

Abstract
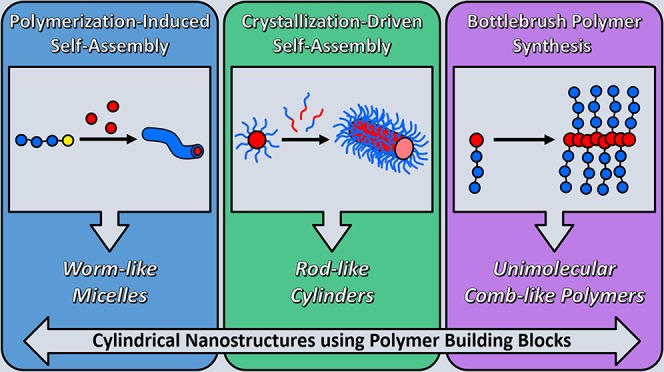
Cylinders are fascinating structures with uniquely high surface area, internal volume, and rigidity. On the nanoscale, a broad range of applications have demonstrated advantageous behavior of cylindrical micelles or bottlebrush polymers over traditional spherical nano-objects. In the past, obtaining pure samples of cylindrical nanostructures using polymer building blocks via conventional self-assembly strategies was challenging. However, in recent years, the development of advanced methods including polymerization-induced self-assembly, crystallization-driven self-assembly, and bottlebrush polymer synthesis has facilitated the easy synthesis of cylindrical nano-objects at industrially relevant scales. In this Perspective, we discuss these techniques in detail, highlighting the advantages and disadvantages of each strategy and considering how the cylindrical nanostructures that are obtained differ in their chemical structure, physical properties, colloidal stability, and reactivity. In addition, we propose future challenges to address in this rapidly expanding field.
Introduction
On the sub-cellular scale, cylindrical nanostructures are ubiquitous in the natural world. These anisotropic constructs fulfill a variety of vital biological functions such as providing cellular structure, dissipating energy through elastic deformation, assisting with cell division, and many others. Among the many remarkable cylindrical nanostructures at work in biological systems are collagen, peptidoglycans, and centrioles. Fibrous proteins comprised of collagen fill the interstitial space between cells. Collagen is the most abundant protein in mammals, comprising up to 35% of the protein content of the entire organism.1 Proteoglycans are often found coincident with collagen, acting as a shock-absorbing material and lubricant in connective tissue and as a viscosity modifier in the mucociliary clearance of lung airways.2 Centrioles produce the spindle fibers that separate chromosomes during cell division. In all cases, these structures are formed through the directed assembly of numerous biomacromolecular components which associate via non-covalent interactions.
The various biological functions of the cylindrical nanostructures highlighted above arise primarily from the unique physical and mechanical properties of their cylindrical shapes. Note that in this Perspective we refer to cylindrical structures as those with an overall cylindrical morphology, which includes both the core and corona volumes. First and most importantly, cylindrical objects have a high aspect ratio and are thus anisotropic.3 Due to their unique geometry, cylinders have large surface areas and increased volume relative to spheres. Consider a sphere of radius r that fits completely within a cylinder with a width of r and a height of 2r. In this case, both the surface area and volume of the cylinder are 1.5 times greater than the sphere. The high surface area and aspect ratio of cylinders make them excellent materials for surface adsorption, and their large volumes are well-suited for encapsulating substantial quantities of molecules, for example drugs. Second, similar to linear polymers, cylinders can interact with one another through entanglement. This property allows for the formation of physically cross-linked gels if the cylindrical objects are present in sufficient concentration. In contrast, even for flexible cylindrical nanostructures, repulsive intermolecular forces such as steric or electronic repulsion can act to prevent entanglement, making such materials excellent viscosity modifiers. Finally, due to their anisotropy, certain cylindrical nanostructures possess differential reactivity on their termini. As we shall discuss below, this differential reactivity can be exploited for further supramolecular assembly.
Inspired by nature, many methods have been developed by synthetic chemists to produce cylindrical nanostructures in the laboratory. Most strategies involve supramolecular self-assembly of compounds such as small molecules,4 peptides,5 or amphiphilic polymers, by taking advantage of the hydrophobic effect—the tendency of hydrophobic substances to aggregate in an aqueous environment to exclude water molecules.6 In this Perspective, we focus on the preparation of cylindrical nanostructures using polymer building blocks. To date, cylindrical nanostructure preparation from polymeric building blocks has focused on the synthesis and self-assembly of amphiphilic block copolymers. Depending on the volume fraction of the hydrophobic/hydrophilic blocks—often described as the packing parameter—the conformational entropy (steric repulsion) of the hydrophilic chains, and the interfacial energy between the hydrophobic domain and water, these amphiphilic polymers assemble into different structures such as spheres, cylinders, or vesicles.7 However, to achieve a pure phase of cylindrical micelles, several iterations of polymer synthesis and self-assembly are often required. Recently, new strategies have been developed to facilitate the formation of pure cylinder morphologies in self-assembled systems (Figure 1). In this Perspective, we highlight two advanced methods to prepare cylindrical nanostructures via aqueous self-assembly: (1) polymerization-induced self-assembly (PISA)8 ,9 and (2) crystallization-driven self-assembly (CDSA).10 In addition, we compare these methods to an alternative approach to prepare cylindrical nanostructures—the synthesis of bottlebrush polymers—which yields cylindrical nanostructures held together by covalent bonds.11 Herein, we review the most recent advances in the aforementioned fields, consider the advantages and disadvantages of each, and provide an evaluation of the future possibilities of these powerful methods of cylindrical nanostructure preparation.
Figure 1.

TEM or AFM images of typical cylindrical nanostructures obtained using PISA or CDSA, or from bottlebrush polymer synthesis. Reproduced with permission from refs (18) (PISA worms; Copyright 2016 Wiley), (10) (CDSA cylinders; Copyright 2010 Springer Nature), and (66) (bottlebrush polymers; Copyright 2015 Springer Nature).
Emerging Applications of Polymeric Cylindrical Nanostructures
The synthesis of cylindrical nanostructures is motivated by their emerging applications as drug delivery vehicles, gel-forming materials, rheology modifiers, and conductive nanowires. In the case of drug delivery vehicles, shape has been identified as a crucial parameter for efficient loading and targeting. Cylindrical architectures have proven to be highly efficient vehicles for drug delivery applications compared to spherical nanoparticles.12 They provide several advantages such as higher loading efficiencies, longer circulation times, and enhanced accumulation, which in turn lead to enhanced active targeting, cellular uptake, and deeper penetration in tumors.13 For example, cylindrical micelles functionalized with near-infrared fluorophore tracers were found to deliver more than double the effective dose of paclitaxel within tumors in mice compared to spherical micelles.14 Bottlebrush polymers loaded with covalently attached drugs have also been exploited for cancer therapy and in vivo imaging; however, to date, no shape comparisons have been made (for example, between hyperbranched polymers or globular graft polymers and cylindrically shaped bottlebrush polymers).15,16 In addition to their applications as drug delivery vehicles, cylindrical architectures have the potential to form physical hydrogels due to inter-structural entanglements.17 Gels prepared by embedding cylindrical nanostructures in a poly(vinyl alcohol) (PVA) matrix have been exploited to avoid the use of toxic organic solvent for red-blood-cell cryopreservation (Figure 2),18 to replace natural mucin, and to encapsulate living cells.19
Figure 2.

(A) Synthetic route for the RAFT aqueous dispersion polymerization of HPMA using a water-soluble PGMA56 macroCTA to form PGMA56-b-PHPMA155 diblock copolymer worms. (B) Representative TEM image of the PGMA56-b-PHPMA155 diblock copolymer worms after drying a dilute aqueous dispersion at 20 °C. (C) The worms, in combination with PVA, exhibit enhanced cryoprotective behavior relative to controls, which was attributed to the inhibition of ice crystal formation in the presence of the worm micelles. Reproduced with permission from ref (18). Copyright 2016 Wiley.
From an industrial perspective, cylindrical structures are of growing interest as lubricants, fuel additives, and coatings. The shape and physical nature of cylindrical structures make them ideal candidates as rheological modifiers compared to high molecular weight linear polymer chains. Cylindrical structures are relatively more rigid and thicker than their linear polymer counterparts. Recent comparisons between high molecular weight polymers and cylindrical micelles in water have shown that cylindrical micelles exhibit improved thickening behavior and result in stiffer materials.20 In the case of bottlebrush polymers, sidechain and backbone length can be tuned to change the rheological behavior and enhance the branch entanglement, which contributes to their interesting rheological profile.21 In the coatings industry, materials are desired which combine high stiffness with high deformability. Hard fillers such as silica particles, carbon black, glass fibers, carbon nanotubes, or natural fibers are often used to stiffen materials without sacrificing deformability. In contrast to this approach, these properties can be achieved by using soft cylindrical nanostructures, which have been shown to increase the stiffness and glass transition temperature of water-based soft acrylic films.22
Bottlebrush polymers have also exhibited promise as photonic crystals. Due to reduced chain entanglement between bottlebrush polymers, particularly in the bulk, and the large domain sizes of the various morphologies, such as lamella, that arise in bottlebrush polymer films, photonic crystals can be easily fabricated with photonic bandgaps that span the entire visible spectrum.23
Finally, the use of cylindrical nanostructures as thermal or electrical conductor wires is also an emerging field. This application takes advantage of the long persistence lengths of the cylindrical nano-objects discussed herein. In particular, polymer-based nanowires offer significant potential for devices, sensors or nerve generation thanks to their anisotropic charge-transfer properties. For example, the Friend and Manners groups developed organic semiconducting nanofibers made of a crystalline poly(di-n-hexylfluorene) core with two different coronas: polyethylene glycol in the center and polythiophene at the ends (Figure 3).24 These cylinders exhibited exciton transfer from the core to the end blocks which occurred along their long axes. This charge transfer across the long dimension of the cylindrical micelle (ca. 200 nm) occurred over a far greater length than is typical for organic semiconductors and could be potentially tuned via the cylinder dimensions to develop new organic photovoltaic devices.
Figure 3.

Schematic illustrating the seeded growth process of PDHF14-b-PEG227. These nanofibers exhibit exciton transfer from the core to the lower-energy polythiophene coronas in the end blocks, which occurs in the direction of the interchain π–π stacking with very long diffusion lengths (>200 nm) and a large diffusion coefficient (0.5 cm2/s). Reproduced with permission from ref (24). Copyright 2018 AAAS.
Advanced Methods To Prepare Cylindrical Nanostructures Using Polymer Building Blocks
Polymerization-Induced Self-Assembly
Conventional block copolymer self-assembly strategies, such as direct dissolution, solvent-switch, and thin-film rehydration are generally conducted under dilute conditions (polymer concentrations ≤1% w/w) and most often require a series of additional laborious and inefficient post-polymerization steps to target certain morphologies.25 In recent years, polymerization-induced self-assembly (PISA) has been established as an attractive alternative self-assembly methodology for reproducible one-pot fabrication of polymeric nano-objects at high solids concentration (10–50% w/w) that provides reliable control over the targeted morphologies and facile access to higher-order structures.9,26
Typically, during the PISA process, in situ self-assembly of amphiphilic block copolymers occurs when a solvophilic homopolymer (stabilizer block), acting as a macroinitiator, is chain-extended using appropriate solvent-soluble monomers that gradually form solvophobic coreblocks (Figure 4). The vast majority of literature reports on PISA to date involve the successful implementation of reversible-deactivation radical polymerization (RDRP) techniques, under either dispersion or emulsion polymerization conditions, using thermally initiated or photoinitiated radical sources. More recently, ring-opening metathesis polymerization (ROMP) has been utilized as a nonradical approach to perform PISA in both organic and aqueous media.27,28
Figure 4.

PISA is conducted via chain extension of a soluble macroCTA with a monomer that produces an insoluble polymer. Pure cylindrical micelle morphologies are obtained at a given weight fraction of the hydrophobic and hydrophilic blocks at a certain concentration. In this example, photo-PISA was conducted in the presence of GOx to remove O2 to prepare cylindrical micelles. Reproduced with permission from ref (26). Copyright 2017 American Chemical Society.
Similar to traditional block copolymer self-assembly, the final morphology obtained through PISA is primarily dictated by the relative volume fractions of the stabilizer and core-forming blocks, a property termed as the packing parameter. The simultaneous chain-extension and self-assembly processes that take place during PISA drive a continuous alteration of the packing parameter of formulations. Synthetic parameters such as monomer concentration and molecular weight of the stabilizer block have been shown to drastically affect the obtained PISA morphologies, while added factors that can influence the packing parameter, and hence the final morphology, include the mobility and degree of solvophobicity of core-forming polymer chains,29 copolymer architecture, and solvent composition.30
The recent rapid development of PISA has facilitated the reproducible synthesis of well-defined block copolymer cylindrical micelles under highly concentrated conditions in a one-pot procedure, overcoming the main existing limitations of conventional self-assembly (low particle concentrations and loading capacities, separate polymerization and self-assembly steps). In addition, pure higher-order morphologies are more readily achieved via PISA by simply tuning the parameters of the polymerization.
Despite the numerous advantages of cylindrical micelle fabrication via PISA, this methodology shares some of the same drawbacks with traditional block copolymer self-assembly, since the development of pure cylinder phases requires the complete construction of morphology diagrams which in almost all cases is a laborious process that coincides with extensive transmission electron microscopy (TEM) imaging. Presumably, this happens due to the fact that the pure cylindrical micelle morphology occupies a very narrow regime of the morphologies diagram, as it is known to be greatly affected by changes in block composition and synthetic conditions.31 A second limiting factor of cylindrical micelles is the fact that they can undergo disassembly upon dilution. This critical aggregation concentration (CAC) can hinder certain applications such as the use of cylindrical micelles as drug delivery vehicles; however, this issue can be circumvented by utilizing highly hydrophobic or glass-forming monomers in the core block or through cross-linking.
Crystallization-Driven Self-Assembly
As mentioned above, typical self-assembly and PISA protocols struggle with achieving pure cylinder phases, often being obtained in very narrow DP and concentration regions. This stems from polymer dispersity, packing parameters, and corona interactions to name a few. However, these disadvantages can be overcome by changing the amorphous core-forming block to a semicrystalline one. Simply put, the two-dimensional folded crystalline lamella of a micelle with a semicrystalline core stabilizes lower curvature structures such as cylindrical micelles.
The addition of a crystallization paradigm facilitates the formation of cylindrical nanostructures while increasing control over the dimensions of the nanoparticles. Detailed studies by the Manners and Winnik groups into the cylinder formation of poly(ferrocenylsilane) (PFS)-containing polymers have been numerous since their initial use in 1998.32 In this seminal study, a series of PFS-b-poly(dimethylsiloxane) copolymers were synthesized by living anionic polymerization. Control over the length of cylindrical nano-objects prepared by CDSA has since been achieved via a seeded-growth approach. As shown in Figure 5, this seeded growth protocol has since been extended to polymer systems using poly(l-lactide) and poly(ε-caprolactone),19,33 poly(3-hexylthiophene),34 polythiophene and oligo(p-phenylenevinylene),35 polyethylene,36 and polyselenophene.37
Figure 5.

(A) CDSA facilitates controlled epitaxial growth of 1D cylinders using a “seeded-growth” protocol. Reproduced with permission from ref (40). Copyright 2016 Springer Nature. (B) Crystallization-driven epitaxial growth of PCL cylinders. Scale bars = 1000 nm. Reproduced with permission from ref (19). Copyright 2017 American Chemical Society. (C) Controlled growth of cylindrical micelles with nP3HT cores. Scale bars = 200 nm. Reproduced with permission from ref (34). Copyright 2011 American Chemical Society. The plots in B and C show the dependence of the average length of the cylinders on the ratio of block copolymer unimers which had been added to seed micelles during the preparation procedure.
Traditionally, researchers have focused on generating increasingly complex and hierarchical structures that utilize the control of the semicrystalline core offered by CDSA. Block co-micelles, scarf-like, dumbbell shaped, non-centrosymmetric, and lenticular micelles are among the many fascinating structures that have been reported to date.38−40 Recent reports have focused on hierarchical self-assembly and/or applications of CDSA cylinders. For example, colloidosomes formed by the assembly of PFS cylinders at an emulsion interface were grown to give concentric layers, each displaying a different functionality (in this case, showcasing different colored dyes).41 This lends itself to the idea that increasingly complex structures could be made by utilizing cylinders as building blocks.
Although CDSA can give unprecedented control over particle length, assessing the solvent parameters which are optimal for the self-assembly can be challenging. One approach may be that of Lazzari et al., using selective solvents based upon Hildebrand solubility parameters.42 They found that a mixture of chloroform and DMF yielded long fiber-like micelles of a polyacrylonitrile-block-polystyrene block copolymer. Conversely, Inam et al. used water/octanol partition coefficients normalized by surface area to calculate the solubility parameters for a PLLA-b-PDMA system instead of the aforementioned Hildebrand solubility parameter.43 Crystallization of polymer micelle cores has displayed an unprecedented degree of control over many systems, as evidenced by a host of morphologies throughout the literature. The future of this technology relies upon finding real and relevant applications that can utilize this control.
Synthesis of Bottlebrush Polymers
Bottlebrush polymers are comprised of numerous polymer side chains that are densely grafted to a macromolecular backbone.11 As a result of this high grafting density, there is a significant degree of interaction between side chains, causing extension of the bottlebrush polymer backbone. In addition to their interesting physical properties, bottlebrush polymers are often large in size—single molecules can exceed lengths of 100 nm. A broad variety of structural diversity can be achieved in bottlebrush polymers. For example, bottlebrush polymers have been prepared with two or more blocks, with gradient structures, with block copolymer side chains, with Janus structures, with variable grafting density, and with different side-chain lengths.44−46 All of these interesting topologies are accessible depending on the synthetic route utilized.
Bottlebrush polymers are typically synthesized via one of four methods (Figure 6). The simplest and perhaps most intuitive strategy for preparing bottlebrush polymers is by covalently grafting the side chains to a polymer backbone that has been decorated with reactive functionalities on each repeat unit (Figure 6A). This process, referred to as grafting-to, has a number of advantages, most of which derive from the fact that the side-chain and backbone polymers are synthesized separately, allowing for precise characterization of these macromolecular constituents prior to bottlebrush synthesis. However, due to steric constraints, bottlebrush polymers produced by grafting-to are often considered to have less than “perfect” grafting density—that is to say that there is less than one side chain per backbone repeat unit.
Figure 6.
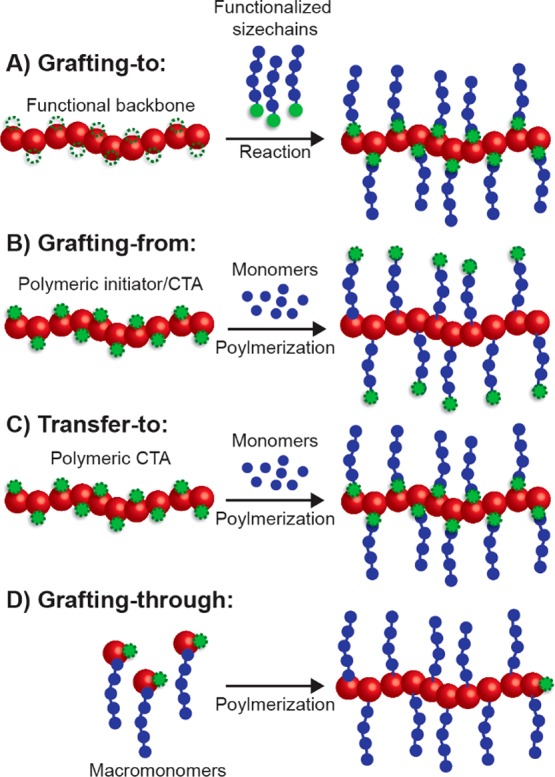
Four routes to prepare bottlebrush polymers: (A) bottlebrush synthesis by grafting functionalized polymers to a functional backbone (grafting-to); (B) grafting-from, involving the polymerization of the side chains from a polymeric initiator/CTA; (C) transfer-to, similar to grafting-from but differing in the attachment and behavior of the CTA moieties; and (D) preparation of bottlebrush polymers via polymerization of macromonomers in the grafting-through method.
A second strategy to prepare bottlebrush polymers is known as grafting-from (Figure 6B). In a grafting-from polymerization, the side chains are grown in situ using a backbone polymer that possesses initiator or chain-transfer agent (CTA) moieties on each repeat unit. This technique is perhaps the best suited to prepare large bottlebrush polymers (i.e., with backbone degrees of polymerization >500). The steric strain between bottlebrush side chains that limits grafting density in grafting-to polymerization is alleviated to some extent during grafting-from due to the fact that the polymerization occurs on the periphery, away from the highly congested area near the bottlebrush backbone. Of the bottlebrush polymer techniques, however, grafting-from theoretically yields the least well-defined polymers. Initiation efficiency, differential polymerization rate, and termination reactions (such as radical coupling and disproportionation in the case of ATRP or RAFT) act to broaden the distribution of side-chain molecular weights.47 Moreover, it is often difficult to characterize this distribution unless the side chains can be decoupled and separated from the bottlebrush backbone. To limit the formation of defects from these side reactions, several approaches have been developed including optimization of the polymerization conditions (to reduce polymerization rate and limit monomer conversion), heterogeneous polymerization,48 and the use of sacrificial initiator/CTA.49
Transfer-to polymerization is a hybrid of the grafting-to and grafting-from strategies that exists for reversibly deactivated radical polymerization mechanisms (Figure 6C). Similar to grafting-from, transfer-to is conducted by polymerizing the side chains using a polymer with CTAs on each repeat unit. These methods differ based on how the CTA is attached to the backbone. To affect grafting-from, the CTA must be attached to the bottlebrush backbone via the R-group. In contrast, the transfer-to mechanism operates if the CTA is coupled to the backbone polymer through the Z-group.50 Instead of the side chains growing from the periphery of the bottlebrush polymer, as is the case with grafting-from, propagation during transfer-to occurs away from the bottlebrush polymer completely. As radicals are produced by the decomposition of the initiator or the fragmentation of the CTA, the side-chain polymer radical becomes decoupled and dissociates from the bottlebrush polymer. It is then free to add monomer units in solution until it returns to a bottlebrush via a chain-transfer reaction. Because the polymer side chains grow in solution away from the bottlebrush polymers, the transfer-to mechanism more resembles the grafting-to method. Consequently, it suffers the same potential disadvantage of “imperfect” grafting density.
The final bottlebrush polymerization strategy is grafting-through (Figure 6D). During a grafting-through polymerization, macromonomers—polymer chains that possess a polymerizable moiety on one chain end—are polymerized to form the bottlebrush polymer. ROMP has been most often utilized for grafting-through due to its rapid polymerization rates and quantitative monomer conversions.51,52 The grafting-through strategy has a few key advantages, most notably the fact that bottlebrush polymers prepared using grafting-through are said to possess “perfect” grafting density in that each macromonomer bears a pendant polymer chain. As the macromonomers are prepared in a separate polymerization step, the bottlebrush polymers produced by grafting-through are often more precise in their structures than those prepared using other methods. In addition, bottlebrush polymers possessing blocks with different side chain lengths or chemistries or with side chains that are themselves block copolymers can be easily prepared using this method, facilitating the preparation of sophisticated structures such as nanotubes53 and Janus particles.54 Notwithstanding, grafting-through is limited in the length (backbone degree of polymerization) of the bottlebrush polymers that can be prepared. This limitation is attributed to entropic factors arising from the reaction of large macromonomers with an even larger bottlebrush polymer but can be overcome to some extent by increasing the rate of the polymerization. For example, Matson and co-workers demonstrated that careful selection of anchor group chemistry could be employed to enhance the polymerization kinetics of macromonomers,55 and the Cheng group achieved rate enhancement through cooperative behavior between α-helical macromonomers.56
Bottlebrush polymers possess a few specific advantages over other cylindrical nanostructures. Due to the fact that these structures are held together by covalent bonds, bottlebrush polymers do not possess a CAC and thus do not lose their structure upon dilution. Second, bottlebrush polymers can be prepared using a wide variety of chemistries including radical polymerization, ring-opening polymerization, and ROMP, which can be employed orthogonally for backbone and/or side-chain synthesis.57 Finally, the dimensions of bottlebrush polymers are perhaps most easily tuned relative to the other methods of cylindrical nanostructure preparation discussed herein (Figure 7). The length of the cylinder is determined by the degree of polymerization of the bottlebrush polymer backbone,58 while the DP of the side chains sets the cylindrical diameter.59,60 By varying these parameters, along with the stiffness of the polymer side chains, a broad range of cylindrical nanostructures can be prepared with variable rigidity/flexibility, dimensions, and functionality.
Figure 7.

Features of bottlebrush polymers. (A) AFM images of bottlebrush polymers with poly(n-butyl acrylate) side chains. The rigidity of the bottlebrush polymer increases with increasing side-chain length. Reproduced with permission from ref (66). Copyright 2015 Springer Nature. (B) Dependence of bottlebrush polymer structure on side-chain grafting density (Z) and the degree of polymerization (Nsc). Reproduced with permission from ref (59). Copyright 2016 AAAS. (C) Dependence of bottlebrush polymer structure on the length of the polymer backbone. The inflection in the plot of maximum dimension as a function of backbone length indicates a transition from globular to cylindrical structures. Reproduced with permission from ref (58). Copyright 2013 American Chemical Society.
Physical and Mechanical Properties of Cylindrical Nanostructures Prepared by PISA or CDSA, or as Bottlebrush Polymers
Cylindrical nanostructures prepared by one of the three methods described above differ in their physical and mechanical properties. Based on route of synthesis, they can have semicrystalline or amorphous cores, be rigid or semi-flexible, be dynamic and undergo molecular exchange or be covalently locked, and possess active or dormant sites at the cylinder termini. Figure 8 highlights some key differences between the cylindrical nanostructures discussed in this Perspective. Importantly, comparisons made in Figure 8 and in the subsequent discussion are made solely on the route of preparation of the cylinders alone, and does not consider the effects of other factors on their physical and mechanical properties. These considerations are discussed in further detail below.
Figure 8.
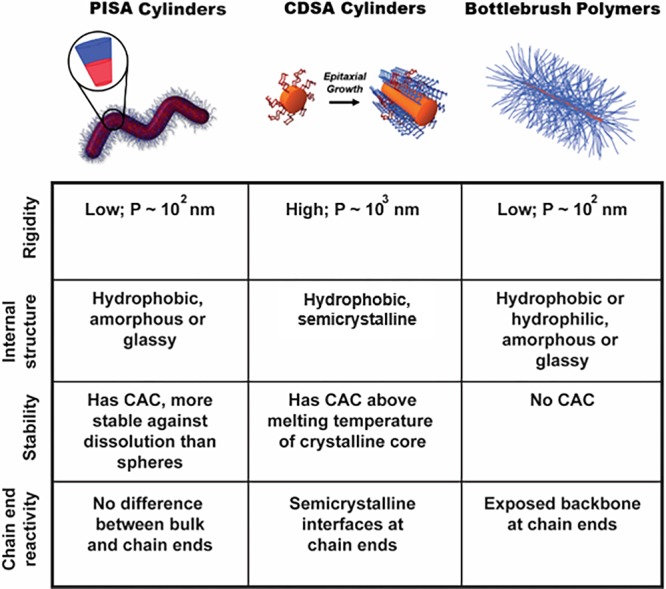
Key differences between the cylindrical nanostructures produced by PISA, CDSA, or bottlebrush polymer synthesis. The variable P signifies the persistence length of the cylindrical constructs. Images reprinted with permission from refs (18) (PISA worms; Copyright 2016 Wiley) and (10) (CDSA cylinders; Copyright 2010 Springer Nature).
Internal Structure
The internal structures of the cylindrical nanostructures highlighted herein vary in their morphology, degree of solvation, and mobility, among other aspects. The first and perhaps most obvious factor that determines internal structure is the fact that cylinders prepared from PISA or CDSA possess core–shell architectures with their cores comprising a solvophobic polymer block. The presence of this solvophobic component is necessary to drive self-assembly. For constructs prepared using PISA, the core can be glassy or amorphous depending on the assembly conditions and the chemistry of the solvophobic block. When glass-forming polymers are used, the core domain can undergo a glass transition temperature (Tg) similar to the bulk polymer, allowing for thermal regulation of the physical properties of the nanoparticle.61 For example, drug release from spherical micelles with glassy core compartments can be accelerated by heating the nanoparticle solution above the Tg (Figure 9A).62 While this study did not explicitly consider drug release from cylindrical micelles, we expect this behavior to hold for nanoparticles of various solution morphologies (i.e., spheres, cylinders, vesicles) that have been prepared using the same core-forming polymer structure. Cylindrical micelles prepared via CDSA possess semicrystalline cores. This adds a second thermal transition—the melting temperature (Tm)—that exists in addition to the Tg (Figure 9B).19
Figure 9.
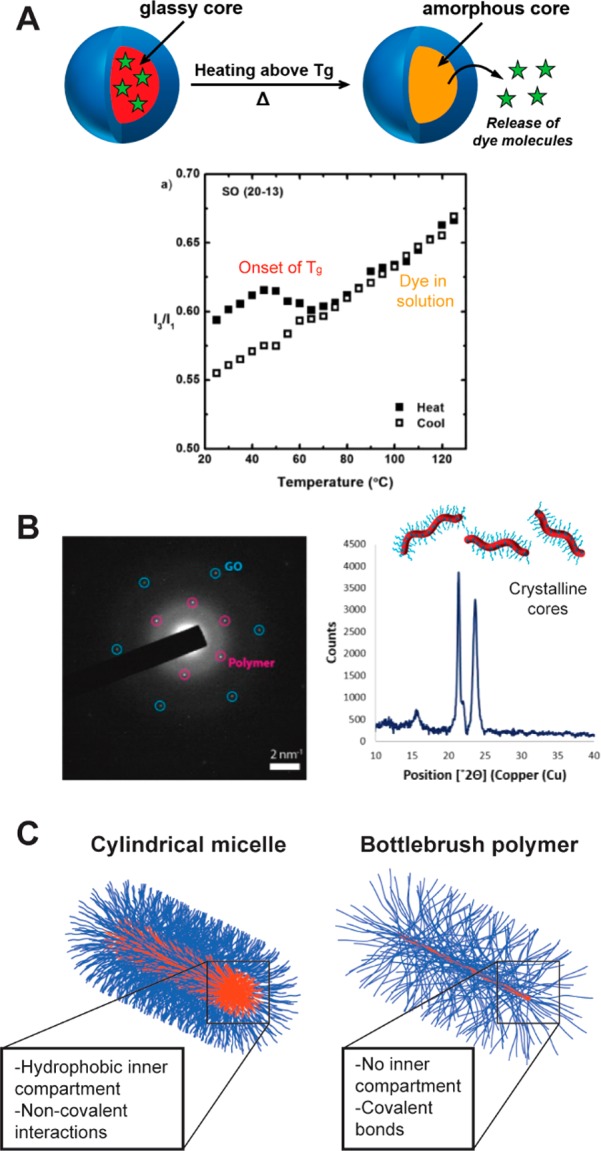
Internal structure of cylindrical micelles can be semicrystalline, glassy, or amorphous. (A) Pyrene release from glassy micelles occurs around and above the Tg of the core block. Reproduced with permission from ref (62). Copyright 2011 Wiley. (B) TEM confirms the crystallinity of cylindrical micelles produced by CDSA. Reproduced with permission from ref (19). Copyright 2017 American Chemical Society. (C) Unlike cylindrical micelles, bottlebrush polymers most likely do not possess an internal hydrophobic compartment.
Bottlebrush polymers differ from the other three types of cylindrical nanostructures due to the fact that they do not necessarily possess an internal hydrophobic compartment (Figure 9C).56 Indeed, EPR studies of spin-labeled bottlebrush polymers demonstrated that nitroxide functionalities near the brush backbone could be easily accessed by small molecule probes (and by extension, solvent molecules).63 Because bottlebrush polymers are comprised of polymers which are covalently attached through the backbone, a self-assembly process is not required to obtain a cylindrical structure.
Rigidity
Cylindrical nanostructures vary in their rigidity based on the chemistry of their constituent polymers, the density of packing of these chains, and the route of preparation. These factors affect the mechanical properties of the resulting material and can influence gelation behavior, rheology, and biological interactions. The following observations with respect to rigidity can be generally applied to the cylindrical nanostructures discussed herein: CDSA cylinders > bottlebrush polymers ∼ cylindrical micelles, with rigidity decreasing across the series. This observation is determined based on the relative persistence lengths of the cylindrical nanostructures produced using these methods, as persistence length is a measure of rigidity.
Cylindrical nanostructures in the form of cylindrical micelles have comparable rigidity to bottlebrush polymers. Persistence lengths can vary from 102 Å to 102 nm, depending on the chemical information coded into the surfactant molecules.64 This sensitivity has been attributed to the interfacial curvature of the micelle as well as the area per hydrophilic headgroup (repulsiveness). In general, electrostatic charge has been shown to be most influential in determining micellar stiffness, with polyelectrolytes exhibiting the largest persistence lengths.3 The mechanical properties of physically cross-linked hydrogels derived from cylindrical micelles also informs about their flexibility, with moduli values typically less than 10 Pa.64 In addition, surfactant molecules (or amphiphilic polymer chains) can be constantly exchanged between cylinder assemblies. This constant breaking and recombination, when occurring on the time scale of relaxation of the gel, generally results in cylindrical micelles exhibiting viscoelastic behavior (as opposed to “gel-like”). As such, factors that affect the CAC of the micelles are also expected to influence the capability of cylindrical solutions to form gels.
In the case of CDSA cylinders, their rigidity stems from the fact that they possess semicrystalline cores. Persistence lengths for these structures can often exceed 1 μm.39 While few examples of hydrogels prepared from CDSA cylinders exist, these typically exhibit storage moduli on the order of 102–103 Pa.65 Because the rigidity of micelles prepared from CDSA is typically greater than those obtained via PISA or traditional polymer self-assembly, as evidenced by higher persistence length values of the cylindrical nanostructures in solution, future research will likely confirm that hydrogels prepared from CDSA cylinders are generally stiffer than PISA hydrogels. However, further study is warranted in this area. Bottlebrush polymers generally possess relatively shorter persistence lengths on the order of 102 nm. Of course, their stiffness depends on the density of grafting, the rigidity of their backbone polymers, and the length and chemistry of the side-chain polymers. In particular, numerous studies have revealed a proportionality between the diameter of the bottlebrush polymer and its persistence length.58,60,66 This relationship originates from mutual repulsion between crowded side chains and is dependent on the number of side chain grafts on each backbone repeat unit. While the solution behavior of bottlebrush polymers has been extensively studied, there exist no reports on the rheology of hydrogels produced via physical cross-linking of bottlebrush polymers. However, gels prepared from intermolecular cross-linking of bottlebrush polymers in the bulk are exceptionally soft, with modulus values on the order of ≤102 Pa.66 It is therefore reasonable to assume that bottlebrush polymer hydrogels would be considerably less stiff than gels prepared from PISA worm-like micelles or CDSA cylinders.
Colloidal Stability
The colloidal stability of the three types of cylindrical nanostructures is determined by a variety of factors, including the capability of their coronae to resist aggregation, their CAC, and the chemical stability of the polymer constituents.67 Here, we focus on how each of the cylindrical structures behave upon dilution. Because cylindrical micelles and CDSA cylinders are formed of individual amphiphilic components, their structures are subject to disassembly below a certain concentration threshold. As with the other properties discussed above, the chemical structure and molecular weight of the constituent amphiphiles are most important in determining CAC. Factors that are known to increase CAC are raising the Tg of the core forming block, increasing the hydrophobicity of this block, and enlarging the size of the unimer. These principles are valid for cylindrical nanostructures prepared by PISA or CDSA.
The solution stability of each of these types of self-assembled cylindrical nanostructures is influenced by additional factors. Cylindrical micelles prepared by PISA have enhanced stability against dilution relative to spherical micelles due to the fact that cylindrical micelles tend to form from “crew-cut” polymer amphiphiles (i.e., those with relatively short hydrophilic segments). Indeed, cylindrical micelles typically exhibit two-phase behavior upon dilution, transitioning from cylinders to spheres before eventually dissociating to their unimer constituents.
In stark contrast, bottlebrush polymers do not possess a CAC due to covalent bonds that constitute their chemical structures. However, bottlebrush polymers are susceptible to degradation by scission of their side chains, as has been demonstrated with poly(n-butyl acrylate) bottlebrush polymers under ultrasonication.68 This tendency of bottlebrush polymer side chains to mechanically cleave may be relevant for applications, especially those involving high shear forces for example in blood vessels or in common industrial processing methods (i.e., extrusion, injection molding, or 3D printing).
Chain-End Reactivity
A final key factor that differentiates cylindrical nanostructures from their spherical analogues is the inherent reactivity of their “chain ends”. Because of their anisotropy, the terminal interfaces of cylinders behave differently than the “bulk” interface. For CDSA cylinders and bottlebrush polymers, the cylinder ends are not fully shielded by the corona chains. This reduced steric bulk at the terminal interfaces renders these regions more hydrophobic and exposes their internal functionalities, which can be utilized as a surface for further crystallization in the case of CDSA cylinders (Figure 10A),69 can undergo chemical reaction, and can facilitate 1D self-assembly of bottlebrush polymers (Figure 10B).70 For bottlebrush polymers, this chain-end reactivity has been evaluated using a nitroxide probe, demonstrating a relatively greater accessibility of the chain-ends to small molecules relative to the bottlebrush polymer backbone.63 While this property is present for CDSA cylinders and bottlebrush polymers, cylindrical PISA micelles typically do not possess reactive chain ends due to the existence of hemispherical end-caps which present a dense corona of hydrophilic chains.71
Figure 10.

Taking advantage of the inherent chain-end reactivity of cylindrical nanostructures. (A) Multiblock cylindrical assemblies prepared via crystallization of each new block from reactive semicrystalline faces at the cylinder ends. Scale bars = 500 nm (TEM) and 2000 nm (LCSM). Reproduced with permission from ref (69). Copyright 2014 Springer Nature. (B) End-to-end self-assembly of bottlebrush polymers occurs through hydrophobic interactions at the bottlebrush chain ends. Scale bar = 100 nm. The inset is from a 500 nm scan. Reproduced with permission from ref (70). Copyright 2011 American Chemical Society.
Conclusions
Cylindrical nanostructures prepared from PISA, CDSA, or bottlebrush polymers have unique properties compared to spherical constructs. Their anisotropic nature leads to enhanced interior volume and external surface area, gelation capability, and chain-end reactivity. Each preparation route yields cylinders with different structures and distinctive properties; therefore, careful consideration is warranted when designing cylinder synthesis for a specific application. For example, CDSA cylinders are ideal for the preparation of nanowires that conduct thermal or electrical energy, and bottlebrush polymers are perhaps most appropriate as drug delivery vehicles owing to their thermodynamic stability.
While much work has gone into understanding the synthesis and properties of the cylindrical structures discussed herein, further studies are warranted. In addition, while these cylinders have been utilized extensively in the preparation of gels, their usefulness in other applications, such as drug delivery, have yet to be fully realized, especially for bottlebrush polymers. Based on our current understanding of the literature, the following points represent the current challenges facing the field of cylindrical nanostructures:
The reactivity of cylindrical chain ends has been observed in multiple systems but is not fully understood. In particular, the 1D assembly of bottlebrush polymers could be exploited for the preparation of stimuli-responsive gels or fibers, and the active chain-end interfaces of CDSA cylinders could be further investigated for the preparation of supramolecular constructs with blocky topologies.
The kinetic and thermodynamic behaviors of cylindrical micelles and CDSA cylinders have not been rigorously established. It is likely the case that these constructs have lower CAC values and reduced unimer exchange rates relative to their spherical analogues. However, structure–property relationships need to be established.
The physical encapsulation capability of bottlebrush polymers with core–shell architectures is virtually unknown. Bottlebrush polymers possess several advantages over the other cylindrical nanostructures for drug delivery. Therefore, their capability to retain and release hydrophobic molecules should be investigated.
While precise control over the size and size distribution of CDSA cylinders has been demonstrated, few reports have considered the placement of useful functionality on different faces/interfaces of the cylinders.
A method for controlling the length of cylindrical micelles prepared using PISA would significantly expand the versatility of these cylindrical nanostructures. Some early progress has been reported in this area,72 but a more general method for programming cylindrical length would further expand their potential applications.
Investigation into the areas mentioned above will facilitate a rapid expansion in the field of cylindrical nanostructures. Indeed, much focus over the past decades has been on the study and deployment of spherical constructs. The numerous advantages of cylindrical nanostructures over their spherical counterparts discussed herein—in particular their larger volume and surface area, enhanced biological retention and cellular internalization, and anisotropy-induced chain end reactivity—make this field an attractive area of study. Better understanding of the synthesis, structure, and properties of cylindrical nanostructures will foster a new dawn in the field of polymer chemistry and could be exploited to answer the evolving challenges of the future.
Acknowledgments
The authors would like to thank the ERC for funding (grant number 615142). B.C. acknowledges funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Grant Agreement No. 703934, FluoroDendriNostic project.
The authors declare no competing financial interest.
References
- Di Lullo G. A.; Sweeney S. M.; Körkkö J.; Ala-Kokko L.; San Antonio J. D. Mapping the Ligand-binding Sites and Disease-associated Mutations on the Most Abundant Protein in the Human, Type I Collagen. J. Biol. Chem. 2002, 277 (6), 4223–4231. 10.1074/jbc.M110709200. [DOI] [PubMed] [Google Scholar]
- Scott J. E. Proteodermatan and Proteokeratan Sulfate (Decorin, Lumican/Fibromodulin) Proteins Are Horseshoe Shaped. Implications for Their Interactions with Collagen. Biochemistry 1996, 35 (27), 8795–8799. 10.1021/bi960773t. [DOI] [PubMed] [Google Scholar]
- Ezrahi S.; Tuval E.; Aserin A. Properties, main applications and perspectives of worm micelles. Adv. Colloid Interface Sci. 2006, 128–130, 77–102. 10.1016/j.cis.2006.11.017. [DOI] [PubMed] [Google Scholar]
- De Greef T. F. A.; Smulders M. M. J.; Wolffs M.; Schenning A. P. H. J.; Sijbesma R. P.; Meijer E. W. Supramolecular Polymerization. Chem. Rev. 2009, 109 (11), 5687–5754. 10.1021/cr900181u. [DOI] [PubMed] [Google Scholar]
- Hendricks M. P.; Sato K.; Palmer L. C.; Stupp S. I. Supramolecular Assembly of Peptide Amphiphiles. Acc. Chem. Res. 2017, 50 (10), 2440–2448. 10.1021/acs.accounts.7b00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesides G.; Mathias J.; Seto C. Molecular self-assembly and nanochemistry: a chemical strategy for the synthesis of nanostructures. Science 1991, 254 (5036), 1312–1319. 10.1126/science.1962191. [DOI] [PubMed] [Google Scholar]
- Discher D. E.; Ahmed F. Polymersomes. Annu. Rev. Biomed. Eng. 2006, 8 (1), 323–341. 10.1146/annurev.bioeng.8.061505.095838. [DOI] [PubMed] [Google Scholar]
- Blanazs A.; Armes S. P.; Ryan A. J. Self-Assembled Block Copolymer Aggregates: From Micelles to Vesicles and their Biological Applications. Macromol. Rapid Commun. 2009, 30 (4–5), 267–277. 10.1002/marc.200800713. [DOI] [PubMed] [Google Scholar]
- Charleux B.; Delaittre G.; Rieger J.; D’Agosto F. Polymerization-Induced Self-Assembly: From Soluble Macromolecules to Block Copolymer Nano-Objects in One Step. Macromolecules 2012, 45 (17), 6753–6765. 10.1021/ma300713f. [DOI] [Google Scholar]
- Gilroy J. B.; Gädt T.; Whittell G. R.; Chabanne L.; Mitchels J. M.; Richardson R. M.; Winnik M. A.; Manners I. Monodisperse cylindrical micelles by crystallization-driven living self-assembly. Nat. Chem. 2010, 2, 566–570. 10.1038/nchem.664. [DOI] [PubMed] [Google Scholar]
- Sheiko S. S.; Sumerlin B. S.; Matyjaszewski K. Cylindrical molecular brushes: Synthesis, characterization, and properties. Prog. Polym. Sci. 2008, 33 (7), 759–785. 10.1016/j.progpolymsci.2008.05.001. [DOI] [Google Scholar]
- Truong N. P.; Quinn J. F.; Whittaker M. R.; Davis T. P. Polymeric filomicelles and nanoworms: two decades of synthesis and application. Polym. Chem. 2016, 7 (26), 4295–4312. 10.1039/C6PY00639F. [DOI] [Google Scholar]
- Huang X.; Teng X.; Chen D.; Tang F.; He J. The effect of the shape of mesoporous silica nanoparticles on cellular uptake and cell function. Biomaterials 2010, 31 (3), 438–48. 10.1016/j.biomaterials.2009.09.060. [DOI] [PubMed] [Google Scholar]
- Christian D. A.; Cai S.; Garbuzenko O. B.; Harada T.; Zajac A. L.; Minko T.; Discher D. E. Flexible Filaments for in Vivo Imaging and Delivery: Persistent Circulation of Filomicelles Opens the Dosage Window for Sustained Tumor Shrinkage. Mol. Pharmaceutics 2009, 6 (5), 1343–1352. 10.1021/mp900022m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowers M. A.; McCombs J. R.; Wang Y.; Paletta J. T.; Morton S. W.; Dreaden E. C.; Boska M. D.; Ottaviani M. F.; Hammond P. T.; Rajca A.; Johnson J. A. Redox-responsive branched-bottlebrush polymers for in vivo MRI and fluorescence imaging. Nat. Commun. 2014, 5, 5460. 10.1038/ncomms6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. A.; Lu Y. Y.; Burts A. O.; Lim Y. H.; Finn M. G.; Koberstein J. T.; Turro N. J.; Tirrell D. A.; Grubbs R. H. Core-clickable PEG-branch-azide bivalent-bottle-brush polymers by ROMP: grafting-through and clicking-to. J. Am. Chem. Soc. 2011, 133 (3), 559–66. 10.1021/ja108441d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreiss C. A. Wormlike micelles: where do we stand? Recent developments, linear rheology and scattering techniques. Soft Matter 2007, 3 (8), 956–970. 10.1039/b705775j. [DOI] [PubMed] [Google Scholar]
- Mitchell D. E.; Lovett J. R.; Armes S. P.; Gibson M. I. Combining Biomimetic Block Copolymer Worms with an Ice-Inhibiting Polymer for the Solvent-Free Cryopreservation of Red Blood Cells. Angew. Chem., Int. Ed. 2016, 55 (8), 2801–2804. 10.1002/anie.201511454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arno M. C.; Inam M.; Coe Z.; Cambridge G.; Macdougall L. J.; Keogh R.; Dove A. P.; O’Reilly R. K. Precision Epitaxy for Aqueous 1D and 2D Poly(ε-caprolactone) Assemblies. J. Am. Chem. Soc. 2017, 139 (46), 16980–16985. 10.1021/jacs.7b10199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Feng Y.; Agrawal N. R.; Raghavan S. R. Wormlike micelles versus water-soluble polymers as rheology-modifiers: similarities and differences. Phys. Chem. Chem. Phys. 2017, 19 (36), 24458–24466. 10.1039/C7CP04962E. [DOI] [PubMed] [Google Scholar]
- Dalsin S. J.; Hillmyer M. A.; Bates F. S. Linear Rheology of Polyolefin-Based Bottlebrush Polymers. Macromolecules 2015, 48 (13), 4680–4691. 10.1021/acs.macromol.5b01153. [DOI] [Google Scholar]
- Albiges R.; Klein P.; Roi S.; Stoffelbach F.; Creton C.; Bouteiller L.; Rieger J. Water-based acrylic coatings reinforced by PISA-derived fibers. Polym. Chem. 2017, 8 (34), 4992–4995. 10.1039/C7PY00302A. [DOI] [Google Scholar]
- Sveinbjörnsson B. R.; Weitekamp R. A.; Miyake G. M.; Xia Y.; Atwater H. A.; Grubbs R. H. Rapid self-assembly of brush block copolymers to photonic crystals. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (36), 14332–14336. 10.1073/pnas.1213055109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X.-H.; Price M. B.; Finnegan J. R.; Boott C. E.; Richter J. M.; Rao A.; Menke S. M.; Friend R. H.; Whittell G. R.; Manners I. Long-range exciton transport in conjugated polymer nanofibers prepared by seeded growth. Science 2018, 360 (6391), 897–900. 10.1126/science.aar8104. [DOI] [PubMed] [Google Scholar]
- Warren N. J.; Mykhaylyk O. O.; Mahmood D.; Ryan A. J.; Armes S. P. RAFT Aqueous Dispersion Polymerization Yields Poly(ethylene glycol)-Based Diblock Copolymer Nano-Objects with Predictable Single Phase Morphologies. J. Am. Chem. Soc. 2014, 136 (3), 1023–1033. 10.1021/ja410593n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J.; Liu D.; Bai Y.; Huang C.; Li X.; He J.; Xu Q.; Zhang L. Enzyme-Assisted Photoinitiated Polymerization-Induced Self-Assembly: An Oxygen-Tolerant Method for Preparing Block Copolymer Nano-Objects in Open Vessels and Multiwell Plates. Macromolecules 2017, 50 (15), 5798–5806. 10.1021/acs.macromol.7b01219. [DOI] [Google Scholar]
- Foster J. C.; Varlas S.; Blackman L. D.; Arkinstall L. A.; O’Reilly R. K. Ring-Opening Metathesis Polymerization in Aqueous Media Using a Macroinitiator Approach. Angew. Chem., Int. Ed. 2018, 57 (33), 10672–10676. 10.1002/anie.201806719. [DOI] [PubMed] [Google Scholar]
- Wright D. B.; Touve M. A.; Thompson M. P.; Gianneschi N. C. Aqueous-Phase Ring-Opening Metathesis Polymerization-Induced Self-Assembly. ACS Macro Lett. 2018, 7 (4), 401–405. 10.1021/acsmacrolett.8b00091. [DOI] [PubMed] [Google Scholar]
- Foster J. C.; Varlas S.; Couturaud B.; Jones J. R.; Keogh R.; Mathers R. T.; O’Reilly R. K. Predicting Monomers for Use in Polymerization-Induced Self-Assembly. Angew. Chem., Int. Ed. 2018, 57 (48), 15733–15737. 10.1002/anie.201809614. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Lv X.; Zhu A.; Zheng J.; Yang Y.; An Z. Morphological Stabilization of Block Copolymer Worms Using Asymmetric Cross-Linkers during Polymerization-Induced Self-Assembly. Macromolecules 2018, 51 (8), 2776–2784. 10.1021/acs.macromol.8b00246. [DOI] [Google Scholar]
- Canning S. L.; Smith G. N.; Armes S. P. A Critical Appraisal of RAFT-Mediated Polymerization-Induced Self-Assembly. Macromolecules 2016, 49 (6), 1985–2001. 10.1021/acs.macromol.5b02602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey J.; Power K. N.; Manners I.; Winnik M. A. Self-Assembly of a Novel Organometallic–Inorganic Block Copolymer in Solution and the Solid State: Nonintrusive Observation of Novel Wormlike Poly(ferrocenyldimethylsilane)-b-Poly(dimethylsiloxane) Micelles. J. Am. Chem. Soc. 1998, 120 (37), 9533–9540. 10.1021/ja981803d. [DOI] [Google Scholar]
- Sun L.; Petzetakis N.; Pitto-Barry A.; Schiller T. L.; Kirby N.; Keddie D. J.; Boyd B. J.; O’Reilly R. K.; Dove A. P. Tuning the Size of Cylindrical Micelles from Poly(l-lactide)-b-poly(acrylic acid) Diblock Copolymers Based on Crystallization-Driven Self-Assembly. Macromolecules 2013, 46 (22), 9074–9082. 10.1021/ma401634s. [DOI] [Google Scholar]
- Patra S. K.; Ahmed R.; Whittell G. R.; Lunn D. J.; Dunphy E. L.; Winnik M. A.; Manners I. Cylindrical Micelles of Controlled Length with a π-Conjugated Polythiophene Core via Crystallization-Driven Self-Assembly. J. Am. Chem. Soc. 2011, 133 (23), 8842–8845. 10.1021/ja202408w. [DOI] [PubMed] [Google Scholar]
- Tao D.; Feng C.; Cui Y.; Yang X.; Manners I.; Winnik M. A.; Huang X. Monodisperse Fiber-like Micelles of Controlled Length and Composition with an Oligo(p-phenylenevinylene) Core via “Living” Crystallization-Driven Self-Assembly. J. Am. Chem. Soc. 2017, 139 (21), 7136–7139. 10.1021/jacs.7b02208. [DOI] [PubMed] [Google Scholar]
- Schöbel J.; Karg M.; Rosenbach D.; Krauss G.; Greiner A.; Schmalz H. Patchy Wormlike Micelles with Tailored Functionality by Crystallization-Driven Self-Assembly: A Versatile Platform for Mesostructured Hybrid Materials. Macromolecules 2016, 49 (7), 2761–2771. 10.1021/acs.macromol.6b00330. [DOI] [Google Scholar]
- Kynaston E. L.; Nazemi A.; MacFarlane L. R.; Whittell G. R.; Faul C. F. J.; Manners I. Uniform Polyselenophene Block Copolymer Fiberlike Micelles and Block Co-micelles via Living Crystallization-Driven Self-Assembly. Macromolecules 2018, 51 (3), 1002–1010. 10.1021/acs.macromol.7b02317. [DOI] [Google Scholar]
- Hudson Z. M.; Boott C. E.; Robinson M. E.; Rupar P. A.; Winnik M. A.; Manners I. Tailored hierarchical micelle architectures using living crystallization-driven self-assembly in two dimensions. Nat. Chem. 2014, 6 (10), 893–898. 10.1038/nchem.2038. [DOI] [PubMed] [Google Scholar]
- Gädt T.; Ieong N. S.; Cambridge G.; Winnik M. A.; Manners I. Complex and hierarchical micelle architectures from diblock copolymers using living, crystallization-driven polymerizations. Nat. Mater. 2009, 8, 144–150. 10.1038/nmat2356. [DOI] [PubMed] [Google Scholar]
- Lunn D. J.; Gould O. E. C.; Whittell G. R.; Armstrong D. P.; Mineart K. P.; Winnik M. A.; Spontak R. J.; Pringle P. G.; Manners I. Microfibres and macroscopic films from the coordination-driven hierarchical self-assembly of cylindrical micelles. Nat. Commun. 2016, 7, 12371. 10.1038/ncomms12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou H.; Li M.; Qiao Y.; Harniman R.; Li X.; Boott C. E.; Mann S.; Manners I. Higher-order assembly of crystalline cylindrical micelles into membrane-extendable colloidosomes. Nat. Commun. 2017, 8 (1), 426–426. 10.1038/s41467-017-00465-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzari M.; Scalarone D.; Vazquez-Vazquez C.; López-Quintela M. A. Cylindrical Micelles from the Self-Assembly of Polyacrylonitrile-Based Diblock Copolymers in Nonpolar Selective Solvents. Macromol. Rapid Commun. 2008, 29 (4), 352–357. 10.1002/marc.200700818. [DOI] [Google Scholar]
- Inam M.; Cambridge G.; Pitto-Barry A.; Laker Z. P. L.; Wilson N. R.; Mathers R. T.; Dove A. P.; O’Reilly R. K. 1D vs. 2D shape selectivity in the crystallization-driven self-assembly of polylactide block copolymers. Chem. Sci. 2017, 8 (6), 4223–4230. 10.1039/C7SC00641A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenyves R.; Schmutz M.; Horner I. J.; Bright F. V.; Rzayev J. Aqueous Self-Assembly of Giant Bottlebrush Block Copolymer Surfactants as Shape-Tunable Building Blocks. J. Am. Chem. Soc. 2014, 136 (21), 7762–7770. 10.1021/ja503283r. [DOI] [PubMed] [Google Scholar]
- Radzinski S. C.; Foster J. C.; Scannelli S. J.; Weaver J. R.; Arrington K. J.; Matson J. B. Tapered Bottlebrush Polymers: Cone-Shaped Nanostructures by Sequential Addition of Macromonomers. ACS Macro Lett. 2017, 6 (10), 1175–1179. 10.1021/acsmacrolett.7b00724. [DOI] [PubMed] [Google Scholar]
- Lin T.-P.; Chang A. B.; Chen H.-Y.; Liberman-Martin A. L.; Bates C. M.; Voegtle M. J.; Bauer C. A.; Grubbs R. H. Control of Grafting Density and Distribution in Graft Polymers by Living Ring-Opening Metathesis Copolymerization. J. Am. Chem. Soc. 2017, 139 (10), 3896–3903. 10.1021/jacs.7b00791. [DOI] [PubMed] [Google Scholar]
- Barner L.; Davis T. P.; Stenzel M. H.; Barner-Kowollik C. Complex Macromolecular Architectures by Reversible Addition Fragmentation Chain Transfer Chemistry: Theory and Practice. Macromol. Rapid Commun. 2007, 28 (5), 539–559. 10.1002/marc.200600805. [DOI] [Google Scholar]
- Min K.; Yu S.; Lee H.-i.; Mueller L.; Sheiko S. S.; Matyjaszewski K. High Yield Synthesis of Molecular Brushes via ATRP in Miniemulsion. Macromolecules 2007, 40 (18), 6557–6563. 10.1021/ma071137d. [DOI] [Google Scholar]
- Zheng Z.; Ling J.; Müller A. H. E. Revival of the R-Group Approach: A “CTA-shuttled” Grafting from Approach for Well-Defined Cylindrical Polymer Brushes via RAFT Polymerization. Macromol. Rapid Commun. 2014, 35 (2), 234–241. 10.1002/marc.201300578. [DOI] [PubMed] [Google Scholar]
- Foster J. C.; Radzinski S. C.; Matson J. B. Graft polymer synthesis by RAFT transfer-to. J. Polym. Sci., Part A: Polym. Chem. 2017, 55 (18), 2865–2876. 10.1002/pola.28621. [DOI] [Google Scholar]
- Xia Y.; Kornfield J. A.; Grubbs R. H. Efficient Synthesis of Narrowly Dispersed Brush Polymers via Living Ring-Opening Metathesis Polymerization of Macromonomers. Macromolecules 2009, 42 (11), 3761–3766. 10.1021/ma900280c. [DOI] [Google Scholar]
- Li Z.; Zhang K.; Ma J.; Cheng C.; Wooley K. L. Facile syntheses of cylindrical molecular brushes by a sequential RAFT and ROMP “grafting-through” methodology. J. Polym. Sci., Part A: Polym. Chem. 2009, 47 (20), 5557–5563. 10.1002/pola.23626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K.; Rzayev J. Well-Defined Organic Nanotubes from Multicomponent Bottlebrush Copolymers. J. Am. Chem. Soc. 2009, 131 (19), 6880–6885. 10.1021/ja901936g. [DOI] [PubMed] [Google Scholar]
- Li Y.; Themistou E.; Zou J.; Das B. P.; Tsianou M.; Cheng C. Facile Synthesis and Visualization of Janus Double-Brush Copolymers. ACS Macro Lett. 2012, 1 (1), 52–56. 10.1021/mz200013e. [DOI] [PubMed] [Google Scholar]
- Radzinski S. C.; Foster J. C.; Chapleski R. C.; Troya D.; Matson J. B. Bottlebrush Polymer Synthesis by Ring-Opening Metathesis Polymerization: The Significance of the Anchor Group. J. Am. Chem. Soc. 2016, 138 (22), 6998–7004. 10.1021/jacs.5b13317. [DOI] [PubMed] [Google Scholar]
- Baumgartner R.; Fu H.; Song Z.; Lin Y.; Cheng J. Cooperative polymerization of α-helices induced by macromolecular architecture. Nat. Chem. 2017, 9, 614–622. 10.1038/nchem.2712. [DOI] [PubMed] [Google Scholar]
- Bolton J.; Rzayev J. Synthesis and Melt Self-Assembly of PS–PMMA–PLA Triblock Bottlebrush Copolymers. Macromolecules 2014, 47 (9), 2864–2874. 10.1021/ma500625k. [DOI] [Google Scholar]
- Pesek S. L.; Li X.; Hammouda B.; Hong K.; Verduzco R. Small-Angle Neutron Scattering Analysis of Bottlebrush Polymers Prepared via Grafting-Through Polymerization. Macromolecules 2013, 46 (17), 6998–7005. 10.1021/ma401246b. [DOI] [Google Scholar]
- Paturej J.; Sheiko S. S.; Panyukov S.; Rubinstein M. Molecular structure of bottlebrush polymers in melts. Sci. Adv. 2016, 2 (11), e1601478 10.1126/sciadv.1601478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecommandoux S.; Chécot F.; Borsali R.; Schappacher M.; Deffieux A.; Brûlet A.; Cotton J. P. Effect of Dense Grafting on the Backbone Conformation of Bottlebrush Polymers: Determination of the Persistence Length in Solution. Macromolecules 2002, 35 (23), 8878–8881. 10.1021/ma0203344. [DOI] [Google Scholar]
- Nicolai T.; Colombani O.; Chassenieux C. Dynamic polymeric micelles versus frozen nanoparticles formed by block copolymers. Soft Matter 2010, 6 (14), 3111–3118. 10.1039/b925666k. [DOI] [Google Scholar]
- Mok M. M.; Lodge T. P. Temperature-based fluorescence measurements of pyrene in block copolymer micelles: Probing micelle core glass transition breadths. J. Polym. Sci., Part B: Polym. Phys. 2012, 50 (7), 500–515. 10.1002/polb.23029. [DOI] [Google Scholar]
- Xia Y.; Li Y.; Burts A. O.; Ottaviani M. F.; Tirrell D. A.; Johnson J. A.; Turro N. J.; Grubbs R. H. EPR Study of Spin Labeled Brush Polymers in Organic Solvents. J. Am. Chem. Soc. 2011, 133 (49), 19953–19959. 10.1021/ja2085349. [DOI] [PubMed] [Google Scholar]
- Raghavan S. R.; Douglas J. F. The conundrum of gel formation by molecular nanofibers, wormlike micelles, and filamentous proteins: gelation without cross-links?. Soft Matter 2012, 8 (33), 8539–8546. 10.1039/c2sm25107h. [DOI] [Google Scholar]
- Huang Z.; Lee H.; Lee E.; Kang S.-K.; Nam J.-M.; Lee M. Responsive nematic gels from the self-assembly of aqueous nanofibres. Nat. Commun. 2011, 2, 459. 10.1038/ncomms1465. [DOI] [PubMed] [Google Scholar]
- Daniel W. F. M.; Burdyńska J.; Vatankhah-Varnoosfaderani M.; Matyjaszewski K.; Paturej J.; Rubinstein M.; Dobrynin A. V.; Sheiko S. S. Solvent-free, supersoft and superelastic bottlebrush melts and networks. Nat. Mater. 2016, 15, 183. 10.1038/nmat4508. [DOI] [PubMed] [Google Scholar]
- Jones M.-C.; Leroux J.-C. Polymeric micelles – a new generation of colloidal drug carriers. Eur. J. Pharm. Biopharm. 1999, 48 (2), 101–111. 10.1016/S0939-6411(99)00039-9. [DOI] [PubMed] [Google Scholar]
- Li Y.; Niu Z.; Burdyńska J.; Nese A.; Zhou Y.; Kean Z. S.; Dobrynin A. V.; Matyjaszewski K.; Craig S. L.; Sheiko S. S. Sonication-induced scission of molecular bottlebrushes: Implications of the “hairy” architecture. Polymer 2016, 84, 178–184. 10.1016/j.polymer.2015.12.044. [DOI] [Google Scholar]
- Hudson Z. M.; Lunn D. J.; Winnik M. A.; Manners I. Colour-tunable fluorescent multiblock micelles. Nat. Commun. 2014, 5, 3372. 10.1038/ncomms4372. [DOI] [PubMed] [Google Scholar]
- Li Z.; Ma J.; Lee N. S.; Wooley K. L. Dynamic Cylindrical Assembly of Triblock Copolymers by a Hierarchical Process of Covalent and Supramolecular Interactions. J. Am. Chem. Soc. 2011, 133 (5), 1228–1231. 10.1021/ja109191z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan N.; Safran S. A. Junctions and end-caps in self-assembled non-ionic cylindrical micelles. Adv. Colloid Interface Sci. 2006, 123–126, 323–331. 10.1016/j.cis.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Boott C. E.; Gwyther J.; Harniman R. L.; Hayward D. W.; Manners I. Scalable and uniform 1D nanoparticles by synchronous polymerization, crystallization and self-assembly. Nat. Chem. 2017, 9, 785–792. 10.1038/nchem.2721. [DOI] [PubMed] [Google Scholar]