivo assay:
The SV40-derived pML113, 114 and 115 vectors (Follonier et al., 2013) were gifts from Massimo Lopes (University of Zurich). For the ori-proximal vectors, a 630 bp fragment encoding CAGAGG tandem repeats (Key Resource Table) was cloned into the pML114 and 115 plasmids using the MCS/BspEI and BssHII sites, creating plasmids with the repeats in two orientations. As controls, randomized sequences of the same nucleotide composition and length were created (Table S5) and similarly subcloned into pML114/115 vectors. For the ori-distal vectors, the repeats were cloned into the BamHI site of pML113, in two orientations. Subconfluent U-2OS cells (ATCC, HTB-96) were transfected with 5μg vector DNA using Xtreme Gene XP (ver. 8) transfection reagent (Roche). To induce replication stress, cells were treated with 0.6 μM Aphidicolin (Sigma; DMSO solvent) 24 hours post-transfection, or 0.5μM VE-822, 21 hours post-transfection, followed by Aphidicolin treatment three hours later. For all experiments, DNA was isolated 48 hours post-transfection using a modified Hirt method, as described (Chandok et al., 2011). Briefly, adherent cells were washed with 1M Tris-buffered saline (50 mM Tris-HCl, pH 7.0 and 150 mM NaCl) followed by lysis in 50 mM Tris–HCl pH 7.0, 20 mM EDTA, 10 mM NaCl, 10% SDS, 0.2 mg/ml proteinase K (Sigma-Aldrich). Chromosomal DNA was precipitated by incubation of lysate overnight in 5M NaCl followed by centrifugation at 27, 200 xg for 50 minutes at 4°C. Resulting supernatants were incubated in the presence of ~0.1 mg/mL proteinase K for 2–3 hours at 55°C. DNA was extracted by phenol-chloroform extraction and precipitated in an equal volume of isopropanol with 0.5 μL polyacryl carrier (Molecular Research Center, Cincinatti, OH). To analyze replication intermediates, the purified DNA was digested with DpnI, EcoRI, and EcoN1 (ori-proximal vectors; New England Biolabs) or DpnI, PpuMI, and SacII (ori-distal vectors; New England Biolabs) for 3 hours, followed by ethanol precipitation. DNA products were resuspended in Tris-EDTA and separated first through a 0.4% TBE agarose gel (1V/cm, 14 hr, room temperature, -EtBr) and second through a 1% TBE agarose gel (4V/cm, 6–8hr, 4˚C, +EtBr) (Friedman and Brewer, 1995). DNA fragments were transferred by capillary action overnight to a Hybond-N+ membrane (Amersham/GE Healthcare). After UV crosslinking, pre-hybridization of the membranes was carried out by incubation with a 0.25M sodium phosphate (pH 7.2)/ 7% SDS/ 100 μg/ml sonicated sperm DNA buffer, and incubation at 65˚C, ≥2 hrs. DNA probes were made by isolating the indicated restriction fragments and using a random labeling method (High Prime; Roche) with α32P dCTP (6000Ci/mmol); Perkin-Elmer) followed by purification using a Microspin G-50 column (GE Healthcare). Membranes were hybridized using 100–200 ng probe at 65˚C overnight (with rotation), washed according to manufacturer’s instructions, and imaged using a Molecular Dynamics STORM 860 Phosphoimager.
Biophysical Characterization of DNA Secondary Structure –
DNA and buffers for structural studies:
All DNA sequences used for biophysical characterization are summarized in Table S3. DNA samples were ordered from IDT (Texas, USA) as HPLC purified samples, dissolved in water at 1 mM final concentration and stored at −80°C. Samples were diluted to the desired concentration into final 10 mM lithium cacodylate buffer pH 7.2 supplemented with 100 mM KCl and 2 mM MgCl2 (100K2Mg buffer). Samples were annealed at 90°C for 5 minutes, cooled slowly to room temperature over the course of 3–4 hrs and equilibrated overnight at 4°C. All samples were examined with circular dichroism (CD) for consistency in folding.
Circular Dichroism wavelength scans:
All experiments were performed on an Aviv 410, Aviv 435, or a Jasco 815 spectropolarimeter with a Peltier heating unit using 1 cm quartz cuvettes. The accuracy of the external temperature probe was ± 0.3 K. Each CD trace was an average of 3 – 5 scans collected from 220 to 330 nm with 1–2 nm bandwidth, 1 nm step, 1 second averaging time at 4°C. CD data were treated as described elsewhere (Nicoludis et al., 2012).
Thermal stability of DNA via CD melting:
Thermal denaturation experiments were collected at the maxima of CD wavelength scans with 2 nm bandwidth, 5 second equilibration time, 1°C step, and 15 or 20 second averaging time. The samples were heated from 4°C to 95°C, maintained at 95°C for less than 5 minutes, and cooled to 4°C at the same rate. The cooling step was included to determine the reversibility of folding/unfolding process. Superposition of melting and cooling data suggested that the folding process was reversible. Melting data were analyzed assuming a two-state model with constant enthalpy, ΔH (heat capacity, Cp = 0) (Ramsay and Eftink, 1994). This model suggests that at any point during melting or cooling only folded and unfolded DNA is present (no intermediates, i.e. two-state system). Starting and final baselines (assuming to be linear), melting temperature (Tm) and enthalpy of unfolding were adjusted to get the best fits. The melting curves were also analyzed by the derivative method. The Tm was determined as the minima or maxima on the first derivative curves. These values were read by eye and are associated with an error of ±0.5°C. When the melting transition was poorly defined, the derivative method was deemed more reliable. Melting and cooling data were processed separately and the hysteresis (difference in Tm between the heating and cooling curves) was determined.
Thermal stability of DNA via UV-vis melting:
Studies were performed on a Cary 300 spectrophotometer thermostated with an external waterbath (temperature accuracy of ±0.3°C). We monitored 260, 295, and 330 nm wavelengths. The latter wavelength was used as a reference to monitor instrument performance; the extinction coefficient of DNA at 330 nm is negligible. Melting studies were completed in 1 cm pathlength quartz cuvettes, data were typically collected from 4°C to 95°C and back to 4°C with 2 nm bandwidth, 1°C temperature step, 2 seconds averaging time, and 0.75 or 1°C/min rate. Melting and cooling curves were collected to determine the reversibility of the melting process. The temperature was measured with the temperature sensor inserted into the cuvette filled with water and placed next to DNA samples. The signal at 330 nm was subtracted from each data set. The resulting data were analyzed as described above. The results from UV-vis study are in great agreement with the results from CD melting study.
Molecularity of DNA structures via UV-vis melting:
Samples were annealed in 100K2Mg buffer at concentrations ranging from 1.4 to 23.6 μM for CA5 and from 0.3 to 6.9 μM for CA10; targeted concentration ratio between the most dilute and the most concentrated sample was ∼20. Samples were placed in quartz cuvettes with 1.0 or 0.2 cm pathlength depending on strand concentration and equilibrated at 4°C for at least 20 minutes. Melting experiments and data analysis was performed as described above.
Polyacrylamide gel electrophoresis:
Native PAGE gels were typically prepared at 12% polyacrylamide in 1xTAC (50 mM Tris Acetate pH 7.3) buffer supplemented with 3 mM MgCl2. Running buffer consisted of 1xTAC with 3 mM MgCl2. Gels were cooled with a water bath and premigrated for 30 minutes at 140 V. Each DNA sample of 10 μL contained ∼3 μg of annealed oligo in 100K2Mg buffer to which 3 μL of 50% w/v sucrose was added immediately prior to loading. Oligothymidylate markers 5’ dTn (where n = 15, 24, 30, 57 or 60, and 90) as well as a 76-nt tRNA were used as internal migration standards. Typically, gels were run for 3–4 hrs at 140–300 V; gel temperature did not exceed 16°C. Gels were stained using Stains-All and de-colored in water under visible light. Gels were visualized on ETNA-NS ChemiBis 3.2 gel visualization device (using lower light, 580 nm filter) or with an iPhone 5 camera.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details can be found in figure legends and in Method Details.
RPA ChIP-Seq –
For ChIP-Seq, two biological replicates normalized by a single input from each experimental condition underwent an irreproducibility rate (IDR) analysis (Landt et al., 2012) from the ENCODE project with the MACS2 peak-calling program (Zhang et al., 2008) to give the final peak list per condition. IDR thresholds of >0.05 were used for self-consistency within each biological replicate and for comparison between biological replicates, and >0.005 for pooled-consistency analysis. Peaks that passed IDR thresholds and that were within a defined region were merged. Merged peaks were further selected for p-value <10−20 and >4-fold signal enrichment over input. The final peak list was generated as a set intersection and subtraction from the DMSO-control peak list.
For REQer analysis, ChIP-Seq reads from two biological replicates of ATRi+aph18hrs were combined and quantified for short tandem repeat presence. This was compared against similarly quantified ChIP-Seq reads from the inputs, allowing for calculation of the fold enrichment of short tandem repeats in ChIP reads over input reads. For these values graphed at the minimum and maximum range of repeat units within a sequencing read (Supplemental Fig. S3B-D), p-values were obtained at 95% confidence interval using the Kolmogorov-Smirnov test between the distributions of ATRi+aph18hrs and its input.
To assess the statistical significance of the overlap of RPL or BrITL peaks with specific genomic features (Table 1 and Fig. 2A), we performed permutation tests in which RPL or BrITL peaks were randomly shuffled across the mouse or human reference genome 100,000 times. We determined the number of overlapping peaks between each set of shuffled RPL or BrITL sites and the associated data set, thereby generating an empirical distribution of the overlap under the null hypothesis that the RPL or BrITL peaks were randomly distributed throughout the genome. The p-value of each comparison is the number of times the overlap between the two datasets occurred more than observed. A small p-value indicates that the observed overlap between RPL or BrITL sites and the associated dataset is statistically significant and is not due to random chance. For box-plot analysis of short tandem repeat length within RPL peaks identified from ATRi+aph18hrs versus the background genome (Fig. 2G), a two-sided Wilcoxon test was performed.
BrITL –
For qPCR graphs, experiments were performed in triplicate and the data are represented as mean +/− SEM. For the significance of breakage associated with transfected sequences (scrambled or (CAGAGG)105) by BrITL qRT-PCR, dot plots were made and a Student’s T-test was performed between the averages of each condition (n=3 for Scrambled, DMSO; n=5 for Scrambled, ATRi+aph; CAGAGG, DMSO; CAGAGG, ATRi+aph), where n is the number of replicates from each condition. Outliers were not included in the calculation of the averages.
For bar graph analysis of repeat count within human BrITL peaks identified from ATRi+aph9hrs versus the count in the background genome (Fig. 7C), the following calculations were performed to obtain statistical values: bedtools shuffle and bedtools getfasta generated randomly selected genomic regions (hg19) (excludes unmappable regions) with equivalent footprint size as the human ATRi+aph9hrs BrITL peaks (5,268,109 iterations). The MISA- and HOMER2-identified motif repeats were then counted throughout each randomly generated peak list. The occurrences of the repeat count being >= or <= than the repeat count in the ATRi+aph9hrs BrITL peaks were recorded. The empirical p-value was then calculated by dividing these occurrences by the number of randomly generated peak sets (5,268,109).
Biophysical Characterization of DNA Secondary Structure –
Each biophysical experiment was done a minimum of three times for each oligo using at least two independently synthesized DNA stocks. Peak molar ellipticity values, melting temperatures, and enthalpies of unfolding were determined as the arithmetic mean of these trials. Errors reported for these values correspond to one standard deviation from the mean. Native gels were run a minimum of two times.
Fork-pausing Experiments –
In vitro assay:
Three independent Pol δHE reactions were performed for each primer-template examined. Differences in the normalized termination probabilities among template sequences and various regions were analyzed statistically using a two-way ANOVA and significance was defined as producing a p-value of < 0.05. The columns in Figure 4C indicate the mean of the three replicate reactions and error bars indicate the SEM. The statistical details can be found in the Results section and in the legend to Figure 4.
Ex vivo assay–
ImageQuant 5.2 software was used to determine the amount of radioactivity in the descending arm of the simple Y arc, relative to radioactivity in the double Y arc extending above the simple Y apex. Background radioactivity was calculated and subtracted for each blot prior to calculating the Replication Fork Barrier (RFB) index. RFB values shown are the average of two or three biological replicates (independent U2OS cell transfections).
KEY RESOURCES TABLE
The table highlights the genetically modified organisms and strains, cell lines, reagents, software, and source data essential to reproduce results presented in the manuscript. Depending on the nature of the study, this may include standard laboratory materials (i.e., food chow for metabolism studies), but the Table is not meant to be comprehensive list of all materials and resources used (e.g., essential chemicals such as SDS, sucrose, or standard culture media don’t need to be listed in the Table). Items in the Table must also be reported in the Method Details section within the context of their use. The number of primers and RNA sequences that may be listed in the Table is restricted to no more than ten each. If there are more than ten primers or RNA sequences to report, please provide this information as a supplementary document and reference this file (e.g., See Table S1 for XX) in the Key Resources Table.
Please note that ALL references cited in the Key Resources Table must be included in the References list. Please report the information as follows:
REAGENT or RESOURCE: Provide full descriptive name of the item so that it can be identified and linked with its description in the manuscript (e.g., provide version number for software, host source for antibody, strain name). In the Experimental Models section, please include all models used in the paper and describe each line/strain as: model organism: name used for strain/line in paper: genotype. (i.e., Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J). In the Biological Samples section, please list all samples obtained from commercial sources or biological repositories. Please note that software mentioned in the Methods Details or Data and Software Availability section needs to be also included in the table. See the sample Table at the end of this document for examples of how to report reagents.
SOURCE: Report the company, manufacturer, or individual that provided the item or where the item can obtained (e.g., stock center or repository). For materials distributed by Addgene, please cite the article describing the plasmid and include “Addgene” as part of the identifier. If an item is from another lab, please include the name of the principal investigator and a citation if it has been previously published. If the material is being reported for the first time in the current paper, please indicate as “this paper.” For software, please provide the company name if it is commercially available or cite the paper in which it has been initially described.
- IDENTIFIER: Include catalog numbers (entered in the column as “Cat#” followed by the number, e.g., Cat#3879S). Where available, please include unique entities such as RRIDs, Model Organism Database numbers, accession numbers, and PDB or CAS IDs. For antibodies, if applicable and available, please also include the lot number or clone identity. For software or data resources, please include the URL where the resource can be downloaded. Please ensure accuracy of the identifiers, as they are essential for generation of hyperlinks to external sources when available. Please see the Elsevier list of Data Repositories with automated bidirectional linking for details. When listing more than one identifier for the same item, use semicolons to separate them (e.g. Cat#3879S; RRID: AB_2255011). If an identifier is not available, please enter “N/A” in the column.
-
○A NOTE ABOUT RRIDs: We highly recommend using RRIDs as the identifier (in particular for antibodies and organisms, but also for software tools and databases). For more details on how to obtain or generate an RRID for existing or newly generated resources, please visit the RII or search for RRIDs.
-
○
Please use the empty table that follows to organize the information in the sections defined by the subheading, skipping sections not relevant to your study. Please do not add subheadings. To add a row, place the cursor at the end of the row above where you would like to add the row, just outside the right border of the table. Then press the ENTER key to add the row. You do not need to delete empty rows. Each entry must be on a separate row; do not list multiple items in a single table cell. Please see the sample table at the end of this document for examples of how reagents should be cited.
TABLE FOR AUTHOR TO COMPLETE
Please upload the completed table as a separate document. Please do not add subheadings to the Key Resources Table. If you wish to make an entry that does not fall into one of the subheadings below, please contact your handling editor. (NOTE: For authors publishing in Current Biology, please note that references within the KRT should be in numbered style, rather than Harvard.)
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-RPA32 (RPA ChIP on MEF 4–3 cells) | Millipore | NA19L-100UG; RRID:AB_565123 |
| Mouse IgG2a Isotype Control from murine myeloma antibody (Pre-clear for RPA ChIP on MEF 4–3 cells) | Sigma Aldrich | M5409; RRID:AB_1163691 |
| Bacterial and Virus Strains | ||
| R408 Helper Phage | Promega | Cat# P2291 |
| SURE 2 Supercompetent Cells | Agilent Technologies | Cat# 200152 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 4-OHT | EMD Chemicals/Calbiochem | 579002–5MG |
| Shield-1 | Laboratory of Tom Wandless, Stanford | N/A |
| ATR45 (ATR inhibitor for MEFs) | Ohio State University (OSU); Charrier et al., 2011 | ATR45 |
| VE-822 (ATR inhibitor for MDA-MB-231 cells) | Selleck Chemicals | Cat# S7102 |
| Aphidicolin (for RPA ChIP and BrITL experiments) | Calbiochem | CAS 38966–21-1 |
| Biotin-16-ddUTP | Enzo Life Sciences | ENZ-42813 |
| TdT | Roche | Cat# 03333566001 |
| Polyethylene Glycol (Avg Mol Wt of 8000) | Sigma | Cat# P5413; CAS 25322–68-3 |
| Aphidicolin (for Fork Pausing experiments) | Sigma | Cat#A0781; CAS 38966–21-1 |
| Equilibrated Phenol, pH 8.0, Ultrapure | Affymetrix/Thermo-Fisher | Cat# AAJ75829AN; CAS 108–95-2 |
| γ32P ATP (6000Ci/mmol) | Perkin-Elmer | Cat# BLU002Z001MC |
| T4 Polynucleotide Kinase | Thermo-Fisher | Cat# 18004010 |
| Recombinant human PCNA | Laboratory of Marietta Lee | Biochemistry 2001 40: 4512–4520 |
| Recombinant yeast RFC | Laboratory of Linda Bloom | The Journal of Biological Chemistry 2012 287: 2203–9 |
| Sequenase 2.0 | Affymetrix/Thermo-Fisher | Cat# 70775Y |
| Exo- Klenow Polymerase | Affymetrix/Thermo-Fisher | Cat# 70057Z |
| Critical Commercial Assays | ||
| NEBNext Library KIt | NEB | E6000S |
| Dynabeads KilobaseBinder kit | Life Technologies | 601–01 |
| Ampure XP Beads purification kit | Beckman Coulter | A63880 |
| Lipofectamine™ 3000 Transfection Reagent | Thermo Fisher Scientific | L3000001 |
| Biotin Chromogenic Detection kit | Thermo Fisher Scientific | K0661 |
| High Prime DNA labeling kit version 10 | Roche Diagnostics | REF: 11585592001 |
| Biotin DecaLabel DNA Labeling Kit | Thermo Fisher Scientific | K0651 |
| Dynabeads Protein A for Immunoprecipitation | Invitrogen | 100–02D |
| Deposited Data | ||
| RPA ChIP-Seq raw and processed files | This paper | GEO: GSE100790 |
| BrITL raw and processed files | This paper | GEO: GSE115623; GSE115624 |
| Mendeley deposited data | This paper | https://data.mendeley.com/datasets/9gwyx4vxt8/draft?a=7789056e-b4d3–4f8b-9735–992304ad2679 |
| Experimental Models: Cell Lines | ||
| MEF 4–3 cells (ATRflox/-Cre-ERT2+) | Smith et al., 2009 | N/A |
| TIM KD MEF 4–3 cells | Smith et al., 2009 | N/A |
| I-PpoI MEF 4–3 cells | This paper | N/A |
| MDA-MB-231 cells | Andy J. Minn, Univ. of Pennsylvania | N/A |
| U-2OS osteosarcoma cells | ATCC | HTB-96 |
| Fetal Bovine Serum | GE Life Sciences/Hyclone | Cat. No. SH 30071.03 |
| Oligonucleotides | ||
| Primers for BrITL qRT-PCR, see below | This paper | N/A |
| Oligos used for biophysical studies, see Table S3 | Integrated DNA Technology | N/A |
| dTn gel electrophoresis ladder: 5’-dT15, 5’-dT24, 5’-dT30, 5’-dT57, 5’-dT60, 5’-dT90 |
Integrated DNA Technology | N/A |
| G94–43mer, PAGE-Purified 5’-GGA TAA CAA TTT CAC ACA GGA AAC AGC TAT GAC CAT GAT TAC G-3’ |
Integrated DNA Technologies | N/A |
| Scrambled and (CAGAGG)105 insert sequences, see below | This paper | N/A |
| PSH1 probe primer set: Forward: 5’-TTCCAGTGATGGAAGCACCC-3’ Reverse: 5’-CTTGGCTTGGCACACAGAAC-3’ |
Integrated DNA Technologies | N/A |
| HFUGW probe primer set (against UBC promoter): Forward: 5’-CATAAGACTCGGCCTTAGAACC-3’ Reverse: 5’-GCCAGAATTTAGCGGACAATTT-3’ |
Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| pBabe-ddIPpoI | Addgene, Goldstein et al., 2013 | #49052 |
| pMIG Bcl-XL | Addgene | #8790 |
| pGEM-3Zf(-) Vector | Promega | Cat# P2261 |
| pML113, pML114, pML115 | Massimo Lopes, Univ. of Zurich | Nature Structural and Molecular Biology 2013 20: 486–494. |
| Software and Algorithms | ||
| Trimmomatic | Bolger et al., 2014 | N/A |
| Fastqc | Leggett et al., 2013 | N/A |
| STAR-2.5.2a Aligner | Dobin et al., 2013 | N/A |
| MACS2 peak-calling | Zhang et al., 2008 | N/A |
| IDR analysis | Landt et al., 2012 | N/A |
| Homer2 motif-finding | N/A | |
| The M-fold Web Server (DNA folding form) | Zuker, 2003 | N/A |
| MISA | Beier et al., 2017 | N/A |
| ImageQuant version 5.2 | GE Healthcare | N/A |
| Other | ||
| Illustra Microspin G-50 column | GE Healthcare | Cat# 27–5330-1 |
| Xtreme Gene XP version 8 | Roche Diagnostics | REF: 06366236001 |
Primers Used in BrITL qRT-PCR, related to Figures 6 and 7
| RPL Peak Genomic Location (mm9) | Distance relative to (CAGAGG)n repeat region | Forward Primer (5' -> 3') | Reverse Primer (5' -> 3') |
| Chr7: 35943530 – 35946581 | 20 kb 5' | GCAAGCCATGGAACTCATCTA | TGGCATGAAGACACCAAGAG |
| 1,260 bp 5' | CCCTGGATGTGGTTGTCATTA | GGAACAGAACTGCTCACAAATG | |
| 220 bp 5' | ACACCCAGTGAACCAAAGTATAG | CCACCAGTCTTGACTTACTCAC | |
| 230 bp 3' | GACAAGTGCCTGAGGGATAAA | ACTCAGGAGACAGAGGGTTAT | |
| 530 bp 3' | AGAATCCTCGGGTGGAAATG | CAGTGGCTCCTGTTGTGTAT | |
| 20 kb 3' | GCTGAGGCAGGGATAGATTTG | AGGCTTGAGAAGAGTTGAAGATAAG | |
| Chr6: 87722438 – 87728725 | 20 kb 5' | CCGACTGACCTGTGCTATTT | CATGTGGTTGCTGGGATTTG |
| 1,450 bp 5' | GTGATTGCCACCAAACCTAATG | CTGGGAAAGGCAGAGAACTAC | |
| 285 bp 5' | CCCAGCACTTGGTAAGTAGAG | AACTCTCGCAGCCGTTTAT | |
| 970 bp 3' | GCCCAAGGCTCTACCATAAA | GCCTGGAACCTACCTTCATAC | |
| 20 kb 3' | CACAGACACGCACACATAGA | TGCTAGGGCCTGAGAATAGA | |
| Chr11:5641800–5644000 | 20 kb 5' | TCCTTCCATTTCTCTCCATTCC | CACGGTCAATTCGGACTTCT |
| 430 bp 5' | TGGGCTCTGCCTACTTACTA | GCCCTCAGAGAAGAAGAGAATG | |
| 115 bp 5' | GGCTGGATTCAAGGCAGTAA | CCTCCCAAGTGCTGAGATTAAA | |
| 575 bp 3' | TGGGCAGCGAAGATGAAAT | GCCAAGCCACTCCCTTATT | |
| 20 kb 3' | GTGCAGAGACTATGGAGGAATG | GCAAAGGATCCTGGGTTGTA | |
| 20 kb 3' | CCTTTGCAGGAAGTAGGTCAA | AGAGATGCCAGGAGCAAATC | |
| RPL Peak Genomic Location (mm9) | Distance relative to (CAGG)n repeat region | Forward Primer (5' -> 3') | Reverse Primer (5' -> 3') |
| Chr9:121838100–121841700 | 20 kb 5' | GTGACTTTATCTCCACCGTACTC | CTACTGGAGTGAACCTGGAATG |
| 460 bp 5' | GCAGCTTGGCTCTGTATCTAT | GTGTGTGTGTTCAGAGGTCTAT | |
| 165 bp 5' | CCTGTCTCTGACACAATGTACTC | CCCAATACACCGCTCCTTT | |
| 230 bp 3' | GCCTCTAGTGATACATCTCCTCTA | GATAAGGCCATCTGCAGTGAT | |
| 360 bp 3' | CAAAGTGCCTCTCGCCTAAA | GCCTGGCTTCACAAGGATAG | |
| 20 kb 3' | CCTTTGCAGGAAGTAGGTCAA | AGAGATGCCAGGAGCAAATC | |
| RPL Peak Genomic Location (mm9) | Distance relative to (CACAG)n repeat region | Forward Primer (5' -> 3') | Reverse Primer (5' -> 3') |
| Chr17: 13737678–13751955 | 20 kb 5’ | CTTTCTGTGTCCGTGTCCTATC | CACCAACCGTAGAATGGGTATC |
| 1,085 bp 5’ | TTCAAGGTGAGAGAGATGGATTG | GCCTTCTCCTGTGCTGTATT | |
| 600 bp 5’ | GAAGTATCAGGCAGCAAGGAA | CTGACTTTGACGGCAGGATAA | |
| 0 bp 3’ | GCTGTCTCTGCTGTCTGTAATG | GCTACTCTTGTCCCATGTTTCC | |
| 500 bp 3’ | GCCTCCAGGTTGCCTAAATA | GCTCTCTGTTGCTATGGTGAA | |
| 1,400 bp 3’ | CCCGCTAATTCCTCTGAAGTC | TCTTCCCTGACTGGTCCTATT | |
| Chr6: 29694103–29706050 | 30 kb 5’ | GTCTTCTGTAGGACCAGAGTTTG | GAAGTCCAGAAGAGAGCATCAG |
| 20 kb 5’ | GCCACAGGTATTCATCCAAGA | GCCTTTATCGCTCAGGTATGT | |
| 1,200 bp 5’ | GCCAGGACTACACAGAGAAAC | TGGAGATCCAACTCCCTCTT | |
| 170 bp 5’ | ATCCACCACCTCCAACAAG | GAATTCCACCCATAGGCTCATA | |
| 190 bp 3’ | AGGTGCAGTGACATGAGTTC | CCTATGGGATCCTGCTATCTCTA | |
| 400 bp 3’ | CCCTAGATGGCTCCTTAATTGT | GAACTCACAGAGATCCACCTTC | |
| 1,015 bp 3’ | CCTCTGCTCACTCCAATCAAT | TGAGATTTGACGCCCTCTTC | |
| 20 kb 3’ | AGTCTTCTCCCACCCTTGA | GGCGCTACCTCCATGTTATT | |
| 30 kb 3’ | CCCTAACTGGTCTGGGATTTG | CACCTTTCACCACAGCACTA | |
| I-PpoI site in rDNA sequence | Distance relative to I-PpoI site | Forward Primer (5' -> 3') | Reverse Primer (5' -> 3') |
| 11.5 kb 3' | CACGCTGTCCTTTCCCTATT | GACAGACCCAAGCCAGTAAA | |
| 7 kb 3' | CTGAGAAACGGCTACCACATC | GCCTCGAAAGAGTCCTGTATTG | |
| 780 bp 3' | ACAGCCTCTGGCATGTTG | GCCAATCCTTATCCCGAAGTTA | |
| 70 bp 3' | CTAGCAGCCGACTTAGAACTG | CAGAAATCACATCGCGTCAAC | |
| 60 bp 5' | CCTACCTACTATCCAGCGAAAC | CTACACCTCTCATGTCTCTTCAC | |
| 120 bp 5' | GGGAAAGAAGACCCTGTTGAG | GGCCTCCCACTTATTCTACAC | |
| 750 bp 5' | GAACGTGAGCTGGGTTTAGA | CTCTCGTACTGAGCAGGATTAC | |
| 20 kb 5' | GAAACCAAAGCGACCTGAAAC | CAGCCATCTTGTCTGCTAACT | |
| HFUGW vector | Distance relative to insert site | Forward Primer (5' -> 3') | Reverse Primer (5' -> 3') |
| 1,764 bp 5’ | TGTACTGGGTCTCTCTGGTTAG | AGCACTCAAGGCAAGCTTTA | |
| 909 bp 5’ | GCTTTGTTCCTTGGGTTCTTG | TTCTGCTGCTGCACTATACC | |
| 338 bp 5’ | GAGTTAGGCAGGGATATTCACC | TCTCTCTCTCCACCTTCTTCTT | |
| 153 bp 3’ | CATAAGACTCGGCCTTAGAACC | GTTCCGCTCTCTGGAAAGAA | |
| 959 bp 3’ | CTAGGGTAGGCTCTCCTGAAT | ACCGGAGCTTCAGCTACTTA | |
| Break Peak Genomic Location (hg19) | Distance relative to repeat region | Forward Primer (5' -> 3') | Reverse Primer (5' -> 3') |
| Chr12:121692970–121693250 | 730 bp 5’ | AGGCAACAGGGCAAGAAT | GAGATCAGCCAGTACACATCAG |
| 220 bp 5’ | AGGCAGACCCACAATCTAATC | CAGGAAGCATCCAGATAGGAAA | |
| 30 kb 3’ | CAGGGACATGGACATGACTAAC | GGCCTTGGGACAGAAGAAATA | |
| Chr6:55491300–55494100 | 20 kb 5’ | GTCTTCAGCTGTCAGTCTCTTC | GCTGAGATGCTGGTCCTTTAT |
| 230 bp 5’ | TCTAATCAGCCTCCAGGGAATA | GAAGCATCCAGCACAAGAGA | |
| 20 kb 3’ | GCACCCACTAATGTGTCATCTA | GAGATCACTTGGACACAGGAAG | |
| Chr6:81329562–81332945 | 30 kb 5’ | AACTCACTCGTTACCACAAGG | GTCTCCACCCAAATCTCATCTC |
| 715 bp 5’ | GGGCAAGAGTCTCATGTGTAT | CTCAGCCTTTCTGGTCTTGT | |
| 520 bp 3’ | TCACCAACACCACTTCCTTATC | CTCCCAGTCATTGTGCTCTC |
Scrambled and (CAGAGG)105 Insert Sequences, related to Figures 4 and 6, and to Supplemental Figures S5 and S6
Scrambled insert sequence:
(scrambled sequence underlined; PacI sites in bold)
TTAATTAAATAGGATCCTCCGGAGCCGGAGCGGCAGAAGAGAGAGAGGAGAGCAGCGAAGGACGAGCGGGCAGCACGGGAGGCGCGAAGACCAGAGACGCGGGACGGAGGAGGCGCGGCGACAGAACGAAGCGAAGCGGGAGGGGCGGGAAGAGGGAGGACGAAGGCAGAAGAAACGCAAAGAACAGAGGGAAGGACGGGCAAAACAACGGGCAGCCCGGGAGGGAAGGAAAGGAGGAGAGAGCGGGAGGGAAGAGAAAAGAGGGCGGAGGCGGCGGCGAGCGGAGGGAGGGCGGGAGAGCGACGGAGAAGAAACGAGGGAGAAGCAGGAGAGGGCCGGAAGGGAGACACAGCCCAAGAGGAAGCGGACGGAAAGAGCGAGCGAAAGCGGAGAGGGAGAACAAGGGAAGACAAGGCGGAAGGGCAGGGCGGGAAGAGGAGGGCGCGCGGAGGAGGCGAAAGGCGGGACAGAGCAGAGACGGAGGGCGAGGGAGGAGAAGGGACGAGAGGCCCCCGGACGGGAGGGCGGAGGCGGAGGGAGAGAAGAGGGCGAACAGGAGAGAGCAGGGAAAGCCGAGGCGAGAGACCGGCGAGAAAGAGGGAACGACGGCCGAGGAAAAGGCGAAGCCGGACGCACAGGGAGAGGCGAAGAGGGCGCGCGGATCCTATTTAATTAA
(CAGAGG)105 insert sequence:
(repeat sequence underlined; PacI sites in bold)
TTAATTAAGAATTCGATATCAAGCTTCTCGAGGGTACCTCCGGAGGGTGCTTTCTGCTTAAAAGTGATGAAAGCCAGCCGGGCTGTAGTGACACACACCTTTAATCCCAGCACTTGGGAGACAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGAGGCAGGTAGGTCTCTATGAGTTGCGCGCGGTACCCTCGAGAAGCTTGATATCGAATTCTTAATTAA
Supplementary Material
Research Highlights:
>500 ATR-dependent sites were identified in the mouse and human genomes
ATR inhibitor-driven fork collapse occurs primarily at structure-forming repeats
Repeat structures include non-B form DNA and hairpins (AT-rich and inverted repeats)
Discrete repeat types accumulate RPA differentially upon fork collapse
Acknowledgements
This project was funded by grants from the NIH (EJB: 2R01AG027376, 1R01CA189743; NS: 5R25CA101871; Y-CT: T32ES019851; LAY: 1R15CA208676). Additional funding was provided through the Pennsylvania Department of Health (EJB), Center of Excellence in Environmental Toxicology (P30ES013508, EJB), Basser Center for BRCA Research (EJB), Abramson Family Cancer Research Institute (EJB), Camille and Henry Dreyfus Teacher-Scholar Award (LAY), and Jake Gittlen Cancer Research Foundation (KAE). We are grateful to Dr. Jean-Louis Mergny (IECB, Bordeaux, France) in whose laboratory some of these studies were conducted (LAY). We would also like thank Dr. J. Brad Chaires for CD/TDS principal component comparisons to existing signatures and Dr. Beverly Emanuel (CHOP) for helpful insights. The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Declaration of Interests
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
Mendeley Deposited Data:
https://data.mendeley.com/datasets/9gwyx4vxt8/draft?a=7789056e-b4d3–4f8b-9735–992304ad2679
Accession Numbers
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO SuperSeries accession number GSE115625.
Subseries linked to GSE115625 are as follows:
RPA-ChIP Seq data of ATR+/− MEFs are deposited under accession number GSE100790.
BrITL data of ATR+/− MEFs are deposited under accession number GSE115624.
BrITL data of MDA-MB-231 cells are deposited under accession number GSE115623.
ENCODE Mouse Datasets –
DNaseI hypersensitivity:
ENCODE Project Consortium, 2012; wgEncodeEM001936; Stamatoyannopoulous Laboratory, ENCODE/UW
H3K4me3 ChIP-Seq:
ENCODE Project Consortium, 2012; wgEncodeEM001455; Ren Laboratory, ENCODE/LICR
H3K27ac ChIP-Seq:
ENCODE Project Consortium, 2012; wgEncodeEM002501; Ren Laboratory, ENCODE/LICR
Replication timing (Repli-Chip):
ENCODE Project Consortium, 2012; wgEncodeEM002974; Gilbert Laboratory, ENCODE/FSU
CTCF ChIP-Seq:
ENCODE Project Consortium, 2012; wgEncodeEM001698; Ren Laboratory, ENCODE/LICR
ENCODE Human Datasets –
H3K9me3 ChIP-Seq:
ENCODE Project Consortium, 2012; wgEncodeEH002875; Farnham Laboratory, ENCODE/SYDH
H3K27ac ChIP-Seq:
ENCODE Project Consortium, 2012; wgEncodeEH002872; Farnham Laboratory, ENCODE/SYDH
H3K27me3 ChIP-Seq:
ENCODE Project Consortium, 2012; wgEncodeEH002922; Farnham Laboratory, ENCODE/SYDH
H3K36me3 ChIP-Seq:
ENCODE Project Consortium, 2012; wgEncodeEH002923; Farnham Laboratory, ENCODE/SYDH
H3K4me3 ChIP-Seq:
ENCODE Project Consortium, 2012; wgEncodeEH000967; Stamatoyannopoulous Laboratory, ENCODE/UW
CTCF ChIP-Seq:
ENCODE Project Consortium, 2012; wgEncodeEH002057; Stamatoyannopoulous Laboratory, ENCODE/UW
References
- Admire A, Shanks L, Danzl N, Wang M, Weier U, Stevens W, Hunt E, and Weinert T (2006). Cycles of chromosome instability are associated with a fragile site and are increased by defects in DNA replication and checkpoint controls in yeast. Genes & Development 20, 159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacolla A, Tainer JA, Vasquez KM, and Cooper DN (2016). Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences. Nucleic Acids Research 44, 5673–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier S, Thiel T, Munch T, Scholz U, and Mascher M (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EJ, and Baltimore D (2000). ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes & Development 14, 397–402. [PMC free article] [PubMed] [Google Scholar]
- Burhans WC, and Weinberger M (2007). DNA replication stress, genome instability and aging. Nucleic Acids Research 35, 7545–7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper AM, Nghiem P, Arlt MF, and Glover TW (2002). ATR regulates fragile site stability. Cell 111, 779–789. [DOI] [PubMed] [Google Scholar]
- Chandok GS, Kapoor KK, Brick RM, Sidorova JM, and Krasilnikova MM (2011). A distinct first replication cycle of DNA introduced in mammalian cells. Nucleic Acids Research 39, 2103–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrier JD, Durrant SJ, Golec JM, Kay DP, Knegtel RM, MacCormick S, Mortimore M, O’Donnell ME, Pinder JL, Reaper PM, et al. (2011). Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. Journal of Medicinal Chemistry 54, 2320–2330. [DOI] [PubMed] [Google Scholar]
- Delihas N (2015). Complexity of a small non-protein coding sequence in chromosomal region 22q11.2: presence of specialized DNA secondary structures and RNA exon/intron motifs. BMC Genomics 16, 785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entezam A, and Usdin K (2008). ATR protects the genome against CGG.CCG-repeat expansion in Fragile X premutation mice. Nucleic Acids Research 36, 1050–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flach J, Bakker ST, Mohrin M, Conroy PC, Pietras EM, Reynaud D, Alvarez S, Diolaiti ME, Ugarte F, Forsberg EC, et al. (2014). Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 512, 198–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follonier C, Oehler J, Herrador R, and Lopes M (2013). Friedreich’s ataxia-associated GAA repeats induce replication-fork reversal and unusual molecular junctions. Nature Structural & Molecular Biology 20, 486–494. [DOI] [PubMed] [Google Scholar]
- Friedman KL, and Brewer BJ (1995). Analysis of replication intermediates by two-dimensional agarose gel electrophoresis. Methods in Enzymology 262, 613–627. [DOI] [PubMed] [Google Scholar]
- Glover TW, Wilson TE, and Arlt MF (2017). Fragile sites in cancer: more than meets the eye. Nature reviews. Cancer 17, 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein M, Derheimer FA, Tait-Mulder J, and Kastan MB (2013). Nucleolin mediates nucleosome disruption critical for DNA double-strand break repair. Proceedings of the National Academy of Sciences of the United States of America 110, 16874–16879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hile SE, and Eckert KA (2004). Positive correlation between DNA polymerase alpha-primase pausing and mutagenesis within polypyrimidine/polypurine microsatellite sequences. Journal of Molecular Biology 335, 745–759. [DOI] [PubMed] [Google Scholar]
- Hile SE, and Eckert KA (2008). DNA polymerase kappa produces interrupted mutations and displays polar pausing within mononucleotide microsatellite sequences. Nucleic Acids Research 36, 688–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini SA, Horton S, Saldivar JC, Miuma S, Stampfer MR, Heerema NA, and Huebner K (2013). Common chromosome fragile sites in human and murine epithelial cells and FHIT/FRA3B loss-induced global genome instability. Genes, Chromosomes & Cancer 52, 1017–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberman JA (1997). Mapping replication origins, pause sites, and termini by neutral/alkaline two-dimensional gel electrophoresis. Methods 13, 247–257. [DOI] [PubMed] [Google Scholar]
- Johnson JE, Smith JS, Kozak ML, and Johnson FB (2008). In vivo veritas: using yeast to probe the biological functions of G-quadruplexes. Biochimie 90, 1250–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Kurahashi H, and Emanuel BS (2012). Chromosomal translocations and palindromic AT-rich repeats. Current Opinion in Genetics & Development 22, 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katou Y, Kanoh Y, Bando M, Noguchi H, Tanaka H, Ashikari T, Sugimoto K, and Shirahige K (2003). S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 424, 1078–1083. [DOI] [PubMed] [Google Scholar]
- Kocman V, and Plavec J (2014). A tetrahelical DNA fold adopted by tandem repeats of alternating GGG and GCG tracts. Nature Communications 5, 5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocman V, and Plavec J (2017). Tetrahelical structural family adopted by AGCGA-rich regulatory DNA regions. Nature Communications 8, 15355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krings G, and Bastia D (2004). swi1- and swi3-dependent and independent replication fork arrest at the ribosomal DNA of Schizosaccharomyces pombe. Proceedings of the National Academy of Sciences of the United States of America 101, 14085–14090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri M, Gustafson TL, Majors ER, and Freudenreich CH (2004). Expanded CAG repeats activate the DNA damage checkpoint pathway. Molecular Cell 15, 287–293. [DOI] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. (2012). ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Research 22, 1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leggett RM, Ramirez-Gonzalez RH, Clavijo BJ, Waite D, and Davey RP (2013). Sequencing quality assessment tools to enable data-driven informatics for high throughput genomics. Frontiers in Genetics 4, 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letessier A, Millot GA, Koundrioukoff S, Lachages AM, Vogt N, Hansen RS, Malfoy B, Brison O, and Debatisse M (2011). Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 470, 120–123. [DOI] [PubMed] [Google Scholar]
- Liang K, and Keles S (2012). Normalization of ChIP-seq data with control. BMC Bioinformatics 13, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobachev KS, Rattray A, and Narayanan V (2007). Hairpin- and cruciform-mediated chromosome breakage: causes and consequences in eukaryotic cells. Frontiers in Bioscience 12, 4208–4220. [DOI] [PubMed] [Google Scholar]
- Mirkin SM (2007). Expandable DNA repeats and human disease. Nature 447, 932–940. [DOI] [PubMed] [Google Scholar]
- Mohanty BK, Bairwa NK, and Bastia D (2006). The Tof1p-Csm3p protein complex counteracts the Rrm3p helicase to control replication termination of Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences of the United States of America 103, 897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil AJ, Kim JC, and Mirkin SM (2017). Precarious maintenance of simple DNA repeats in eukaryotes. BioEssays 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoludis JM, Barrett SP, Mergny JL, and Yatsunyk LA (2012). Interaction of human telomeric DNA with N-methyl mesoporphyrin IX. Nucleic Acids Research 40, 5432–5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HT, and Zoghbi HY (2007). Trinucleotide repeat disorders. Annual Review of Neuroscience 30, 575–621. [DOI] [PubMed] [Google Scholar]
- Pope BD, Ryba T, Dileep V, Yue F, Wu W, Denas O, Vera DL, Wang Y, Hansen RS, Canfield TK, et al. (2014). Topologically associating domains are stable units of replication-timing regulation. Nature 515, 402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay G, and Eftink MR (1994). A multidimensional spectrophotometer for monitoring thermal unfolding transitions of macromolecules. Biophysical Journal 66, 516–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzankina Y, Pinzon-Guzman C, Asare A, Ong T, Pontano L, Cotsarelis G, Zediak VP, Velez M, Bhandoola A, and Brown EJ (2007). Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell 1, 113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Cortez D, and Cimprich KA (2017). The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nature reviews. Molecular Cell Biology 18, 622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, and de Lange T (2009). Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138, 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KD, Fu MA, and Brown EJ (2009). Tim-Tipin dysfunction creates an indispensible reliance on the ATR-Chk1 pathway for continued DNA synthesis. The Journal of Cell Biology 187, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilard RK, Jacques PE, Laramee L, Cheng B, Galicia S, Bataille AR, Yeung M, Mendez M, Bergeron M, Robert F, et al. (2010). Systematic identification of fragile sites via genome-wide location analysis of gamma-H2AX. Nature Structural & Molecular Biology 17, 299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JA, Marzahn MR, O’Donnell M, and Bloom LB (2012). Replication factor C is a more effective proliferating cell nuclear antigen (PCNA) opener than the checkpoint clamp loader, Rad24-RFC. The Journal of Biological Chemistry 287, 2203–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Narayanan V, Lobachev KS, and Mirkin SM (2008). Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proceedings of the National Academy of Sciences of the United States of America 105, 9936–9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh E, Wang X, Lee MY, and Eckert KA (2013). Mechanism of replicative DNA polymerase delta pausing and a potential role for DNA polymerase kappa in common fragile site replication. Journal of Molecular Biology 425, 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt HD, Laister RC, Martin SR, Arrowsmith CH, and West SC (2017). The SMX DNA Repair Tri-nuclease. Molecular Cell 65, 848–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Zhang P, Liu L, and Lee MY (2001). A novel PCNA-binding motif identified by the panning of a random peptide display library. Biochemistry 40, 4512–4520. [DOI] [PubMed] [Google Scholar]
- Yamane A, Robbiani DF, Resch W, Bothmer A, Nakahashi H, Oliveira T, Rommel PC, Brown EJ, Nussenzweig A, Nussenzweig MC, et al. (2013). RPA accumulation during class switch recombination represents 5’−3’ DNA-end resection during the S-G2/M phase of the cell cycle. Cell Reports 3, 138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Minocherhomji S, Chan KL, Palmai-Pallag T, Chu WK, Wass T, Mankouri HW, Liu Y, and Hickson ID (2013). MUS81 promotes common fragile site expression. Nature Cell Biology 15, 1001–1007. [DOI] [PubMed] [Google Scholar]
- Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, Sandstrom R, Ma Z, Davis C, Pope BD, et al. (2014). A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biology 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Saini N, Sheng Z, and Lobachev KS (2013). Genome-wide screen reveals replication pathway for quasi-palindrome fragility dependent on homologous recombination. PLoS Genetics 9, e1003979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Shishkin AA, Nishida Y, Marcinkowski-Desmond D, Saini N, Volkov KV, Mirkin SM, and Lobachev KS (2012). Genome-wide screen identifies pathways that govern GAA/TTC repeat fragility and expansions in dividing and nondividing yeast cells. Molecular Cell 48, 254–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Meng X, Zhang S, Lee EY, and Lee MY (2012). Characterization of human DNA polymerase delta and its subassemblies reconstituted by expression in the MultiBac system. PloS One 7, e39156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Research 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.