

Abstract
Rationale:
Hyponatremia is one of the most common electrolyte disorders in clinic. Due to the complicated etiology and the nonspecific clinical manifestations, the diagnosis of hyponatremia is a complicated process. A variety of clinical disorders can cause inappropriately increased antidiuretic hormone (ADH) secretion, leading to inappropriate water retention and consequent hyponatremia. The most common cause of hyponatremia in hospital inpatients is syndrome of inappropriate antidiuretic (SIADH). The action of glucocorticoid against pituitary posterior lobe can reduce the secretion of ADH. However, the effect of hormone on diuretic hormone during treatment has been less reported.
Patient concerns and diagnosis:
The patient in this case report was misdiagnosed as anterior pituitary hypofunction because of the long-term glucocorticoid therapy was effective in this patient, and the patient was finally diagnosed as SIADH after reassessment. The patient is a 76-year-old male with long-term symptomatic hyponatremia after traumatic brain injury (TBI). The patient has been consistently diagnosed as anterior pituitary hypofunction. Based on the diagnosis, glucocorticoid replacement therapy was administered. The serum sodium of the patient gradually increased to normal level after hydrocortisone intravenous injection but dropped again after switch to hydrocortisone oral administration. Through examination and analysis of the patient status during the five-time hospitalization, syndrome of inappropriate antidiuretic hormone (SIADH) was considered.
Interventions:
Water intake limitation and oral furosemide and antisterone were administered after glucocorticoid therapy was stopped.
Outcome:
The serum sodium level of the patient gradually increased and maintained within normal range based on his clinical follow-up.
Lessons:
For hyponatremia with effective glucocorticoid treatment, SIADH should still be excluded.
Keywords: anterior pituitary hypofunction, glucocorticoid therapy, hyponatremia, syndrome of inappropriate antidiuretic hormone, traumatic brain injury
1. Introduction
Hyponatremia, characterized by a serum sodium concentration <135 mmol/L, is a common disorder of water and electrolyte imbalance encountered in 15%–30% of hospitalized patients.[1] Hyponatremia can lead to extensive clinical symptoms from mild to severe and even death. However, the complicated pathogenesis and nonspecific clinical manifestations of hyponatremia could lead to misdiagnosis. Hyponatremia caused by different pathogeneses requires distinct treatments. Therefore, finding the exact pathogenesis is crucial for making treatment plans. At present, the literature reports about SIADH mainly were case reports, and generally focus on the etiology. Most of the literatures do not make further comparison of the similarities and differences of diagnosis and treatment, especially the effect of hormone on ADH is seldom reported. Therefore, in this paper we make a detailed presentation of a case in which a SIADH patient's blood sodium returns to normal after the use of glucocorticoid. In the present report, we expound the whole therapeutic process of the patient in detail and review relevant literatures to understand the effect of glucocorticoid on ADH, and elaborate emphasis need to be paid attention to in the diagnosis of SIADH.
2. Case description
A 76-year-old male was hospitalized in the neurosurgery department and endocrinology department because of repeated debilitation for 8 months after traumatic brain injury (TBI) (December 11, 2012–September 23, 2013).
2.1. The first hospitalization (December 11, 2012–February 5, 2013)
The patient suffered from traumatic brain injury (TBI) after a car accident on December 11, 2012. Accompanied by an episode of unconsciousness, he was immediately sent to the emergency department in our hospital. The head computer tomography (CT) showed cerebral contusion and laceration, subdural edema, traumatic subarachnoid hemorrhage, and parietal fracture. Emergency operation was immediately performed in the neurosurgery department, and preoperative serum sodium was 137 mmol/L (reference range 136–146 mmol/L). Drowsiness was noted on the third day after the operation, with serum sodium at 124.6 mmol/L; the symptoms indicative of hyponatremia was relieved after intravenous injection of sodium, but serum sodium rapidly decreased when sodium supplementation stopped. On January 18, 2013, the patient presented with fatigue and anorexia, the serum sodium was 112.2 mmol/L and serum cortisol was 9.9 μg/dL (reference range: 8.7–22.4 μg/dL), but the serum adrenocorticotrophic hormone (ACTH) and pituitary magnetic resonance (MR) were not performed. The surgeon considered that the patient had an anterior pituitary hypofunction. 150 mg hydrocortisone was administered intravenously, and the serum sodium increased to 122 mmol/L the following day. The symptoms were improved after intravenous hydrocortisone treatment, serum sodium increased to 126.2 mmol/L. After discharge, prednisone 10 mg/d was orally administered instead.
2.2. The second hospitalization (March 17, 2013–March 25, 2013)
When the patient deliberately stopped taking glucocorticoid on March 10, 2013, debilitation recurred. On March 17, he went back to the hospital for consultation, and his serum sodium level decreased to 121.4 mmol/L. He was then admitted with a preliminary diagnosis of “anterior pituitary hypofunction.” Hydrocortisone therapy was provided and serum sodium level increased from 123.4 mmol/L to 136 mmol/L. Patient's condition consequently improved. Moreover, oral administration of 10 mg/day prednisone was prescribed, and he was discharged.
2.3. The third hospitalization (March 31, 2013–April 10, 2013)
On March 20, 2013, the patient experienced debilitation and poor appetite again. The serum sodium level was 116.7 mmol/L, which prompted for his re-admission to receive hydrocortisone therapy. After the intravenous treatment with hydrocortisone, the symptoms of fatigue and poor appetite were improved and the serum sodium levels returned to normal; he was discharged from hospital and advised prednisone 10 mg/d replacement therapy orally.
2.4. The fourth hospitalization (June 3, 2013–June 13, 2013)
After discharge, the patient suffered debilitation again. We find that this is because he has intentionally increased the oral dosage of prednisone from 15 mg/day to 45 mg/day gradually. On June 1, 2013, he experienced nausea and poor appetite. Hence, he was hospitalized for the fourth time. His symptoms were relieved after hydrocortisone therapy, and he was discharged with 40 mg/day hydrocortisone administration orally.
2.5. The fifth hospitalization (August 23, 2013–September 23, 2013)
On July, 2013, edema gradually appeared on both lower extremities of the patient. Along with debilitation and poor appetite, he was hospitalized again on August 23, 2013. He was physically examined and obtained the following results: body temperature = 36.7°C, pulse rate = 84 beats/min, respiratory rate = 20 breaths/min, and blood pressure = 145/80 mm Hg. In addition, his mental state was relatively normal and remained cooperative throughout the physical examination. However, he had moon-shaped face, scattered bruises were noted all over his body, and his right cephalic was sunken. Nonetheless, manifestation of goiter was not evident on both sides. Breath sounds of both lungs were clear without wet and dry rale. Heart rate was 84 beats/min with regular rhythm. His abdomen was flat and soft without purple striaes or subcutaneous varicose vein of the abdominal wall, pain, and rebound tenderness. His liver and spleen, however, cannot be thoroughly assessed. Edema of both lower extremities existed, and muscle force of four extremities reached 4+ grade. Laboratory examinations after hospitalization are listed in Table 1.
Table 1.
Laboratory characteristics after hospitalization.
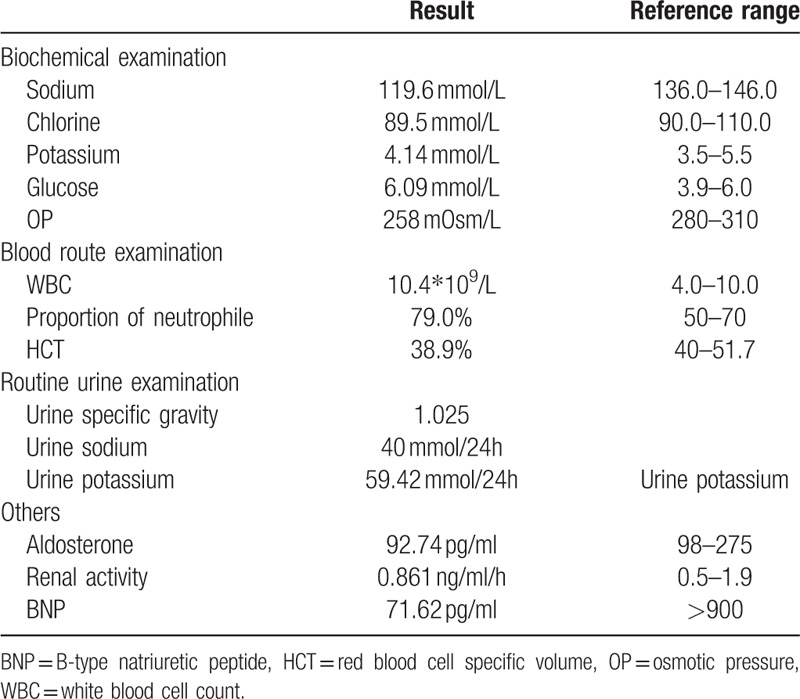
We reviewed the pituitary hormone examination (seen in Table 2) and pituitary MR (Fig. 1) that the patient carried out during the fifth hospitalization, and no signs of abnormality were found. The series of examinations mentioned did not support the diagnosis of anterior pituitary hypofunction. Although the patient's serum sodium level increased after intravenous injection of hydrocortisone, his serum sodium dropped again without clear inducement when switched to oral administration of prednisone, which went against the diagnosis of anterior pituitary hypofunction.
Table 2.
Pituitary hormone examination results.

Figure 1.
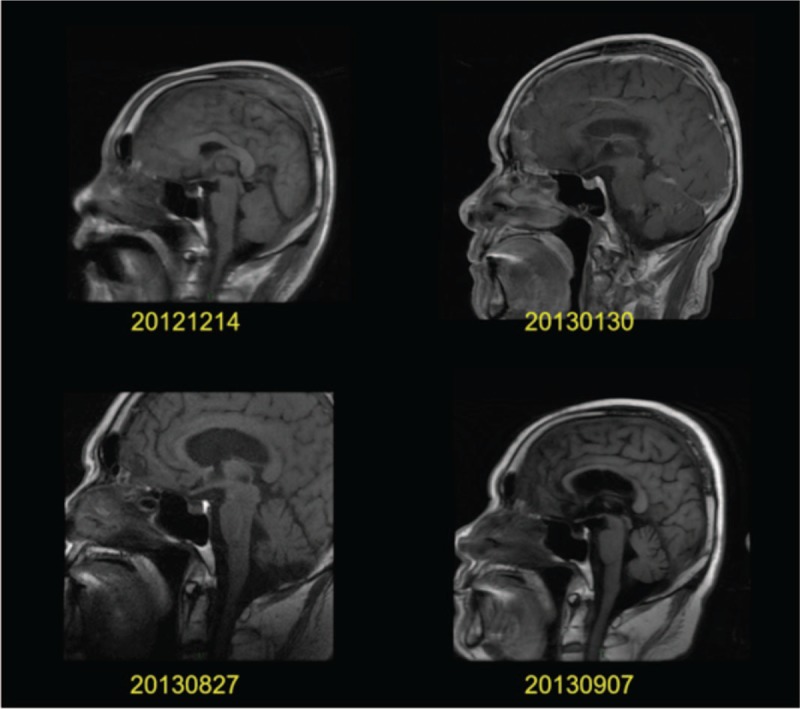
Pituitary magnetic resonance during hospitalization: pituitary magnetic resonance imaging showed low signal in the anterior pituitary and high signal in the posterior pituitary. No abnormalities were found in the morphology of the pituitary.
The laboratory examination results suggested that the patient had hypotonic hyponatremia accompanied by apparent elevated urine sodium level (>30 mmol/L). Considering the above examination results and the repeated abnormal serum sodium after using glucocorticoids did not support anterior pituitary hypofunction, SIADH was considered, so glucocorticoid therapy was stopped. Water intake limitation and furosemide diuresis therapy were provided. As a result, the serum sodium level increased gradually. However, during furosemide diuresis treatment, serum potassium level was low. Based on the above conditions, antisterone combined diuresis was administered, and serum sodium level maintained normal and stable.
2.6. Follow-up (October 14, 2013-May 7, 2017)
After the patient was discharged, he was followed up every 2 weeks to 1 month in the endocrinology department. The therapeutic dosage of antisterone was gradually reduced from 20 mg/day at the beginning to 0 mg/day, and was discontinued at June 30, 2015. The therapeutic dose of furosemide was gradually reduced from 120 mg/day at the beginning to 0 mg/day, and was discontinued on January 17, 2017. During the follow-up visit, in addition to one time we noticed that the serum sodium concentration decreased after a reduction of furosemide and then increased after the restoration of furosemide treatment; the serum sodium level was stable, and there was no decrease of serum sodium concentration after drug withdrawal (Fig. 2).
Figure 2.
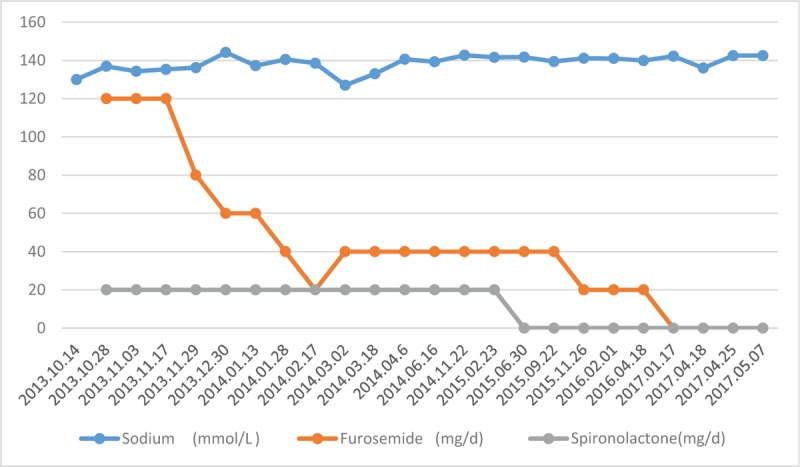
Clinical follow-up treatment: during the follow-up period, the serum sodium concentration was stable, and there was no decrease in the serum sodium after drug withdrawal. Despite the serum sodium decreased after a furosemide reduction, when restoring furosemide treatment, the serum sodium level remained stable. There was still no decrease in serum sodium concentration after the withdrawal.
3. Discussion
The patient was hospitalized due to intractable hyponatremia after craniocerebral trauma. According to the clinical practice guideline on diagnosis and treatment of hyponatremia,[2] it is necessary to determine the plasma osmolality of patients first. In this case, the patient showed hypotonic hyponatremia. At the same time, the urine sodium level was high (>30 mmol/L). CSWS and SIADH are the two most common clinical types causing neurosurgical hyponatremia.[3] After excluding the anterior pituitary hypofunction, we should focus on cerebral salt-wasting syndrome (CSWS) and SIADH. The main manifestations of the former are hyponatremia, increased urine sodium level, and hypovolemia, whereas those of the latter are hyponatremia, increased urine sodium level, and normal or slightly increased blood volume. Hence, the difference between the two mainly lies in the blood volume. The blood pressure of the patient exceeded normal level with normal B-type natriuretic peptide (BNP) and hematocrit and without manifestations of insufficient blood volume, such as hypotension, polycardia, dry skin, mucosa, and so on. Therefore, CSWS was not the correct diagnosis. SIADH should be considered.
As the most common pathogenesis of normal blood volume-type hyponatremia in clinical practice,[1] SIADH nearly accounts for a half of hyponatremia cases in some research.[4,5] SIADH, discovered by Schwartz[6] in two patients with bronchogenic carcinoma together with hyponatremia, is a clinical syndrome with increased in vivo moisture, delusional hyponatremia, and elevated urine sodium level and urine osmotic pressure due to excessive secretion of antidiuretic hormone (ADH). Among currently published literature, diagnostic criteria for SIADH[7] include the following: (1) low plasma osmotic pressure (<275 mOsm/kg); (2) urine concentration (urine osmotic pressure <100 mOsm/kg); (3) urine sodium >30 mmol/L; (4) clinically normal blood volume; and (5) exclusion of hypothyroidism and secondary adrenocortical hypofunction. To this day, the pathophysiological mechanism responsible for SIADH after TBI remain unclear.[8] The possible cascade mechanism is that damage to the supraoptic nucleus and the paraventricular nucleus of the hypothalamus results in abnormal release of pituitary vasopressin in the pituitary nerve lobe, which leads to decreased clearance of free water, then resulting in increased water retention and extracellular capacity, finally hyponatremia happened.
The feature of this case was intractable hyponatremia, and increased serum sodium level after glucocorticoid therapy. In general, the daily cortisol secretion in healthy adults is 9–11 mg (m2)−1·d−1.[9] Patients accepted >20 mg/d hydrocortisone treatment are exposed in a higher risk of adverse metabolism.[10] This patient was admitted to hospital by refractory hyponatremia after craniocerebral trauma, and was administered long-term glucocorticoid replacement therapy. However, his serum sodium level still fluctuated greatly and hyponatremia recurred repeatedly. Glucocorticoid side effects emerged. Therefore, we need to re-evaluate the patient's clinical data and denied the diagnosis of hypopituitarism. The cause of such clinical manifestation might be that some corticoid steroids (e.g., hydrocortisone and prednisone) have evident mineral corticoid activity, which may influence the clinical regulation of body fluids and electrolytes. Kinoshita et al[11] held that hyponatremia is caused by glucocorticoids that promote sodium reabsorption by kidney tubules, though this view lacked theoretical basis. Based on retrospective analysis of diagnosis and treatment process of five hyponatremic patients with hypopituitarism, Oelkers[12] speculated that glucocorticoids corrected hyponatremia by inhibiting ADH secretion. Papanek et al[13] verified this speculation by administering cortisol on dogs and presented that the inhibiting mechanism of glucocorticoids for ADH might be explained as follows: (1) glucocorticoids indirectly inhibit ADH secretion by inhibiting corticotrophin-releasing hormone (CRH); and (2) glucocorticoids directly inhibit ADH by affecting the hypothalamic supraoptic nucleus or paraventricular nucleus to synthesize or secrete ADH. Study of Erkut et al[14] indicated that in hypothalamic nerve cells, CRH and ADH expression levels are affected by glucocorticoids. Moreover, the authors found in clinical practice that not all SIADH patients experience increased serum sodium after taking glucocorticoids. Another 36-year-old female patient in our hospital was admitted due to recurrence of hyponatremia. After intravenous injection of 100 to 150 mg hydrocortisone per day for 2 weeks, serum sodium did not increase. The patient was diagnosed with cervical cancer concurrent with SIADH during follow-up diagnosis and treatment process. The serum sodium level of this patient did not increase after taking glucocorticoids, which indicated that ADH activity was too high due to the tumor and, consequently, glucocorticoids were unable to exert the anti-ADH effect.
SIADH is usually recognized as an early complication of TBI, it rarely persists or recurs. The published reports about recurrent hyponatremia on chronic SIADH caused by TBI are few. Dick et al[15] reported a case of brain injury after 4 years of SIADH causing persistent symptomatic hyponatremia, and Chang et al[16] also reported a case of a 48-year-old man with recurrent symptomatic hyponatremia in TBI-associated SIADH. In this case, the patient from appearing hyponatremia to stopping the treatment of diuretics, underwent a total of 53 months (December 2012 to May 2017). This indicates that the duration of TBI-related SIADH varies, and it is necessary for us to pay more attention to the sodium changes during follow-up.
4. Conclusion
Hyponatremia caused by “secondary adrenocortical hypofunction” can be effectively treated by glucocorticoids and restores serum sodium level to normal. However, the probability of SIADH should still be considered. Glucocorticoids have distinct manifestations in correcting hyponatremia caused by SIADH. We have observed the followings: in the treatment of SIADH caused by head trauma, intravenous administration of glucocorticoids could normalize serum sodium level but oral administration of glucocorticoids could not; for SIADH caused by tumor, glucocorticoids probably could not inhibit ADH activity and normalize serum sodium level. The above relevant speculations should be further verified in a follow-up experimental study. In addition, the duration of TBI-related SIADH varies, and it is necessary for us to pay more attention to the sodium changes during follow-up.
Author contributions
Data curation: Lijun Huang.
Investigation: Qiaoan Zheng, Faming Su, Maoshan Liang.
Project administration: Xianmei Chen.
Supervision: Ge Wu.
Writing – original draft: Huaqian Li.
Writing – review & editing: Xiaoming Chen.
Footnotes
Abbreviations: ACTH = adrenocorticotrophic hormone, ADH = antidiuretic hormone, BNP = B-type natriuretic peptide, CRH = corticotrophin-releasing hormone, CSWS = cerebral salt-wasting syndrome, CT = computer tomography, CTS = cortisol, FSH = follicle-stimulating hormone, FT3 = serum free triiodothyronine, FT4 = serum free thyroxine, HCT = red blood cell specific volume, LH = luteinizing hormone, OP = osmotic pressure, SIADH = syndrome of inappropriate antidiuretic hormone, T = testosterone, TBI = traumatic brain injury, TSH = thyroid-stimulating hormone, WBC = white blood cell count.
HL and LH contributed equally to this work.
Ethical review: The ethical approval was not provided because our hospital stipulates case reports that do not belong to medical research and hence do not require ethical review. The patient has agreed that we use all or any part of his information in the case report and has signed the informed consent.
The authors report no conflicts of interest
References
- [1].Verbalis JG, Goldsmith SR, Greenberg A, et al. Diagnosis, evaluation, and treatment of hyponatremia: expert panel recommendations. Am J Med 2013;126Suppl 1:S1–42. [DOI] [PubMed] [Google Scholar]
- [2].Spasovski G, Vanholder R, Allolio B, et al. Clinical practice guideline on diagnosis and treatment of hyponatraemia. Eur J Endocrinol 2014;170:G1–47. [DOI] [PubMed] [Google Scholar]
- [3].Rahman M, Friedman WA. Hyponatremia in neurosurgical patients: clinical guidelines development. Neurosurgery 2009;65:925–35. discussion 935–6. [DOI] [PubMed] [Google Scholar]
- [4].Holland-Bill L, Christiansen CF, Heide-Jørgensen U, et al. Hyponatremia and mortality risk: a Danish cohort study of 279508 acutely hospitalized patients. Eur J Endocrinol 2015;173:71–81. [DOI] [PubMed] [Google Scholar]
- [5].Peri A, Giuliani C. Management of euvolemic hyponatremia attributed to SIADH in the hospital setting. Minerva Endocrinol 2014;39:33–41. [PubMed] [Google Scholar]
- [6].Schwartz WB, Bennett W, Curelop S, et al. A syndrome of renal sodium loss and hyponatremia probably resulting from inappropriate secretion of antidiuretic hormone. Am J Med 1957;23:529–42. [DOI] [PubMed] [Google Scholar]
- [7].Cuesta M, Garrahy A, Thompson CJ. SIAD: practical recommendations for diagnosis and management. J Endocrinol Invest 2016;39:991–1001. [DOI] [PubMed] [Google Scholar]
- [8].Kirkman MA, Albert AF, Ibrahim A, et al. Hyponatremia and brain injury: historical and contemporary perspectives. Neurocrit Care 2013;18:406–16. [DOI] [PubMed] [Google Scholar]
- [9].Kraan GP, Dullaart RP, Pratt JJ, et al. The daily cortisol production reinvestigated in healthy men. The serum and urinary cortisol production rates are not significantly different. J Clin Endocrinol Metab 1998;83:1247–52. [DOI] [PubMed] [Google Scholar]
- [10].Filipsson H, Monson JP, Koltowska-Häggström M, et al. The impact of glucocorticoid replacement regimens on metabolic outcome and comorbidity in hypopituitary patients. J Clin Endocrinol Metab 2006;91:3954–61. [DOI] [PubMed] [Google Scholar]
- [11].Kinoshita Y, Tominaga A, Arita K, et al. Post-operative hyponatremia in patients with pituitary adenoma: post-operative management with a uniform treatment protocol. Endocr J 2011;58:373–9. [DOI] [PubMed] [Google Scholar]
- [12].Oelkers W. Hyponatremia and inappropriate secretion of vasopressin (antidiuretic hormone) in patients with hypopituitarism. N Engl J Med 1989;321:492–6. [DOI] [PubMed] [Google Scholar]
- [13].Papanek PE, Raff H. Physiological increases in cortisol inhibit basal vasopressin release in conscious dogs. Am J Physiol 1994;266(t 2):R1744–51. [DOI] [PubMed] [Google Scholar]
- [14].Erkut ZA, Pool C, Swaab DF. Glucocorticoids suppress corticotropin-releasing hormone and vasopressin expression in human hypothalamic neurons. J Clin Endocrinol Metab 1998;83:2066–73. [DOI] [PubMed] [Google Scholar]
- [15].Dick M, Catford SR, Kumareswaran K, et al. Persistent syndrome of inappropriate antidiuretic hormone secretion following traumatic brain injury. Endocrinol Diabetes Metab Case Rep 2015;2015:150070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chang CH, Liao JJ, Chuang CH, et al. Recurrent hyponatremia after traumatic brain injury. Am J Med Sci 2008;335:390–3. [DOI] [PubMed] [Google Scholar]