

Summary
TCRs recognize cognate pMHCs to initiate T cell signaling and adaptive immunity. Mechanical force strengthens TCR-pMHC interactions to elicit agonist specific catch bonds to trigger TCR signaling, but the underlying dynamic structural mechanism is unclear. We combined steered molecular dynamics (SMD) simulation, single-molecule biophysical approaches, and functional assays to collectively demonstrate that mechanical force induces conformational changes in pMHCs to enhance pre-existing contacts and activates new interactions at the TCR-pMHC binding interface to resist bond dissociation under force, resulting in TCR-pMHC catch bonds and T cell activation. Intriguingly, cancer-associated somatic mutations in HLA-A2 that may restrict these conformational changes suppressed TCR-pMHC catch bonds. Structural analysis also indicated that HLA polymorphism might also alter the equilibrium of these conformational changes. Our findings not only reveal critical roles of force-induced conformational changes in pMHCs for activating TCR-pMHC catch bonds but also have implications for T cell-based immunotherapy.
Keywords: TCR, MHC, Mechano-regulation, Antigen recognition, Catch bond, Molecular dynamics, Single molecule
In brief
Wu et al. report that a dynamic structural mechanism of mechano-chemical coupling for TCR antigen recognition, that is, mechanical force-induced conformational changes in the agonist peptide-MHC-I allosterically activate TCR-pMHC-I catch bonds to determine TCR antigen recognition and to trigger T-cell signaling.
Graphical Abstract
Introduction
αβ T cell receptors (TCRs) on T cells recognize peptide-loaded major histocompatibility complexes (pMHCs) on antigen-presenting cells (APCs) to initiate T cell signaling and adaptive immunity. Randomly distributed monomeric TCRs on T cell’s microvilli sensitively recognize rare or even a single cognate pMHC on APCs and trigger TCR signaling (Brameshuber et al., 2018; Cai et al., 2017; Huang et al., 2013; Rossboth et al., 2018; van der Merwe and Dushek, 2011). Once engaging with cognate antigens on infected or transformed APCs, TCRs rapidly activate cytotoxic T lymphocytes (CTLs) to form a dynamic immunological synapse (IS) with and to kill APCs (Grakoui et al., 1999; Huse, 2017). Such exceptional sensitivity and discrimination power of TCRs is indispensable for anti-cancer immunity and crucial for most T cell-based immunotherapies. Yet the molecular mechanism of how TCRs recognize cognate pMHCs in such a dynamic microenvironment remains poorly understood.
The dynamic nature of T-cell antigen recognition inevitably integrates mechanical force with the biochemistry of TCR-pMHC interactions to activate T cell functions (Huse, 2017; Kim et al., 2009; Ma et al., 2008; O’Connor et al., 2012; Saitakis et al., 2017; van der Merwe and Dushek, 2011). With recent applications of biophysical techniques, cumulative evidences have demonstrated that force not only counter-intuitively activated agonist-specific TCR-pMHC catch bonds on both CD8+ (Das et al., 2015; Liu et al., 2014) and CD4+ T cells (Hong et al., 2015), or even on pre-TCR expressing immature thymocytes (Das et al., 2016; Mallis et al., 2015), but also plays an essential role in triggering proximal TCR signaling (Bashour et al., 2014; Hu and Butte, 2016; Liu et al., 2016), in inducing the thymocyte negative selection (Hong et al., 2018) and in potentiating CTL’s killing of target cells (Basu et al., 2016). Force-induced TCR-pMHC catch bonds amplify the discriminative power of TCRs for agonists and antagonists by increasing their differences on two-dimensional (2D) bond lifetimes under an optimal force, and rapid accumulation of these catch bonds trigger intracellular Ca2+ signaling in T cells (Liu et al., 2014). Given ultra-fast rebinding rates of TCR-pMHC interactions that are confined in the plasma membrane of T-cell/APC contacts, the force-dependent 2D dissociation rate of TCR-pMHC bonds has emerged as a crucial parameter in TCR recognition of cognate antigens and TCR triggering (Dushek et al., 2009; Lever et al., 2014; Sibener et al., 2018).
Though vitally important, TCR-pMHC catch bonds remain enigmatic. It is still unclear how a tensile force exerting on the TCR-pMHC bond could make the bond stronger. Fortunately, since the first crystal structure of TCR-pMHC-I complex was solved two decades ago (Garboczi et al., 1996), ~100 human TCR-HLA and ~30 mouse TCR-H-2 complex structures have been determined, significantly advancing our understanding of how TCRs recognize various antigens at the atomic level. With a few exceptions, most TCR-pMHC (MHC referring to MHC class I hereafter unless noted otherwise) complexes have a diagonal docking geometry, in which MHC α1 and α2 domains mainly contact TCR Vβ and Vα respectively. Complementarity-determining region 3 (CDR3) loops of TCR’s variable domains sensitively recognize the key residues or “hotspots” on the peptide and undergo subtle conformational changes upon pMHC binding (Degano et al., 2000; Rudolph et al., 2006). These structures revealed poor shape complementarity at the TCR-pMHC binding interface (Degano et al., 2000; Rossjohn et al., 2015), and a universal structural understanding of TCR antigen recognition and discrimination is still elusive. In the mouse 2C TCR, no significant conformational difference was found between the super-agonist SIYR-MHC-2C TCR complex (Figure 1A) and the self dEV8-MHC-TCR complex despite their large differences in activation potencies (Degano et al., 2000; Garcia et al., 1998). Similarly, the complexed structure of the human 1G4 TCR with the tumor epitope NY-ESO-1157–165 (9C) presented by human leukocyte antigen-A2 (HLA-A2) is almost identical to that of a more potent pMHC with only a cysteine-to-valine substitution at peptide position 9 (9V) (Chen et al., 2005). Considering the dynamic nature of TCR antigen recognition, force-regulated dynamics of the TCR-pMHC complex structure may offer intrinsic mechanistic insights for TCR antigen recognition and discrimination.
Figure 1. Force-induced strong engagement between agonist peptide and TCR determines pMHC/2C TCR catch bonds and 2C T cell responses.

(A) In SMD simulations, force was applied along the longitudinal axis (gray arrow) of the 2C TCR/R4-MHC complex to simulate BFP experiments. (B-D) Structural comparisons of the H-bond network in the vicinity of the 4th residue of the peptides R4 (B), EVSV (C) or L4 (D), at the TCR/pMHC binding interface under force or no force. Defined H-bonds are indicated as dashed blue lines. (E-H) Time-courses of the numbers of H-bonds for R4 (E) and L4 (G), and their occuring frequencies for R4 (F) and L4 (H). (I) Schematics of BFP experiments. (J-K) Force-dependent lifetimes of single TCR bonds with R4- (J), L4-, or EVSV-MHCs (K). (L) IL-2 productions of 2C T cells stimulated with serial concentrations of surface-coated R4-, L4- or EVSV-MHC. Error bars in (J-L) represent SEM. See also Figure S1.
In the present study, we took an integrated approach to decipher the dynamic structural mechanism of TCR-pMHC catch bonds by combining SMD simulations and single-molecule biophysical methods. Our findings revealed a dynamic mechano-chemical coupling mechanism for TCR-pMHC catch bonds and TCR antigen recognition, potentially shedding lights on T-cell based immunotherapies for cancers.
Results
Strong pre-existing or force-enhanced engagement between the agonist peptide and the TCR determines TCR-pMHC catch bonds and T cell activation
We used the 2C TCR as a model system due to the availability of TCR-pMHC complex structures. The 2C TCR has been reported to function differentially in recognizing the super-agonist SIYR (or R4), the weak agonist dEV8, and the antagonist EVSV peptides loaded on the H-2Kb (Degano et al., 2000; Garcia et al., 1998). To reveal the dynamic structural basis for agonist-specific pMHC/2C TCR catch bonds, we first ran large-scale free molecular dynamics (MD, no force) and SMD (with force) simulations with crystal structures of the R4-MHC/2C TCR and dEV8-MHC/2C TCR complexes and a modeled structure of EVSV-MHC/2C TCR. Multiple independent pulling trials were performed for each complex. At first, we noticed that before force application, both the R4 and dEV8 peptides but not the EVSV peptide maintained H-bonds with TCR CDR loops mainly through their “hotspot” residues (ArgP4 in the R4 peptide and LysP4 in the dEV8 peptide, Table S1) that had been identified to be critical for 2C TCR antigen recognition (Degano et al., 2000; Garcia et al., 1998). Strikingly, upon application of force normal to the TCR-pMHC binding interface (Figure 1A), we found that the frequencies of H-bond formations between the functional “hotspot” ArgP4 (R4) and LysP4 (dEV8) residues and TCR CDR loops (Figures 1B, 1E, 1F, S1A-S1C) increased, but not for the EVSV peptide (Figures 1C, S1B, and S1C). Specifically, ArgP4 (R4) formed one H-bond on average with the TCR (Figures 1E and 1F) in the absence of force, but subtly rotated to from two H-bonds on average with the TCR (Figures 1E and 1F) after force was applied. For example, ArgP4 formed H-bonds with Y50 on the CDR2 loop of the TCRβ subunit in the absence of force (Figure 1B, upper panel); while force induced formations of two more H-bonds between ArgP4 and the main chain carbonyl oxygen of G96 in the CDR1β loop and S102 in the CDR3α loop (Figure 1B, lower panel). In addition, Q65 on the MHCα chain switched to form an H-bond with E56 in the TCRβ (Figure 1B). Similarly, force enhanced H-bond formations between LysP4 (dEV8) and the CDR3α loop (Figures S1A and S1B). In contrast, ValP4 in the EVSV peptide did not form any H-bond with the TCR regardless of force application (Figure 1C). Therefore, the agonist pMHC has a stronger association with TCR CDR loops through its critical “hotspot” residue of the peptide than the antagonist pMHC, and this association can be further strengthened by mechanical force, revealing forced amplification of structural differences between agonist and antagonist pMHCs.
We next hypothesized that this “hotspot” residue was required for forming TCR-pMHC catch bonds. We then mutated ArgP4 in the R4 peptide to LeuP4 in silico (referred to as L4 hereafter). As expected, we did not observe any H-bond formation between LeuP4 and CDR loops regardless of force application in any simulation (Figures 1D, 1G, and 1H). Using a biomembrane force probe (BFP, Figure 1I), we confirmed previously reported 2C TCR catch bonds with the super-agonist R4-MHC (Figures 1J, S1D, and S1E) (Liu et al., 2014). The weak agonist dEV8-MHC also formed catch bonds with the 2C TCR (Figure S1E), but the antagonist EVSV-MHC only formed slip bonds with the 2C TCR (Figures 1K and S1D), consistent with our previously reported agonist-specific catch bonds of the OT-1 TCR system (Liu et al., 2014). As expected, the “hotspot” mutation L4-MHC abolished R4-MHC/2C TCR catch bonds, converting to slip bonds with the 2C TCR (Figures 1K, S1D, and S1E).
To test the functional importance of this “hotspot” in the R4 peptide, we next examined the triggering potencies of R4-, EVSV- and L4-MHC peptides on T-cell activation in vitro. We found that R4-MHC but not EVSV- or L4-MHC robustly stimulated IL-2 release (Figure 1L) from 2C hybridomas, showing stronger potency of R4-MHC. We further confirmed stronger potency of R4-MHC than that of L4-MHC on activating primary 2C T cells in vitro with stronger proliferation and interferon gamma (IFN-γ) production (Figures S1F and S1G).
Collectively, our data demonstrated that the strong pre-existing or force-enhanced engagement between the “hotspot residue” of the agonist peptide and the TCR determined agonist-TCR-pMHC catch bonds, which correlated well with potencies in activating T cells.
Force-induced H-bonds at the TCR and MHC binding interface contributed to TCR-pMHC catch bonds
Having identified this critical functional “hotspot” residue ArgP4 in the R4 peptide for R4-MHC/2C TCR catch bonds, we next investigated how a MHC molecule participated in forming agonist-specific TCR-pMHC catch bonds. Focusing on the TCR-MHC interaction, surprisingly, we found large conformational changes of the MHC in both R4- and dEV8-MHC/2C TCR simulations (Figures 2A and 2B; Movies S1 and S2) but not in EVSV- or L4-MHC/2C TCR simulations (Figures 2C and 2D; Movies S3 and S4). Our simulations revealed that pulling force gradually extended and rotated the R4- or dEV8- but not EVSV- or L4-MHC’s binding interface with the 2C TCR to become almost parallel to the force direction along with unexpected partial dissociation of the β2m from the MHC α chain (Figures 2A-2D and S2A; Movies S1 and S2). The extension of MHC α chain was initiated by the forced extension of the linker between the MHC α1α2 domain and the α3 domain (Figure 2A, 44 ns; Figure 2B, 40 ns), followed by forced break-up of the intramolecular association either between α1α2 and β2m domains (α1α2/β2m) (Figure 2A, 100 ns; Figure 2B, 90 ns; Movies S1 Trace-1 and S2) or between β2m and α3 domains (β2m/α3) (Figure S2A; Movie S1 Trace-2). These break-ups led to a large extension of the MHC from 7 nm (α1α2/β2m or β2m/α3 dissociation only, Figures 2A, 2B and S2A; Movie S1) to 12 nm (α3 partially unfolded in addition to α1α2/β2m or β2m/α3 dissociation, Figures S2B and S2C; Movie S1 Trace-3). In contrast, the TCR was rigid and stable during mechanical pulling in all SMD simulations with a backbone root-mean-square deviation (RMSD) of <4Å (Figures S2D and S2E); the only notable change on the TCR was the ~2 nm extension on the C-terminal linker regions of both α and β subunits (Figure S2D). α1α2/β2m or α3/β2m partial dissociation further allowed force to rotate the TCR-pMHC binding interface towards the pulling direction (Figure 2A, 100 ns, 122 ns and 127.7 ns; Figure 2B, 90 ns, 110 ns and 115 ns; Figure 2E). We quantified the domain rotation as the angle between the line linking the two ends of the peptide and the pulling direction. This angle was ~90° at small forces and increased to ~115° after α1α2/β2m or α3 /β2m partial dissociation at large forces, and further increased to ~135° before the dissociation of R4- or dEV8-MHC from the 2C TCR (Figure 2E; Movies S1 and S2). In addition, we found that the total number of H-bonds between the TCR and the pMHC increased for R4-, dEV8-MHC, had no change for EVSV-MHC, but decreased for L4-MHC under force compared with force free condition (Figure 2F).
Figure 2. Force induced two distinct dissociation pathways of TCR-pMHC bonds for agonists and lower-potency ligands.

(A-D) Sequential snapshots of SMD simulations of the dissociation of R4- (A), dEV8- (B), L4-(C) or EVSV-MHC (D) from the 2C TCR under force (gray arrow). (E) Time-course of the angle between the peptide (R4, dEV8, EVSV or L4) and the direction of force in SMD simulations. (F) The distributions of H-bond numbers between TCR and pMHC for the peptide (R4, dEV8, EVSV or L4) in the presence (red square,) or absence (blue circle) of force. Distributions were fitted by the Gaussian. See also Figure S2.
We next asked whether such extension and rotation of the MHC could further allosterically strengthen TCR-pMHC binding under force. Analyzing trajectories of R4-MHC/2C TCR simulations in details (Figure 3A and Table S1), we indeed found that force increased the formation frequencies of some pre-existed H-bonds and also induced the formation of new H-bonds at the TCR and MHC binding interface (Figure S3A and Table S1). Especially, the formation frequencies of H-bonds, including residues N30, E56 and Q72 of the TCR β and residues K146, Q72, and R79 of the MHC, were enhanced the most upon force application (Figure S3A and Table S1); and the formation frequencies between TCRβ CDR1 N30 and MHC α2 K146 (Figure 3B), TCRβ CDR2 E56 and MHC α1 Q72 (Figure 3C), and TCRβ Q72 and MHC α1 R79 (Figure 3D) showed higher force-enhancing effect (Figures 3E and S3A; Table S1). For example, force increased the frequency of forming H-bonds between TCRβ N30 and MHC K146 from ~10% to ~24% (Figures 3E, S3A, and S3B; Table S1), and between TCRβ E56 and MHC Q72 from ~1% to ~15% (Figures 3E, S3A, and S3B; Table S1). More strikingly, following the rotation of the MHC α1α2 domain, force induced H-bond formation between TCRβ Q72 and MHC R79 that did not exist in the absence of force (Figures 3D, 3E, and S3B; Table S1). Thus, these force-enhanced or force-induced H-bonds at the TCR-MHC binding interface before or after the MHC conformational change further resisted the dissociation of the TCR-pMHC bond under force, facilitating the formation of TCR-pMHC catch bonds.
Figure 3. Force-strengthened H-bonds between MHC and TCR contribute to TCR-pMHC catch bonds and T cell responses.

(A-D) The location of residues involved in force-strengthtened H-bonds in SMD simulations (A). Representative snapshots of H-bond formations between TCRβ N30 and MHC K146 (B), between TCRβ E56 and MHC Q72 (C), and between TCRβ Q72 and MHC R79 (D) under force or no force. H-bonds are indicated as dashed blue lines. (E) The frequencies of forming H-bonds for above paired residues under force or no force. N.D., not detected. (F) Force-dependent bond lifetimes of WT or mutated 2C TCRs binding with WT or mutated R4-MHCs measured by the BFP. (G) IL-2 production from WT or mutated 2C hybridomas that were stimulated with WT or mutated R4-MHCs. Error bars in (F and G) represent SEM. See also Figure S3.
We next used the BFP to test whether these force-enhanced or force-induced H-bonds at the TCR-MHC binding interface could affect TCR-pMHC catch bonds. The aforementioned residues on R4-MHC and 2C TCR were substituted by alanine. Not surprisingly, all single mutations suppressed R4-MHC/2C TCR catch bonds (Figures S3C and S3D). We next further tested the effects of paired mutations. R4-MHC/2C TCR catch bonds were suppressed more significantly in paired mutations, as evidenced by smaller optimal forces and decreased peak bond lifetimes (Figure 3F). These data demonstrated that force-enhanced or force-induced H-bonds at the TCR-MHC binding interface contributed to TCR-pMHC catch bonds.
We next assessed the functional consequences of these paired mutants. We found that these paired mutations suppressed IL-2 release from 2C hybridomas to various extent (Figure 3G). Compared to the amount of IL-2 release activated by the wild-type (WT) R4-MHC/2C TCR interaction, the TCRβ-E56A/R4-MHC Q72A and WT 2C TCR/MHC R79A reduced IL-2 production by half, while the TCRβ-N30A/MHC-K146A completely abolished IL-2 production, suggesting that the TCRβ-N30/MHC-K146 interaction was essential for T-cell activation. Since 2C hybridomas expressing different mutated 2C TCRs at the same level (Figure S3F) have comparable abilities to secrete IL-2 (Figure S3G), we conclude that perturbing these force-enhanced or force-induced H-bonds at the TCR-pMHC binding interface impairs T-cell function by suppressing TCR-pMHC catch bonds.
Collectively, these results reveal that force allosterically reshapes the interaction network at the TCR-MHC binding interface by mechanically inducing MHC conformational changes to strengthen pre-existing H-bonds or to allow the formation of more H-bonds to elicit agonist-specific TCR-pMHC catch bonds and activate T cell functions.
Validation of MHC conformational change with in vitro single-molecule manipulation
We next used single-molecule stretching experiments to further confirm force-induced MHC conformational changes. It was firstly evident as a sudden extension increase when we stretched single TCR-pMHC complexes (Figures 4A and 4B) either in the force ascending phase (Figure 4A) or the distance-clamping phase (Figures S4A and S4B) during lifetime measurements with the BFP. The extension increase was ~16 nm (Figure 4C), similar to that previously reported on pulling single N15-TCR-pMHC complexes using an optical trap (Das et al., 2015; Das et al., 2016) and also in agreement with ~12 nm extension increase from the pMHC (Figure S2C) plus ~2 nm extension from the TCR (Figure S2D) in our SMD simulations and ~2–4 nm spatial resolution of the BFP (Chen et al., 2008). The occurring probabilities of these sudden extension increases were significantly lower for L4 and EVSV (3- and 6-fold, respectively) than those for R4 (Figure 4D). A disulfide-bond-locked MHC mutant (referred as C-C-locked MHC) that locks β2m onto the MHC α chain (Figures S4C-S4F) also significantly reduced the occurring probability of sudden extension changes (Figure 4D). Moreover, the C-C-locked R4-MHC mutant also suppressed R4-MHC/2C TCR catch bonds (Figure 4E) by reducing the optimal bond lifetime by ~2 folds, suggesting the force-induced rotation and extension of pMHCs contributed to TCR catch bonds.
Figure 4. Direct measurements of force-induced MHC conformational change.

(A-B) A sudden extension increase occurred in force ascending phase for the 2C TCR/R4-pMHC interaction measured by the BFP. (C) Histogram of sudden extension increases was fitted by Gaussian. (D) The occurring probabilities of sudden extension increases in BFP lifetime measurements of R4-, C-C locked R4-, L4- or EVSV-MHCs binding with 2C TCRs on hybridomas. (E) Force-dependent lifetimes of the 2C TCR binding with R4- and C-C locked R4- MHCs measured by the BFP. (F) Schematics of single-molecule MT experiments with WT or C-C locked MHC. (G-L) The relative bead height changes when cyclically pulling WT (G and H) or C-C locked (I) OVA-MHC in the MT. A sudden increase of the relative bead height occurred on the loading phase for WT OVA-MHC (G and H). The occurring probabilities of first three pulling cycles (J) and total relative bead height changes (K) of pulling WT or C-C locked MHC. Histogram of sudden bead height changes when pulling WT OVA-MHC was fitted by Gaussian (L). *** P <0.001, ** P <0.01. Error bars in (E, J, and K) represent SEM. See also Figure S4.
Force-induced MHC conformational changes were also directly observed with single-molecule pulling assay on magnetic tweezers (MT; Figures 4F and S4G-S4L). When cyclically stretching the complex of OVA-H-2Kb binding with an anti-OVA-H-2Kb mAb with the MT, the sudden increases of relative bead height (SIRBH, a signature for MHC conformational change) in the force loading phase were evident (Figures 4G and 4H). While pulling the C-C-locked OVA-H-2Kb (Figure 4I), the occurring probability of the SIRBH reduced significantly (Figure 4J). Moreover, the total relative bead height when pulling C-C-locked MHC till ~25 pN (~71 nm) was ~13 nm shorter than that when pulling WT MHCs (~84 nm) (Figure 4K). The SIRBH when stretching WT MHCs were normally distributed around ~13 nm (Figure 4L), in close agreement with the ~10 nm extension increase in SMD simulations (Figures S2C and S2D) considering ~2 nm spatial resolution of the MT in the vertical direction (van Loenhout et al., 2012). Collectively, our single-molecule stretching measurements confirmed our observations in SMD simulations that force indeed induced MHC conformational changes that partially and allosterically contributed to formations of TCR-pMHC catch bonds.
Force-induced HLA-A2 conformational change regulates human TCR-pHLA-A2 catch bonds
Having characterized mouse TCR-pMHC catch bonds and revealed their dynamic structural basis of the mechano-chemical coupling, we next asked whether such dynamic regulation was conserved in human TCR-pHLA-A2 interactions. We first carried out SMD simulations with the 9C-HLA-A2/1G4 TCR complex. We also noticed similar force-induced conformational changes of the pHLA-A2 to that of mouse H-2Kb, leading to formations of H-bonds at the pHLA-A2/1G4 TCR binding interface to stabilize the TCR-pMHC interaction under force (Figures 5A-5C; Movie S5; Table S1). Similar to the 2C TCR system, force also induced the large extension and rotation of pHLA-A2 but through the break-up of the β2m/α3 intramolecular association (Figures 5A, 5B). Force also enhanced pre-existing H-bond stability and induced more H-bond formations (Table S1), especially for H-bonds involving residues E19 and R75 of the HLA-A2 (Figure 5C and Table S1).
Figure 5. Force-induced MHC conformational changes and TCR-pMHC catch bonds are conserved in human TCR binding with pHLA-A2, and TCR-pMHC catch bonds are suppressed by cancer-associated somatic mutations on HLA-A2.

(A) SMD simulations of 9C-HLA-A2 in complex with 1G4 TCR showed focrce-induced MHC conformaitonal changes. (B) The time course curve of the angle between the peptide and the direction of force in SMD simulations. (C) Representative snapshots for H-bonds between TCRβ R67, T70 and MHC E19 (top), between TCRβ G50 and MHC R75 (bottom) under force and no force, zoomed in from purple and red dashed box respectively in (A). (D) Cancer-associated somatic mutations (A236T and F8V in HLA-A2) induce H-bonds or salt bridges between α and β2m subunits, zoomed in from magenta and gray dashed box respectively in (A). H-bonds are indicated as blue-dashed lines. (E-F) Force-dependent lifetimes of WT, simulation-predicted mutatnts (E19A or R75A) (E), or cancer-associated mutants (F8V or A236T) (F) of 9C-HLA-A2 binding with 1G4 TCR by the BFP. Error bars represent in (E and F) SEM. See also Figure S5.
These SMD simulation results predicted catch bonds in the 9C-HLA-A2/1G4 TCR interaction and suggested key residues contributing to catch-bond formation. To test these predictions, we measured 9C-HLA-A2/1G4 TCR bond lifetimes under force with the BFP and revealed that 1G4 TCR indeed formed catch-slip bonds with 9C-HLA-A2 with a peak bond lifetime of ~1 s at ~10 pN force (Figure 5E). The trend and optimal force were similar to those in 2C or OT-I TCR catch bonds (Liu et al., 2014) but with longer peak bond lifetimes. Preventing force-induced H-bond formations between two SMD-predicted residues with single alanine substitution (E19A or R75A) suppressed 9C-HLA-A2/1G4 TCR catch bonds, while maintaining catch-slip translational bonds (Figures 5E and S5A), suggesting that other residues at the TCR-pMHC interfaces may also contribute to catch bond formations. Moreover, we also found that the occurring probability of sudden extension increases was ~13% before the dissociation of pHLA-A2 from the 1G4 TCR under force (Figure S5D). Further analysis showed that sudden extension increases occurred in both the ascending phase (~40%) and the clamping phase (~60%) (Figure S5E), which was different from those when pulling the mouse pMHC/2C TCR complex but consistent with the results when pulling the mouse pMHC-N15-TCR with an optical trap (Das et al., 2015).
Cancer-associated somatic mutations in HLA-A2 suppress pHLA-A2/1G4 TCR catch bonds
The relationship between HLAs and cancers has been intensively investigated and cancer-associated somatic mutations in HLA-A2 have been identified. Particularly, some somatic mutations occur at the α1α2/β2m binding interface (e.g., HLA-A2 F8V) or at the α3/β2m binding interface (e.g., HLA-A2 A236T) (Figures 5A and S5B) (Shukla et al., 2015). Molecular modeling of the F8V mutation suggested that its presence may lead to two more H-bonds at the α1α2/β2m binding interface (between β2m E53 and α1 Q32, and β2m E53 and α1 R48) in addition to the β2m E53 and α1 R35 H-bond in the WT structure (Figure 5D, top panels), thus the F8V mutation may potentially restrict α1α2/β2m dissociation and HLA-A2 conformational changes. As residue A236 was located near the α3/β2m binding interface, the A236T mutation may enable a H-bond between T236 and the main chain carbonyl oxygen of R12 in β2m domain (Figure 5D, bottom panels), which may also stabilize the α/β2m association. These findings led us to hypothesize that HLA-A2 F8V or A236T might suppress 9C-HLA-A2/1G4 TCR catch bonds by preventing the conformational change of the pHLA-A2. BFP experiments with these cancer-associated somatic mutations indeed revealed significantly shortened peak bond lifetimes at ~10 pN comparing with the WT MHC, while also maintaining catch-slip translational bonds (Figures 5F and S5C), suggesting that other unknown residues at the TCR-pMHC interface may contribute to catch bond formations. Interestingly, these cancer-associated mutations (F8V and A236T) slightly reduced the ratio of extension changes on the 9C-HLA-A2/1G4 TCR complex (Figure S5D) and slightly increased the percentage of sudden extension change in the clamping phase (Figure S5E), implying that these two cancer-associated mutants of HLA-A2 restricted HLA-A2 conformational changes to some extent.
Discussion
We have used an integrated approach to demonstrate that mechanical force induced dynamic mechano-chemical coupling to sequentially change agonist pMHC conformations, which was essential for activating both mouse and human TCR-pMHC catch bonds, amplifying TCR antigen discrimination, and triggering T-cell functions. Restricting pMHC conformational changes by cancer-associated somatic mutations suppressed TCR-pMHC catch bonds, this inspired us to comprehensively re-examine cancer-associated mutations or subtle polymorphic changes of HLA class I in cancer patients for effective T cell-based immunotherapies in future (Chowell et al., 2018).
Dynamic structural model of mechano-chemical coupling for TCR-pMHC catch bonds
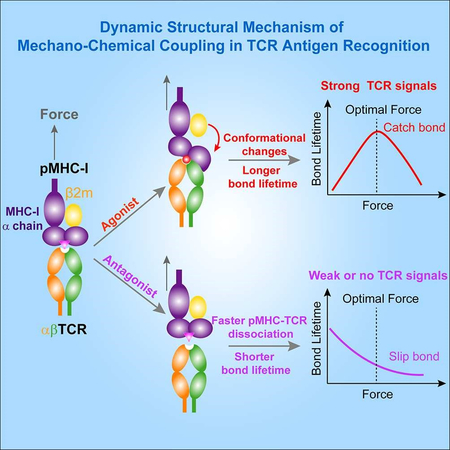
Our present findings not only extend previous findings of agonist-specific TCR-pMHC catch bonds from mouse (Das et al., 2015; Kim et al., 2009; Liu et al., 2014) to human TCRs but also allow us to propose a dynamic structural model of mechano-chemical coupling for TCR-pMHC catch bonds (Figure 6), providing an unconventional force-regulated structural basis for TCR antigen recognition. This dynamic model mainly consists of three consecutive steps of force-induced conformational changes in the pMHC (Figure 6, upper panels). First, TCR antigen recognition is initiated by CDR loops (especially CDR3 loops) of the TCR to engage the functional “hotspot” residue(s) in the agonist peptide (e.g., ArgP4 for R4-H-2Kb and MetP4/TrpP5 for 9C-HLA-A2). The strong engagement of the “hotspot” residue(s) with CDR loops jump-starts mechano-chemical coupling to dynamically regulate TCR/pMHC dissociation. Second, initially strong or mechanically strengthened peptide-TCR engagement helps force to further efficiently pull the MHC α1α2 domains toward CDR loops, inducing better complementarity via forming more interactions at the TCR-pMHC binding interface than those observed in the crystal structures. Third, such force-strengthened TCR-pMHC interactions allow forced partial separation of β2m from MHC α chain, leading to the extension and/or rotation of MHC α1α2 domain toward the TCR. This pronounced extension and/or rotation of the α1α2 domain allosterically induces more interactions at the TCR-pMHC binding interfaces. These three consecutive steps of force-induced conformational changes of the pMHC ultimately lead to agonist-specific TCR-pMHC catch bonds, conferring TCR with power to sensitively discriminate cognate antigens from the ocean of non-stimulatory antigens. Non-stimulatory or antagonist pMHC does not have these conformational changes due to initial weak engagement between the peptide and the TCR. Thereby, force accelerates the dissociation of the weak pMHC from the TCR, resulting in slip-bonds only and the inability to trigger T cells (Figure 6, lower panels).
Figure 6.

A dynamic structural model of mechano-chemical coupling for TCR catch bonds and TCR antigen recognition.
Force-induced MHC conformational changes facilitate TCR antigen recognition and initial triggering
Combining SMD simulations and single-molecule biophysical studies, we demonstrated that mechanical force indeed induced MHC conformational changes, which may be beneficial for TCR initial triggering in two aspects. First, MHC conformational changes activate TCR-pMHC catch bonds, providing an extra layer of discrimination power for TCR antigen recognition (discussed above) and possibly promoting rapid propagation of conformational changes from TCR CDR loops to CD3 tails for rapid and efficient triggering of TCR signaling. For example, strengthened TCR-pMHC binding on TCR CDR loops may allow force to transmit to and change conformations of constant regions of the αβTCR (Natarajan et al., 2017), which may further induce TCRα transmembrane domain structural movement to regulate the topological rearrangement of αβTCR/CD3 complex (Brazin et al., 2018) and alter the conformations of CD3 tails (Natarajan et al., 2016; Xu et al., 2008). These serial conformational changes may ultimately lead to successful CD3 phosphorylation by Lck. Second, such 10–20 nm extension of a TCR-pMHC complex under force reduces its size difference from CD45, thus possibly preventing CD45 exclusions from TCRs during TCR initial triggering according to the kinetic segregation model (Davis and van der Merwe, 2006). This might be beneficial for TCR initial triggering as CD45 was reported to be essential for early TCR proximal signaling by regulating Lck activation (Courtney et al., 2017; Dustin and Depoil, 2011). This hypothesis actually is supported by a recent finding (Cai et al., 2017) that no profound CD45 exclusion was observed on TCR pre-enriched microvillar tips of T cells during the very early stage of TCR searching for cognate antigens on APCs. While at the later stage of IS formation, CD45 was excluded from TCRs in the center of supramolecular activation clusters (Cai et al., 2017; Chang et al., 2016; Choudhuri et al., 2005). We believe at the later stage force on individual TCR/pMHC complex may reduce due to re-distribution of force on larger clustered TCR-pMHC complexes. This reduction of force may help β2m rapidly rebinding to MHCα chain, preventing complete loss of β2m, promoting durable peptide presentation by MHCs (Otten et al., 1992), and also restoring the shorter length of the TCR-pMHC complex to facilitate CD45 exclusions (Choudhuri et al., 2005).
Although these force-free 2D affinities of mutants also correlate well with T-cell functions, consistent with previously reported ones for OT-I and 2B4 TCRs (Huang et al., 2010; Huppa et al., 2010), we present the following reasons to argue for a more critical role of 2D effective off-rate (force-dependent koff) for TCR initial triggering. TCRs on resting T cells recently were found to highly enrich on small tips of microvilli whose diameter is ~70–100nm (Cai et al., 2017; Jung et al., 2016). These small microvillous tips and special 3D topology of microvilli may trap TCR-pMHC complexes when the T cell contacts the APC, preventing TCR-pMHC complexes to diffuse away from those tips. Given this, the TCR-pMHC rebinding rate in such confined 2D microdomains has been speculated to increase tremendously, maybe much faster than previously reported 2D or 3D association rates. The difference of 2D TCR-pMHC rebinding rates between agonist and antagonist might become similar such that 2D TCR-pMHC dissociation rate under force should become a critical parameter for triggering TCR initial signaling (Lever et al., 2014; Sibener et al., 2018). This hypothesis actually is partially supported by a computational study with a rapid rebinding model (Dushek et al., 2009), phenotypic analysis of T cell activation (Lever et al., 2014), and our finding that rapid accumulation of TCR-pMHC catch bonds triggered TCR signaling (Liu et al., 2014), but still requires more experimental evidences to confirm.
Suppressive effects of somatic mutations in HLA-A2 on TCR catch bonds and implications for T-cell based immunotherapies
Our dynamic structural model for TCR-pMHC catch bonds may be relevant in explaining why some cancer patients do not respond to T-cell based immunotherapies (e.g., anti-PD-1 immune checkpoint blockade, ICB) (Chowell et al., 2018). Is it because TCR-pMHC engagement or binding strength, the first critical signal in the anti-tumor responses of CD8+ tumor-infiltrating T cells (TILs), is impaired due to subtle polymorphic changes or somatic mutations in class I HLA genes? If this is the case, even if PD-1/PD-L1 binding is blocked by therapeutic antibodies, the weakened TCR-pMHC binding under force may fail to activate the cytotoxicity of CD8+ TILs. Importantly, previous studies have identified numerous cancer-associated somatic mutations in class I HLA genes, and loss-of-function mutations are located in all functional domains of class I HLA genes (Figure S5B) (Shukla et al., 2015). They speculated that the acquisition of these HLA mutations without abrogation of HLA expressions may provide a complementary mechanism for immune-surveillance escape. Our finding provides a piece of supporting evidence that cancer-associated somatic mutations (F8V and A236T in HLA-A2) suppressed TCR-pMHC catch bonds, potentially impairing the TCR triggering on CD8+ TILs. Moreover, evidences showed that melanoma patients with HLA-B44 supertype has significantly longer survival than patients with HLA-B62 (including HLA-B15) supertype, which suggests that HLA class I genotype influences the immune checkpoint blockade (ICB) efficacy (Chowell et al., 2018). Related to our findings, we propose that HLA polymorphisms might tightly couple with the mechanical regulation of the pMHC conformational changes and TCR-pMHC catch bonds to regulate TCR recognition of neoantigens, TCR triggering, and CD8+ TIL activation in tumor. With MD simulations, we found that HLA-B44 α1α2 domain seems to more loosely contact with β2m than HLA-B15’s due to less H-bonds formed between HLA-B44 and β2m (Figures S5F-S5I). Especially, Q32 in HLA-B15 can form H-bond with β2m D53, while L32 in HLA-B44 cannot (Figures S6G and S6H). As a result, α1α2 domain of HLA-B15 can form more H-bonds with β2m than that of HLA-B44 (Figure S6I). These analyses imply that α1α2/β2m interface of HLA-B15 may more tightly couple with β2m than HLA-B44 to resist force-induced conformational changes in α1α2 domain, which may impair TCR and neoantigen binding under force and TIL’s activation. Thus, we postulate that the 2D force-dependent dissociation kinetics of TCR-pMHC bindings might be an effective parameter to predict and assess the efficacy T cell-based immunotherapies.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Wei Chen (jackweichen@zju.edu.cn).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Cultures
2C hybridoma and 1G4 T cell were cultured at 37°C and 5% CO2 in RPMI 1640 medium supplemented with 100 U/mL streptomycin, 100 ug/mL penicillin and 5% fetal bovine serum. The cells were authenticated.
Naïve 2C T cells from 2C transgenic mice were cultured in complete RPMI media (RPMI 1640, 10% heat-inactivated FBS, 4 mM L-glutamine, 100 mg/mL gentamicin and 20 mM 2-ME) at 37°C with 5% CO2.
Mice
2C Transgenic mice and C57BL/6 mice (purchased from Jackson Laboratory) were bred, housed, and used with approval from the Institutional Animal Care and Use Committee-approved protocol of the Emory University Department of Animal Resources facility (IUCAC Number: DAR-2000870–061414).
METHOD DETAILS
Preparation of biotinylated RBCs and pMHC-coated RBCs and glass beads
Human red blood cells (RBCs) were isolated from the whole blood of healthy volunteers obtained by finger-prick according to protocols approved by the Ethical Review Board of Zhejiang University. The procedures to prepare biotinylated RBCs and pMHC-coated glass beads were as described previously (Liu et al., 2014). Briefly, fresh RBCs were first washed twice with coating buffer (0.1 M NaHCO3, 0.1 M Na2CO3, pH 8.5, ~180 mOsm) and then covalently linked to biotin-PEG3500-SGA (JenKem, China) by incubation for 30 min at room temperature (RT) at a concentration of 1.5 mg/mL. The biotinylated RBCs were then treated with 5 mg/mL nystatin (Aladdin, China) for 1 h at 4°C in N2 buffer (265.2 mM KCl, 38.8 mM NaCl, 0.94 mM KH2PO4, 4.74 mM Na2HPO4, 27 mM sucrose; pH 7.2, 588 mOsm) and stored in N2 buffer for BFP experiments.
To prepare pMHC-coated glass beads, borosilicate glass beads (2 μm in diameter, ThermoFisher) were first modified with mercapto-propyl-trimethoxysilane (Aladdin) and then coupled to streptavidin-maleimide (Sigma-Aldrich) (Chen et al., 2008). Finally, streptavin-coated beads were incubated with the desired concentrations of biotinylated pMHCs for 30 min at RT and resuspended in HBS with 0.5% BSA to achieve 10–15% adhesion frequency in BFP experiments for measuring TCR-pMHC bond lifetimes under force.
Preparation of 2C T hybridomas and 1G4 T cells
Wild-type 2C TCR α and β genes were obtained from 2C hybridomas (kindly provdied by Dr. David Kranz at UIUC, USA) using RT-PCR and cloned into the pHAGE vector with the IRES system (kindly provided by Dr. Qiming Sun, Zhejiang University). The mutant pHAGE-2C TCR constructs (E56A and N30A) were made by serial PCR and the Hieff Clone™ Plus One Step Cloning Kit (YEASEN, China). All constructs were further confirmed by sequencing (Genscript, China). To generate WT and mutated 2C TCR expressing cell lines, the constructed pHAGE-2C TCR plasmids were co-transfected with psPAX2 and pMD2.G (kindly provided by Dr. Qiming Sun, Zhejiang University) into HEK 293T cells using polyethyleneimine (PEI, Polysciences, Inc.) for the production of 2C TCR lentivirus. The virus supernatants were then harvested and used to transduce 2C TCR genes into both TCR and coreceptor CD8 and CD4 deficient 58α−β− hybridomas by the co-culture at 37°C for 72 hrs to produce 2C TCR expressing hybridomas. Finally, the 2C hybridomas were stained with anti-TCR-PE mAb (clone H57–597, BD) and then sorted by fluorescence-activated cell sorting (FACS; Aria II, BD) to obtain cell lines that stably expressed 2C TCRs. Using the same method as described above, we prepared 1G4 TCR expressing T cells with TCR deficient J76 cells.
Expression and Purification of pMHCs
cDNAs of WT and mutant H-2Kb, HLA-A*02:01 (HLA-A2) and human β2m (YouBio, China) were cloned into the expression vector pET28a by PCR and using Hieff Clone™ Plus One Step Cloning Kit (YEASEN). All constructs were confirmed by sequencing (Genscript). All WT and mutated pMHCs were expressed in BL21 (DE3) Escherichia coli cells (Vazyme, China), refolded, and purified according to published protocols (Garboczi et al., 1992). Briefly, H-2Kb, HLA-A2, and human β2m chains were each produced in inclusion bodies by BL21 (DE3) cells and dissolved in 8M urea. To produce pMHC complexes, purified H-2Kb (WT or mutated), and mouse β2m from inclusion bodies and a peptide of interest (SIYRYYGL or R4, SIYLYYGL or L4, or SIINFEKL or OVA, EQYKFYSV or dEV8, RGYVYQEL or EVSV; synthesized by ChinaPeptides, China) were refolded in a folding buffer (100 mM Tris HC1, pH 8, 400 mM L-arginine, 2 mM EDTA, 5 mM reduced glutathione, 0.5 mM oxidized glutathione, and 0.5mM phenylmethylsulfonyl fluoride (PMSF; YEASEN, China)) at 4°C for 24–36 hrs. The folded pMHCs were then concentrated and site-specifically biotinylated by homemade BirA (50 μg/mL) in PBS (pH7.4) containing 100 mM biotin, 10 mM ATP and 10 mM MgCl2 for 1 h at 30°C. The product was then purified via gel filtration (Superdex 75 column, GE, USA) on AKTA Pure (GE). Appropriate peak fractions were collected and tested for biotinylation with streptavidin using an SDS-PAGE gel shift assay. WT and mutated HLA-A2 complexed with human β2m and 9C (SLLMWITQC, synthesized by ChinaPeptides) peptide of the tumor antigen NY-ESO-1157–165 were produced in the same procedure.
Measurements of IL-2 Production
Biotinylated pMHCs at serial concentrations were incubated at 37°C for 1 h in a 96-well plate pre-coated with 50 μg/mL streptavidin. As a positive control, one well in the plate was coated with 5 μg/mL purified anti-mouse CD3ε antibody (145–2C11, Biolegend). 2C hybridomas (5×104) were added to each well containing 200 μL complete Dulbecco’s modified Eagle’s medium (DMEM) (5% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin) and incubated at 37°C with 5% CO2 for 18 h, then the supernatants were harvested and analyzed for IL-2 Cytometric Bead Array assay (CBA; BD) according to the manufacturer’s manual.
2C T cell activation
For in vitro 2C T cell proliferation, spleens from 2C TCR transgenic mice were processed into single-cell suspensions in 2 mL sterile Hanks’ balanced salt solution (Corning Mediatech, USA) according to the Institutional Animal Care and Use Committee-approved protocol from Emory University, Atlanta, GA. 2C cells were plated into 96-well flat-bottom plate at 6×105 cells/well with a series of peptide concentrations in complete RPMI medium (RPMI 1640, 4 mM L-glutamine, 100 mg/mL gentamicin from Corning, 10% heat-inactivated FBS from Life Technologies, and 20 mM 2-mercaptoethanol from Sigma-Aldrich) and incubated at 37°C with 5% CO2. After 48 h, 3H-thymidine (0.4 mCi per well) was added to the culture medium for 24 h before the cells were harvested onto a filtermat (PerkinElmer, USA) using the FilterMate 196 harvester (Packard, USA). Incorporation of 3H-thymidine was analyzed with the 1450 Microbeta TriLux microplate liquid scintillation counter (PerkinElmer).
For IFN-γ secretion, splenocytes from 2C TCR-transgenic mice were processed into a single cell suspension as noted above. 2C splenocytes were plated at 5×106 cells/mL in complete RPMI media and stimulated with 10 nM SIYRYYGL peptide for 5 days at 37°C in 5% CO2. Stimulated 2C cells were purified using a Ficoll gradient (Mediatech) and 1.5×105 live 2C cells were added to a single well in a flat-bottomed 96 well plate along with 6×105 of irradiated splenocytes from C57BL/6 mice in complete RPMI media. To these wells the respective 2C peptide dilutions (100 μM-0.001 nM) were added and incubated for 18 h at 37°C in 5% CO2. Plates were spun down and supernatant was collected, frozen, and thawed. Supernatant IFN-γ concentration was measured with the IFN gamma ELISA Ready-SET Go! (eBioscience) kit as per manufacturer protocol. Plates were read on a Microplate Autoreader (Biotek Synergy HT) at 450 nm wavelength. Samples were normalized to the maximum absorbance value in the dilution, fit to a log (peptide dilution) vs normalized response curve (Prism Graph Pad) and EC50 values were calculated.
Biomembrane Force Probe (BFP)
The BFP setup has been described in detail (Chen et al., 2008; Liu et al., 2014). Briefly, a biotinylated RBC was aspirated using a probe micropipette (Figure 1I), and a glass bead coated with pMHC and streptavidin was attached to the apex of the RBC to form a highly-sensitive force probe (Chen et al., 2008). On the opposite side, a T cell was aspirated using a target micropipette and its movement was driven by a linear piezoelectric actuator (P-753 LISA, Physic Instrument, Germany) with sub-nanometer precision. The bead and the T cell were aligned in a chamber filled with DMEM with 0.5% BSA and observed under an inverted microscope (Nikon TiE) through two video cameras. One camera (GC1290, Allied Vision, Canada) captured real-time images at 30 frames per second (fps), while the other (GE680, Allied Vision, Canada) recorded at a higher speed of 1,600 fps, focusing on the RBC-bead contact interface as the region of interest. Bead position was tracked in real-time with 3-nm displacement resolution using a customized image analysis program (Chen et al., 2008) implemented in LabView (National Instruments, TX). The BFP spring constant was set to ~0.3 pN/nm, which was determined by the radius of and the suction pressure inside the probe pipette that held the RBC, the radius of the spherical portion of the RBC outside of the probe pipette, and the contact area between the bead and the RBC (Chen et al., 2008).
Lifetime measurement by the Bond Lifetime Assay
We used the bond lifetime assay (Liu et al., 2014) on the BFP to measure TCR-pMHC bond lifetimes under force at the single-molecule level. Briefly, a 2C or 1G4 T cell captured by the target pipette was driven by our customized LabVIEW program to approach and contact the pMHC-coated glass bead with a 20-pN impingement force for 0.1 s to allow TCR and pMHC bond formation. The target pipette was then retracted at 1,000 pN/s to a desired force, held until bond dissociation, and returned to the original position to start the next cycle. A bond lifetime was measured from the moment the force loaded to the desired level to the moment of bond dissociation. To ensure that most adhesion events were mediated by a single bond, adhesion frequency (number of adhesion events divided by total number of contacts) was controlled to be ≤15% by adjusting the density of pMHC coating on the beads (Liu et al., 2014). All experiments were performed in DMEM containing 0.5% BSA at RT.
Thermal Fluctuation Assay
A 2C or 1G4 T cell on a target pipette was brought into contact with a bead coated with pMHCs for 0.1 s, retracted, and held at zero force for 15 s. During the holding period, bond association and dissociation events were identified from reduction and resumption, respectively, of bead fluctuations. Bond lifetime was measured as the time from fluctuation reduction to resumption (Chen et al., 2008).
Magnetic Tweezers (MT) Setup and Force Calibration
Our MT was built in-house on an inverted microscope (Ti-S, Nikon) (Figure S4G). The experimental chamber was positioned on a horizontal moving stage (Ti-SSR, Nikon) mounted onto the microscope. The vertical position (height, H) of a pair of permanent magnets (N38, Neodymium iron boron) above the chamber was controlled by a moving electrical stage (M404.8PD, Physic Instrument) to vary the magnetic force. The magnetic forces were routinely calibrated by Brownian fluctuations. Briefly, a magnetic bead (M270, 2.8 μm in diameter, ThermoFisher, Cat#65801D) was linked to a λ-DNA tether (N3011S, 48,502 bp, NEB) via biotin-streptavidin chemistry which in turn covalently anchored it to the bottom coverslip. Thermal fluctuations of the bead in the direction along the magnetic field were recorded for a series of magnet heights (H) corresponding to a series of DNA extensions (l). For each and (l) pair, force (F) was calculated according to the equipartition theorem (Chen et al., 2011):
| (1) |
where kB is the Boltzmann constant, T is the absolute temperature, r is the radius of the magnetic bead (1.4 μm), and is the Brownian motion variance. A single exponential equation was fitted to the resulting data set of (H, F) (Figure S4H). The fitted empirical relationship was used for force calculation in MT experiments by recording the magnet heights (Chen et al., 2011).
Single-molecule experiments with magnetic tweezers
MT experiments were performed in a flow chamber constructed with two cover-slips (ThermoFisher) in a sandwich configuration (Figure S4I). First, cover-slips were cleaned with 10% Decon90 (Decon Laboratories Ltd, UK) in deionized distilled water (ddH2O) with two rounds of 30-min ultrasonication (100 Hz) and then followed by 5-min incubation in 75% alcohol and rinsing twice with ddH2O. After that, they were boiled in 1% H2O2 and 3% NH4OH (Sinopharm Group, Co. Ltd, China) for 10 min, and then washed twice with ddH2O before incubation with 2% aminopropyltriethoxysilane (APTES, Macklin, China, Cat#C10068618) in methanol on a shaking table for 60 min. After cleaning twice with methanol and ddH2O, the cover-slips were dried in a 65°C air-drying oven for 30 min and then stored in a vacuum box until use. To make a flow chamber, a APTES-coated cover-slip and another nonfunctional coverslip were separated by two narrow strips of Parafilm on both sides and then placed on an 85°C plate with slight pressure for 10 s for better sealing, and then silica gel was injected to make room for solution exchange (Figure S4I). The prepared chamber was washed twice with 200 μL PBS (Sangon Biotech), followed by incubation with 100 μL 2% glutaraldehyde in PBS for 30 min. After washing, 100 μL 2.5 μg/mL anti-OVA H-2Kb mAb (clone 25-D1.16, ThermoFisher, Cat#14–5743-81) in PBS was captured on the glass surface by glutaraldehyde cross-linking, and after 5 min, 50 μL amino-coated polystyrene beads (2×106/mL) (3.0 μm in diameter, Big Goose (Tianjin) Technology Co. Ltd, Cat#PSN-03000) in PBS was quickly added to the chamber. After 2-h incubation at RT, the chamber was washed twice with 200 μL PBS and then blocked with 100 μL 1% BSA (Sigma) for 2 h at RT. Meanwhile, 100 μL BSA-blocked M270 magnetic beads (5×106/mL, ThermoFisher) was incubated with 0.5 ng/mL pMHC for 30 min. Finally, these magnetic beads were added to the chamber to form tethers for 10 min before the MT experiments (Yao et al., 2016). The prepared chamber was observed under a 100× oil objective (NA1.25, Nikon) and LED light (MCWHL5, Thorlabs). Once a fixed polystyrene bead (white, for drifting elimination) and a fluctuant M270 magnetic bead (yellow, for applying force to pMHC) were found near each other (Chen et al., 2011), a customized program based on the QI algorithm (van Loenhout et al., 2012) was used to test whether a pMHC molecule was successfully linked to the magnetic bead (Chen et al., 2011). A single pMHC molecule was stretched/relaxed by cycles of force loading/releasing phases at a constant force loading rate of 1 pN/s. A waiting time (30 s) between every 2 force cycles was set to allow the stretched pMHC to fully refold (Figures S4J and S4K). To record the details of MHC conformational changes, a high-speed CCD camera (GE680, Allied Vision, Canada) was used to track the three-dimensional positions of the polystyrene and magnetic beads. The Z (vertical) position of the magnetic bead subtracted from that of the polystyrene bead was treated as the relative bead height change (Chen et al., 2011). A sudden extension increase without significant force change during the force loading phase was recorded (Figure S4J), signifying as a MHC conformational change (Chen et al., 2011; Yao et al., 2016).
Molecular Dynamics (MD) and Steered Molecular Dynamics (SMD) Simulation System setup
The structures of mouse 2C TCR complexed with superagonist R4-H-2Kb (PDB code: 1G6R) and dEV8-H-2Kb (PDB code: 2CKB), and that of human 1G4 TCR complexed with agonist 9C-HLA-A2 (PDB code: 2BNR) were used as the starting point in our simulations. 2C-R4-H-2Kb structure contains TCRα residues Gln1 to Cys213, TCRβ residues Glu1 to Cys247, MHCα residues Gly1 to Trp274, β2m residues Ile1 to Met99 and SIYR peptide: SIYRYYGL. 2C-dEV8-H-2Kb structure contains TCRα residues Gln1 to Cys213, TCRβ residues Glu1 to CyS247, MHCα residues Gly1 to Trp274, β2m residues Ile1 to Met99, and dEV8 peptide: EQYKFYSV. 9C-HLA-A2/1G4 TCR structure contains TCRα residues Gln2 to Ser204, TCRβ residues Gly2 to Asp242, MHCα residues Gly1 to Pro276, β2m residues Met0 to Met99, and 9C peptide: SLLMWITQC. High resolution structures of peptide loaded HLA-B44 (PDB code 3KPM) and HLA-B15 (PDB code 1XR9) were used for simulations comparing HLA-B15 and HLA-B44 conformation dynamics.
2C-R4-H-2Kb and 2C-dEV8-H-2Kb structures include the Cys residues (TCRα residue Cys213 and TCRβ residue Cys247) on the C-termini of the TCRα and β subunits which formed interchain disulfide bond to stablize the structure and provide anchor to the connecting peptide and the transmembrane regions of the two subunits. But for 9C-HLA-A2/1G4 TCR structure, the required Cys residues on both subunits and several residues on N-terminal to the Cys on α subunit are not solved. Because this disulfide bond may affect the force distribution on the two subunits, it should be included in simulations with force. To enable this disulfide bond in 9C-HLA-A2/1G4 TCR structure, one residue Cys243 on TCRβ subunit, and 5 residues ( Pro205, Glu206, Ser207, Ser208, Cys209) on TCRα subunit should be added. We use ModLoop (Fiser and Sali, 2003) with manual adjustments to add these loop residues. ModLoop added loops between two ends with solved/known coordinates, thus to use ModLoop, we first generated a disufide bond model (two cysteine residues connected with disulfide bond) and manually attached one Cys residue to Asp242 residue of TCRβ subunit and treated it as Cys243, another Cys residue was treated as residue Cys209 of TCRα subunit, and then the positions of four other residues (Pro205, Glu206, Ser207, Ser208) in TCRα subunit were modeled with ModLooop linking residues Ser204 and Cys209. We manually placed five different Cys209/Cys243 configurations and three loop models were generated for each configuration. Finally, all 15 models were compared and the structure contains two H-bonds between the added loop and the resting part of TCR (residues Glu206 and Ser207 bond with residue Gln125 of TCRα) was selected. We also removed residue Met0 from β2m and manually added the position of Cα atoms for Lys1 residue at the N-terminal of TCRα by considering the position of this residue in another WT 9C-HLA-A2/1G4 TCR complex structure (PDB code: 2F54) (Dunn et al., 2006). Structure 2BNR instead of 2F54 was used as starting structure for our 1G4 simulations because 2BNR has higher resolution.
Models for EVSV and L4 were generated based on the R4 complex structure with the MUTATE plugin in VMD (visual molecular dynamics) (Humphrey et al., 1996). For L4 (SIYLYYGL), only one residue Arg4 in SIYR peptide was mutated to a Leu. For EVSV (RGYVYQEL), MUTATE was applied to five residues (S1R, I2G, R4V, Y6Q, and G7E) on R4 structure.
The mouse R4, EVSV, dEV8, and L4 structures, 9C-HLA-A2/1G4 TCR struture, were rotated to make its long axis (the line linking C-terminal of MHC and C-terminal of TCR subunits) parallel to x axis and the direction of the peptide parallel to y axis. These structrues, and structures for HLA-B15 and HLA-B44 were further processed with VMD PSFGEN plugen to add hydrogen atoms and other missing atoms. The resulted systems were solvated,in 256×96×96 Å3 (for TCR/pMHC complex systems, includes extra water in x direction to enable extension in forced SMD simulaitons) and 108×96×96 Å3 (for MHC only systems) rectagular water boxes with TIP3P water model. Na+ and Cl− ions were than added to these solvated systems to neutralize the systems and maintain salt concentration at ~150 mM. The final systems contained ~0.24 million atoms in total for TCR/pMHC systems and ~0.1 million atoms in total for MHC only systems.
Energy minimization, equilibration and molecular dynamics simulations
Energy minimizations and molecular dynamics simulations were performed with NAMD (Phillips et al., 2005) using the CHARM22 force field for proteins (MacKerell et al., 1998) under periodic boundary conditions. For 1G4 system, an energy minization step where only the added loop can move are performed first. Then all systems were refined with energy minimization in five steps: (1) fixed all heavy atoms; (2) fixed protein heavy atoms (except for those from mutated/modeled residues and/or with guessed coordinates); (3) fixed protein backbone atoms; (4) fixed protein Cα atoms; (5) freed all atoms. The energy-minimized systems were heated to 310°K and equilibrated for 10 ns at 1-fs time steps. Then 100 ns production simulations were carried out with 2-fs time steps under rigid bond algorithms. In these processes, the temperature of the systems was maintained at 310°K with Langevin dynamics and the pressure was controlled at 1 atm with the Nosé-Hoover Langevin piston method. Particle Ewald Mesh summation was used for electrostatic calculation and a 12 Å cutoff was used for short-range non-bounded interactions.
Steered molecular dynamics simulations
Representative snapshots of the production runs for each TCR/pMHC system were used as initial conformation for steered molecular dynamics (SMD) simulations. Before the forces were applied, these snapshots were first simulated under temperature and pressure control with 1fs time step for 5 ns and free dynamics simulations for another 5 ns for relaxation. The final confirguations were used for the following SMD simulaitons. The SMD simulations were carried out with the SMD module in NAMD package. During SMD simulation, the C-terminal Cα atom of MHC (Cα atom of residue Trp274 for mouse H-2Kb and residue Pro276 for HLA-A2) was constrained at its initial position with a spring with spring constant ~ 1400pN/nm, and the C-terminal Cα atoms of TCRα and β subunits (i.e., Cα atoms of the TCRα residue Cys213 and TCRβ residue Cys247 for mouse 2C TCR, and TCRα residue Cys209 and TCRβ residue Cys243 for human 1G4 TCR) were pulled with a dummy spring with spring constant ~70 pN/nm which moves at ~0.1 nm/ns velocity. The SMD simulations continued till the pMHC and TCR molecules were completely separated.
All the simulation trajectories were analyzed with VMD (Humphrey et al., 1996). Snapshots of selected critical frames were photographed in Chimera (UCSF) (Pettersen et al., 2004).
QUANTIFICATION AND STATISTICAL ANALYSIS
All significance values were obtained from unpaired two-sample Student’s t-tests in Prism 6 (GraphPad Software, Inc.).
Supplementary Material
Movie S1: Related to Figure 2. SMD simulation trajectories for pulling the R4-H-2Kb/2C TCR complex. The C-terminal residue of H-2Kb was harmonically constrained and the C-terminus of TCR was pulled by a dummy spring moving at ~0.1 nm/ns with a spring constant of ~70 pN/nm. The color codes were the same as that in Figure 2A, 2C TCRα (green), 2C TCRβ (orange), MHC H-2Kb α (purple), MHC H-2Kb β2m (yellow), and R4 peptide (cyan). Three different pathways of conformational changes occurred in the pMHC.
Movie S2: Related to Figure 2. SMD simulation trajectory for pulling the dEV8-H-2Kb/2C TCR complex under the same conditions as Movie S1. The color codes are the same as in Movie S1.
Movie S3: Related to Figure 2. SMD simulation trajectory for pulling the EVSV-H-2Kb/2C TCR complex under the same conditions as Movie S1. The color codes are the same as in Movie S1.
Movie S4: Related to Figure 2. SMD simulation trajectory for pulling the L4-H-2Kb/2C TCR complex under the same conditions as Movie S1. The color codes are the same as in Movie S1.
Movie S5: Related to Figure 5. SMD simulation trajectory for pulling the 9C-HLA-A2/1G4 TCR complex under the same conditions as Movie S1. The color codes are the same as in Figure 5A: 1G4 TCRα (light blue), 2C TCRβ (purple), MHC HLA-A2 α (green), MHC HLA-A2 β2m (yellow), 9C peptide (red).
Table S1: Interaction network analysis for the simulations of mouse 2C TCR systems and human 1G4 TCR system under force or no force. Related to Figures 1, 2, 3 and 5. The frequencies of H-bonds and hydrophobic contacts between TCR residues and the peptides (or MHCα, or HLA-A2) were listed. An H-bond is assumed if the distance for at least one pair of acceptor and donor atoms between the two residues is closer than 3.5Å. A hydrophobic contact is assumed if the distance for at least one pair of carbon atoms between the two residues is closer than 4Å. Results for the conditions under force or no force are provided and compared. Interactions with increased formation frequencies under force condition were colored red. The tables are provided as an excel file, and only these interaction pairs with frequency greater than 1% in at least one condition (with or without force) were included.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-TCR β Chain (Clone H57–597) | BD | Cat#553172 |
| Mouse monoclonal anti-CD3ε mAb (clone 145–2C11) | Biolegend | Cat#100302l |
| Human monoclonal anti-β2m (clone TU99) | BD | Cat#551337 |
| Mouse monoclonal anti-OVA H-2Kb (clone 25-D1.16) | Thermo Fisher Scientific | Cat#14–5743–81 |
| Bacterial and Virus Strains | ||
| BL21 (DE3) Escherichia coli cells | Vazyme | Cat#C504–03 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Peptide: SIINFEKL (OVA) | ChinaPeptides | N/A |
| Peptide: SIYRYYGL (SIYR, R4) | ChinaPeptides | N/A |
| Peptide: SIYLYYGL (SIYL, L4) | ChinaPeptides | N/A |
| Peptide: RGYVYQEL (EVSV) | ChinaPeptides | N/A |
| Peptide: EQYKFYSV (dEV8) | ChinaPeptides | N/A |
| Peptide: SLLMWITQC (NY-ESO-1,9C) | ChinaPeptides | N/A |
| Protein: OVA-MHC | This paper | N/A |
| Protein: R4-MHC | This paper | N/A |
| Protein: R4-MHC (Q72A) | This paper | N/A |
| Protein: R4-MHC (K146A) | This paper | N/A |
| Protein: L4-MHC | This paper | N/A |
| Protein: EVSV-MHC | This paper | N/A |
| Protein: dEV8-MHC | This paper | N/A |
| Protein: R4-MHC C-C Lock | This paper | N/A |
| Protein: OVA-MHC with double biotin | This paper | N/A |
| Protein: OVA-MHC C-C Lock with double biotin | This paper | N/A |
| Protein: 9C-HLA-A2 | This paper | N/A |
| Protein: 9C-HLA-A2 (F8V) | This paper | N/A |
| Protein: 9C-HLA-A2 (A236T) | This paper | N/A |
| Protein: 9C-HLA-A2 (E19A) | This paper | N/A |
| Protein: 9C-HLA-A2 (R75A) | This paper | N/A |
| Protein: R4-MHC (H-2Kb D227K) | the NIH Tetramer Core Facility at Emory University, USA | N/A |
| Protein: L4-MHC (H-2Kb D227K) | the NIH Tetramer Core Facility at Emory University, USA | N/A |
| Protein: dEV8-MHC (H-2Kb D227K) | the NIH Tetramer Core Facility at Emory University, USA | N/A |
| Protein: EVSV-MHC (H-2Kb D227K) | the NIH Tetramer Core Facility at Emory University, USA | N/A |
| Critical Commercial Assays | ||
| Cytometric Bead Array (CBA) assay | BD | Cat#558297 |
| 2 × Phanta Max Master Mix | Vazyme | Cat#P515–03 |
| Hieff Clone™ Plus One Step Cloning Kit | YEASEN | Cat#10911ES62 |
| The QuantiBRITE PE beads (PE Fluorescence Quantitation Kit) | BD | Cat#340495 |
| Deposited Data | ||
| 2C TCR/R4-H-2Kb | PDB | 1G6R |
| 2C TCR/dEV8-H-2Kb | PDB | 2CKB |
| 9C-HLA-A2/1G4 TCR | PDB | 2BNR |
| Experimental Models: Cell Lines | ||
| HEK293T | Qiming Sun | N/A |
| 58α−β− hybridoma | Xun Zeng | N/A |
| 2C hybridoma | This paper | N/A |
| 2C N30A hybridoma | This paper | N/A |
| 2C E56A hybridoma | This paper | N/A |
| J76 T cell | Jie Sun | N/A |
| 1G4 T cell | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | JAX: 000664 |
| Mouse: 2C | Brian Evavold at Emory University, USA | N/A |
| Oligonucleotides | ||
| 2C TCR cDNA | David Kranz lab at UIUC, USA | N/A |
| HLA-A2 cDNA | Xun Zeng | N/A |
| H2K1 cDNA | YouBio | Cat#G132036 |
| Human β2m cDNA | YouBio | Cat#G115212 |
| SpyCather cDNA | Yi Cao at Nanjing University, China | N/A |
| Recombinant DNA | ||
| Plasmid: pET28-H2K1-AVI | This paper | N/A |
| Plasmid: pET28-H2K1 (Q72A)-AVI | This paper | N/A |
| Plasmid: pET28-H2K1 (K146A)-AVI | This paper | N/A |
| Plasmid: pET28-H2K1 (G120C/C121S)-AVI | This paper | N/A |
| Plasmid: pET28-hβ2m | This paper | N/A |
| Plasmid: pET28-hpβm-(GGGGS)3-AVI | This paper | N/A |
| Plasmid: pET28-hβ2m (I1 C)-AVI | This paper | N/A |
| Plasmid: pET28-hβ2m (I1C)-(GGGGS)3-AVI | This paper | N/A |
| Plasmid: pHAGE-(2C) TCRα-IRES-(2C) TCRβ | This paper | N/A |
| Plasmid: pHAGE-(2C) TCRα-IRES-(2C) TCRβ-N30A | This paper | N/A |
| Plasmid: pHAGE-(2C) TCRα-IRES-(2C) TCRβ-E56A | This paper | N/A |
| Plasmid: pET28-HLA-A2-AVI | This paper | N/A |
| Plasmid: pET28-HLA-A2 (F8V)-AVI | This paper | N/A |
| Plasmid: pET28-HLA-A2 (A236T)-AVI | This paper | N/A |
| Plasmid: pET28-HLA-A2 (E19A)-AVI | This paper | N/A |
| Plasmid: pET28-HLA-A2 (R75A)-AVI | This paper | N/A |
| Plasmid: pET28–6×His-SpyCather | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad | https://www.graphpad.com/ |
| FlowJo | FLOWJO | https://www.flowjo.com/ |
| Chimera | UCSF CHIMERA | https://www.cgl.ucsf.edu/chimera/ |
| Labview | National Instruments | http://www.ni.com |
| QI algorithm | Marijn T. J. van Loenhout et al., 2012 | |
| VMD (Visual Molecular Dynamics) | University of Illinois at Urbana-Champaign | http://www.ks.uiuc.edu/Research/vmd/ |
| NAMD | University of Illinois at Urbana-Champaign | http://www.ks.uiuc.edu/Research/namd/ |
| CHARMM Force Field | Alex Mackerell lab at School of Pharmacy, University of Maryland | http://mackerell.umaryland.edu/charmm_ff.shtml |
| ModLoop (Modeling of Loops in Protein Structures) | Andrej Sali’s Lab at University of California San Fransisco | https://modbase.compbio.ucsf.edu/modloop/ |
| Other | ||
| Streptavidin coated magnetic bead | Thermo Fisher Scientific | Cat#65801D |
| Amino coated polystyrene bead | Big goose (Tianjin) technology co. LTD | Cat#PSN-03000 |
| Glass bead | Thermo Fisher Scientific | Cat#9002 |
Highlights.
Force-enhanced peptide-TCR H-bonds determine TCR-pMHC-I catch bonds.
Force-strengthened TCR-MHC binding interface contributes to TCR-pMHC-I catch bonds.
Force-induced MHC-I rotation allosterically enhance TCR-pMHC-I catch bonds.
Tumor associated somatic mutations in HLA-A2 impair TCR-pHLA-A2 catch bonds.
ACKNOWLEDGEMENTS
We thank Drs. D. Kranz, Y. Cao, and Q. Sun for kindly providing us reagents, Drs. H. Chen, S. Le and J. Yan for kind supports on MT, Drs. J. Wan, W.L. Liu, H. Hu, D. Neculai, H.P. Wang, M. Krummel and I. Bruce for insightful comments and suggestions, Drs. N. Aswin and M. Krogsgaard for helping with reagents in the early stage of the project, core facilities in Zhejiang University School of Medicine for technical supports, especially X. Song for FACS supports, and the NIH Tetramer Core Facility at Emory University for providing pMHC monomers. This work was supported by grants from the National Basic Research Program of China (2015CB910800 to W.C. and 2014CB910202 to J.L.), the National Science Foundation of China (31470900 and 31522021 to W.C.; 11672317 and 31222022 to J.L.), the Fundamental Research Funds for the Central Universities (2015XZZX004-32 to W.C.), the National Institutes of Health of America (U01CA214354 and R01AI124680 to C. Zhu. and R01AI096879 to B.E.). The computational resources were provided by the National Supercomputing Center Tianjin Center and HPC-Service Station at the Center for Biological Imaging of the Institute of Biophysics.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bashour KT, Gondarenko A, Chen H, Shen K, Liu X, Huse M, Hone JC, and Kam LC (2014). CD28 and CD3 have complementary roles in T-cell traction forces. Proc. Natl. Acad. Sci. U S A 111, 2241–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu R, Whitlock BM, Husson J, Le Floc’h A, Jin W, Oyler-Yaniv A, Dotiwala F, Giannone G, Hivroz C, Biais N, et al. (2016). Cytotoxic T Cells Use Mechanical Force to Potentiate Target Cell Killing. Cell, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brameshuber M, Kellner F, Rossboth BK, Ta H, Alge K, Sevcsik E, Gohring J, Axmann M, Baumgart F, Gascoigne NRJ, et al. (2018). Monomeric TCRs drive T cell antigen recognition. Nat. Immunol 19, 487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazin KN, Mallis RJ, Boeszoermenyi A, Feng Y, Yoshizawa A, Reche PA, Kaur P, Bi K, Hussey RE, Duke-Cohan JS, et al. (2018). The T Cell Antigen Receptor alpha Transmembrane Domain Coordinates Triggering through Regulation of Bilayer Immersion and CD3 Subunit Associations. Immunity 49, 829–841 e826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai E, Marchuk K, Beemiller P, Beppler C, Rubashkin MG, Weaver VM, Gerard A, Liu TL, Chen BC, Betzig E, et al. (2017). Visualizing dynamic microvillar search and stabilization during ligand detection by T cells. Science 356, eaal3118–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang VT, Fernandes RA, Ganzinger KA, Lee SF, Siebold C, McColl J, Jonsson P, Palayret M, Harlos K, Coles CH, et al. (2016). Initiation of T cell signaling by CD45 segregation at ‘close contacts’. Nat. Immunol 17, 574–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Fu H, Zhu X, Cong P, Nakamura F, and Yan J (2011). Improved high-force magnetic tweezers for stretching and refolding of proteins and short DNA. Biophys. J 100, 517–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Zarnitsyna VI, Sarangapani KK, Huang J, and Zhu C (2008). Measuring Receptor-Ligand Binding Kinetics on Cell Surfaces: From Adhesion Frequency to Thermal Fluctuation Methods. Cell Mol. Bioeng 1, 276–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhuri K, Wiseman D, Brown MH, Gould K, and van der Merwe PA (2005). T-cell receptor triggering is critically dependent on the dimensions of its peptide-MHC ligand. Nature 436, 578–582. [DOI] [PubMed] [Google Scholar]
- Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, Kuo F, Kendall SM, Requena D, Riaz N, et al. (2018). Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 359, 582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney AH, Amacher JF, Kadlecek TA, Mollenauer MN, Au-Yeung BB, Kuriyan J, and Weiss A (2017). A Phosphosite within the SH2 Domain of Lck Regulates Its Activation by CD45. Mol. Cell. 67, 498–511 e496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das DK, Feng Y, Mallis RJ, Li X, Keskin DB, Hussey RE, Brady SK, Wang JH, Wagner G, Reinherz EL, et al. (2015). Force-dependent transition in the T-cell receptor beta-subunit allosterically regulates peptide discrimination and pMHC bond lifetime. Proc. Natl. Acad. Sci. U S A 112, 1517–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das DK, Mallis RJ, Duke-Cohan JS, Hussey RE, Tetteh PW, Hilton M, Wagner G, Lang MJ, and Reinherz EL (2016). Pre-T Cell Receptors (Pre-TCRs) Leverage Vbeta Complementarity Determining Regions (CDRs) and Hydrophobic Patch in Mechanosensing Thymic Self-ligands. J. Biol. Chem 291, 25292–25305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degano M, Garcia KC, Apostolopoulos V, Rudolph MG, Teyton L, and Wilson IA (2000). A functional hot spot for antigen recognition in a superagonist TCR/MHC complex. Immunity 12, 251–261. [DOI] [PubMed] [Google Scholar]
- Dunn SM, Rizkallah PJ, Baston E, Mahon T, Cameron B, Moysey R, Gao F, Sami M, Boulter J, Li Y, et al. (2006). Directed evolution of human T cell receptor CDR2 residues by phage display dramatically enhances affinity for cognate peptide-MHC without increasing apparent cross-reactivity. Protein Sci. 15, 710–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dushek O, Das R, and Coombs D (2009). A role for rebinding in rapid and reliable T cell responses to antigen. PLoS. Comput. Biol 5, e1000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin ML, and Depoil D (2011). New insights into the T cell synapse from single molecule techniques. Nat. Rev. Immunol 11, 672–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiser A, and Sali A (2003). ModLoop: automated modeling of loops in protein structures. Bioinformatics 19, 2500–2501. [DOI] [PubMed] [Google Scholar]
- Garboczi DN, Ghosh P, Utz U, Fan QR, Biddison WE, and Wiley DC (1996). Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature 384, 134–141. [DOI] [PubMed] [Google Scholar]
- Garboczi DN, Hung DT, and Wiley DC (1992). HLA-A2-peptide complexes: refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc. Natl. Acad. Sci. U S A 89, 3429–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, and Dustin ML (1999). The immunological synapse: a molecular machine controlling T cell activation. Science 285, 221–227. [DOI] [PubMed] [Google Scholar]
- Hong J, Ge C, Jothikumar P, Yuan Z, Liu B, Bai K, Li K, Rittase W, Shinzawa M, Zhang Y, et al. (2018). A TCR mechanotransduction signaling loop induces negative selection in the thymus. Nat. Immunol 19, 1379–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J, Persaud SP, Horvath S, Allen PM, Evavold BD, and Zhu C (2015). Force-Regulated In Situ TCR-Peptide-Bound MHC Class II Kinetics Determine Functions of CD4+ T Cells. J. Immunol 195, 3557–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu KH, and Butte MJ (2016). T cell activation requires force generation. J. Cell Biol 213, 535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Brameshuber M, Zeng X, Xie J, Li QJ, Chien YH, Valitutti S, and Davis MM (2013). A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4(+) T cells. Immunity 39, 846–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Zarnitsyna VI, Liu B, Edwards LJ, Jiang N, Evavold BD, and Zhu C (2010). The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature 464, 932–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K (1996). VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–38. [DOI] [PubMed] [Google Scholar]
- Huppa JB, Axmann M, Mortelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, Klein LO, Schutz GJ, and Davis MM (2010). TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature 463, 963–U143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse M (2017). Mechanical forces in the immune system. Nat. Rev. Immunol 17, 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Y, Riven I, Feigelson SW, Kartvelishvily E, Tohya K, Miyasaka M, Alon R, and Haran G (2016). Three-dimensional localization of T-cell receptors in relation to microvilli using a combination of superresolution microscopies. Proc. Natl. Acad. Sci. U S A 113, E5916–E5924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ST, Takeuchi K, Sun ZY, Touma M, Castro CE, Fahmy A, Lang MJ, Wagner G, and Reinherz EL (2009). The alphabeta T cell receptor is an anisotropic mechanosensor. J. Biol. Chem 284, 31028–31037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever M, Maini PK, van der Merwe PA, and Dushek O (2014). Phenotypic models of T cell activation. Nat. Rev. Immunol 14, 619–629. [DOI] [PubMed] [Google Scholar]
- Liu B, Chen W, Evavold BD, and Zhu C (2014). Accumulation of dynamic catch bonds between TCR and agonist peptide-MHC triggers T cell signaling. Cell 157, 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Blanchfield L, Ma VP, Andargachew R, Galior K, Liu Z, Evavold B, and Salaita K (2016). DNA-based nanoparticle tension sensors reveal that T-cell receptors transmit defined pN forces to their antigens for enhanced fidelity. Proc. Natl. Acad. Sci. U S A 113, 5610–5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Sharp KA, Janmey PA, and Finkel TH (2008). Surface-anchored monomeric agonist pMHCs alone trigger TCR with high sensitivity. PLoS. Biol 6, e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. (1998). All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Mallis RJ, Bai K, Arthanari H, Hussey RE, Handley M, Li Z, Chingozha L, Duke-Cohan JS, Lu H, Wang JH, et al. (2015). Pre-TCR ligand binding impacts thymocyte development before alphabetaTCR expression. Proc. Natl. Acad. Sci. U S A 112, 8373–8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan A, Nadarajah V, Felsovalyi K, Wang W, Jeyachandran VR, Wasson RA, Cardozo T, Bracken C, and Krogsgaard M (2016). Structural Model of the Extracellular Assembly of the TCR-CD3 Complex. Cell Rep. 14, 2833–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan K, McShan AC, Jiang J, Kumirov VK, Wang R, Zhao H, Schuck P, Tilahun ME, Boyd LF, Ying J, et al. (2017). An allosteric site in the T-cell receptor Cbeta domain plays a critical signalling role. Nat. Commun 8, 15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor RS, Hao X, Shen K, Bashour K, Akimova T, Hancock WW, Kam LC, and Milone MC (2012). Substrate rigidity regulates human T cell activation and proliferation. J. Immunol 189, 1330–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otten GR, Bikoff E, Ribaudo RK, Kozlowski S, Margulies DH, and Germain RN (1992). Peptide and beta 2-microglobulin regulation of cell surface MHC class I conformation and expression. J. Immunol. 148, 3723–3732. [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, and Schulten K (2005). Scalable molecular dynamics with NAMD. J. Comput. Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossboth B, Arnold AM, Ta H, Platzer R, Kellner F, Huppa JB, Brameshuber M, Baumgart F, and Schutz GJ (2018). TCRs are randomly distributed on the plasma membrane of resting antigen-experienced T cells. Nat. Immunol 19, 821–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph MG, Stanfield RL, and Wilson IA (2006). How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 24, 419–466. [DOI] [PubMed] [Google Scholar]
- Saitakis M, Dogniaux S, Goudot C, Bufi N, Asnacios S, Maurin M, Randriamampita C, Asnacios A, and Hivroz C (2017). Different TCR-induced T lymphocyte responses are potentiated by stiffness with variable sensitivity. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, Stevens J, Lane WJ, Dellagatta JL, Steelman S, et al. (2015). Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat. Biotechnol 33, 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibener LV, Fernandes RA, Kolawole EM, Carbone CB, Liu F, McAffee D, Birnbaum ME, Yang X, Su LF, Yu W, et al. (2018). Isolation of a Structural Mechanism for Uncoupling T Cell Receptor Signaling from Peptide-MHC Binding. Cell 174, 672–687 e627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Merwe PA, and Dushek O (2011). Mechanisms for T cell receptor triggering. Nat. Rev. Immunol 11, 47–55. [DOI] [PubMed] [Google Scholar]
- van Loenhout MT, Kerssemakers JW, De Vlaminck I, and Dekker C (2012). Non-bias-limited tracking of spherical particles, enabling nanometer resolution at low magnification. Biophys. J 102, 2362–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu CQ, Gagnon E, Call ME, Schnell JR, Schwieters CD, Carman CV, Chou JJ, and Wucherpfennig KW (2008). Regulation of T Cell Receptor Activation by Dynamic Membrane Binding of the CD3 epsilon Cytoplasmic Tyrosine-Based Motif. Cell 135, 702–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao M, Goult BT, Klapholz B, Hu X, Toseland CP, Guo Y, Cong P, Sheetz MP, and Yan J (2016). The mechanical response of talin. Nat. Commun 7, 11966. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1: Related to Figure 2. SMD simulation trajectories for pulling the R4-H-2Kb/2C TCR complex. The C-terminal residue of H-2Kb was harmonically constrained and the C-terminus of TCR was pulled by a dummy spring moving at ~0.1 nm/ns with a spring constant of ~70 pN/nm. The color codes were the same as that in Figure 2A, 2C TCRα (green), 2C TCRβ (orange), MHC H-2Kb α (purple), MHC H-2Kb β2m (yellow), and R4 peptide (cyan). Three different pathways of conformational changes occurred in the pMHC.
Movie S2: Related to Figure 2. SMD simulation trajectory for pulling the dEV8-H-2Kb/2C TCR complex under the same conditions as Movie S1. The color codes are the same as in Movie S1.
Movie S3: Related to Figure 2. SMD simulation trajectory for pulling the EVSV-H-2Kb/2C TCR complex under the same conditions as Movie S1. The color codes are the same as in Movie S1.
Movie S4: Related to Figure 2. SMD simulation trajectory for pulling the L4-H-2Kb/2C TCR complex under the same conditions as Movie S1. The color codes are the same as in Movie S1.
Movie S5: Related to Figure 5. SMD simulation trajectory for pulling the 9C-HLA-A2/1G4 TCR complex under the same conditions as Movie S1. The color codes are the same as in Figure 5A: 1G4 TCRα (light blue), 2C TCRβ (purple), MHC HLA-A2 α (green), MHC HLA-A2 β2m (yellow), 9C peptide (red).
Table S1: Interaction network analysis for the simulations of mouse 2C TCR systems and human 1G4 TCR system under force or no force. Related to Figures 1, 2, 3 and 5. The frequencies of H-bonds and hydrophobic contacts between TCR residues and the peptides (or MHCα, or HLA-A2) were listed. An H-bond is assumed if the distance for at least one pair of acceptor and donor atoms between the two residues is closer than 3.5Å. A hydrophobic contact is assumed if the distance for at least one pair of carbon atoms between the two residues is closer than 4Å. Results for the conditions under force or no force are provided and compared. Interactions with increased formation frequencies under force condition were colored red. The tables are provided as an excel file, and only these interaction pairs with frequency greater than 1% in at least one condition (with or without force) were included.