

Abstract
The G2/M checkpoint inhibits mitotic entry upon DNA damage thereby preventing segregation of broken chromosomes and preserving genome stability. The tumor suppressor proteins BRCA1, PALB2 and BRCA2 constitute a BRCA1-PALB2-BRCA2 axis that is essential for homologous recombination (HR)-based DNA double strand break repair. Besides HR, BRCA1 has been implicated in both the initial activation and the maintenance of the G2/M checkpoint, while BRCA2 and PALB2 have been shown to be critical for its maintenance. Here we show that all 3 proteins can play a significant role in both checkpoint activation and checkpoint maintenance, depending on cell type and context, and that PALB2 links BRCA1 and BRCA2 in checkpoint response. The BRCA1-PALB2 interaction can be important for checkpoint activation, whereas the PALB2-BRCA2 complex formation appears to be more critical for checkpoint maintenance. Interestingly, the function of PALB2 in checkpoint response appears to be independent of CHK1 and CHK2 phosphorylation. Following ionizing radiation, cells with disengaged BRCA1-PALB2 interaction show greatly increased chromosomal abnormalities due apparently to combined defects in HR and checkpoint control. These findings provide new insights into DNA damage checkpoint control and further underscore the critical importance of the proper cooperation of the BRCA and PALB2 proteins in genome maintenance.
Keywords: BRCA1, PALB2, BRCA2, G2/M checkpoint
Introduction
Upon genotoxic insults, eukaryotic cells activate an elaborate DNA damage response (DDR), which consists of DNA repair pathways that repair the lesions, as well as signaling pathways that control cell cycle progression and gene expression7, 28. The DDR, owing to its essential role in maintaining genome stability, is critical for tumor suppression7, 15. The importance of the DDR for tumor suppression is exemplified by hereditary breast and ovarian cancer (HBOC), for which over a dozen “susceptibility” genes, such as BRCA1 and BRCA2, have been identified, with most of them having a function in the repair of and/or signaling in response to DNA damage, particularly DNA double strand breaks (DSBs)1, 9, 36.
BRCA1 and BRCA2 encode very large proteins that play critical roles in the faithful repair of DSBs by homologous recombination (HR)24, 29, 35. In addition to breast and ovarian cancer, germline mutations in the two genes may also cause increased risks of developing pancreatic, prostate and stomach cancers6. PALB2 was discovered as a major BRCA2 binding protein that controls its intra-nuclear localization and stability, tethers it to the chromatin, recruits it to DNA damage sites and enables its function in HR37. Importantly, PALB2 also directly binds BRCA1 and links BRCA1 and BRCA2 in the HR pathway32, 44, 45. Consistent with its ‘BRCA3’-like molecular functions, PALB2 has been established as a BRCA-type tumor suppressor that is also mutated in breast, ovarian, pancreatic, prostate and stomach cancers21, 34, 36.
As part of the DDR, normal cells activate cell cycle checkpoints to slow down or halt cell cycle progression. The G2/M checkpoint, conserved from yeast to mammals, arrests cells in the G2 phase after DNA damage and minimizes segregation of damaged chromosomes into daughter cells20. BRCA1 has long been implicated in both the activation and the maintenance of this checkpoint under various settings8, 30, 39, 41, and BRCA2 and PALB2 were more recently found to be among the most critical factors that maintain the checkpoint following DNA damage induced by ionizing radiation (IR)8, 23. However, it is currently unclear whether BRCA2 and PALB2 can also function in checkpoint activation under certain conditions, whether the three proteins function together in checkpoint control and, if so, how they work together. In this study, we analyzed the checkpoint function of these proteins in multiple cell types and assessed the importance of the BRCA1-PALB2 and PALB2-BRCA2 interactions in checkpoint activation and maintenance in different contexts. We also assessed the extent of genome instability induced by IR in cells with disengaged endogenous BRCA1-PALB2 interaction.
Results
Comparative analysis of BRCA1, PALB2 and BRCA2 in the G2/M checkpoint response
Although BRCA1, BRCA2 and PALB2 have all been reported to play a role in the G2/M checkpoint, a comparative analysis of all 3 proteins in checkpoint response has not been conducted. To understand their relative importance in this aspect, we used siRNAs to deplete the 3 proteins in parallel in U2OS cells and compared the effects on the checkpoint response following two different doses of IR, 3 and 10 Gy, by measuring the number of cells that stained positive for phospho-histone H3 (ser10), a marker of condensed chromosomes in mitotic cells17, 39. As shown in Fig. 1A, following 3 Gy of IR, control siRNA-treated cells showed an almost complete loss of mitotic cells at 1 hr after IR. The checkpoint was maintained for at least 6 hr, and by 24 hr after IR, mitosis had largely resumed, indicative of checkpoint recovery. After 10 Gy of IR, an even stronger checkpoint response was observed, as the cells had barely started to recover even at 24 hr. Compared with control siRNA-treated cells, cells depleted of each of the 3 proteins showed equally efficient checkpoint activation in response to each dose of IR; however, these cells all showed earlier recovery from the checkpoint. Specifically, after 3 Gy of IR, mitosis started to resume within 3 hr in BRCA2- and PALB2-depleted cells and within 6 hr in BRCA1-depleted cells; after 10 Gy of IR, mitotic entry started within 6 hr in BRCA2- and PALB2-depleted cells and well within 24 hr in BRCA1-depleted cells. These results are consistent with previous reports that all 3 proteins are dispensable for the activation but required for the maintenance of the checkpoint in U2OS cells8, 23. Depletion of PALB2 led to a substantial loss of BRCA2 (Fig. 1B), consistent with our previous report that PALB2 stabilizes BRCA237.
Figure 1.
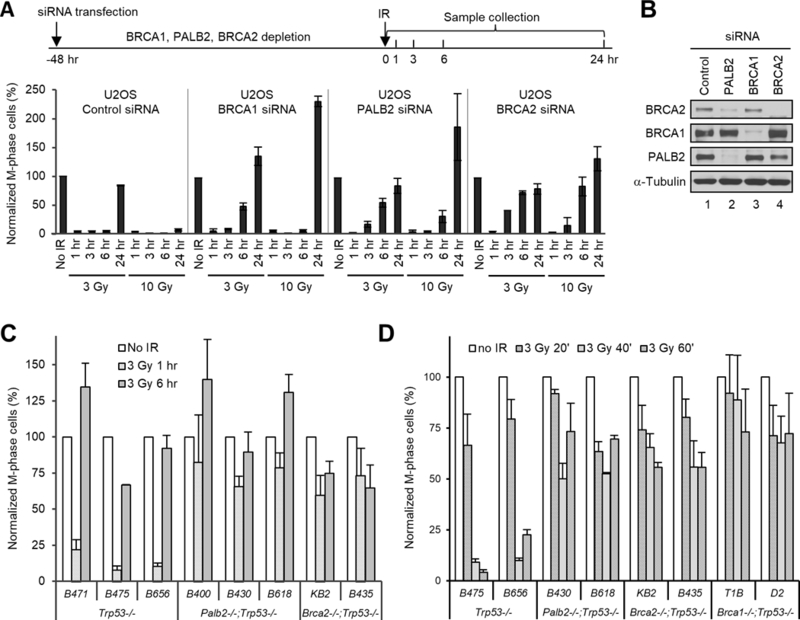
Effects of BRCA1, BRCA2 or PALB2 deficiencies on the G2/M checkpoint response in human and mouse tumor cells. (A) G2/M checkpoint responses in U2OS cells depleted of BRCA1, PALB2 and BRCA2. Cells were treated with control, BRCA1, PALB2 and BRCA2 siRNAs for 48 hr and then irradiated with 3 or 10 Gy of IR; cells were collected at indicated time points and their mitotic indexes were measured. Upper panel, schematic diagram of the timeline of the experiment; lower panel, relative mitotic indexes of the above cells at different time points after IR. (B) Representative western blots showing the depletion of BRCA1, PALB2 and BRCA2 proteins in A. (C,D) Relative mitotic indexes of the different mouse tumor cell lines before and after IR (3 Gy). The numbers of mitotic cells were normalized against that of unirradiated cells for each siRNA treatment or cell line. Values shown are the means of relative mitotic indexes from 2 (A and C) or 3 (D) independent experiments; error bars represent standard errors of the mean (SEMs).
Checkpoint response in Brca1, Brca2 and Palb2-deficient mouse mammary tumor cells
To better understand the function of these proteins in checkpoint control, we analyzed the checkpoint response in a panel of mouse mammary tumor cell lines that are deficient in p53 alone, p53 and PALB2, or p53 and BRCA2, which were derived from mammary tumors from Trp53, Palb2/Trp53, and Brca2/Trp53 conditional knockout (CKO) mice14, 16. Co-deletion of Trp53 greatly facilitates mammary tumor development in Palb2 and Brca2 CKO mouse models and subsequent generation of tumor-derived cell lines. The cells were treated with the more therapeutically relevant dose of IR (3 Gy), and checkpoint responses were analyzed 1 and 6 hr after treatment. As shown in Fig. 1C, while the 3 Trp53−/− (control) cell lines showed 78–92% reductions in the number of mitotic cells at 1 hr after IR, mitotic activity was only reduced by 17–34% in the 3 Palb2−/−;Trp53−/− cell lines and by 27–40% in the 2 Brca2−/−;Trp53−/− cell lines, suggesting a possible defect in checkpoint activation. By 6 hr after IR, mitotic activity had largely recovered to pre-IR levels in the Trp53−/− cells, while Palb2−/−;Trp53−/− cells showed a further increase and Brca2−/−;Trp53−/− cells remained approximately at the 1 hr levels.
To rule out the possibility that checkpoint activation in the double null cells was extremely transient and had significantly recovered by 1 hr after IR, we further measured mitotic activity in these as well as 2 newly generated Brca1−/−;Trp53−/− cell lines at 20, 40 and 60 min after IR. Our results showed that for all of the cells lines, it took approximately 40–60 min for mitotic index to reach their respective lowest points, and all of the double null cells showed much higher mitotic activity than that of control cells at 40 min and, again, 60 min after IR (Fig. 1C). These observations suggest that not only BRCA1 but also PALB2 and BRCA2 can also play an important role in checkpoint activation and that the precise role of BRCA and PALB2 proteins in G2/M checkpoint response may be cell type or context dependent.
Roles of p53 and MLH1 in the G2/M checkpoint
p53 is a critical cell cycle regulator that has been implicated in G2/M checkpoint control33. Given the observed difference in checkpoint activation in U2OS (p53-wt) and the mouse tumor cells (p53-null), we asked if p53 status would dictate the consequences of BRCA1/2 and PALB2 deficiency in the G2/M checkpoint. To this end, we used siRNAs to deplete each of the proteins in an isogenic pair of p53-wt and p53-null HCT116 colon cancer cells5 and measured checkpoint activation after 3 Gy of IR. Notably, even in the p53-wt cells, loss of each of the three proteins led to a significant defect in checkpoint activation (Fig. 2A). Thus, the role of BRCA1, BRCA2 and PALB2 in promoting G2/M checkpoint activation is not restricted to only mouse cells or p53-null cells. In addition, either in the presence or absence of BRCA1/2 or PALB2, the checkpoint activation defect was more pronounced in the p53-null cells than p53-wt cells, suggesting that p53 indeed contributes to checkpoint activation in HCT116 cells and that the checkpoint-promoting activities of p53 and BRCA/PALB2 proteins may be additive.
Figure 2.
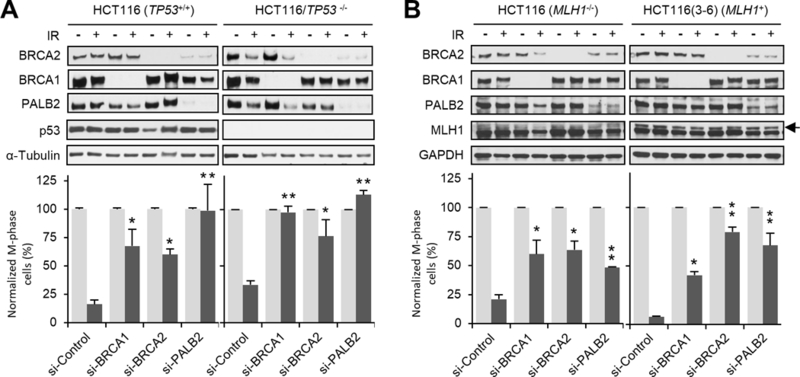
Role of p53 and MLH1 in BRCA- and PALB2-mediated G2/M checkpoint activation. (A) Isogenic p53 wt and null HCT116 cells were depleted of BRCA1, BRCA2 or PALB2, and checkpoint activation was analyzed 1 hr after 3 Gy of IR. Upper panels: representative immunoblots showing levels of the indicated proteins. Lower panels: relative mitotic indexes of each cell type and condition. (B) Isogenic HCT116 and HCT116:3–6 cells were treated and analyzed as in A. Data shown are means ± SEMs of the relative mitotic indexes from n>3 independent experiments. Statistical significance was calculated with one-way ANOVA. *, P<0.05; **, P<0.01.
Since HCT116 cells are deficient in the mismatch repair protein MLH1, which also has been implicated in G2/M checkpoint control11, we asked whether the lack of MLH1 sensitizes HCT116 cells to the loss of BRCA1/2 and PALB2 with respect to checkpoint activation. Checkpoint activation was analyzed in naïve (p53-wt) HCT116 cells and genetically-matched, MLH1-reconstituted HCT116:3–6 cells11 after knockdown of each of the three genes. Consistent with the previous report, re-expression of MLH1 led to more effective checkpoint activation in cells treated with control siRNA (Fig. 2B). However, this effect was not observed when BRCA2 or PALB2 were depleted. Therefore, BRCA2 and PALB2 proteins promote G2/M checkpoint activation in HCT116 cells in a manner that is largely independent of p53 and MLH1.
PALB2 function in checkpoint activation is independent of CHK1 and CHK2 activation
In a separate approach to study the G2/M checkpoint function of PALB2, we tested checkpoint activation in a previously described panel of SV40-transformed human fibroblasts with various PALB2 statuses38. These include FEN5280 (derived from a normal individual with wt PALB2), EUFA1341 (derived from a Fanconi anemia patient with biallelic germline mutations in PALB2), and EUFA1341 cells reconstituted with wt PALB2 (Fig. 3A). U2OS cells were also used, as a reference. Similar to U2OS cells depleted of PALB2 (Fig. 1B), EUFA1341 cells had reduced amount of BRCA2 as compared with either FEN5280 or U2OS cells. Upon expression of exogenous PALB2, the amount of BRCA2 was restored, again demonstrating the key role of PALB2 in maintaining BRCA2 stability. At 1 hr after 3 Gy of IR, FEN5280 cells showed a 61% drop in mitotic index, whereas the drop was 34% in EUFA1341 cells (Fig. 3A). Similar to FEN5280 cells, EUFA1341 cells reconstituted with wt PALB2 displayed a 66% reduction of mitotic cells. These results again indicate that PALB2 plays a significant role in checkpoint activation at least in some contexts. Both FEN5280 and the PALB2-reconsituted EUFA1341 cells showed less effective checkpoint activation compared with U2OS cells, which could be due to expression of the SV40 large T antigen, which inactivates p53 and RB, both being regulators of the cell cycle.
Figure 3.
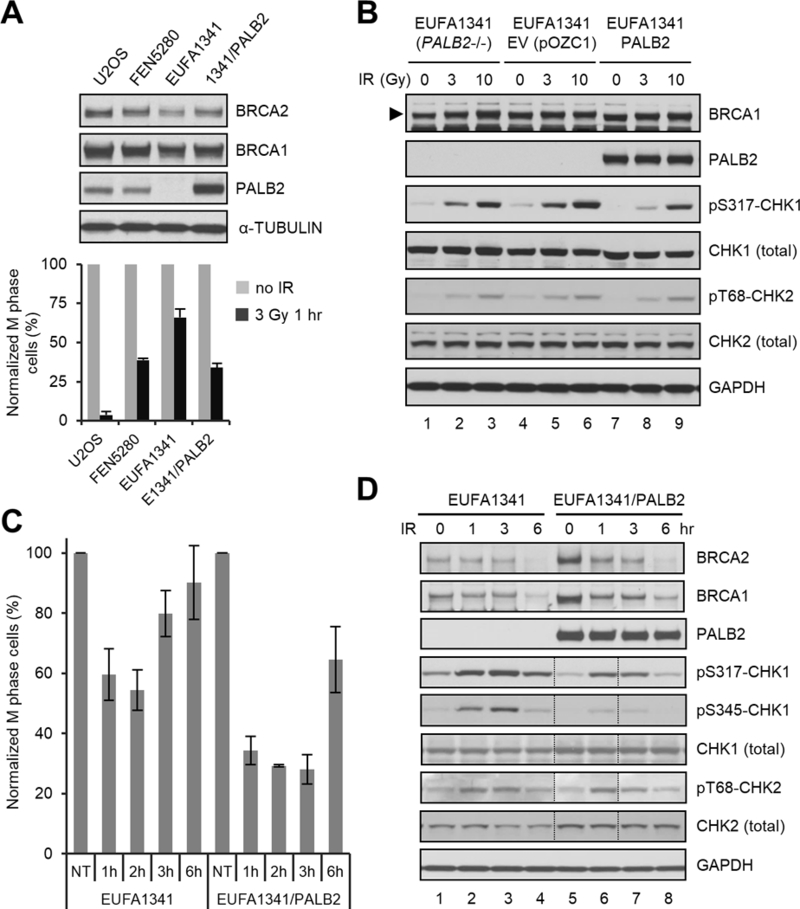
G2/M checkpoint defect of PALB2-deficient human fibroblasts and its rescue by re-expression of wt PALB2. (A) G2/M checkpoint activation in U2OS, FEN5280, EUFA1341, and EUFA1341 cells reconstituted with wt PALB2. Upper panel, representative western blots showing expression levels of PALB2, BRCA1 and BRCA2 in the cells; lower panel, relative mitotic indexes of the cells before and 1 hr after 3 Gy of IR. Data shown are means ± SEMs of the relative mitotic indexes from n>3 independent experiments. (B) Dose-dependent CHK1 and CHK2 phosphorylation in blank, empty vector-harboring and PALB2-reconstituted EUFA1341 cell lines. Cells were collected before or 1 hr after 3 or 10 Gy of IR; total and phosphorylated proteins were analyzed by western blotting. (C) Checkpoint maintenance in blank and reconstituted EUFA1341 cells. Cells were treated with 3 Gy of IR and mitotic cells were measured before and at 1, 2, 3 and 6 hr after IR. Data shown are means ± SEMs of the relative mitotic indexes from 3 independent experiments. (D) Kinetics of CHK1 and CHK2 phosphorylation in blank and reconstituted EUFA1341 cells. Cells were treated with 3 Gy of IR and collected at indicated time points; total and phosphorylated proteins were analyzed by western blotting. The lanes between dotted vertical lines were loaded in the wrong order, which were reversed back using Adobe Photoshop.
The G2/M checkpoint response is commonly attributed to the activation of apical DNA damage response kinases ATM and ATR, which phosphorylate and activate their downstream checkpoint kinases CHK2 and CHK1, respectively, to inhibit cell cycle progression3. To test whether the absence of PALB2 would lead to defective ATM/ATR activation, we compared the phosphorylation status of CHK1 and CHK2 in blank, vector-harboring and PALB2-reconstituted EUFA1341 cells. As shown in Fig. 3B, both blank and vector-harboring cells showed weak phosphorylation of CHK1-S317 and CHK2-T68 before IR, suggesting weak but constitutive activation of ATM and ATR due presumably to increased endogenous DNA damage as a result of PALB2 deficiency. Indeed, these phosphorylation events were even weaker in PALB2-reconstituted cells, consistent with the role of PALB2 in DNA damage repair and recovery of stalled DNA replication forks25. One hour after IR, CHK1 and CHK2 phosphorylation was induced in a dose-dependent manner in all 3 cell lines. While CHK2 phosphorylation was comparable in all 3 lines, CHK1 phosphorylation varied, with the PALB2-reconstituted cells displaying the lowest level of pS317- CHK following both low (3 Gy) and high (10 Gy) doses of radiation.
To gain a fuller understanding of the G2/M checkpoint response in these cells, we measured the mitotic indexes of the blank and PALB2-reconstituted EUFA1341 cells at different time points following 3 Gy of IR. As shown in Fig. 3C, mitotic activity of blank EUFA1341 cells dropped to its lowest level at about 2 hr after IR and then started to recover, whereas the reconstituted cells not only showed more robust checkpoint activation but also maintained the checkpoint for at least 3 hr. Again, phosphorylation of CHK2 at T68 was comparable in the two cells, whereas CHK1 phosphorylation at both S317 and S345 was weaker in the reconstituted cells (Fig. 3D), despite the stronger checkpoint response in them. These results suggest that the role of PALB2 in the G2/M checkpoint is likely independent of CHK1 and CHK2 phosphorylation.
Requirements of BRCA1-PALB2 and PALB2-BRCA2 interactions for effective checkpoint response in human cells
PALB2 directly interacts with BRCA1 via its N-terminal coiled-coil (CC) motif and with BRCA2 through its C-terminal WD repeat domain, thereby linking the two BRCA proteins in HR32, 45. Based on the crystal structure of the PALB2 WD repeat domain, an artificially generated mutation (A1025R) was identified to severely impair BRCA2 binding to PALB226. Recently, we also identified a breast cancer-associated missense mutation in PALB2 (L35P) that disrupts its binding to BRCA1 and completely abrogates HR activity13. To test whether the interactions between PALB2 and BRCA1 or BRCA2 are required for a checkpoint response, we generated EUFA1341 cells stably expressing L35P and A1025R mutants of PALB2 (Fig. 4A). As a control, we re-generated cells expressing the wt protein in parallel. These cells were subjected to 3 Gy of IR along with blank EUFA1341 cells, and their mitotic indexes were measured at different time points (Fig. 4B). Checkpoint activation in the newly generated wt PALB-expressing cells was not as robust as in the previously generated cells (compare Fig. 4B with Fig. 3A and C). Instead, the new cells showed a similar reduction of mitotic index to that of blank cells at 1 hr after IR. However, the mitotic index of these cells continued to decrease until around 3 hr after IR, when the blank cells had almost fully recovered. Rather than contradicting the afore-described role of PALB2 in checkpoint activation, this finding indicates that checkpoint activation was slower in these newly generated cells and that the previous batch of cells could have adapted to exogenous PALB2 expression better over more passages. Under the same condition, cells expressing the L35P mutant showed clear defects in both activation and maintenance of the checkpoint. In cells expressing the A1025R mutant, however, checkpoint activation was similar to cells expressing the wt protein, whereas the maintenance of the checkpoint was evidently compromised. Taken together, these results suggest that the BRCA1-PALB2 interaction can play a key role in both checkpoint activation and maintenance, whereas the binding of BRCA2 to PALB2 mainly contributes to checkpoint maintenance.
Figure 4.
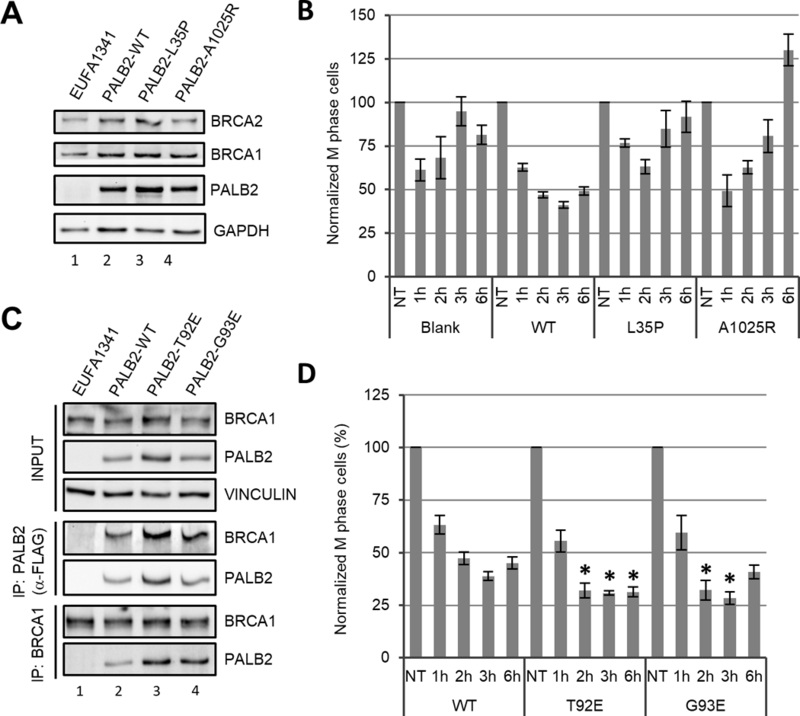
Roles of BRCA1, BRCA2 and KEAP1 binding to PALB2 in G2/M checkpoint response. (A,B) PALB2, BRCA1 and BRCA2 expression levels (A) and checkpoint response (B) in EUFA1341 cells reconstituted with wt, BRCA1 binding mutant (L35P) or BRCA2 binding mutant (A1025R) of PALB2. (C,D) PALB2-BRCA1 interaction (C) and checkpoint response (D) in EUFA1341 cells reconstituted with wt or KEAP1 binding mutants (T92E and G93E) of PALB2. For B and D, data shown are means ± SEMs of the relative mitotic indexes from ≥3 independent experiments. Statistical significance was calculated with one-way ANOVA comparing the values of cells expressing PALB2-T92E and G93E to cells expressing the wt protein at the same time point. *, P<0.05.
We previously found that PALB2 directly interacts with KEAP1, an adaptor protein for a CUL3-based E3 ubiquitin ligase22. More recently, it was reported that KEAP1 mediates the ubiquitination of PALB2 on multiple lysine residues in its N-terminal CC motif27. The same study showed that these ubiquitination events does not appear to cause PALB2 degradation but instead hinders the binding of BRCA127. To test if KEAP1-mediated ubiquitination of PALB2 and the associated reduction in BRCA1 binding impact G2/M checkpoint regulation, we generated stable EUFA1341 cells expressing two mutants of PALB2, T92E and G93E, both defective in KEAP1 binding22. Another new control cell line expressing wt PALB2 was generated in parallel. Consistent with the above report, stronger association of BRCA1 with the mutant PALB2 proteins was found in reciprocal co-immunoprecipitation (co-IP) assays (Fig. 4C). When checkpoint response was analyzed, cells expressing the mutant proteins showed modestly but significantly more robust checkpoint activation (Fig. 4D). These data lend further support to the role of the BRCA1-PALB2 interaction in checkpoint activation.
Critical role of BRCA1-PALB2 interaction in checkpoint response and genome stability in mouse cells
Given the strong and stable association between BRCA2 and PALB2, it is not surprising for the two proteins to function together in checkpoint response. By comparison, the interaction between BRCA1 and PALB2 appears to be much weaker (as judged by co-IP), or perhaps transient. To further understand the role of the BRCA1-PALB2 complex formation in checkpoint control, we analyzed checkpoint response in cells from a Palb2-cc6 knockin mutant mouse strain which contains a 3-aa mutation (24LKK26 → 24AAA26) in the PALB2 CC domain that disengages the endogenous PALB2-BRCA1 interaction31. First, we generated mouse embryonic fibroblasts (MEFs) from wt, heterozygous and homozygous mutant mice and measured checkpoint activation (Fig. 5). When cultured under normal conditions, homozygous mutant MEFs progressively undergo senescence after passage 331. At passage 2, however, their growth rate is similar to wt cells and the abundance of mutant PALB2 protein is comparable to the wt protein (Fig. 5A). After 3 Gy of IR, homozygous mutant MEFs showed greatly impaired checkpoint activation, whereas heterozygous cells behaved similarly to wt cells (Fig. 5B). Next, we isolated splenic B cells from the wt and homozygous mutant mice. The abundance of PALB2 in the mutant B cells was similar to that in the wt cells (Fig. 5C); however, the mutant cells displayed a strong defect in both checkpoint activation and checkpoint maintenance after IR (Fig. 5D). Again, CHK1 phosphorylation was normal in the mutant cells (Fig. 5C).
Figure 5.
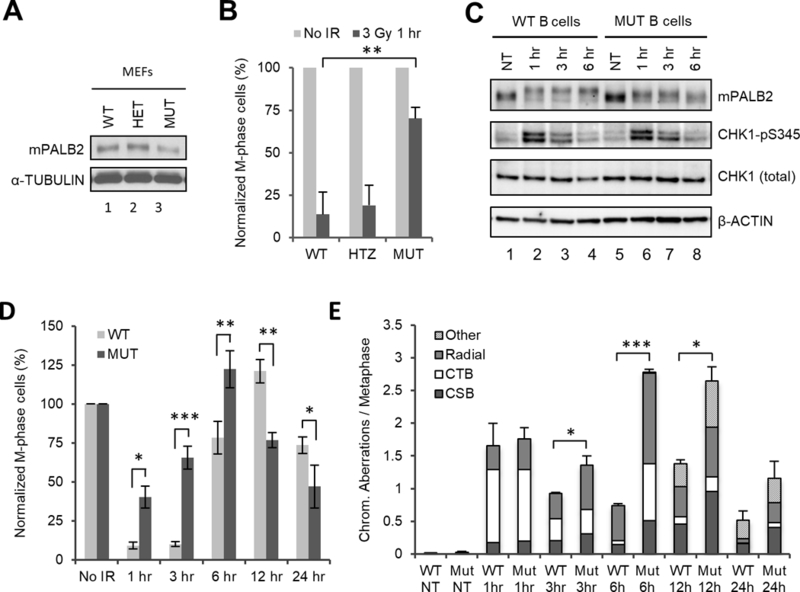
Defective checkpoint response and increased IR-induced chromosomal abnormalities in mouse cells with abrogated BRCA1-PALB2 interaction. (A,B) PALB2 expression levels (A) and G2/M checkpoint activation (B) in MEFs from wt (WT), heterozygous (HET) and homozygous (MUT) Palb2-cc6 knockin mutant mice. (C,D) CHK1 phosphorylation (C) and checkpoint response (D) in splenic B cells isolated from wt and homozygous mutant mice after 3 Gy of IR. Data shown are the means ± SEMs from 4 independent experiments. Statistical significance was calculated with one-way ANOVA. *, P<0.05; **, P<0.01, ***, P<0.001. (E) Chromosomal abnormalities in wt and Palb2 mutant B cells at indicated time points after 3 Gy of IR. Cells were arrested for 1 hr with colcemid prior to collection at each time point (for the 1 hr time point, colcemid was added 10–15 min after IR and incubated for 1 hr). CTB, chromatid break; CSB, chromosomal break. Data shown are the means ± standard deviations (STDVs) from 3 independent experiments. Statistical significance was calculated with two-tailed student’s t test. *, P<0.05; **, P<0.01, ***, P<0.001.
Finally, we analyzed the amount of DNA damage in the cells at different time points after IR. For this purpose, splenic B cells were treated with 3 Gy of IR, mitotic cells were arrested with colcemid for 1 hr before cell collection at each time point, and chromosomal abnormalities were analyzed by combined fluorescent in situ hybridization (FISH) of telemetric DNA and 4,6-Diamidino-2-phenylindole (DAPI) staining of metaphase spreads. As shown in Fig. 5E, similar induction of chromosomal aberrations was detected in both wt and mutant cells at 1 hr after IR. The major type of aberration was chromatid breaks (CTB), consistent with the notion that cells undergoing mitotis during the first hour post IR were mainly the ones that had completed DNA replication, i.e. in M or possibly late G2 phase, at the time of irradiation. Additionally, some radial formation and chromosomal breaks (CSB) were also detected. At 3 hr after IR, the level of overall chromosomal aberrations had decreased in both cells, due largely to the reduction of cells with chromatid breaks, implying the completion of mitosis by cells that were in M or G2 phases at the time of IR. At this time, radial formation and chromosomal breaks appeared to have increased in the mutant cells as compared with those at 1 hr, while little difference in this regard was observed in wt cells. By 6 hr after IR, a further decrease of chromatid breaks and an increase in radial formation were observed in the wt cells, whereas all 3 types of aberrations, especially radial chromosomes, had increased in the mutant cells. These findings indicate that wt cells resumed mitosis only after the majority of DSBs are repaired, whereas the mutant cells continued to enter mitosis with still a substantial amount of damage. The consistently higher levels of radial chromosomes in the mutant cells likely reflect increased usage of non-homologous recombination (NHEJ) or perhaps even heterologous recombination as a result of severely diminished HR activity. The 12 hr time point was marked with the emergence of “other” types of chromosomal aberrations, mostly consisting of dicentric chromosomes, in both cells; at the same time, all 4 types of abnormality were more frequent in the mutant cells. By 24 hr after IR, overall levels of chromosomal abnormality had decreased substantially in both cells as compared with those at 12 hr, with the mutant cells still having more chromosomal aberrations than wt cells and both cells having much more aberrations than their basal levels before IR. Together, these results demonstrate the critical role of the BRCA1-PALB2 complex formation in genome stability maintenance and an inability of cells with a broken BRCA1-PALB2 link to mount an effective G2/M checkpoint even with higher levels of DNA damage than wt cells.
Discussion
BRCA1 has been shown to contribute to G2/M checkpoint activation in a number of studies conducted under various settings39–43. More recently, two separate siRNA screens found BRCA2 and PALB2 (FLJ21816) to be among the most critical players in the maintenance of this checkpoint8, 23. BRCA1 also scored positive in the screens, but its function appeared to be less critical. Both studies were conducted using U2OS cells, in which all 3 proteins were found to be dispensable for checkpoint activation8, 23. In this study, the above findings were confirmed in U2OS cells (Fig. 1A). However, we also found that all 3 proteins can play a significant role in checkpoint activation in other cell types of both human and mouse origins. Therefore, the exact role of these proteins in this checkpoint response is cell type or context dependent. Furthermore, checkpoint response was compromised when either of the BRCA1-PALB2 or BRCA2-PALB2 interactions were disrupted (Figs. 4 and 5). This finding indicates that the three proteins may function in a common pathway to promote checkpoint control, with PALB2 acting as a nexus between the two BRCA proteins, much like the way they function in HR32, 37, 45. Interestingly, however, the checkpoint function of these proteins is likely independent of their HR function, since depletion of RAD51 did not produce any effect on checkpoint response23.
BRCA1 functions upstream to promote the recruitment of PALB2 and, in turn, BRCA2 to DNA damage sites32, 45. Since loss of neither PALB2 nor BRCA2 affects BRCA1 abundance or localization32, 37, 45, the factor among the 3 proteins that directly communicates with the checkpoint machinery is unlikely to be BRCA1. Instead, BRCA1 probably acts as a facilitator of the checkpoint function of PALB2/BRCA2 by directing the optimal positioning of the latter in damaged chromatin. Additionally, BRCA1 may also contribute to checkpoint response by facilitating end resection4, 10, which generates single stranded DNA (ssDNA) required for ATR activation. BRCA2 and PALB2 form a tight complex with high stoichiometry. Approximately 50% of the proteins are complexed with each other, and most, if not all, chromatin-associated BRCA2 is bound to PALB232, 37. Therefore, they likely function as a complex in checkpoint control. Which of the two directly interacts with the checkpoint signaling machinery to promote checkpoint response is currently unclear. However, when BRCA2 is depleted or when the PALB2-BRCA2 interaction is disrupted by PALB2 A1025R mutation, much of PALB2 would still remain at DNA damage sites, yet checkpoint is defective. Moreover, BRCA2 has been shown to be a substrate for the polo-like kinase PLK1, a major driver of mitosis18, 19. As such, BRCA2 is more likely to be the direct player, although there are other possible scenarios that cannot be ruled out. Consistent with the role of PALB2 in sustaining BRCA2 stability as we reported before37, loss of PALB2 significantly reduced BRCA2 protein amount (Figs. 1A, 2A, 2B and 3A), indicating that the impact of PALB2 loss on checkpoint response could consist of both direct, if any, and indirect effects. The precise mechanisms of all 3 proteins in checkpoint response still await further investigation.
The HR function of BRCA1, BRCA2 and PALB2 is widely thought to be essential for their tumor suppressive activity. Still, these proteins also play important roles in multiple other cellular processes, including transcriptional regulation, cell cycle checkpoint control, cell division, and oxidative stress response, etc. It is not clear if their HR function is solely responsible for tumor suppression or whether any of the “other” functions above are also required. With respect to the G2/M checkpoint function, the role of these proteins in the DDR is counterintuitive - on the one hand, loss of the proteins results in more DNA damage accumulation upon genotoxic insults or endogenous DSB formation; on the other hand, the absence of these proteins permits continued cell division with excessive DNA damage, not to mention the products of misrepair such as radial and dicentric chromosomes. As an example, at 6 hr after IR, the dividing Palb2 mutant cells harbored approximately 7 times the amount of DNA breaks (CTB and CSB combined) and 2.5 times the amount of radial chromosomes relative to wt cells (Fig. 5E). It is currently unclear whether the DNA breaks carried-over are repaired after mitosis. If so, repair is likely via non-homologous end joining (NHEJ) or single strand annealing (SSA), which are both error-prone, otherwise segments of chromosomes would be lost during the next round of cell division. In either case, the G2/M checkpoint defect associated with BRCA1/2 and PALB2 mutations or loss can be expected to exacerbate genomic instability. As long as the mutant cells survive and proliferate, they are likely to accumulate mutations and chromosomal abnormalities at a much faster pace than normal cells, eventually leading to transformation and tumor development.
Materials and Methods
Cell lines and cultures
U2OS and 293T cells were purchased from ATCC. Isogenic TP53 wt and null HCT116 cells5, isogenic HCT116 and HCT116:3–6 cells11 and SV-40 transformed EUFA1341 and FEN5280 fibroblasts38 were described before. EUFA1341 cell lines expressing wt or mutant PALB2 proteins were generated as described38. All above cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1X Penicillin-Streptomycin (Pen-Strep). Mouse tumor cell lines were generated as described before14 from mammary tumors that developed in Palb2F/F;Trp53F/F;Wap-cre, Brca1F/F;Trp53F/F;Wap-cre and Brca2F/F;Trp53F/F;Wap-cre conditional knockout mice (Huo, et al., unpublished). The Brca2−/−;Trp53−/− KB2 cells were described before12. These mouse cells were cultured in DMEM/F12 (1:1) medium supplemented with 5 μg/ml insulin, 5 ng/ml EGF, 5 ng/ml Cholera toxin and 1 x Pen-Strep. Mouse embryonic fibroblasts (MEFs) and splenic B-lymphocytes from wt and Palb2-cc6 knockin mice were isolated and cultured as described31. All cells were cultured at 37oC in a humidified incubator with 5% CO2.
siRNAs and transfections
The sense sequences of siRNAs used are as follows: Control (5’-UUCGAACGUGUCACGUCAAdTdT-3’); BRCA1 #296 (5’- GGAACCUGUCUCCACAAAGdTdT-3’); BRCA1 #4641 (5’- GCAGAUAGUUCUACCAGUAdTdT-3’); BRCA2 #1949 (5’- GAAGAAUGCAGGUUUAAUAdTdT-3’); BRCA2 #2618 (5’- GCUCAAAGGUAACAAUUAUdTdT −3’); PALB2 #1493 (5’-UCAUUUGGAUGUCAAGAAAdTdT-3’) and PALB2 #2693 (5’- GCAUAAACAUUCCGUCGAAdTdT-3’). The siRNAs were custom synthesized by Sigma Genosys. Transfections were carried out using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions, at a final concentration of 10 nM. BRCA1, BRCA2 and PALB2 siRNAs were used as pools of the two.
Gamma irradiation
Ionizing radiation was delivered by a Cesium 137 irradiator from Gammacell 40 Exactor (Best Theratronics, Ottawa, Ontario, Canada) at a dose rate of 1.026 Gy/min.
Quantification of M-phase cells
Cells were seeded into 6-well plates at a density of either 1 × 106 cells per well for analysis on the following day or 0.2 × 106 cells per well if they were to be first subjected to siRNA transfection (for 48 hr). Cells were γ-irradiated, allowed to recover for indicated time periods and then collected and fixed overnight in ice-cold 70% ethanol. Fixed cells were permeabilized with 0.25% Triton-X100 in 1X phosphate-buffered saline (PBS) on ice for 15 min, stained for 2 hr with rabbit anti-phospho-histone H3 (Ser10) Ab (#9701, Cell Signaling) in PBS containing 1% bovine serum albumin (BSA), followed by staining with Fluorescein (FITC)-conjugated AffiniPure goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories) in PBS/1% BSA for 30 min. Phospho-H3 positive cells were quantified by fluorescence-activated cell sorting (FACS) on a Cytomics FC-500 flow cytometer (Beckman-Coulter) with laser excitation at 488 nm. Prior to FACS analysis, propidium iodide (PI) (Sigma, P4170) was added to a final concentration of 10 μg/mL to stain DNA. For each sample, 1.5 × 104 events were scored.
Western blotting and immunoprecipitation (IP)
To analyze protein expression levels in mouse tumor cell lines and EUFA1341 cell lines, cells were plated at a density of 1.0 × 106 per well in 6-well plates and collected 24 hr later. Cells were lysed in NETNG-300 buffer (300 mM NaCl, 1 mM EDTA, 20 mM Tris-HCl [pH 7.5], 0.5% Non-Idet P, 10% Glycerol) containing Complete® protease inhibitor cocktail (Roche). To analyze BRCA1-PALB2 complex formation in EUFA1341 cells reconstituted with wt and mutant PALB2 proteins, the endogenous BRCA1 was IPed using anti-BRCA1 (Santa Cruz, sc-6954) coupled with protein A beads and the exogenous PALB2 proteins were IPed with anti-FLAG M2 agarose beads (Sigma) for 3 hr at 4°C. Proteins in cell lysates or immunoprecipitates were resolved on 4–12% Tris-Glycine gels (Invitrogen), transferred onto nitrocellulose membranes and probed with relevant antibodies following standard procedures. Immobilon Western Chemiluminescent HRP substrate (EMD Millipore) was used for developing the blots.
Human and mouse PALB2 proteins were detected using rabbit polyclonal M11 and NB3 antibodies described previously31. Monoclonal anti-mouse BRCA1 antibody, against amino acids 160–300 of mouse BRCA1, was described before2. Other antibodies used included anti-HA (H3663, Sigma), BRCA1 (EMD Millipore, #07–434), BRCA2 (OP95, EMD Millipore), TP53 (Santa Cruz, DO-1) and RAD51 (Santa Cruz, H92), phospho-CHK1 S317 (Cell Signaling, #2344), phospho-CHK1 S345 (Cell Signaling, #2348), CHK1 (Bethyl Labs, A300–161A), α-Tubulin (Sigma, T9026), β-Actin (Santa Cruz, AC-15) and GAPDH (Santa Cruz, sc25778).
B cell chromosome spreads
For analysis of metaphase chromosomes, activated B cells were arrested with 100 ng/mL colcemid (Sigma) for 1 hr prior to collection at each time point. This was followed by treatment with hypotonic solution (0.075 M KCl) and fixation with 3:1 methanol/acetic acid. Telomere-FISH analysis was performed with Cy3-labeled telomere peptide nucleic acid probe (Panagene). 50–55 images were analyzed per sample.
Statistical analyses
In all experiments, the numbers of mitotic cells were normalized against unirradiated controls for each cell line or siRNA treatment. Statistical analyses were performed using one-way ANOVA with GraphPad Prism6 or two-tailed student’s t test with Microsoft Excel, as indicated in the figure legends.
Supplementary Material
Acknowledgements
We thank Drs. J. Jonkers (Netherlands Cancer Institute) and D. Boothman (UT Southwestern Medical Center) for provision of the KB2 mouse mammary tumor cells and isogenic HCT116 cells, respectively. This work was supported by the National Institutes of Health (R01CA138804 and R01CA188096 to B.X., R01CA190858 to S.F.B. and R01CA169182 to S.G.). G. Vincelli was supported by a postdoctoral fellowship and S. Misenko by a predoctoral fellowship from the New Jersey Commission for Cancer Research (NJCCR).
Footnotes
Conflict of interest
The authors declare no conflicts of interest.
References
- 1.Apostolou P, Fostira F. Hereditary breast cancer: the era of new susceptibility genes. BioMed research international 2013; 2013: 747318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barlow JH, Faryabi RB, Callen E, Wong N, Malhowski A, Chen HT et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 2013; 152: 620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol 2007; 19: 238–245. [DOI] [PubMed] [Google Scholar]
- 4.Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010; 141: 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science (New York, NY 1998; 282: 1497–1501. [DOI] [PubMed] [Google Scholar]
- 6.Cavanagh H, Rogers KM. The role of BRCA1 and BRCA2 mutations in prostate, pancreatic and stomach cancers. Hereditary cancer in clinical practice 2015; 13: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40: 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cotta-Ramusino C, McDonald ER 3rd,, Hurov K, Sowa ME, Harper JW, Elledge SJ. A DNA damage response screen identifies RHINO, a 9–1-1 and TopBP1 interacting protein required for ATR signaling. Science (New York, NY 2011; 332: 1313–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Couch FJ, Nathanson KL, Offit K. Two decades after BRCA: setting paradigms in personalized cancer care and prevention Science (New York, NY 2014; 343: 1466–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cruz-Garcia A, Lopez-Saavedra A, Huertas P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep 2014; 9: 451–459. [DOI] [PubMed] [Google Scholar]
- 11.Davis TW, Wilson-Van Patten C, Meyers M, Kunugi KA, Cuthill S, Reznikoff C et al. Defective expression of the DNA mismatch repair protein, MLH1, alters G2-M cell cycle checkpoint arrest following ionizing radiation. Cancer Res 1998; 58: 767–778. [PubMed] [Google Scholar]
- 12.Evers B, Schut E, van der Burg E, Braumuller TM, Egan DA, Holstege H et al. A high-throughput pharmaceutical screen identifies compounds with specific toxicity against BRCA2-deficient tumors. Clin Cancer Res 2010; 16: 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foo TK, Tischkowitz M, Simhadri S, Boshari T, Zayed N, Burke KA et al. Compromised BRCA1-PALB2 interaction is associated with breast cancer risk. Oncogene 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huo Y, Cai H, Teplova I, Bowman-Colin C, Chen G, Price S et al. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer discovery 2013; 3: 894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461: 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet 2001; 29: 418–425. [DOI] [PubMed] [Google Scholar]
- 17.Juan G, Traganos F, James WM, Ray JM, Roberge M, Sauve DM et al. Histone H3 phosphorylation and expression of cyclins A and B1 measured in individual cells during their progression through G2 and mitosis. Cytometry 1998; 32: 71–77. [DOI] [PubMed] [Google Scholar]
- 18.Lee M, Daniels MJ, Venkitaraman AR. Phosphorylation of BRCA2 by the Polo-like kinase Plk1 is regulated by DNA damage and mitotic progression. Oncogene 2004; 23: 865–872. [DOI] [PubMed] [Google Scholar]
- 19.Lin HR, Ting NS, Qin J, Lee WH. M phase-specific phosphorylation of BRCA2 by Polo-like kinase 1 correlates with the dissociation of the BRCA2-P/CAF complex. The Journal of biological chemistry 2003; 278: 35979–35987. [DOI] [PubMed] [Google Scholar]
- 20.Lobrich M, Jeggo PA. The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat Rev Cancer 2007; 7: 861–869. [DOI] [PubMed] [Google Scholar]
- 21.Lu C, Xie M, Wendl MC, Wang J, McLellan MD, Leiserson MD et al. Patterns and functional implications of rare germline variants across 12 cancer types. Nature communications 2015; 6: 10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma J, Cai H, Wu T, Sobhian B, Huo Y, Alcivar A et al. PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Molecular and cellular biology 2012; 32: 1506–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Menzel T, Nahse-Kumpf V, Kousholt AN, Klein DK, Lund-Andersen C, Lees M et al. A genetic screen identifies BRCA2 and PALB2 as key regulators of G2 checkpoint maintenance. EMBO reports 2011; 12: 705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nature reviews 2010; 11: 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murphy AK, Fitzgerald M, Ro T, Kim JH, Rabinowitsch AI, Chowdhury D et al. Phosphorylated RPA recruits PALB2 to stalled DNA replication forks to facilitate fork recovery. J Cell Biol 2014; 206: 493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oliver AW, Swift S, Lord CJ, Ashworth A, Pearl LH. Structural basis for recruitment of BRCA2 by PALB2. EMBO reports 2009; 10: 990–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orthwein A, Noordermeer SM, Wilson MD, Landry S, Enchev RI, Sherker A et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature 2015; 528: 422–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes & development 2011; 25: 409–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer 2012; 12: 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shabbeer S, Omer D, Berneman D, Weitzman O, Alpaugh A, Pietraszkiewicz A et al. BRCA1 targets G2/M cell cycle proteins for ubiquitination and proteasomal degradation. Oncogene 2013; 32: 5005–5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simhadri S, Peterson S, Patel DS, Huo Y, Cai H, Bowman-Colin C et al. Male fertility defect associated with disrupted BRCA1-PALB2 interaction in mice. The Journal of biological chemistry 2014; 289: 24617–24629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci U S A 2009; 106: 7155–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene 2001; 20: 1803–1815. [DOI] [PubMed] [Google Scholar]
- 34.Tischkowitz M, Xia B. PALB2/FANCN: recombining cancer and Fanconi anemia. Cancer Res 2010; 70: 7353–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Venkitaraman AR. Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science (New York, NY 2014; 343: 1470–1475. [DOI] [PubMed] [Google Scholar]
- 36.Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci U S A 2011; 108: 18032–18037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell 2006; 22: 719–729. [DOI] [PubMed] [Google Scholar]
- 38.Xia B, Dorsman JC, Ameziane N, de Vries Y, Rooimans MA, Sheng Q et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat Genet 2007; 39: 159–161. [DOI] [PubMed] [Google Scholar]
- 39.Xu B, Kim S, Kastan MB. Involvement of Brca1 in S-phase and G(2)-phase checkpoints after ionizing irradiation. Molecular and cellular biology 2001; 21: 3445–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu X, Weaver Z, Linke SP, Li C, Gotay J, Wang XW et al. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol Cell 1999; 3: 389–395. [DOI] [PubMed] [Google Scholar]
- 41.Yarden RI, Pardo-Reoyo S, Sgagias M, Cowan KH, Brody LC. BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat Genet 2002; 30: 285–289. [DOI] [PubMed] [Google Scholar]
- 42.Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science (New York, NY 2003; 302: 639–642. [DOI] [PubMed] [Google Scholar]
- 43.Yu X, Chen J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Molecular and cellular biology 2004; 24: 9478–9486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang F, Fan Q, Ren K, Andreassen PR. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol Cancer Res 2009; 7: 1110–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B et al. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol 2009; 19: 524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.