

Abstract
Type I interferon (IFN-I) signaling paradoxically impairs host immune responses during many primary and secondary bacterial infections. Lack of IFN-I receptor reduces bacterial replication and/or bacterial persistence during infection with several bacteria. However, the mechanisms that mediate the adverse IFN-I effect are incompletely understood. Here, we show that Usp18, an interferon-stimulated gene that negatively regulates IFN-I signaling, is primarily responsible for the deleterious effect of IFN-I signaling during infection of mice with Listeria monocytogenes or Staphylococcus aureus. Mechanistically, USP18 promoted bacterial replication by inhibiting antibacterial tumor necrosis factor-α (TNF-α) signaling. Deleting IFNAR1 or USP18 in CD11c-Cre+ cells similarly reduced bacterial titers in multiple organs and enhanced survival. Our results demonstrate that inhibiting USP18 function can promote control of primary and secondary bacterial infection by enhancing the antibacterial effect of TNF-α, which correlates with induction of reactive oxygen species (ROS). These findings suggest that USP18 could be targeted therapeutically in patients to ameliorate disease caused by serious bacterial infections.
INTRODUCTION
Primary and secondary bacterial infections are major public health concerns across the globe given their prevalence and accelerated development of widespread resistance to antibiotic therapies. Modulating host responses has the distinct advantage of exerting less selective pressure on bacterial populations and potentially being broadly effective against multiple bacterial species. Type I interferon (IFN-I) signaling has been reported to both contribute to protection and increase susceptibility to bacterial infections (1–3). Moreover, elevated IFN-I production after viral infection has been causally linked with promoting host susceptibility to secondary bacterial infections (4–9), some of which can result in severe morbidity and mortality. Despite a causal link between IFN-I signaling and susceptibility to bacterial infection, the cellular and molecular mechanisms by which IFN-I signaling promotes primary and secondary bacterial infection are not well defined.
The IFN-I family contains multiple members, mainly divided into two groups (IFN-αs and IFN-β) (10). All members share the same receptor IFNAR, which consists of two subunits, IFNAR1 and IFNAR2. Signaling through IFNAR leads to the phosphorylation of two receptor-associated kinases: tyrosine kinase 2 (TYK2) and Janus kinase 1 (JAK1). These kinases recruit signal transducer and activator of transcription 1 (STAT1) and STAT2 that form either homo- or heterodimers and translocate to the nucleus to originate the transcription of IFN-I-stimulated genes (ISGs) (11). One of the most highly induced ISGs is ubiquitin-specific protease 18 [USP18 or UBP43 (ubiquitin binding protein 43)] (12). USP18 itself inhibits IFNAR signaling in a negative feedback loop by binding to IFNAR2 and displacing JAK1 (13). Recently, it was also demonstrated that USP18 can directly interact with STAT2 to form a complex that inhibits IFN-I ligand binding to IFNAR2 (14, 15). USP18 has an additional functional domain that exerts isopeptidase activity and removes the ubiquitin-like modifier ISG15 from conjugated proteins (12). A mutation of USP18 within the Cys box at position 61 in mice [Usp18C61/C61 transgenic mice (16, 17)] or at position 64 in humans abolishes the isopeptidase activity of the protein, whereas a mutation at site 361 in mice [Usp18ity9 mice (18)] or at site 365 in human abolishes both the IFNAR antagonistic and isopeptidase functions of Usp18 (table S1). Although both mutations have been extensively demonstrated to contribute to promoting virus infection (16, 17), the role of Usp18 in bacterial infection has not been rigorously studied. Using a mutant Usp18 mouse model, Usp18ity9, two groups demonstrated that mutant animals were more susceptible to Salmonella typhimurium and Mycobacterium tuberculosis than wild-type (WT) controls (17, 18). However, in a separate study, it was demonstrated that knockout mice lacking Usp18 exhibited enhanced control of S. typhimurium compared with Usp18-sufficient controls (19). The discrepancy between the above studies suggests that specific domains in the USP18 protein may differentially regulate bacterial infection.
Despite the crucial role of IFN-I during bacterial infection, studies to date have only reported on the consequences of the absence of IFN-I in bacterial control (3). Moreover, given the ability of IFN-I to induce hundreds of ISGs, whether a single specific ISG contributes to the negative effect of IFN-I has yet to be reported in either bacterial or viral infection (20). In this study, we wanted to further explore how IFN-I mechanistically increases the susceptibility to both primary and secondary bacterial infection with a focus on the role of specific ISGs.
RESULTS
Usp18 is an important IFN-I-induced gene that promotes L.m. infection
We set out to determine the cellular subsets that require IFN-I signaling and define whether specific ISGs promote bacterial infection. To answer these questions, we used a mouse model of Listeria monocytogenes (L.m.) infection, which has been previously reported to require IFN-I signaling for optimal L.m. replication and dissemination (4–9). L.m. has been described to enter both macrophages and dendritic cells (DCs) early after infection, and these cell types are important for L.m. infection and distribution (21–23). By assessing ISGs, we found that DCs are more responsive to IFN-I than macrophages after L.m. infection with elevated expression levels of various ISGs (Fig. 1A and fig. S1). The up-regulation of these ISGs required IFNAR signaling because blocking IFNAR1 with a neutralizing antibody reduced their expression to levels similar to uninfected mice (Fig. 1A and fig. S1). L.m. infection of mice that lack the IFN-I receptor mainly on DCs (Ifnarfl/fl CD11c-Cre) resulted in significantly reduced bacterial titers in liver and spleen and reduced titers in kidney and lung 4 days after infection (Fig. 1B). In contrast to DCs, infection of Ifharfl/fl LysM-Cre mice that lack IFNAR1 receptor on macrophages and monocytes resulted in similar bacterial titers in liver, spleen, lung, and kidney as Cre-negative littermate controls (fig. S2). Together, our results demonstrate that IFN-I signaling in CD11c+ DCs, rather than macrophages, is crucial to amplify L.m. infection.
Fig. 1. Usp18 is an important IFN-I-induced gene that promotes L.m. infection.
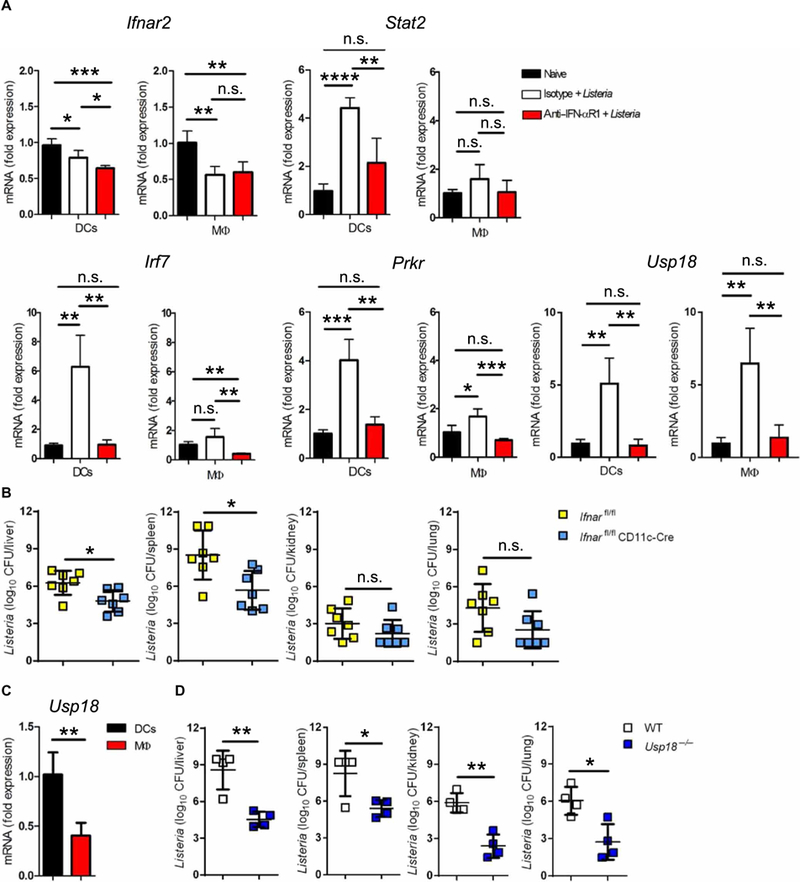
(A) C57BL/6 WT mice were treated with 1 mg of anti-IFN-αR1 or isotype control antibody. The next day, mice were infected with 4000 colony-forming units (CFU) L.m. for 24 hours. DCs and macrophages were sorted by fluorescence-activated cell sorting (FACS), and indicated genes were measured with quantitative reverse transcription PCR (qRT-PCR). RQs were determined with the equation RQ = 2−ΔΔCt (n = 4). Data are representative of at least two independent experiments. (B) Ifnarfl/fl CD11c-Cre mice and littermate controls were infected with 4000 CFU L.m. After 4 days, bacterial titers were measured in the liver, spleen, kidney, and lung (n = 7). Data are pooled from two independent experiments. (C) DCs and macrophages were sorted by FACS from naive spleens of C57BL/6 mice. Usp18 mRNA expression was measured by qRT-PCR. RQs were determined with the equation RQ = 2−ΔΔCt relative to DCs′ average value (n = 4). Data are representative of at least two independent experiments. (D) WT or Usp18−/− mice were infected with 4000 CFU L.m. After 4 days, bacterial titers were measured in the indicated organs (n = 4). Data are representative of at least two independent experiments. Statistical significance was determined by Student′s t test (A to D). n.s., not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Given the requirement of IFN-I signaling in CD11c+ DCs to promote L.m. infection, we sought to identify whether a specific ISG downstream of IFN-I signaling in DCs might explain this phenomenon. Upon analysis of the ISGs up-regulated during L.m. infection, ubiquitin-specific peptidase 18 (Usp18) was elevated in both DCs and macrophages (Fig. 1A). This finding was unexpected because we found here, and in our previous work (24), that Usp18 transcripts were higher in DCs compared with macrophages from uninfected animals (Fig. 1C), although Usp18 was induced in both cell types after infection with L.m. (Fig. 1A and fig. S3). Thus, we hypothesized that USP18 itself may play a negative role downstream of IFN-I signaling during L.m. infection. To directly assess the role of USP18 during L.m. infection, we infected Usp18-deficient mice with L.m. and analyzed bacterial titers after 4 days. We found that Usp18−/− mice were more resistant to L.m. infection, demonstrated by significantly reduced bacterial titers in multiple organs (Fig. 1D). This finding was particularly unexpected because all other ISGs measured were significantly more elevated in Usp18-deficient mice due to lack of its inhibitory effect on IFN-I signaling (fig. S4). Our data strongly suggest that Usp18 is an important ISG capable of promoting L.m. infection.
Deletion of Usp18 in CD11c-Cre+ cells enhances the resistance to L.m.
We next asked whether USP18 signaling was required specifically in CD11c+ DCs to promote L.m. infection. To address this question, we used Usp18fl/fl mice crossed to the CD11c-Cre strain (25). Similar to Ifnarfl/fl CD11c-Cre mice, L.m. infection in mice that lack Usp18 mainly in DCs resulted in significant reductions in bacterial titers in all investigated organs compared with Cre-negative littermate controls (Fig. 2A). However, by reducing the dose of infection 10-fold, we found that bacterial titers in Usp18fl/fl CD11c-Cre and littermate control mice were similar (fig. S5), probably due to low levels of IFN-I induced by the lower L.m. inoculum. To check whether the absence of Usp18 only in CD11c-Cre+ cells can enhance the survival of mice after lethal L.m. infection, we infected Usp18fl/fl CD11c-Cre and littermate control mice with a lethal dose of L.m. Mice lacking Usp18 in CD11c-Cre+ cells exhibited significantly reduced mortality compared with littermate controls after lethal L.m. infection (Fig. 2B). In contrast, lack of Usp18 in LysM-Cre+ cells increased rather than reduced L.m. titers (Fig. 2C), suggesting that Usp18 is a major ISG in CD11c-Cre+ cells, which acts to promote L.m. infection. Consistent with this finding, blocking IFN-I signaling in control mice reduced L.m. to the same level as in Usp18fl/fl CD11c-Cre mice treated with isotype control antibody (Fig. 2D). However, blocking IFN-I in Usp18fl/fl CD11c-Cre mice reduced L.m. titers only in liver but not in other examined tissues (Fig. 2D). This phenotype cannot be explained by reduction of DCs in Usp18-deficient mice because numbers of DCs were equal in Usp18fl/fl CD11c-Cre mice compared with controls early after infection, and DC numbers were even higher in Usp18fl/fl CD11c-Cre than in WT mice 3 days after infection (fig. S6). Moreover, after 2 days of infection, we noticed a reduction in L.m. titers specifically in DCs lacking USP18 (fig. S7). To examine whether Usp18 expression promotes initial bacterial infection or dissemination/amplification, we measured bacterial loads early after infection. Bacterial uptake was measured up to 60 min after infection. Neither Usp18−/− nor Usp18fl/fl CD11c-Cre mice showed any difference in L.m. uptake in comparison with littermate control mice (fig. S8, A and B). In addition, L.m. titers were similar after 24 hours of infection in all examined tissues (fig. S8, C and D), which indicates that the negative effect of USP18 takes place after initial establishment of infection and promotes bacterial amplification or dissemination. To test the role of USP18 on the control of another IFN-I-sensitive bacteria, we chose Staphylococcus aureus strain (502A) whose replication benefits from the presence of IFN-I signaling (Fig. 2E) (26). In addition, it has been reported that CD11c+ cells are central coordinators of host immune response during infection with S. aureus (27), and thus we tested the role of IFN-I signaling in CD11c+ DCs. Similar to L.m., Ifnarfl/fl CD11c-Cre mice displayed significantly reduced S. aureus bacterial loads compared with littermate controls (Fig. 2F). Moreover, deletion of Usp18 specifically in CD11c-Cre+ cells resulted in significant reductions in bacterial titers compared with littermate controls (Fig. 2G). Together, our data demonstrate that the probacterial USP18 effect is observed with an additional IFN-I-sensitive Gram-positive bacterial species and suggest that inhibition of USP18 may help to control other IFN-sensitive bacterial infections.
Fig. 2. Deletion of USP18 in CD11c-Cre+ cells enhances the resistance to L.m.
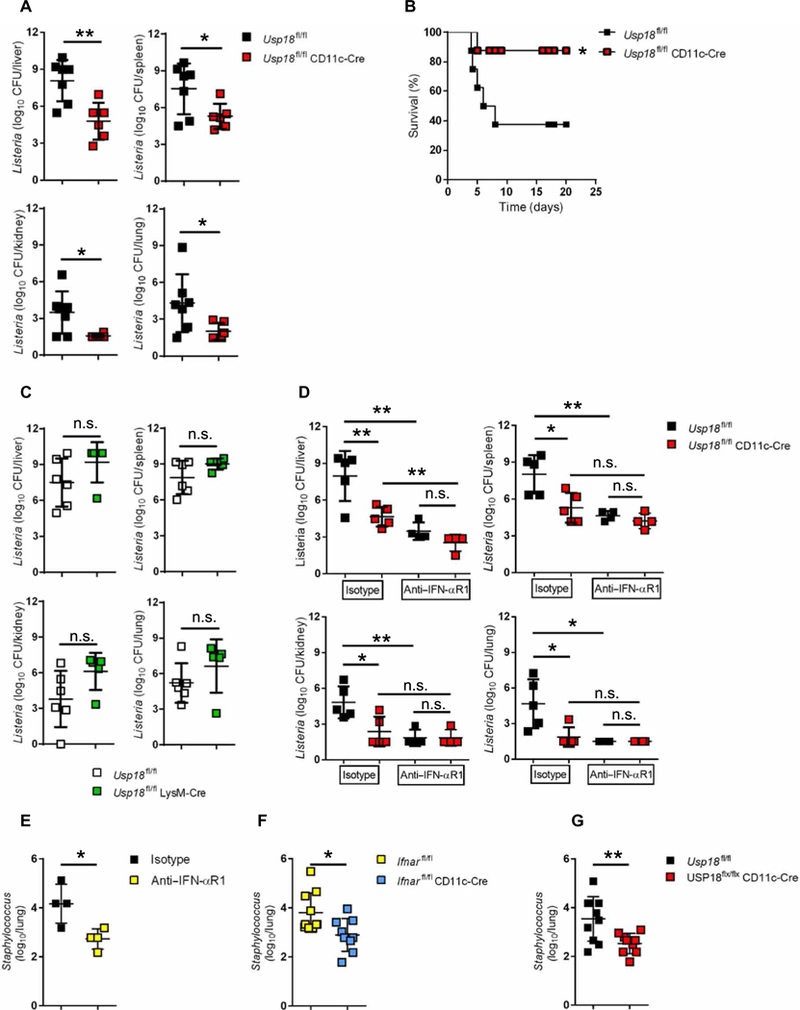
(A) Usp18fl/fl CD11c-Cre mice and littermate controls were infected with 4000 CFU L.m. After 4 days, titers of bacteria were measured in the indicated organs (n = 6 to 7). (B) Survival curve of Usp18fl/fl CD11c-Cre and littermate control mice infected with 7000 CFU L.m. (n = 8). (C) Usp18fl/fl LysM-Cre mice and littermate controls were infected with 4000 CFU L.m. After 4 days, bacterial titers were measured in the indicated organs (n = 5 to 6). (D) Usp18fl/fl CD11c-Cre mice and littermate controls were treated intraperitoneally with 1 mg of anti-IFN-αR1 or isotype control antibody. The next day, mice were infected with 4000 CFU L.m. After 4 days, titers of bacteria were measured in the indicated organs (n = 4 to 5). Data are representative of at least two independent experiments. (E) C57BL/6 WT mice were treated with 1 mg of anti-IFN-αR1 or isotype control antibody. The next day, mice were infected with 2 × 108 CFU S. aureus. After 2 days, titers of bacteria were measured in the lungs (n = 4). (F) Ifnarfl/fl CD11c-Cre mice or (G) Usp18fl/fl CD11c-Cre mice and littermate controls were infected with 2 × 108 CFU S. aureus. After 2 days, titers of bacteria were measured in the lungs (n = 9 to 10). Statistical significance was determined by Student′s t test (A and C to G) and log-rank (Mantel-Cox) test (B). n.s., not significant; *P < 0.05; **P < 0.01. Data are pooled from two independent experiments (A to C, F, and G).
Probacterial effect of USP18 is independent of its isopeptidase activity or an effect on cytokine production
We investigated the mechanism by which Usp18 expression in DCs promotes L.m. infection. As mentioned above, USP18 has two well-described activities: One enables binding to the intracellular domain of the IFNAR2 subunit and attenuates JAK-STAT signaling downstream of IFN-I binding, and the other exerts isopeptidase function and removes ISG15 from conjugated proteins (12). To analyze the contribution of the protease activity of USP18, we infected WT and Usp18C61A/C61A knock-in mice (16), which express only a catalytically inactive USP18 protein, with L.m. and bacterial titers were measured. Usp18C61A/C61A mice showed similar or even elevated bacterial titers compared with WT controls, which suggests that the USP18 effect on L.m. infection is independent of its isopeptidase activity (Fig. 3A). To further exclude the role of ISGylation/deISGylation in mediating the function of USP18, we used Ube1L−/− mice that lack the ability to conjugate ISG15 to its substrates (28). Ube1L−/− mice exhibited similar bacterial titers in the investigated organs (Fig. 3B). This suggests that neither ISGylation nor deISGylation is important to control L.m. infection. Given that USP18 displays a strong ability to negatively regulate IFN-I signaling (12, 29), we reasoned that the absence of Usp18 expression may promote cytokine production, particularly elevation of early IFN-γ and tumor necrosis factor-α (TNF-α), cytokines known to promote control of L.m. infection (30, 31). However, the levels of antibacterial cytokines (IFN-γ and TNF-α) were reduced in knockout mice compared with littermate controls (Fig. 3C), suggesting that the IFN-I antagonistic effect of USP18 is likely not at play in controlling L.m. infection.
Fig. 3. Probacterial effect of USP18 is independent of its isopeptidase activity or cytokine production.
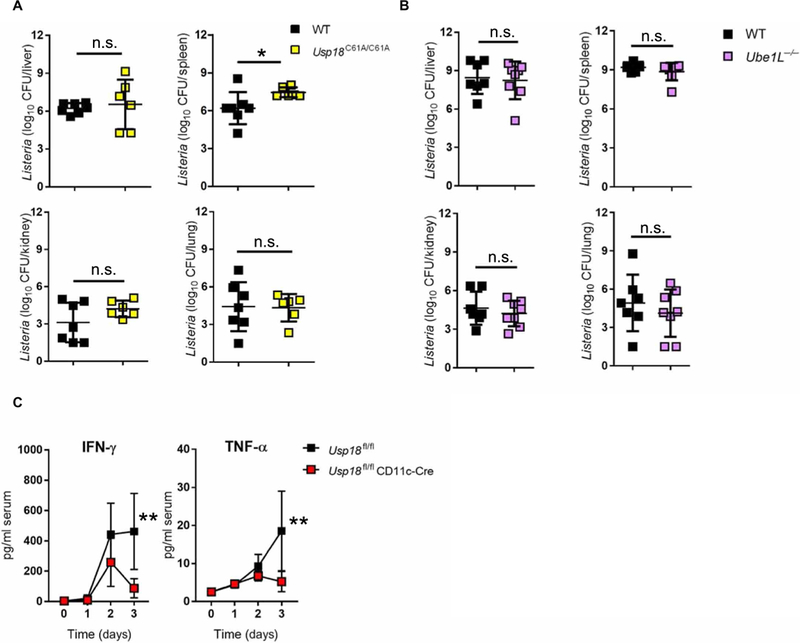
(A) WT and Usp18C61A/C61A mice were infected with 4000 CFU L.m. After 4 days, titers of bacteria were measured in the indicated organs (n = 6 to 7). (B) WT and Ube1L−/− mice were infected with 4000 CFU L.m. After 4 days, titers of bacteria were measured in the indicated organs (n = 7 to 8). (C) Usp18fl/fl CD11c-Cre mice and littermate controls were infected with 4000 CFU L.m. IFN-γ and TNF-α production were measured in the serum at the indicated time points (n = 4 to 6). Statistical significance was determined by Student’s t test (A and B) and two-way ANOVA (C). n.s., not significant; *P < 0.05; **P < 0.01. Data are pooled from two independent experiments (A to C).
USP18 inhibits the antibacterial effect of TNF-α
Previous studies showed that silencing USP18 can enhance TNF-α signaling (32). The USP18 effect was demonstrated to be executed by its interaction with TAK1 and NEMO (21) (fig. S9A), two proteins involved in TNF-induced nuclear factor κB (NF-κB) signaling. Human USP18 variants with point mutations in the IFNAR2 binding domain L365F or isopeptidase domain C64A (12) were still able to interact with TAK1 and NEMO (fig. S9A). In addition, overexpression of human USP18 in HeLa cells reduced NF-κB phosphorylation after TNF-α treatment (fig. S9B). Moreover, overexpression of USP18L365F or USP18C64A in HeLa cells still resulted in inhibition of NF-κB phosphorylation after TNF-α treatment (fig. S9B), which strongly suggests that these domains are not essential for regulating TNF-α signaling. To check whether DCs that lack USP18 exhibit increased TNF-α signaling, we treated bone marrow–derived DCs generated from Usp18fl/fl CD11c-Cre and littermate control mice with TNF-α. We noticed elevated phosphorylation levels of NF-κB p65 in Usp18-deficient DCs (fig. S9C). Our results suggest that USP18 attenuates TNF-α signaling through an unknown mechanism that is independent of the IFNAR2 binding and isopeptidase domains.
We next asked whether the antibacterial effect of TNF-α is enhanced in Usp18-deficient DCs. Treatment of bone marrow–derived DCs generated from Usp18fl/fl CD11c-Cre mice with TNF-α showed higher levels of iNOS expression than WT DCs (Fig. 4A). In addition, the generation of reactive oxygen species (ROS) was enhanced in Usp18-deficient DCs after TNF-α treatment both in vitro and in vivo (Fig. 4B and fig. S10). Moreover, we observed a significant increase in the generation of ROS in DCs of Usp18fl/fl CD11c-Cre mice in comparison with littermate controls after L.m. infection (Fig. 4C). To assess whether enhanced TNF-α signaling in Usp18-deficient DCs translated to better bacterial control, we infected DCs in vitro with L.m. in the presence or absence of TNF-α. Usp18-deficient DCs showed stronger efficiency in killing bacteria after TNF-α treatment than WT DCs, with ~2-log reductions in bacterial titers in Usp18-deficient compared with WT DCs (Fig. 4D). To further confirm our finding, we treated THP-1 monocytes that overexpressed USP18 or control THP-1 monocytes with TNF-α and infected them with L.m. USP18 overexpression significantly blunted the antibacterial effect of TNF-α (Fig. 4E). Next, we treated Usp18fl/fl CD11c-Cre mice with anti–TNF-α neutralizing antibody during L.m. infection. Blocking TNF-α restored titers in Usp18fl/fl CD11c-Cre mice to similar levels as littermate controls (Fig. 4F and fig. S11A). Moreover, treatment of mice with 10-fold less anti–TNF-α antibody still inhibited bacterial control in Usp18fl/fl CD11c-Cre animals (fig. S11B). We hypothesized that USP18 may also prevent the phosphorylation of JAK1 after IFN-γR signaling, similarly to how USP18 inhibits IFNAR2 signaling. However, Usp18fl/fl CD11c-Cre mice still exhibited enhanced control of L.m. infection in multiple organs even after blocking IFN-γ compared with Usp18fl/fl control mice (Fig. 4G and fig. S11C), which strongly suggests that USP18 does not inhibit the antibacterial effect of IFN-γ and that the enhanced ability of Usp18-deficient DCs to control L.m. is highly dependent on intact TNF-α signaling.
Fig. 4. USP18 inhibits the antibacterial effect of TNF-α.
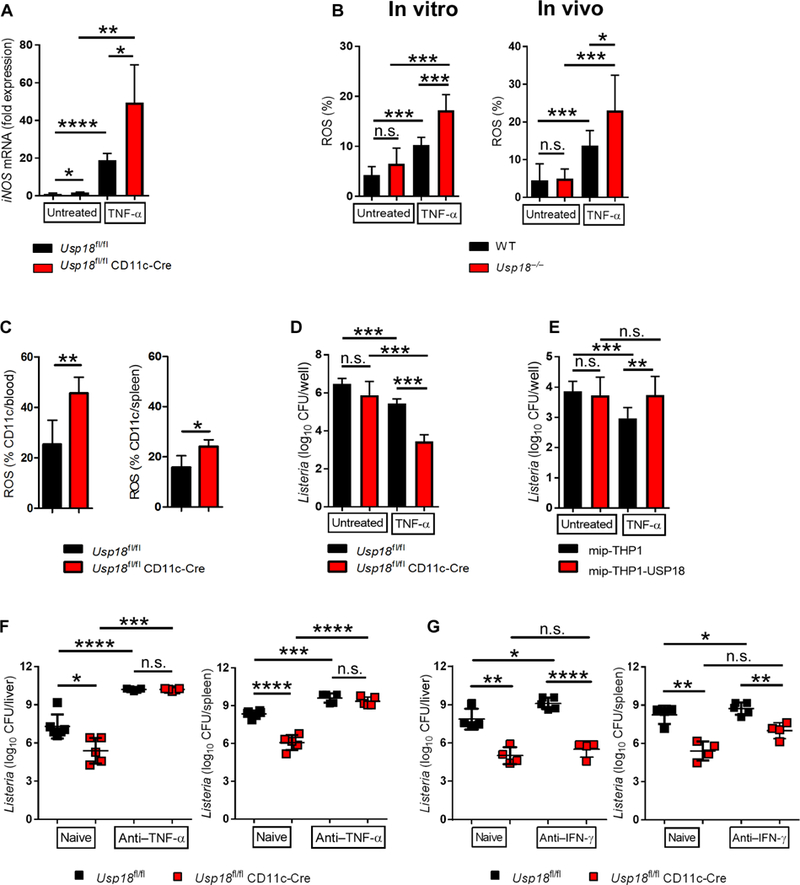
(A) Bone marrow-derived DCs from Usp18fl/fl CD11c-Cre mice and littermate controls were treated with 100 ng of recombinant mouse TNF-α for 24 hours or left untreated. iNOS expression was measured with qRT-PCR. RQs were determined with the equation RQ = 2−ΔΔCt (n = 5 to 6). Data are representative of at least two independent experiments. (B) Bone marrow-derived DCs from Usp18−/− and control mice were treated with 100 ng of recombinant mouse TNF-α for 30 min or left untreated (in vitro experiment), or Usp18−/− and control mice were treated with 50 ng of TNF-α for 30 min or left untreated (in vivo experiment). ROS was measured in DCs (n = 6 in vitro, n = 7 to 9 in vivo). (C) Usp1fl/fl CD11c-Cre mice and littermate controls were infected with 4000 CFU L.m. ROS was measured in DCs on day 1 in blood or day 3 in spleen (n = 4 to 6). Data are representative of at least two independent experiments. (D) Bone marrow–derived DCs from Usp18fl/fl CD11c-Cre mice and littermate controls were infected with L.m. [multiplicity of infection (MOI), 10] in the presence or absence of TNF-α. After 6 hours, intracellular bacterial titers were measured (n = 6). Data are representative of at least two independent experiments. (E) THP-1 control (mip-THP-1) or USP18-overexpressing monocytes (mip-THP-1-USP18) were infected with L.m. (MOI, 10) in the presence or absence of TNF-α. After 6 hours, intracellular bacterial titers were measured (n = 8). (F) Usp18fl/fl CD11c-Cre mice and littermate controls were treated intraperitoneally with 500 μg of anti-TNF-α antibody on day −1, 1, and 2 or left untreated. The next day, mice were infected with 4000 CFU L.m. After 4 days, titers of bacteria were measured in the indicated organs (n = 5). Data are representative of at least two independent experiments. (G) Usp18fl/fl CD11c-Cre mice and littermate controls were treated with 250 μg of anti-IFN-γ antibody on day −1, 1, and 2 or left untreated. The next day, mice were infected with 4000 CFU L.m. After 4 days, titers of bacteria were measured in the indicated organs (n = 4 to 5). Data are representative of at least two independent experiments. Statistical significance was determined by Student′s t test (A to G). n.s., not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Data are pooled from two independent experiments (B and E).
The probacterial role of IFN-I during superinfection is USP18-dependent
Elevated IFN-I production after viral infection has been causally linked with promoting host susceptibility to secondary bacterial infections (2, 3). Given the requirement of IFNAR1 signaling in DCs to promote primary L.m. infection, we asked whether IFNAR1, specifically on CD11c+ DCs, could promote bacterial superinfection. To address this question, we infected Ifnarfl/fl CD11c-Cre or Cre-negative littermates with acute lymphocytic choriomeningitis virus (LCMV), which induces high levels of IFN-I within 24 to 48 hours (33). After 2 days, mice were superinfected with L.m., and bacterial titers were measured 4 days after L.m. infection. In agreement with a previous report (5), L.m. superinfection after primary LCMV infection resulted in significantly elevated L.m. titers in liver, spleen, kidney, and lung in littermate control mice (Fig. 5A). The absence of IFNAR1 expression in DCs was sufficient to reduce L.m. titers to similar levels as detected in Ifnarfl/fl CD11c-Cre mice infected with L.m. alone (Fig. 5A). This result strongly suggests that LCMV-derived IFN-I exerts a DC-intrinsic negative role during viral-bacterial superinfection. Next, we asked whether Usp18 deficiency in DCs would also promote control of L.m. after superinfection with LCMV. Usp18fl/fl CD11c-Cre mice were more resistant to L.m. infection as compared with littermate controls with lower bacterial titers in liver, spleen, kidney, and lung (Fig. 5B). Despite stronger IFN-I signaling in DCs from Usp18fl/fl CD11c-Cre mice (fig. S12), bacterial titers in LCMV-L.m. super-infected Usp18fl/fl CD11c-Cre mice were even lower than observed in single infected littermate control mice (Fig. 5B). Moreover, L.m. titers in spleen, kidney, and lung were comparable in primary and secondary infected Usp18fl/fl CD11c-Cre mice. Analogous to viral infection, induction of IFN-I production by treating mice with polyinosinic:polycytidylic acid (poly I:C) at the same time as L.m. infection increased bacterial titers in control mice (Fig. 5C). Titers were similar in spleen, kidney, and lung of Usp18fl/fl CD11c-Cre mice whether they received poly I:C or control injections (Fig. 5C). Although bacterial titers were increased in liver of poly I:C-treated Usp18fl/fl CD11c-Cre mice, titers remained lower than untreated littermate controls and were significantly reduced compared with poly I:C-treated littermate controls (Fig. 5C).
Fig. 5. The probacterial role of IFN-I during superinfection is USP18-dependent.
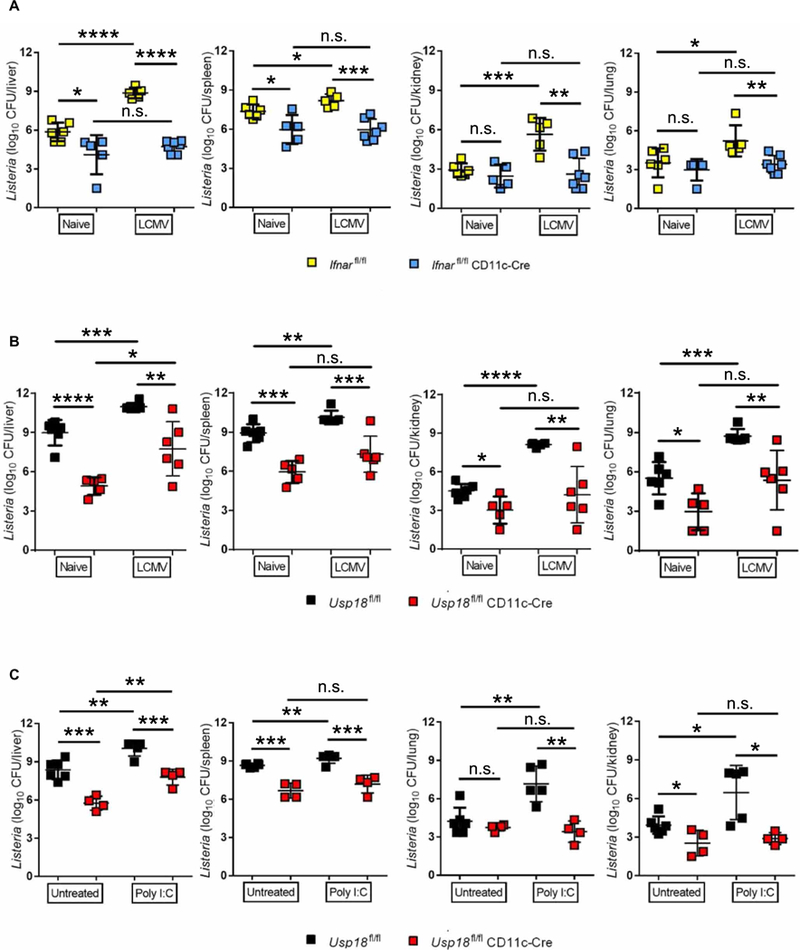
(A) lfnarfl/fl CD11c-Cre and littermate control mice or (B) Usp18fl/fl CD11c-Cre mice and littermate controls were either infected with 2 × 106 plaque-forming units LCMV-WE or left uninfected. After 2 days, mice were infected with 4000 CFU L.m. Bacterial titers were measured in the indicated organs on day 4 [n = 5 to 7 (A), n = 5 to 6 (B)]. (C) Usp18fl/fl CD11c-Cre mice and littermate controls were infected with 4000 CFU L.m. with or without 200 μg of poly I:C. Bacterial titers were measured in the indicated organs on day 3 (n = 4 to 6). Statistical significance was determined by Student′s t test (A to C). n.s., not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Data are pooled from two independent experiments.
DISCUSSION
In summary, we define Usp18 as an essential ISG downstream of IFN-I signaling that promotes susceptibility to both primary and secondary bacterial infection through attenuating antibacterial TNF-α signaling and subsequent ROS production. Further, we demonstrate that USP18 attenuation of TNF-α signaling does not require either the deISGylation or IFN-I antagonistic functions of USP18 and most likely occurs via an unidentified domain on USP18. Our results are unexpected given that the absence of USP18 augments IFN-I signaling (29, 34) and is associated with prolonged JAK-STAT signaling, which, if the current paradigm is correct, should promote rather than prevent L.m. infection. Moreover, because IFN-I signaling has been reported to promote bacterial infection by multiple pathogens, including M. tuberculosis, Francisella tularensis, S. typhimurium, and S. aureus (3), therapeutically targeting USP18 could be applicable and transformative for treatment of these diseases without attenuating IFN-I signaling, which could be detrimental to control endogenous and acquired viral infections. We support this notion by demonstrating that the probacterial effect of USP18 is involved during infection with Gram-positive bacteria: Usp18 deficiency in DCs resulted in reduced bacterial titers after S. aureus infection (Fig. 2G). Moreover, IFN-I signaling has been reported to be protective during many bacterial infections, such as Helicobacter pylori, Streptococcus pyogenes, or Legionella pneumophilla (3). Given that Usp18 deficiency results in increased IFN-I signaling, it would be interesting to test how Usp18 deletion or inhibition affects these bacterial infections. However, it is note-worthy to mention that Usp18 expression may be required in different cell subsets to exert probacterial effects, which will likely depend on the bacterial species being studied, and this should be considered when testing additional bacterial infections.
To study the role of IFN-I on DCs during L.m. infection, we used Ifnarfl/fl CD11c-Cre and Usp18fl/fl CD11c-Cre mouse models. Although CD11c is expressed on multiple cell types including activated T cells (35), we believe that effects on bacterial infection in our models using CD11c-Cre mainly result from DC deletion due to our focus on early times after infection. Moreover, we exclude the role of other innate cells (neutrophils, monocytes, and macrophages) by using LysM-Cre mice that displayed similar L.m. titers as Cre-negative littermate controls. In addition to understanding the specific cell types that require USP18 to mediate its probacterial effects, whether USP18 can be targeted pharmacologically has not been addressed to date. Thus, it is unclear whether USP18 could be inhibited therapeutically to control primary and secondary bacterial infections without promoting overt pathology due to its IFN-I antagonistic functions. In humans, mutations that result in USP18 deletion lead to interferonopathy, severe pathological manifestations, and premature death (36). This suggests that targeting USP18 could provoke life-threatening consequences. However, congenital deletion of USP18 at birth may not mimic pharmacological inhibition. This is emphasized by the identification of patients who lack ISG15 and who also have significantly reduced expression of USP18 due to the ability of ISG15 to stabilize USP18 protein expression (37). It is noteworthy to mention that these patients live to adulthood and their cells are less susceptible for viral infection (38). These studies suggest that USP18 can be inhibited without causing life-threatening pathological manifestations.
An additional open question is how USP18 regulates TNF-α signaling in innate immune cells. A previous study demonstrated that USP18 can bind to and regulate proteins downstream of TNF-α signaling (32). Moreover, USP18 has been shown to act both alone and in combination with other molecules. For instance, in addition to STAT2-USP18 complex, it has been demonstrated that USP18 can recruit an additional ubiquitin-specific peptidase, USP20, to deubi-quinate STING independently of the enzymatic domain of USP18 (39). Therefore, it is possible that USP18 may also regulate TNF-α signaling by recruiting other USP proteins. These findings highlight the importance of studying whether USP18 has additional functional domain(s) and whether USP18 can inhibit TNF-α signaling by itself or in combination with other protein(s).
Collectively, our results suggest that inhibition of USP18 function could be extended to other medically important bacterial infections that are IFN-I-sensitive. Our data, coupled to the known proviral role of USP18 (16, 33, 40, 41), suggest that this protein could be pharmacologically targeted to help control both viral and bacterial infections. Moreover, targeting host USP18 will have the distinct advantage of exerting less selective pressure on viral or bacterial populations and thereby will likely attenuate the development of antibiotic resistance.
MATERIALS AND METHODS
Study design
In this study, we aimed to determine the mechanism by which IFN-I signaling can influence bacterial infection. To do that, we used different transgenic mice and two strains of bacteria, L.m. and S. aureus. First, we determined which cell types are more responsive to IFN-I during L.m. infection by using conditional knockout mice of IFNAR (Ifnarfl/fl CD11c-Cre and Ifnarfl/fl LysM-Cre). We hypothesized that one ISG (USP18) in DCs is responsible dominantly in increasing susceptibility to L.m. To check that, we also used conditional knockout mice of Usp18 (Usp18fl/fl CD11c-Cre, Usp18fl/fl LysM-Cre). We confirmed our finding by treating Usp18-deficient mice with IFNAR blocking antibody before infection. To determine which functional domain of USP18 is responsible for its probacterial role, we infected knock-in mice that have a mutation in the isopeptidase domain of Usp18 (Usp18C61A/C61A) and Ube1L−/− mice. These strains have normal IFN-I signaling but lack the ability of deISGylation or ISGylation, respectively. We also investigated the ability of USP18 to inhibit antibacterial signaling of TNF-α. We measured ROS after TNF-α treatment in vitro and in vivo in Usp18-deficient DCs. In addition, we analyzed the ability of USP18 to inhibit bactericidal effect of TNF-α. Finally, we investigated the role of USP18 during secondary bacterial infection by using viral infection (LCMV) or during induction of IFN-I with a synthetic double-stranded RNA (poly I:C).
Mice, bacteria, and virus
C57BL6/J, Ifnar−/−, Ifnarfl/fl CD11c-Cre, Ifnarfl/fl LysM-Cre, Usp18−/−, Usp18fl/fl CD11c-Cre, Usp18fl/fl LysM-Cre, Usp18C61A/C61A, and Ube1L−/− male and female mice (8 to 12 weeks of age) were used. Usp18fl/fl mice were provided by M. Prinz, and Usp18C61A/C61A mice were provided by K.-P. Knobeloch. Usp18−/− and Ube1L−/− mice were provided by D.-E. Zhang, crossed with C57BL6/J for four generations, and then crossed further with 129SVE-M129S6/SvEvTac purchased from Taconic Laboratory for more than four generations; and mated heterozygous × heterozygous with WT littermates were used as controls for all experiments. CD11c- and LysM-Cre mice were purchased from the Jackson Laboratory. Ifnar−/− mice were obtained from M. Oldstone (The Scripps Research Institute, La Jolla, CA), and Ifnarfl/fl mice were obtained from H. Virgin (Washington University, St. Louis). Except Usp18−/− mice, all strains were maintained on a C57BL6/J background. Mice were maintained in pathogen-free conditions, and handling conformed to the requirements of the National Institutes of Health and the Scripps Research Institute Animal Research Committee. L.m. [American Type Culture Collection (ATCC), strain 43251] bacteria were gift of K. Pfeffer (Institute of Medical Microbiology and Hospital Hygiene, Heinrich-Heine-University Düsseldorf, Düsseldorf, Germany) and were maintained in heart infusion agar. S. aureus (strain 27217) were purchased from ATCC and maintained in heart infusion agar. LCMV strain WE was originally obtained from F. Lehmann-Grube (Heinrich Pette Institute, Hamburg, Germany) and propagated in L929 cells. For all experiments, both LCMV-WE and L.m. were injected by the intravenous route at the indicated dose per mouse. For S. aureus, mice were infected by the intranasal route.
Cluster assay
For quantification of bacteria, organs were homogenized and titrated on brain heart agar plate. For in vitro studies, cells were incubated with L.m. for 1 hour at 37°C, 5% CO2, and cells were then treated with gentamicin (10 mg/ml) to kill extracellular bacteria. After 5 hours, cells were washed and lysed with distilled water + 1% Triton X-100 (Sigma, MO, USA) and titrated on brain heart agar plate.
Antibody and poly I:C treatment
Mice were treated intraperitoneally with 1 mg of anti-IFN-αR-1 clone MAR1–5A3 (Leinco, MO, USA), isotype immunoglobulin 1 (Leinco), 500 or 50 μg of anti-TNF-α clone XT3.11 (Bio X Cell, NH, USA) (42), or 250 μg of anti-IFN-γ clone XMG1.2 (Bio X Cell) (43). Mice were injected with 200 μg of poly I:C (Tocris, Bristol, UK).
Generation of bone marrow–derived DCs and transfer experiments
Primary DCs were generated by isolating bone marrow from femurs and tibias of mice and eliminating erythrocytes. DCs were generated by culturing bone marrow cells in very low endotoxin–Dulbecco’s modified Eagle’s medium (VLE-DMEM; Sigma, MO, USA) including 10% (v/v) fetal bovine serum (FBS; Thermo Fisher Scientific, MA, USA), 0.1% (v/v) β-mercaptoethanol (β-ME; Invitrogen, CA, USA), and granulocyte-macrophage colony-stimulating factor (GM-CSF; 10 ng/ml; made in house) (24). Briefly, supernatant from X63-GM-CSF cell line was collected and titrated to obtain the best working concentration.
Eukaryotic cell lines
HeLa cells were purchased from ATCC-CCL2 and cultured in VLE-DMEM (Sigma) including 10% (v/v) FBS (Thermo Fisher Scientific) and 0.1% (v/v) β-ME (Invitrogen).
Fluorescence-activated cell sorting
Spleens were digested with collagenase from Clostridium histolyticum type IV (Sigma), deoxyribonuclease I from bovine pancreas grade II (Roche, IN, USA), and trypsin inhibitor type II-S soybean. Cells were washed with phosphate-buffered saline (Thermo Fisher Scientific) and stained with anti-CD11c (clone N418; BioLegend, CA, USA), anti-CD11b (clone M1/70; BioLegend), anti-CD8a (clone 53–6.7; BioLegend), anti-Ly6G (clone RB6–8C5; BioLegend), and anti-Ly6C (clone HK1.4; BioLegend). For measuring intracellular proteins, anti-phospho-NF-κB p65 (Ser536; clone 93H1; Cell Signaling, MA, USA) was used. For ROS measurement, cells were incubated with dihydrorhodamine 123 (100 μM; AnaSpec, CA, USA) for 30 min at 37°C.
Total RNA extraction, complementary DNA synthesis, and quantitative real-time polymerase chain reaction
RNA was isolated from splenocytes or sorted cells with TRIzol (Thermo Fisher Scientific), as described by the manufacturer’s protocol. The RNA was reverse-transcribed into complementary DNA with the QuantiTect Reverse Transcription Kit (Qiagen, Germany). Gene expression analysis was performed with assays from Qiagen: glyceraldehyde-3-phosphate dehydrogenase (GAPDH, QT01658692), Stat1 (QT00162183), Stat2 (QT00160216), Irf7 (QT00245266), Prkr (QT00162715), Usp18 (QT00167671), Ifnar1 (QT00105112), Ifnar2 (QT00102340), Oas1 (QT01056048), Mx1 (QT01743308), Isg15 (QT02274335), and iNOS (QT01547980). Relative quantities (RQs) were determined with the equation RQ = 2−ddCt.
Enzyme-linked immunosorbent assay
Multiplex enzyme-linked immunosorbent assay (ELISA) (Millipore, CA, USA) was performed to detect TNF-α and IFN-γ in serum samples.
Transfection and USP18 overexpression
pEBG-hUBP43 was a gift from D.-E. Zhang (Addgene, plasmid 12455)(28). The plasmid was used in conjunction with the polymerase chain reaction (PCR)-based GeneArt Site-Directed Mutagenesis System (Life Technologies, Carlsbad, CA) to create USP18C64A mutant plasmid. The primers used to construct the mutant were 5′-CAACATTGGACAGACCGCCTGCCTTAACTCCTTG-3′ and 5′-CAAGGAGTTAAGGCAGGCGGTCTGTCCAATGTTG-3′. Successful recombinants were selected on LB plates containing ampicillin. Plates were kept overnight at 37°C, and colonies were picked, cultured, and purified for sequencing (Eton Bio, San Diego, CA) to check for the desired substitution of cysteine residue 61 to alanine. L365F mutant of USP18 is cloned into pCMV3-FLAG vector. pWZL Neo Myr Flag MAP3K7 (TAK1) was a gift from W. Hahn and J. Zhao (Addgene, plasmid 20520). NEMO is cloned into pCAG vector. HeLa cells were transfected with 4 μg of plasmid or empty vector using Lipofectamine 3000 (Invitrogen) for 24 hours. Cells were treated with human TNF-α for 15 min. To generate THP-1 cells that overexpress USP18, THP-1 cells were infected with MIP-USP18 retrovirus and selected by puromycin.
Immunoprecipitation
Cells were lysed in binding buffer containing 25 mM tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, and 0.5% NP-40 for coimmuno-precipitation assays. The cell lysates were centrifuged (15,000 rpm) at 4°C for 5 min. All lysis buffers in this study contain proteinase and phosphatase inhibitors (Roche). Soluble fractions were precleared by protein G–Sepharose at 4°C for 15 min. Precleared cell lysates were immunoprecipitated for 1 hour with the anti-FLAG M2 agarose (Sigma). After three washes, immunocomplexes were eluted by boiling for 5 min.
Statistics
Data are expressed as means ± SD. Unpaired two-tailed Student’s t test was calculated using GraphPad Prism to detect statistically significant differences between groups. Significant differences between several groups were detected by two-way analysis of variance (ANOVA). Log-rank (Mantel-Cox) test was used for survival study. The level of statistical significance was set at n.s. (not significant), *P < 0.05, **P < 0.01, ***P < 0.001, or ****P < 0.0001.
Supplementary Material
Acknowledgments:
We thank M. Wilson, M. Carillo, and J. Van Leeuwen from the Department of Animal Resources and FACS Core in Scripps Research Institute for technical support.
Funding: N.S. was supported by a Deutsche Forschungsgemeinschaft (DFG) fellowship (SH 1140/2–1). This work was supported by the NIH (R01 AI123210 to J.R.T. and R01 CA177305 to D.-E.Z.) and the DFG (KN 590/7–1 to K.-P.K.).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data supporting the findings of this work are included within the article.
REFERENCES AND NOTES
- 1.Davidson S, Maini MK, Wack A, Disease-promoting effects of type I interferons in viral, bacterial, and coinfections. J. Interferon Cytokine Res. 35, 252–264 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Decker T, Muller M, Stockinger S, The yin and yang of type I interferon activity in bacterial infection. Nat. Rev. Immunol. 5, 675–687 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Boxx GM, Cheng G, The roles of type I interferon in bacterial infection. Cell Host Microbe 19, 760–769 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, Perry AK, Nguyen BO, Lane TF, Taniguchi T, Miller JF, Cheng G, Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J. Exp. Med. 200, 437–445 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Navarini AA, Recher M, Lang KS, Georgiev P, Meury S, Bergthaler A, Flatz L, Bille J, Landmann R, Odermatt B, Hengartner H, Zinkernagel RM, Increased susceptibility to bacterial superinfection as a consequence of innate antiviral responses. Proc. Natl. Acad. Sci. U.S.A. 103, 15535–15539 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Auerbuch V, Brockstedt DG, Meyer-Morse N, O’Riordan M, Portnoy DA, Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J. Exp. Med. 200, 527–533 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carrero JA, Calderon B, Unanue ER, Listeriolysin O from Listeria monocytogenes is a lymphocyte apoptogenic molecule. J. Immunol. 172, 4866–4874 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Henry T, Kirimanjeswara GS, Ruby T, Jones JW, Peng K, Perret M, Ho L, Sauer J-D, Iwakura Y, Metzger DW, Monack DM, Type I IFN signaling constrains IL-17A/F secretion by γδ T cells during bacterial infections. J. Immunol. 184, 3755–3767 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rayamajhi M, Humann J, Penheiter K, Andreasen K, Lenz LL, Induction of IFN-αβ enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J. Exp. Med. 207, 327–337 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Platanias LC, Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, Sanmamed MF, Le Bon A, Melero I, Direct effects of type I interferons on cells of the immune system. Clin. Cancer Res. 17, 2619–2627 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Honke N, Shaabani N, Zhang DE, Hardt C, Lang KS, Multiple functions of USP18. Cell Death Dis. 7, e2444 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malakhova OA, Yan M, Malakhov MP, Yuan Y, Ritchie KJ, Kim KI, Peterson LF, Shuai K, Zhang DE, Protein ISGylation modulates the JAK-STAT signaling pathway. Genes Dev. 17, 455–460 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arimoto K-I, Löchte S, Stoner SA, Burkart C, Zhang Y, Miyauchi S, Wilmes S, Fan J-B, Heinisch JJ, Li Z, Yan M, Pellegrini S, Colland F, Piehler J, Zhang D-E, STAT2 is an essential adaptor in USP18-mediated suppression of type I interferon signaling. Nat. Struct. Mol. Biol. 24, 279–289 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arimoto K-I, Miyauchi S, Stoner SA, Fan J-B, Zhang D-E, Negative regulation of type I IFN signaling. J. Leukoc. Biol. 103, 1099–1116 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Ketscher L, Hannß R, Morales DJ, Basters A, Guerra S, Goldmann T, Hausmann A, Prinz M, Naumann R, Pekosz A, Utermöhlen O, Lenschow DJ, Knobeloch KP, Selective inactivation of USP18 isopeptidase activity in vivo enhances ISG15 conjugation and viral resistance. Proc. Natl. Acad. Sci. U.S.A. 112, 1577–1582 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dauphinee SM, Richer E, Eva MM, McIntosh F, Paquet M, Dangoor D, Burkart C, Zhang D-E, Gruenheid S, Gros P, Behr M, Malo D, Contribution of increased ISG15, ISGylation and deregulated type I IFN signaling in Usp18 mutant mice during the course of bacterial infections. Genes Immun. 15, 282–292 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richer E, Prendergast C, Zhang DE, Qureshi ST, Vidal SM, Malo D, N-ethyl-N-nitrosourea-induced mutation in ubiquitin-specific peptidase 18 causes hyperactivation of IFN-αß signaling and suppresses STAT4-induced IFN-γ production, resulting in increased susceptibility to Salmonella typhimurium. J. Immunol. 185, 3593–3601 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim KI, Malakhova OA, Hoebe K, Yan M, Beutler B, Zhang D-E, Enhanced antibacterial potential in UBP43-deficient mice against Salmonella typhimurium infection by up-regulating type I IFN signaling. J. Immunol. 175, 847–854 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM, A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aoshi T, Carrero JA, Konjufca V, Koide Y, Unanue ER, Miller MJ, The cellular niche of Listeria monocytogenes infection changes rapidly in the spleen. Eur. J. Immunol. 39, 417–425 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neuenhahn M, Kerksiek KM, Nauerth M, Suhre MH, Schiemann M, Gebhardt FE, Stemberger C, Panthel K, Schröder S, Chakraborty T, Jung S, Hochrein H, Rüssmann H, Brocker T, Busch DH, CD8α+ dendritic cells are required for efficient entry of Listeria monocytogenes into the spleen. Immunity 25, 619–630 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Verschoor A, Neuenhahn M, Navarini AA, Graef P, Plaumann A, Seidlmeier A, Nieswandt B, Massberg S, Zinkernagel RM, Hengartner H, Busch DH, A platelet- mediated system for shuttling blood-borne bacteria to CD8α+ dendritic cells depends on glycoprotein GPIb and complement C3. Nat. Immunol. 12, 1194–1201 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Honke N, Shaabani N, Zhang D-E, Iliakis G, Xu HC, Häussinger D, Recher M, Löhning M, Lang PA, Lang KS, Usp18 driven enforced viral replication in dendritic cells contributes to break of immunological tolerance in autoimmune diabetes. PLOS Pathog. 9, e1003650 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldmann T, Zeller N, Raasch J, Kierdorf K, Frenzel K, Ketscher L, Basters A, Staszewski O, Brendecke SM, Spiess A, Tay TL, Kreutz C, Timmer J, Mancini GM, Blank T, Fritz G, Biber K, Lang R, Malo D, Merkler D, Heikenwälder M, Knobeloch K-P, Prinz M, USP18 lack in microglia causes destructive interferonopathy of the mouse brain. EMBOJ. 34, 1612–1629 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parker D, Planet PJ, Soong G, Narechania A, Prince A, Induction of type I interferon signaling determines the relative pathogenicity of Staphylococcus aureus strains. PLOS Pathog. 10, e1003951 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schindler D, Gutierrez MG, Beineke A, Rauter Y, Rohde M, Foster S, Goldmann O, Medina E, Dendritic cells are central coordinators of the host immune response to Staphylococcus aureus bloodstream infection. Am. J. Pathol. 181, 1327–1337 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Kim KI, Yan M, Malakhova O, Luo J-K, Shen M-F, Zou W, de la Torre JC, Zhang D-E, Ube1L and protein ISGylation are not essential for alpha/beta interferon signaling. Mol. Cell. Biol. 26, 472–479 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.François-Newton V, Magno G de Freitas Almeida, Payelle-Brogard B, Monneron D, Pichard-Garcia L, Piehler J, Pellegrini S, Uze G, USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon α response. PLOS ONE 6, e22200 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dussurget O, Bierne H, Cossart P, The bacterial pathogen Listeria monocytogenes and the interferon family: Type I, type II and type III interferons. Front. Cell. Infect.Microbiol. 4, 50 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mizuki M, Nakane A, Sekikawa K, Tagawa Y-I, Iwakura Y, Comparison of host resistance to primary and secondary Listeria monocytogenes infections in mice by intranasal and intravenous routes. Infect. Immun. 70, 4805–4811 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang Z, Xian H, Hu J, Tian S, Qin Y, Wang R-F, Cui J, USP18 negatively regulates NF-κB signaling by targeting TAK1 and NEMO for deubiquitination through distinct mechanisms. Sci. Rep. 5, 12738 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shaabani N, Khairnar V, Duhan V, Zhou F, Tur RF, Haussinger D, Recher M, Tumanov AV, Hardt C, Pinschewer D, Christen U, Lang PA, Honke N, Lang KS, Two separate mechanisms of enforced viral replication balance innate and adaptive immune activation. J. Autoimmun. 67, 82–89 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Burkart C, Arimoto K-I, Tang T, Cong X, Xiao N, Liu Y-C, Kotenko SV, Ellies LG, Zhang D-E, Usp18 deficient mammary epithelial cells create an antitumour environment driven by hypersensitivity to I FN-l and elevated secretion of Cxcl10. EMBO Mol. Med. 5, 1035–1050 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qualai J, Li L-X, Cantero J, Tarrats A, Fernández MA, Sumoy L, Rodolosse A, McSorley SJ, Genesca M, Expression of CD11c is associated with unconventional activated T cell subsets with high migratory potential. PLOS ONE 11, e0154253 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meuwissen MEC, Schot R, Buta S, Oudesluijs G, Tinschert S, Speer SD, Li Z, van Unen L, Heijsman D, Goldmann T, Lequin MH, Kros JM, Stam W, Hermann M, Willemsen R, Brouwer RWW, Van IJcken WFJ, Martin-Fernandez M, de Coo I, Dudink J, de Vries FAT, Bertoli Avella A, Prinz M, Crow YJ, Verheijen FW, Pellegrini S, Bogunovic D, Mancini GMS, Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J. Exp. Med. 213, 1163–1174 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, Volpi S, Li Z, Sanal O, Mansouri D, Tezcan I, Rice GI, Chen C, Mansouri N, Mahdaviani SA, Itan Y, Boisson B, Okada S, Zeng L, Wang X, Jiang H, Liu W, Han T, Liu D, Ma T, Wang B, Liu M, Liu J-Y, Wang QK, Yalnizoglu D, Radoshevich L, Uzé G, Gros P, Rozenberg F, Zhang S-Y, Jouanguy E, Bustamante J, Garcia-Sastre A, Abel L, Lebon P, Notarangelo LD, Crow YJ, Boisson-Dupuis S, Casanova JL, Pellegrini S, Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature 517, 89–93 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Speer SD, Li Z, Buta S, Payelle-Brogard B, Qian L, Vigant F, Rubino E, Gardner TJ, Wedeking T, Hermann M, Duehr J, Sanal O, Tezcan I, Mansouri N, Tabarsi P, Mansouri D, Francois-Newton V, Daussy CF, Rodriguez MR, Lenschow DJ, Freiberg AN, Tortorella D, Piehler J, Lee B, Garcia-Sastre A, Pellegrini S, Bogunovic D, ISG15 deficiency and increased viral resistance in humans but not mice. Nat. Commun. 7, 11496 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang M, Zhang M-X, Zhang Q, Zhu G-F, Yuan L, Zhang D-E, Zhu Q, Yao J, Shu H-B, Zhong B, USP18 recruits USP20 to promote innate antiviral response through deubiquitinating STING/MITA. Cell Res. 26, 1302–1319 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Honke N, Shaabani N, Cadeddu G, Sorg UR, Zhang D-E, Trilling M, Klingel K, Sauter M, Kandolf R, Gailus N, van Rooijen N, Burkart C, Baldus SE, Grusdat M, Löhning M, Hengel H, Pfeffer K, Tanaka M, Häussinger D, Recher M, Lang PA, Lang KS, Enforced viral replication activates adaptive immunity and is essential for the control of a cytopathic virus. Nat. Immunol. 13, 51–57 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Ritchie KJ, Hahn CS, Kim KI, Yan M, Rosario D, Li L, de la Torre JC, Zhang DE, Role of ISG15 protease UBP43 (USP18) in innate immunity to viral infection. Nat. Med. 10, 1374–1378 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Baeyens A, Saadoun D, Billiard F, Rouers A, Gregoire S, Zaragoza B, Grinberg-Bleyer Y, Marodon G, Piaggio E, Salomon BL, Effector T cells boost regulatory T cell expansion by IL-2, TNF, OX40, and plasmacytoid dendritic cells depending on the immune context. J. Immunol. 194, 999–1010 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Clever D, Roychoudhuri R, Constantinides MG, Askenase MH, Sukumar M, Klebanoff CA, Eil RL, Hickman HD, Yu Z, Pan JH, Palmer DC, Phan AT, Goulding J, Gattinoni L, Goldrath AW, Belkaid Y, Restifo NP, Oxygen sensing by T cells establishes an immunologically tolerant metastatic niche. Cell 166, 1117–1131.e14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.