

Abstract
INTRODUCTION:
Primary liver tumors and liver metastasis currently represent the leading cause of cancer-related deaths. The liver intimately cross-talks with the gut and performs many essential functions related to digestion, metabolism of nutrients, and clearance of bacterial metabolites. Diseased livers are often associated with altered gut bacterial composition, or dysbiosis, and it has been suggested that gut bacterial products contribute to malignant transformation of hepatocytes. The liver is exposed to the gut microbiome through the portal vein and is an immunological organ that is heavily populated by immune cells. Emerging studies have shown that gut commensal bacteria are important regulators of antitumor immunity. Although it has been established that the gut microbiome influences the efficacy of cancer immunotherapy, the role of gut bacteria in antitumor surveillance in the liver is poorly understood.
RATIONALE:
The liver is exposed to gut bacterial metabolites and products by way of blood from the intestine, which comprises 70% of the whole liver blood supply. Changes in the gut microbiome may affect immune cell function in the liver, and commensal bacteria can mediate the metabolism of primary into secondary bile acids, which recirculate back into the liver through the enterohepatic circulation. Given that bile acids are known to be involved in liver cancer development, we focused on the role of bile acids in immunosurveillance of tumors growing in the liver. We altered gut bacteria and examined changes of hepatic immune cells and antitumor immunity directed against liver tumors. Uncovering how the gut microbiome uses bile acids to shape immunity to liver cancer may have future therapeutic applications.
RESULTS:
Using one primary liver model and three liver metastasis models, we found that altering commensal gut bacteria induced a liver-selective antitumor effect. A selective increase of hepatic CXCR6+ natural killer T (NKT) cells was observed, independent of mouse strain, gender, or presence of liver tumors. The accumulated hepatic NKT cells showed an activated phenotype and produced more interferon-γ upon antigen stimulation. In vivo studies using both antibody-mediated cell depletion and NKT-deficient mice confirmed that NKT cells mediated the inhibition of tumor growth in the liver. Further investigation showed that NKT cell accumulation was regulated by the expression of CXCL16, the solo ligand for CXCR6, on liver sinusoidal endothelial cells, which form the lining of liver capillaries and the first barrier for the blood coming from the gut entering the liver. Primary bile acids increased CXCL16 expression, whereas secondary bile acids showed the opposite effect. Removing gram-positive bacteria by antibiotic treatment with vancomycin, which contains the bacteria mediating primary-to-secondary bile acid conversion, was sufficient to induce hepatic NKT cell accumulation and decrease liver tumor growth. Feeding secondary bile acids or colonization of bile acid-metabolizing bacteria, reversed both NKT cell accumulation and inhibition of liver tumor growth in mice with altered gut commensal bacteria. In nontumor liver tissue from human patients with primary liver cancer, primary bile acid cheno-deoxycholic acid (CDCA) levels correlated with CXCL16 expression, whereas an inverse correlation was observed with secondary bile acid glycolithocholate (GLCA), suggesting that the finding may apply to humans.
CONCLUSION:
We describe a mechanism by which the gut microbiome uses bile acids as messengers to control a chemokine-dependent accumulation of hepatic NKT cells and antitumor immunity in the liver, against both primary and metastatic liver tumors. These findings not only have possible implications for future cancer therapeutic studies but also provide a link between the gut microbiome, its metabolites, and immune responses in the liver.
Primary liver tumors and liver metastasis currently represent the leading cause of cancer-related death. Commensal bacteria are important regulators of antitumor immunity, and although the liver is exposed to gut bacteria, their role in antitumor surveillance of liver tumors is poorly understood. We found that altering commensal gut bacteria in mice induced a liver-selective antitumor effect, with an increase of hepatic CXCR6+ natural killer T (NKT) cells and heightened interferon-γ production upon antigen stimulation. In vivo functional studies showed that NKT cells mediated liver-selective tumor inhibition. NKT cell accumulation was regulated by CXCL16 expression of liver sinusoidal endothelial cells, which was controlled by gut microbiome-mediated primary-to-secondary bile acid conversion. Our study suggests a link between gut bacteria-controlled bile acid metabolism and liver antitumor immunosurveillance.
Graphical Abstract
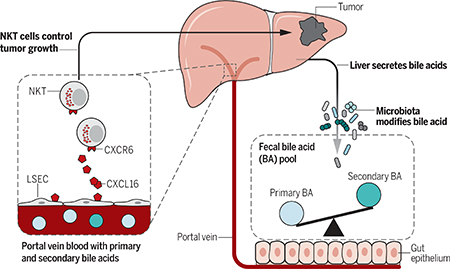
Gut microbiome modulates liver cancer through bile acid-regulated NKT cells. Gut microbiome uses bile acids as a messenger to regulate chemokine CXCL16 level on liver sinusoidal endothelial cells (LSEC) and thus controls the accumulation of CXCR6+ hepatic NKT cells. The accumulated NKT cells have an activated phenotype and inhibit liver tumor growth.
The gut microbiome has emerged as a critical factor regulating antitumor immunity controlling the efficacy of chemo- and immunotherapies (1–6). It is noteworthy that the liver is exposed to bacterial components and metabolites through the portal vein, and profound effects of the gut microbiome on hepatocellular carcinoma (HCC) have been described (7, 8). Secondary hepatic malignancies (liver metastases) account for 95% of all hepatic cancers, and the liver is the most common site for organ metastasis in the body (9). To evaluate how the gut microbiome shapes antitumor immunity in the liver, we investigated the effects of gut commensal bacteria on both primary HCC and liver metastasis in mouse models.
Alterations in the gut microbiome suppress liver tumors in multiple mouse models
Spontaneous HCC was induced using MYC transgenic mice as described before (10). An antibiotic cocktail (ABX, consisting of vancomycin, neomycin, and primaxin) was added to drinking water to deplete gut commensal bacteria (3). The antibacterial efficacy of ABX was confirmed, and the cocktail was not toxic to the liver (fig. S1, A to C). Consistent with previous findings, fewer and smaller HCC were found in ABX-treated MYC mice (Fig. 1A and fig. S1, D and E). We extended our studies to a subcutaneous implantation model (EL4 thymoma) to study potential systemic effects. ABX treatment did not affect the growth of subcutaneous EL4 tumors in syngeneic C57BL/6 mice (fig. S1F). In contrast, fewer spontaneous liver metastases were seen in mice with large subcutaneous EL-4 tumors upon ABX treatment (Fig. 1B). To confirm this liver-selective antitumor effect, we used an intrasplenic tumor injection model (11) and found a robust decrease of B16 liver metastasis (Fig. 1C). Unlike in the liver, lung metastases were increased by ABX when the same B16 tumor cells were injected into the tail vein (fig. S1, G and H). Similar results were observed in BALB/c mice using A20 tumors (12) (Fig. 1D and fig. S1, I and J). These findings suggest that modulating gut commensal bacteria can specifically modify growth kinetics of intrahepatic tumors.
Fig. 1. Depleting gut commensal bacteria inhibits liver tumor growth in multiple mouse models.

(A) MYC transgene was turned on at the age of 4 weeks. MYC-ON mice were fed with ABX or regular water. Ten weeks later, mice were killed and liver surface tumor nodules were counted. Representative liver images are shown. Data represent mean ± SEM of two pooled experiments. n = 5 for H2O, 6 for ABX. P < 0.05, Student’s t test. (B) C57BL/6 mice were treated with ABX or H2O for 3 weeks before receiving subcutaneous EL4 tumor cell injection. Four weeks later, liver metastases were determined. Representative images of five pooled experiments are shown. n = 17 for H2O, 12 for ABX. P < 0.05, Chi-square test. (C) ABX- or H2O-pretreated C57BL/6 mice were given intrasplenic B16 tumor cell injection. One and a half weeks later, liver metastases were measured. Representative images are shown. Data represent mean ± SEM of five pooled experiments n = 18 for H2O, 15 for ABX. P < 0.05, Student’s t test. (D) BALB/c mice were treated with ABX or H2O for 3 weeks. Then mice received A20 tumor cell tail vein injection. Three weeks later, liver metastases were counted. Representative images are shown. Data represent mean ± SEM of two pooled experiments. n = 8, P < 0.05, Student’s t test.
Hepatic NKT cell accumulation precedes tumor inhibition
To explore the mechanism behind tumor suppression, we studied immune cell subsets in EL4-tumor-bearing mice on ABX treatment. A prominent expansion of hepatic natural killer T (NKT) cells and CD8+ T cells was found (Fig. 2A and fig. S2A), whereas no changes were found in other immune subsets (B cells, CD4+ T, NK, γ/δ T cells and G-MDSC). The accumulation of hepatic NKT cells, but not CD8+ T cells, was also observed in ABX-treated MYC mice bearing HCC (Fig. 2B and fig. S2B), and splenic NKT cells remained unchanged, suggesting a liver-specific effect (fig. S2C).
Fig. 2. Hepatic NKT cells increase after depleting gut commensal bacteria.

(A) ABX- or H2O-pretreated C57BL/6 mice were given subcutaneous EL4 tumor injection. Two and a half weeks later, liver infiltrating immune cells were measured. Data represent mean ± SEM of three pooled experiments. n = 15 for EL4-H2O, 20 for EL4-ABX. P < 0.05, two-way analysis of variance (ANOVA). (B) Hepatic NKT, CD4, and CD8 T cell levels of MYC mice described in Fig. 1A. Data represent mean ± SEM of two pooled experiments. P < 0.05, two-way ANOVA. (C and D) Hepatic NKT cells in tumor-free C57BL/6 mice were fed with ABX or H2O. Data represent mean ± SEM of two pooled experiments. n = 9 for H20,10 for ABX. P < 0.05, Student’s t test. (E) Hepatic NKT cells in tumor-free BALB/c mice fed with ABX or H2O. Data represent mean ± SEM of two pooled experiments. n = 5, P < 0.05, Student’s t test. (F) Representative CXCR6 staining in hepatic NKT cells from three independent experiments. (G) Representative NKT cell staining in CXCR6+ liver infiltrating mononuclear cells from three independent experiments. (H) Composition of CXCR6+ liver infiltrating mononuclear cells in tumor-free C57BL/6 mice fed with ABX or H2O. DN T: double negative T cells. Data represent pooled results from three experiments. (I) Levels of CXCR6+ liver-infiltrating cells in tumor-free C57BL/6 mice fed with ABX or H2O. Data represent mean ± SEM of two pooled experiments. n = 9 for H20,10 for ABX. P < 0.05, Student’s t test. (J and K) IFN-γ and CD69 levels of hepatic NKT cells after in vivo stimulation by injecting aGalCer-loaded A20 tumor cells (Gal/A20) into vancomycin (Vanco) or H2O-fed BABL/c mice. Data represent mean ± SEM of two pooled experiments. n = 9, P < 0.05, Student’s t test.
To understand how the gut microbiome mediates hepatic NKT cell accumulation, tumor-free mice were used. ABX-treated C57BL/6 or BALB/c mice had more absolute and relative hepatic NKT cells than untreated mice, which was not observed in the spleen and was independent of gender (Fig. 2, C to E, and fig. S2, D, E, G, and H).
The chemokine receptor CXCR6 mediates NKT cell survival and accumulation in the liver (13), and we found that all hepatic NKT cells expressed CXCR6 (Fig. 2F) as expected. NKT cells make up the majority of hepatic CXCR6+ cells (~75%) (Fig. 2G), which did not change alter ABX treatment (Fig. 2H). ABX treatment caused a ~2-fold increase of CXCR6+ cells in the liver of C57BL/6 and BALB/c mice (Fig. 2I). CXCR6 is also expressed on T cells (14), and we observed that both hepatic CXCR6+CD62LlowCD44hi effector memory CD4+ and CD8+ T cells increased after ABX treatment (fig. S2, J to L). The observation that most NKT cells were effector memory CD44hiCD62Llow cells (fig. S3A) prompted us to check activation status. Almost all hepatic NKT cells were the activated CD69hi subtype in both tumor-bearing (fig. S3, B and C) and tumor-free mice (fig. S3D). CD25 and 4–1BB, two additional activation markers, did not change in hepatic NKT cells after ABX treatment (fig. S3, E and F). The observed higher CD69 expression but no change in 4–1BB activation level suggests that the NKT cells in ABX-treated mice are more active (Fig. 2K and fig. S3, J to K). NKT cells can exert diverse functions by rapidly releasing cytokines after activation. Thus, cytokine expression was measured from NKT cells after in vivo antigen-specific stimulation by injecting mice with α-galactosylceramide (aGalCer)-loaded tumor cells. Higher interferon-γ (IFN-γ) was detected in hepatic NKT cells from mice that received ABX treatment, whereas tumor necrosis factor (TNF) levels did not change (Fig. 2J and fig. S3, G to I). IFN-γ production by NKT cells has been shown to be instrumental for NKT-initiated tumor immunity. These data suggest that depleting gut commensal bacteria endows hepatic NKT cells with a stronger antitumor function. No change was observed for in vivo cytotoxicity of NKT cells after ABX treatment (fig. S3, L and M).
Finally, we studied NKT subpopulations based on the expression of transcriptional factors (15). The majority of hepatic NKT cells were NKT1, and the levels of NKT subsets did not change (fig. S3N). Interestingly, PLZF, which is required for NKT development (16), significantly decreased in the liver after ABX treatment (fig. S3O), which was not apparent in the spleen (fig. S3, P and Q).
Regulatory T cells (Tregs) are important modulators in tumor progression. The gut microbiome has been reported to affect Tregs (17); however, we found no change in Foxp3+CD4+ subsets in either the liver or spleen of ABX-treated C57BL/6 mice (fig. S3, R and S). Taken together, our results showed that altering the gut microbiome caused accumulation of hepatic NKT and effector memory CD4+ or CD8+ T cells. The activated status of NKT cells leads to higher levels of IFN-γ upon encounter with antigen-loaded tumor cells, thereby fostering a tumor-rejecting environment.
Hepatic NKT cells mediate tumor inhibition
Antibody-mediated cell depletion was performed to investigate the specific function of individual immune cell populations controlling liver tumor immunity in ABX-treated mice. ABX-pretreated C57BL/6 mice were given intrasplenic injection of B16 tumor cells. T cell depletion was performed 1 day before tumor injection (Fig. 3A). Removing all the three major hepatic T cell subsets (CD4+ T, CD8+ T, and CD4+ NKT cells) completely reversed the inhibition of liver metastasis caused by elimination of gut commensal bacteria (Fig. 3, B to D, and fig. S4, A to C), whereas depleting CD8+ T cells alone only had minor effects.
Fig. 3. Hepatic NKT cells mediate the inhibition of liver metastasis in mice.

(A to D) B16 tumor cells were intrasplenically injected into mice pretreated with ABX or H2O. One day before tumor injection, mice were given intraperitoneal injection of a combination of antibodies to CD4 (anti-CD4) (500 μg per mouse) and anti-CD8 (200 mg per mouse) or anti-CD8 alone (200 μg per mouse). Liver surface metastatic nodules were counted. Representative images are shown. Data represent mean ± SEM of two pooled experiments. n = 16 for H2O, 14 for ABX, 13 for anti-CD4 + anti-CD8, 5 for anti-CD8. P < 0.05, one-way ANOVA. (E and F) Loss of hepatic NKT abrogated the inhibition of liver metastasis caused by ABX. CXCR6−/−, CD1d−/−, or wild-type mice pretreated with ABX or H2O were given EL4 tumor cell tail vein injections. Liver surface metastatic nodules were counted. Data represent mean ± SEM of two pooled experiments. n = 10 for Wt-H2O, 7 for Wt-ABX, 4 for CXCR6−/−-H2O, 5 for CXCR6−/−-ABX, 5 for CD1d−/−-H2O, 4 for CD1d−/−-ABX. P < 0.05, two-way ANOVA.
The antitumor activity of NKT cells can cause direct killing of CD1d-expressing tumors (18–20), and we noted that all three (B16, EL4, and A20) of our tumor models tested expressed CD1d (fig. S4D). In addition, NKT cells can recognize tumor antigen from CD1dneg tumors through cross-presentation by professional antigen-presenting cells (21). To investigate the role of NKT cells in CD1d-expressing tumors, we used CD1d-knockout mice (which completely lack NKT cells) (22) and CXCR6-knockout mice (which have a selective NKT deficiency in the liver) (13). We confirmed loss of hepatic NKT cells in these mice (fig. S4E) and then induced liver tumors by intravenous injection of EL4 tumor cells (23). Depleting gut microbiome reduced EL4 liver tumor burden in wild-type mice, but no reduction in liver tumor size was found in either CD1d-knockout or CXCR6-knockout mice alter ABX treatment (Fig. 3, E and F). Intrasplenic injection of B16 tumor cells was repeated in CDld-knockout or CXCR6-knockout mice, and similar results were observed (fig. S4, F to H). These findings suggest that hepatic NKT cells are necessary for effects on tumor growth in the liver induced by changes in the gut microbiome.
A bile acid/CXCL16/CXCR6 axis controls hepatic NKT accumulation
Virtually all hepatic NKT cells express CXCR6 (Fig. 2F), and ABX treatment increased the accumulation of CXCR6+ cells into the liver (Fig. 2I), whereas CXCR6 mean fluorescence intensity did not change on NKT cells (fig. S5A). Therefore, CXCL16, the only ligand for CXCR6 (24), was further studied. As expected, higher Cxcl16 mRNA levels were found in the liver of ABX-treated mice (Fig. f4A). This increase of Cxcl16 mRNA was not detected in the lung (fig. S5B). Liver sinusoidal endothelial cells (LSECs) have been reported to be the major source of CXCL16 production in the liver (13). To identify the source of CXCL16, we isolated LSECs from ABX-treated mice. Figure 4B shows that there was an almost 2-fold increase of Cxcl16 mRNA in LSECs from ABX-treated mice. Consistent with the previous report, LSECs have a much higher basal level of Cxcl16 mRNA, and ABX treatment did not affect Cxcl16 mRNA expression in non-LSECs. The increase of CXCL16 protein in LSECs was confirmed by immunohistochemistry staining (Fig. 4C). In addition, forced CXCL16 expression in the liver increased hepatic NKT levels (fig. S5C). Together, these results suggest that ABX treatment causes LSECs to produce more CXCL16 and recruits NKT cells to the liver.
Fig. 4. Altered bile acids mediate CXCL16 up-regulation in LSECs, NKT accumulation and liver tumor inhibition in mice.

(A) CXCL16 mRNA expression levels in liver tissues from ABX- or H2O-treated tumor-free C57BL/6 mice. Data represent mean ± SEM of two pooled experiments. n = 9 for H2O, 10 for ABX. P < 0.05, Student’s t test. (B) Primary LSECs were isolated from mice treated with ABX or H2O. CXCL16 mRNA levels were measured by real-time polymerase chain reaction (PCR). Data represent mean ± SEM of three pooled experiments. n = 9, P < 0.05, two-way ANOVA. (C) Immunohisto-chemistry staining of CXCL16 and LYVE-1 in liver sections from ABX- or H2O- treated mice. CXCL16+ (red)/LYVE+ (green) LSECs are highlighted by arrows and are shown in more detail in the insert. Scale bar, 50 μm. (D) Mice were fed with a 2% cholestyramine (CHOL) or control diet (Con). Hepatic NKT, CXCR6+, CD4 T, and CD8 T cells were measured. Data represent mean ± SEM of two pooled experiments. n = 5 for control, 9 for cholestyramine diet. P < 0.05, two-way ANOVA. (E) Liver bile acids profile of ABX- or H2O-treated mice. Data represent mean ± SEM of two pooled experiments. n = 9, P < 0.05, Student’s t test. (F and G) Isolated LSECs were treated with different bile acids (F) or a combination of T-β-MCA with T-ω-MCA or ω-MCA (G). Data represent mean ± SEM of two pooled experiments. n = 5, P < 0.05, one-way ANOVA. (H) ABX-treated mice were given three times oral gavage of CDCA or ω-MCA three times at the dose of 6 mg per 15 g body weight. Hepatic NKT cell levels from different treatments were measured. Data represent mean ± SEM of two pooled experiments. n = 5, P < 0.05, two-way ANOVA. (I) A20 liver metastasis in vancomycin (Vanco) or H2O-treated mice receiving LCA or CDCA. As indicated, one group of LCA-treated mice also received hydrodynamic injection to force CXCL16 expression in the liver. Data represent mean ± SEM of two pooled experiments. n = 15 for Vanco + LCA, others n = 10. P < 0.05, one-way ANOVA. (J) EL4 liver metastasis in ABX-treated CXCR6−/− mice with or without ω-MCA feeding. Data represent mean ± SEM of two pooled experiments. n = 5.
CXC16 has both cell-surface and secreted forms; the cell-surface form has been identified as a scavenger receptor for phosphatidylserine and lipoprotein and is involved in lipid metabolism (25). Gut commensal bacteria are well known to mediate bile acid metabolism in the intestine, and the gut microbiome reportedly regulates bile acid composition (26). Therefore, we searched for a possible link between bile acids and CXCL16. First, we used cholestyramine, a bile acid sequestrant, to block the enterohepatic circulation, thus reducing bile acid levels in the liver (27). Cholestyramine treatment increased hepatic NKT and CXCR6+ cells, but not CD4+ T or CD8+ T cells (Fig. 4D and fig. S5D). In parallel, Cxcl16 mRNA was up-regulated in the liver (fig. S5E). This result suggests that bile acids are involved in the accumulation of NKT cells in the liver through CXCL16 regulation. To identify the bile acids involved in NKT cell regulation, the liver bile acid profile was determined. As reported (26), control mouse liver contains the highest amount of primary bile acid taurocholic acid (TCA), followed by primary bile acid tauro-β-muricholic acid (T-β-MCA), and secondary bile acid tauro-ω-muricholic acid (T-ω-MCA) (Fig. 4E). ABX treatment did not affect liver TCA but significantly increased primary bile acids T-β-MCA and β-MCA. Gut commensal bacteria convert primary bile acids into secondary bile acids (28). As expected, secondary bile acids T-ω-MCA, taurodeoxycholic acid (TDCA), ω-MCA, taurolithocholic acid (TLCA), and tauroursodeoxycholic acid (TUDCA) were reduced in ABX-treated mice (Fig. 4E).
To connect these findings, we studied the effect of bile acids on CXCL16 expression in primary murine LSECs in vitro. Secondary bile acid ω-MCA decreased cxcl16 mRNA expression, whereas the primary bile acid T-β-MCA induced Cxcl16 mRNA (Fig. 4F). Because cholestyramine treatment reduces both primary and secondary bile acids and caused NKT accumulation, we tested the possibility that primary and secondary bile acids have opposing effects on CXCL16 regulation. Indeed, the secondary bile acid compromised the primary acid-induced Cxcl16 mRNA up-regulation, and even impaired cxcl16 mRNA expression below the untreated baseline levels. (Fig. 4G). When mice were fed ω-MCA, a secondary bile acid, hepatic NKT accumulation was reversed, whereas feeding chenodeoxycholic acid (CDCA), a primary bile acid, enhanced NKT cell accumulation (Fig. 4H). These data demonstrate that primary and secondary bile acids influence hepatic NKT cell recruitment and have opposing effects in vivo.
Next, bile acids were fed to mice to investigate the effect on liver tumor growth. As expected, secondary bile acids LCA or ω-MCA reversed the inhibition of liver tumor growth caused by antibiotic treatment, and primary bile acid CDCA further enhanced tumor inhibition (Fig. 4I and fig. S5, F to H). Enforced CXCL16 expression in the liver (Fig. 4I and fig. S5F) or depletion of CD4+ NKT cells (fig. S5G) eliminated the effect of secondary bile acids on liver tumor growth (Fig. 4I and fig. S5, F and G), suggesting that this effect was NKT cell mediated. Similarly, secondary bile acids failed to affect liver tumor development in liver-NKT deficient (CXCR6−/−) mice (Fig. 4J) but still preserved the function to block the accumulation of transferred wild-type NKT cells caused by antibiotic treatment (fig. S5I). Therefore, depleting commensal bacteria with antibiotics leads to a model where primary bile acids that induce CXCL16 are preserved, whereas secondary bile acids that inhibit CXCL16 are reduced. This leads to up-regulation of CXCL16 in LSECs and accumulation of NKT cells in the liver.
Clostridium species regulates liver NKT accumulation in mice
Although ABX treatment efficiently reduced gut commensal bacteria load, it did not result in complete elimination of gut bacteria (fig. S1A). To rule out the possibility that the remaining bacteria mediate NKT accumulation, we repeated the experiment in germ-free mice. Whereas more hepatic NKTs were found in germ-free mice when compared with the matched SPF control mice (Fig. 5A and fig. S5, J and K) (29), no change was seen in TLR4-knockout mice (fig. S5L). Similarly, Cxcl16 mRNA levels were higher in the liver of germ-free mice (Fig. 5B).
Fig. 5. Clostridium species influence bile acid-controlled NKT cell accumulation.

(A) Hepatic NKT cell levels from germ-free mice or matched SPF mice were measured. Data represent mean ± SEM of two pooled experiments. n = 5, P < 0.05, Student’s t-test. (B) cxcl16 mRNA expression in liver tissue from germ-free or SPF mice. Data represent mean ± SEM of two pooled experiments. n = 8 for SPF, 7 for GF. P < 0.05, Student’s t-test. (C) Naive C57BL/6 mice were fed with vancomycin (Vanco), neomycin (Neo) or cefoparazone (Cefo). Hepatic NKT levels were determined. Data represent mean ± SEM of three pooled experiments. n = 18 for H2O, 14 for vancomycin, 14 for neomycin, 10 for cefoperazone. P < 0.05, one-way ANOVA. (D and E) Mice were treated with vancomycin for 1 week and then gavaged with C. scindens or vehicle (cessation). Twenty-four hours after C. scindens gavage, 16S rRNA sequencing analysis of stool samples was performed. The relative abundance of OTUs in the fecal bacterial are shown (D). Time-course study of hepatic NKT levels was performed (E). Data represent mean ± SEM of two pooled experiments. n = 10 for H2O, D0, C. scindens D4, and Cessation D4; 5 for C. scindens D2, Cessation D2, C. scindens D7 and Cessation D7 P < 0.05, two-way ANOVA. (F and G) A20 liver tumors were induced in mice treated with vancomycin or H2O. Mice were colonized with C. scindens or control C. innocuum as illustrated in (F). Cumulative liver tumor counts are shown in (G). Data represent mean ± SEM of two pooled experiments. n = 10 for H2O and Vanco, n = 20 for C. scindens and C. innocuum. P < 0.05, one-way ANOVA. (H) SK-HEP1 cells were treated with different bile acids. CXCL16 mRNA levels were measured by real-time PCR. Data represent mean ± SEM of three pooled experiments. n > 10, P < 0.05, one-way ANOVA. (I) Correlation between primary bile acid CDCA and CXCL16 mRNA expression in nontumor liver tissues from hepatocellular carcinoma or cholangiocarcinoma patients of the TIGER cohort. Pearson correlation coefficient test was performed.
Next, we tried to identify the commensal bacteria responsible for the observed liver NKT accumulation. Because the ABX antibiotic cocktail used here contains three antibiotics with a different activity spectrum, individual antibiotic treatment was performed to narrow down the targeting bacteria. Vancomycin alone was sufficient to increase hepatic NKT cells, whereas neomycin had a marginal effect (Fig. 5C and fig. S5M). A clear increase of liver NKT cells was seen in mice fed with cefoperazone, but no significant changes in hepatic CD4+ or CD8 T+ cells were observed (fig. S5, N and O). Interestingly, both vancomycin and cefoperazone target grampositive bacteria and have been reported to increase primary bile acids and deplete secondary bile acids in the gut (30), which is consistent with our finding that bile acids also change in the liver and regulate NKT cell accumulation.
The 7α-dehydroxylation reaction is the most quantitatively important process performed by the gut bacteria in the production of secondary bile acids (28) and is restricted to grampositive bacteria of the Clostridium cluster XIV (31). We found that vancomycin, which increased liver NKT cells, depleted Clostridium, whereas neomycin, which had little effect on liver NKT, had little effect on Clostridium (fig. S5P). To explore a role for Clostridium species on liver NKT cell accumulation, we used C. scindens, which is commonly found in both mice and humans (32) and has a conserved bai (bile acid inducible) gene operon for the 7α-dehydroxylation reaction (28, 33). Mice were fed with vancomycin for 1 week to induce hepatic NKT cells, then vancomycin was stopped, and the mice were given C. scindens or vehicle (fig. S6A). The successful colonization with C. scindens was confirmed (fig. S6B). One day after oral gavage, fecal bacterial were analyzed by 16S ribosomal (rRNA) sequencing (Fig. 5D and fig. S6, C and D). Continuous vancomycin treatment caused a reduction of Clostridiales and Bacteroidales but an expansion of Verrucomicrobiales compared with the water control group. An early recovery of Bacteroidales was observed after vancomycin cessation. Gavage of C. scindens increased the Clostridiales population. A time course study showed that hepatic NKT levels started to drop between day 2 and day 4 after vancomycin withdrawal (Fig. 5E), suggesting recovery of gut commensal bacteria. Clostridium cluster XIV recovered in parallel (fig. S6E). Colonization of C. scindens induced a rapid reduction of hepatic NKT cells (Fig. 5E) but did not affect other immune cells (fig. S6, F and G). As expected, colonization of mice with C. scindens, but not cessation of ABX treatment, resulted in a reduction of primary bile acids and recovery of secondary bile acids on day 2 (fig. S6H). Thus, our results suggest that bile acid-converting Clostridium species such as C. scindens are involved in the regulation of hepatic NKT cell accumulation. We next tested the effect of C. scindens on liver tumor growth. As expected, more liver tumors were found in C. scindens-colonized mice than mice kept on vancomycin or C. innocum-colonized mice (Fig. 5, F and G), suggesting that gut bacteria capable of metabolizing bile acids can regulate growth of liver tumors.
Bile acids control liver CXCL16 expression in humans
We extended our study to human samples to determine the effect of bile acids on CXCL16 mRNA expression of human liver sinusoidal endothelial cells. Similar to the mouse study, primary bile acids CDCA and TCA induced CXCL16 mRNA expression in SK-HEP1 cells (Fig. 5H). Next, the correlation between bile acids and CXCL16 expression was tested in nontumor liver tissues from HCC or cholangiocarcinoma patients of the Thailand Initiative for Genomics and Expression Research in Liver Cancer (TIGER-LC) cohort (34). Primary bile acid CDCA levels correlated with CXCL16 expression (Fig. 5I), whereas inverse correlation was seen for the secondary bile acid glycolithocholate (GLCA) (fig. S6I). The primary/secondary ratio was associated with CXCL16 increase (fig. S6J), indicating that the opposing effect of bile acids on CXCL16 expression also exists in humans.
Mucosal-associated invariant (MAIT) cells are enriched in the liver and comprise 20 to 50% of hepatic lymphocytes in humans (35). Interestingly, CXCR6 is expressed on MAIT cells (35), which raises the question whether liver MAIT cells can also be controlled by gut bacteria through CXCL16 regulation. MAIT cells recognize bacterial derivatives and are involved in inflammatory liver diseases such as nonalcoholic steatohepatitis (NASH). Besides antitumor function, NKT cells have been reported as important regulators of autoimmune responses (36). Our study suggests that gut commensal bacteria and bile acids could be potential targets for controlling liver autoimmune diseases. In summary, we have describe a mechanism by which the gut microbiome uses bile acids as messengers to control the accumulation of hepatic NKT cells and thereby antitumor immunity in the liver of mice. These findings not only have possible implications for future cancer therapeutic studies in humans but also provide a link between the gut microbiome, its metabolites, and immune responses in the liver.
Materials and methods
Murine studies
SPF C57BL/6 and BALB/c mice were purchased from Charles River. CXCR6-knockout mice were purchased from Jackson laboratory. CD1d-knockout mice, LAP-tT and TRE-MYC mice have been previously described (10). Germ-free mice were provided by R. Goldszmid (Cancer and inflammation program, NIH). Newly purchased four-week old C57BL/6 or BALB/c mice were randomized into 5 mice/cage and housed for one week to normalize gut microbiome. Then mice were assigned into H2O or ABX treatment groups. Mice in the ABX group received three-antibiotic cocktail in the drinking water containing vancomycin (Hospira, 0.5 g/L), neomycin (VETone, 0.5 g/L) and pri-maxin (Merck & CO, 0.5 g/L) as previously reported (3). In some experiments mice were given single antibiotic water, and cefoperazone (MP Biomedicals) was given at the concentration of 0.5 g/L. Fresh antibiotic water was replaced every other day. After 3 weeks of ABX pretreatment, mice were challenged with different tumor cell lines. B16-F1 and A20 cells were purchased from ATCC. EL4 cells were used as described (37). 1 × 106 EL4 tumor were given by subcutaneous or tail vein injection, 3 × 105 B16-F1 tumor cells were given by intrasplenic injection as described before (11), and 1 × 106 A20 tumor cell were given by tail vein injection. In some experiments mice were fed with a 2% cholestyramine diet made by Research Diets Inc (New Brunswick, NJ). Mice were treated with 500 μg anti-CD4 (clone GK15, BioxCell) or 200 mg anti-CD8 (clone 2.43, BioxCell) 24 hours before receiving tumor injection for depletion studies. For in vivo NKT cell stimulation, 1 × 106 aGalCer-loaded A20 tumor cells in the combination of brefeldi A (500 ug/mouse) were given by tail vein injection, and mice were sacrificed 3 hours after injection. aGalCer-loading was performed by incubate A20 cells with 1 μg/ml aGalCer overnight followed by 3 times of washing. At the experimental end points, mice were sacrificed for organ harvest. All experiments were conducted according local institution guidelines and approved by the Animal Care and Use Committee of the National Institutes of Health, Bethesda, USA.
Flow cytometry
Cells were surface-labeled with the indicated antibodies for 15 min at 4°C. Intracellular staining using a Foxp3/transcription factor staining buffer set (eBioscience) was used according to the manufacturer’s instructions. Flow cytometry was performed on BD LSRFortessa platform and results were analyzed using FlowJo software version 9.3.1.2 (TreeStar). Dead cells were excluded by using live/dead fixable near-IR dead cell staining kit (ThermoFisher scientific). The following antibodies were used for flow cytometry analysis: anti-TCRb-BV510 (clone H57–587, Biolegend), PBS57/CD1d-tetramer-APC (NIH core facility), anti-CXCR6-FITC (clone SA051D1, Biolegend), anti CD3-PE (clone 17A2, Biolegend), anti-CD4-PE (clone RM4–5, Biolegend), anti-CD4- Alexa Fluor 700 (clone GK1.5, Biolegend), anti-CD8- BV210 (clone 53–6.7 Biolegend), anti-CD19-PerCP/Cy5.5 (clone eBio1D3, eBioscience), anti-CD49b (clone DX5, eBioscience), anti-TCRg/d-PE, (clone GL3, BD pharmigen), anti-CD11b-BV421 (clone M1/70, Biolegend), anti-Ly6G-Alexa Fluor 700 (clone 1A8, Biolegend), anti-Ly6C-AP (clone HK1.4, Biolegend), anti-CD44-PE/Cy7 (clone IM7, eBioscience), anti-CD62L-PerCP/Cy5.5 (MEL-14, Biolegend), anti-CD69-Pacific blue (clone H1.2F3, Biolegend), anti-CD25-FITC (clone 7D4, BD pharmigen), anti-4–1BB-PE (clone 17B5, Biolegend), anti-Foxp3-Alexa Fluor 488 (clone 22F6, Biolegend), anti-Tbet-Pacific Blue (clone 4B10, Biolegend), anti-RORg-PE (clone B2D, eBioscience), anti-PLZF-PerCP/Cy5.5 (clone 9E12, Biolegend), and anti-CD1d-PE (clone 1B1, eBioscience), anti- IFNg-PE (clone XMG1.2, BD Biosciences), anti-TNFa-PerCP/Cy5.5 (clone MP6-XT22, Biolegend). The following markers were used for identifying different immune cell subsets: TCRb+CD1d- Teteramer+ for NKT cells, CD3+CD4hi for hepatic CD4+ T cells, CD3+CD8+ for CD8+ T cells, CD3− CD19+ for B cells, CD3−CD49b+ for NK cells, CD3+TCRg/d+ for g/d T cells, CD11b+Ly6G+Ly6Clo for G-MDSC. Absolute numbers were calculated by multiplying frequencies obtained from flow cytometry by the total live mononuclear cell count, and then divided by liver weight.
In vivo cytotoxcicity assay
Splenocytes isolated from naïve C57BL/6 mice were loaded with aGalCer (1 μg/ml) then labeled with high dose of CFSE as target cells. Unloaded cells were labeled with low dose of CFSE as control cells. Then CFSEhi target cell and CFSElo controls cells were mixed at about 1:1 ratio. 107 mixed cells were injected intravenously into ABX or H2O-treated C57BL/6 mice. Sixteen hours later, mice were killed and cytotoxicity was analyzed by flow cytometry. r = (%CFSElo / %CFSEhi); ro is the ratio of mixed cells without injection; % cytotoxicity = [1- (r0/r) ]×100.
Immunohistochemistry and quantification
Immunohistochemistry was performed on 3 μm sections obtained from formalin-fixed paraffin-embedded liver tissues of H2O-treated (n = 5) or ABX-treated (n = 5) mice using the Opal 5-color IHC Kit (PerkinElmer, Waltham, MA, USA) according to the manufacturer’s instructions. The following primary antibodies were used: anti-CXCL16 (Bioss Antibodies, Woburn, MA, USA; bs-1441R, rabbit polyclonal, 1/4.000, Opal 620), anti- LYVE1 (Abcam, Cambridge, UK; rabbit polyclonal, 1/15.000, Opal 520). Slides were evaluated using the Vectra® 3 automated, high-throughput quantitative pathology imaging system (PerkinElmer) and the inForm software (PerkinElmer) for segmentation and quantification of CXCL16+/LYVE1+ cells.
Hepatic bile acid profiling
Fresh mice liver tissue was snap frozen in liquid nitrogen and then kept at −80°C. Hepatic bile acid composition was measured at West Coast Metabolomics Center at UC Davis using the targeted metabolite analysis service.
Liver sinusoidal endothelial cell preparation and bile acids treatment
Primary mice liver sinusoidal endothelia cells were isolated as previously described (38). Briefly, mice were CO2 euthanized, and then the portal vein was cannulated and the liver was perfused with 0.05% collagenase in Ca2+ deprived medium. Liver cells were dissociated and parenchymal cells were killed by incubation in 0.04% collagenase in Gey’s balanced salt solution at 37°C for 15 min. Then density gradient centrifugation was performed using Nycodenz solution at the final solution of 1.089 g/cm3. LSECs were isolated using anti-LSEC microbeads (Miltenyi) according to the manufacturer’s instructions. LSECs or the human SK-HEP1 cell line (ATCC, HTB-52) were treated with different bile acids for 24 hours. Gene expression was analyzed by real-time PCR. TCA, CDCA, DCA, and TDCA were purchased from Sigma. T-b-MCA, w-MCA, and T- w-MCA were purchased from Steraloids Inc.
In vivo bile acids feeding
Mice were kept on ABX cocktail and fresh ABX was replaced every other day. Mice were fed with bile acids by oral gavage 48, 24 and 16 hours prior to sacrifice. For A20 tumor bearing mice, w-MCA, LCA or CDCA were given 3 times/week. Bile acids were dissolved in corn oil. w-MCA and CDCA were given at the dose of 6 mg/15 g body weight, LCA was given at the dose of 1 mg/15 g body weight.
Adoptive transfer of NKT cells
Donor NKT-rich cells were isolated from livers of wild-type C57BL/6 mice by autoMACs sorting of NK1.1+ cells. Half million of NKT-rich cells were transferred into CXCR6−/− mice by tail vein injection. Two days later mice were scarified and the liver accumulation of transferred NKT cells were measured by flow cytometry as TCRb+CD1d-Teratmer+CXCR6+ population.
Gut colonization with Clostridium scindens
Mice were fed with vancomycin in drinking water (Hospira, 0.5 g/L) for one week. Fresh antibiotic water was replaced every other day. One week later vancomycin was stopped, and the mice were given oral gavage of 109 C. scindens or vehicle (anaerobic glycerol) every day for 5 days. C. scindens was purchased from ATCC (35704), and grown under anaerobic conditions. One day after gavage, the colonization of C. scindens were confirmed by real-time PCR using primers specific for C. scindens. For A20 tumor study, BALB/c mice were fed with vancomycin for one week. Then vancomycin was stopped, and A20 tumor (1 × 106 cells) were injected intravenously. Mice were given oral gavage of 109 C. scindens or C. innocuum (ATCC 14501) every day for 4 days. Then mice were given a second round of vancomycin treatment for 4 days, followed by additional four days of oral gavage of C. scindens or C. innocuum. Fourteen days after tumor injection, mice were sacrificed and liver tumor burden was measured.
16S rRNA sequencing and analysis
Mouse stool DNA extraction and 16S V4 region amplification were performed on the liquid handling robots (Eppendorf, epMotion5075 and epMotion 5073). The V4 region of the 16S rDNA gene (515F-806R) was sequenced; generating partially overlapping, paired-end reads on the Illumina MiSeq platform. After quality control filtering; a total of 3,979,728 reads were processed with an average of 132,657 reads per sample. The demultiplexed FASTQ files containing the 16S rRNA gene sequences were filtered for chimeric sequences using the USEARCH (version 8.1.1831) utility’s UCHIME implementation and the ‘gold’ database (version microbiomeutil-r20110519). The reads were then binned into Operational Taxonomic Units (OTUs) at 97% similarity using USEARCH’s cluster_otus command. The OTUs thus obtained were classified and aligned using QIIME (1.9.1) scripts. The assign_taxonomy.py script was used to assign taxonomy using the default RDP method and the default GreenGenes database. This provided insight into the larger trends at higher taxonomic levels (such as order Clostridiales). The 16S rRNA sequencing data was deposited into Sequence Read Archive (SRA) public database with the accession number SRP136953.
Human studies
Nontumor specimens derived from a set of 142 patients of the TIGER-LC Consortium were used in this study (34). Transcript expression was measured using the Affymetrix Human Transcrip- tome Array 2.0. Data has been deposited into the Gene Expression Omnibus (GEO) public database at NCBI (GEO Series GSE76297). A total of 718 biochemical metabolite species were measured by Metabolon’s Discover HD4 Platform. All expression and metabolite data were log2 transformed. Pearson correlation analysis was performed using GraphPad Prism 7 to determine correlation between CXCL16 gene expression and selected metabolites. Due to the missing information caused by detection limitations, 85 valid patient data were used to correlate CDCA and CXCL16 expression.
Statistical analysis
The sample sizes for animal studies were guided by previous murine studies in our laboratory. Statistical analysis was performed with Graph-Pad Prism 7 (GraphPad Software). The significant differences between groups were calculated by Student’s unpaired t test, one-way, or two-way ANOVA (Tukey’s and Bonferroni’s multiple comparison test). Welch’s corrections were used when variances between the groups were unequal. P < 0.05 was considered as statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank the NIH Tetramer Core Facility at Emory University for the CD1d-tetramer. Funding: T.F.G., M.T., J.A.B., X.W.W., and G.T. were supported by the Intramural Research Program of the NIH, NCI (ZIA BC 011345, ZO1 SC 004020, and Z01 BC 010876). C.M.T. was supported by R35GM119438. T.L. was supported by the Deutsche Forschungsgemeinschaft DFG (SFB/TRR57, project Q1).
Footnotes
Competing interests: C.M and T.F.G are inventors on a patent application (U.S. Patent Application no. 62/578,176) submitted by the National Cancer Institute that covers the use of bile acids to treat and/or prevent adverse liver conditions The authors declare no other competing interests.
Data and materials availability: All data presented in this manuscript are available in the main text and the supplementary materials.
Accession numbers: The transcript expression data of nontumor specimens of the TIGER-LC Consortium were deposited into the Gene Expression Omnibus (GEO) public data set with accession number GSE76297. The 16S rRNA sequencing data was deposited into the Sequence Read Archive (SRA) public database with accession number SRP136953.
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/360/6391/eaan5931/suppl/DC1
Materials and Methods
Figs. S1 to S6
References
REFERENCES AND NOTES
- 1.Viaud S et al. , The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 342, 971–976 (2013). doi: 10.1126/science.1240537; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vétizou M et al. , Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350, 1079–1084 (2015). doi: 10.1126/science.aad1329; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iida N et al. , Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 342, 967–970 (2013). doi: 10.1126/science.1240527; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Routy B et al. , Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97 (2018). doi: 10.1126/science.aan3706; pmid: [DOI] [PubMed] [Google Scholar]
- 5.Matson V et al. , The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359, 104–108 (2018). doi: 10.1126/science.aao3290; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gopalakrishnan V et al. , Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103 (2018). doi: 10.1126/science.aan4236; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshimoto S et al. , Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101 (2013). doi: 10.1038/nature12347; pmid: [DOI] [PubMed] [Google Scholar]
- 8.Dapito DH et al. , Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 21, 504–516 (2012). doi: 10.1016/j.ccr.2012.02.007; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Disibio G, French SW, Metastatic patterns of cancers: Results from a large autopsy study. Arch. Pathol. Lab. Med 132, 931–939 (2008). pmid: [DOI] [PubMed] [Google Scholar]
- 10.Ma C et al. , NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 531, 253–257 (2016). doi: 10.1038/nature16969; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eggert T et al. , Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 30, 533–547 (2016). doi: 10.1016/j.ccell.2016.09.003; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marabelle A et al. , Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J. Clin. Invest 123, 2447–2463 (2013). doi: 10.1172/JCI64859; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geissmann F et al. , Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLOS Biol. 3, e113 (2005). doi: 10.1371/journal.pbio.0030113; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim CH et al. , Bonzo/CXCR6 expression defines type 1-polarized T-cell subsets with extralymphoid tissue homing potential. J. Clin. Invest 107, 595–601 (2001). doi: 10.1172/JCI11902; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee YJ, Holzapfel KL, Zhu J, Jameson SC, Hogquist KA, Steady-state production of IL-4 modulates immunity in mouse strains and is determined by lineage diversity of iNKT cells. Nat. Immunol 14, 1146–1154 (2013). doi: 10.1038/ni.2731; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pobezinsky LA et al. , Let-7 microRNAs target the lineage-specific transcription factor PLZF to regulate terminal NKT cell differentiation and effector function. Nat. Immunol 16, 517–524 (2015). doi: 10.1038/ni.3146; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atarashi K et al. , Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331, 337–341 (2011). doi: 10.1126/science.1198469; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L, Targeting natural killer cells and natural killer T cells in cancer. Nat. Rev. Immunol 12, 239–252 (2012). doi: 10.1038/nri3174; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terabe M, Berzofsky JA, The role of NKT cells in tumor immunity. Adv. Cancer Res 101, 277–348 (2008). doi: 10.1016/S0065-230X(08)00408-9; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terabe M, Berzofsky JA, in Encyclopedia of Immunobiology, Ratcliffe MJH, Ed. (Elsevier, 2016), vol. 4, pp. 460–469. [Google Scholar]
- 21.Wu DY, Segal NH, Sidobre S, Kronenberg M, Chapman PB, Cross-presentation of disialoganglioside GD3 to natural killer T cells. J. Exp. Med 198, 173–181 (2003). doi: 10.1084/jem.20030446; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sonoda KH, Exley M, Snapper S, Balk SP, Stein-Streilein J, CD1-reactive natural killer T cells are required for development of systemic tolerance through an immune-privileged site. J. Exp. Med 190, 1215–1226 (1999). doi: 10.1084/jem.190.9.1215; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smyth MJ et al. , Differential tumor surveillance by natural killer (NK) and NKT cells. J. Exp. Med 191, 661–668 (2000). doi: 10.1084/jem.191.4.661; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balkwill F, Cancer and the chemokine network. Nat. Rev. Cancer 4, 540–550 (2004). doi: 10.1038/nrc1388; pmid: [DOI] [PubMed] [Google Scholar]
- 25.Shimaoka T et al. , Molecular cloning of a novel scavenger receptor for oxidized low density lipoprotein, SR-PSOX, on macrophages. J. Biol. Chem 275, 40663–40666 (2000). doi: 10.1074/jbc.C000761200; pmid: [DOI] [PubMed] [Google Scholar]
- 26.Sayin SI et al. , Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 17, 225–235 (2013). doi: 10.1016/j.cmet.2013.01.003; pmid: [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Klaassen CD, Effects of feeding bile acids and a bile acid sequestrant on hepatic bile acid composition in mice. J. Lipid Res 51, 3230–3242 (2010). doi: 10.1194/jlr.M007641; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Staley C, Weingarden AR, Khoruts A, Sadowsky MJ, Interaction of gut microbiota with bile acid metabolism and its influence on disease states. Appl. Microbiol. Biotechnol 101, 47–64 (2016). pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olszak T et al. , Microbial exposure during early life has persistent effects on natural killer T cell function. Science 336, 489–493 (2012). doi: 10.1126/science.1219328; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Theriot CM, Bowman AA, Young VB, Antibiotic-Induced Alterations of the Gut Microbiota Alter Secondary Bile Acid Production and Allow for Clostridium difficile Spore Germination and Outgrowth in the Large Intestine. MSphere 1, e00045–15 (2016). doi: 10.1128/mSphere.00045-15; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ridlon JM, Kang DJ, Hylemon PB, Bile salt biotransformations by human intestinal bacteria. J. Lipid Res 47, 241–259 (2006). doi: 10.1194/jlr.R500013-JLR200; pmid: [DOI] [PubMed] [Google Scholar]
- 32.Buffie CG et al. , Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–208 (2015). doi: 10.1038/nature13828; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhowmik S et al. , Structure and functional characterization of a bile acid 7a dehydratase BaiE in secondary bile acid synthesis. Proteins 84, 316–331 (2016). doi: 10.1002/prot.24971; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chaisaingmongkol J et al. , Common Molecular Subtypes Among Asian Hepatocellular Carcinoma and Cholangiocarcinoma. Cancer Cell 32, 57–70.e3 (2017). doi: 10.1016/j.ccell.2017.05.009; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kurioka A, Walker LJ, Klenerman P, Willberg CB, MAIT cells: New guardians of the liver. Clin. Transl. Immunology 5, e98 (2016). doi: 10.1038/cti.2016.51; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Kaer L, alpha-Galactosylceramide therapy for autoimmune diseases: Prospects and obstacles. Nat. Rev. Immunol 5, 31–42 (2005). doi: 10.1038/nri1531; pmid: [DOI] [PubMed] [Google Scholar]
- 37.Ma C et al. , Anti-Gr-1 antibody depletion fails to eliminate hepatic myeloid-derived suppressor cells in tumor-bearing mice. J. Leukoc. Biol 92, 1199–1206 (2012). doi: 10.1189/jlb.0212059; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Limmer A et al. , Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat. Med 6, 1348–1354 (2000). doi: 10.1038/82161; pmid: [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.